
Role of Ape1 in Impaired DNA Repair Capacity in Battery Recycling Plant Workers Exposed to Lead

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. Sample Collection
2.3. Blood Lead Concentration
2.4. Delta-Aminolevulinic Acid Dehydratase Activity (δ-ALAD)
2.5. Lipid Peroxidation
2.6. DNA Damage (Comet Assay)
2.7. Irradiation Procedure
2.8. DNA Repair Capacity
2.9. cDNA Expression Array
2.10. Reverse-Transcriptase-Polymerase Chain Reaction (RT-PCR)
- APE1-F-TAATTCTCTATCTCTGCCCC
- APE1-R-CAGTAATTCCCCGAAGCCTT
- GAPDH-F-AAACGACCCCTTCATTGACCT
- GAPDH-R-ATCTTAGTGGGGTCTCGCTC
2.11. Cell Protein Extractions
2.12. Western Blot for Ape1
2.13. Ape1 Functional Assay
2.14. Statistical Analysis
3. Results
3.1. Lead Exposure and Oxidative Stress Markers
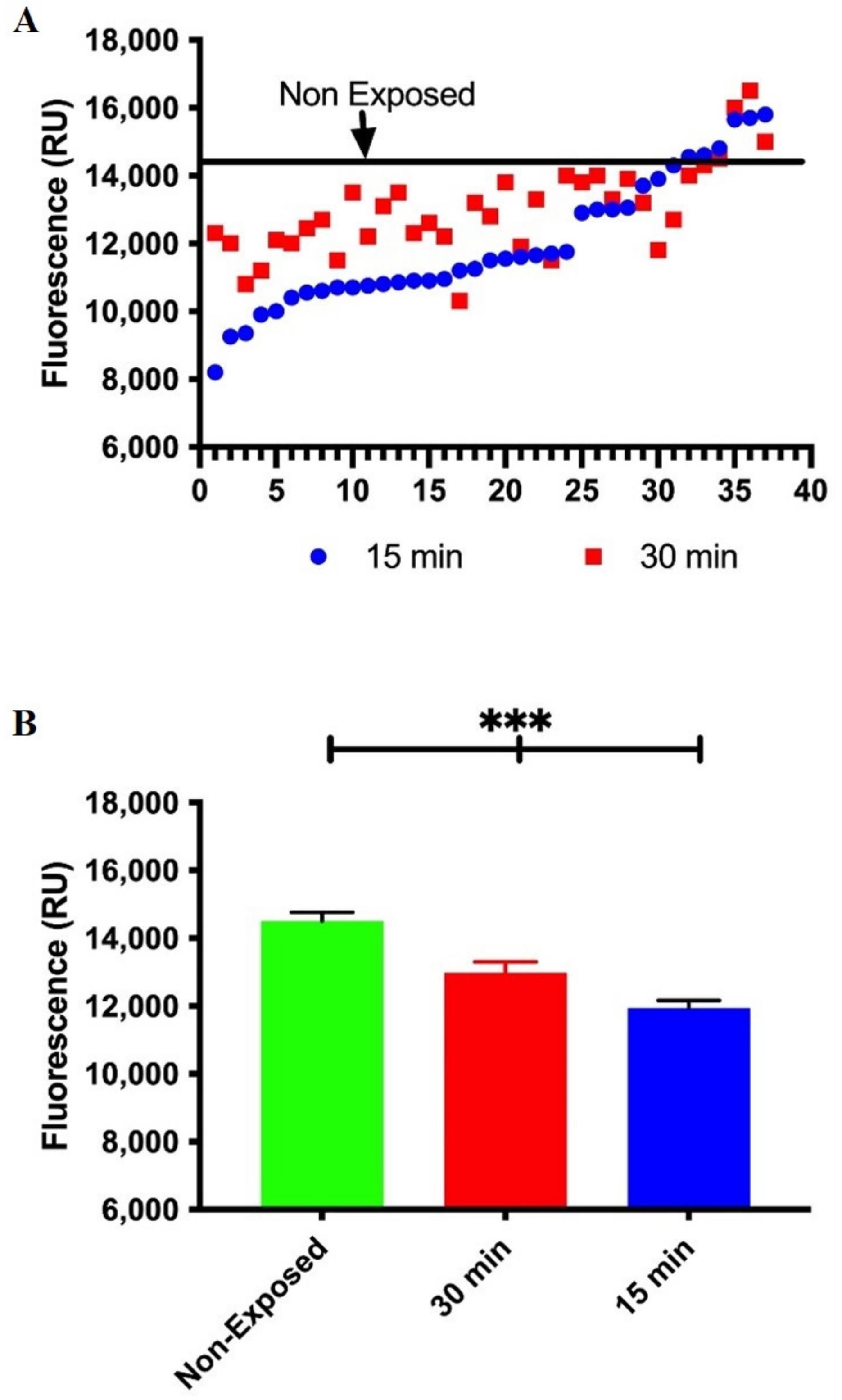
3.2. Occupational Lead Exposure Decreases DNA Repair Capacity
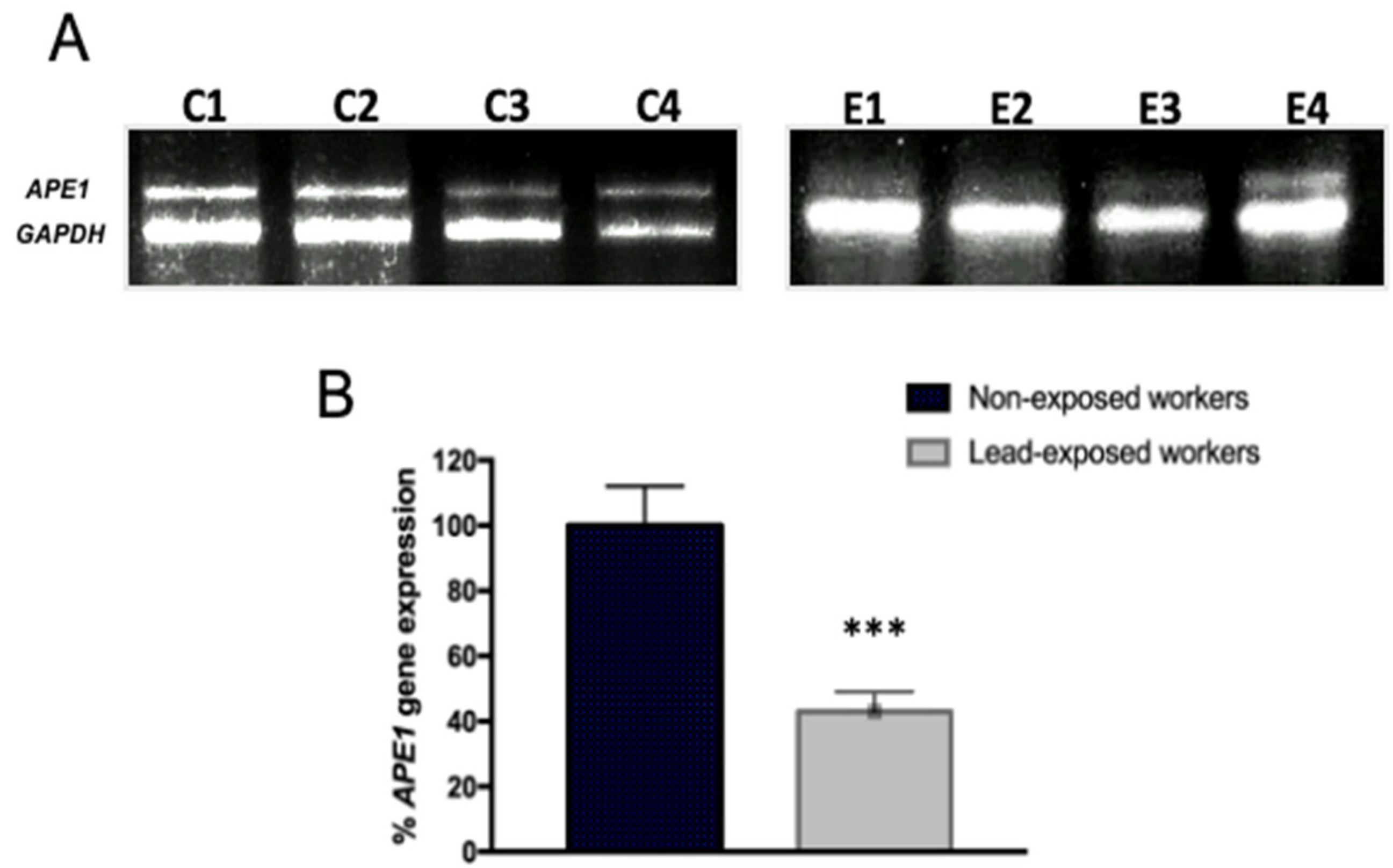
3.3. Expression Profile of Toxicity and Stress Genes in Workers Exposed to Lead
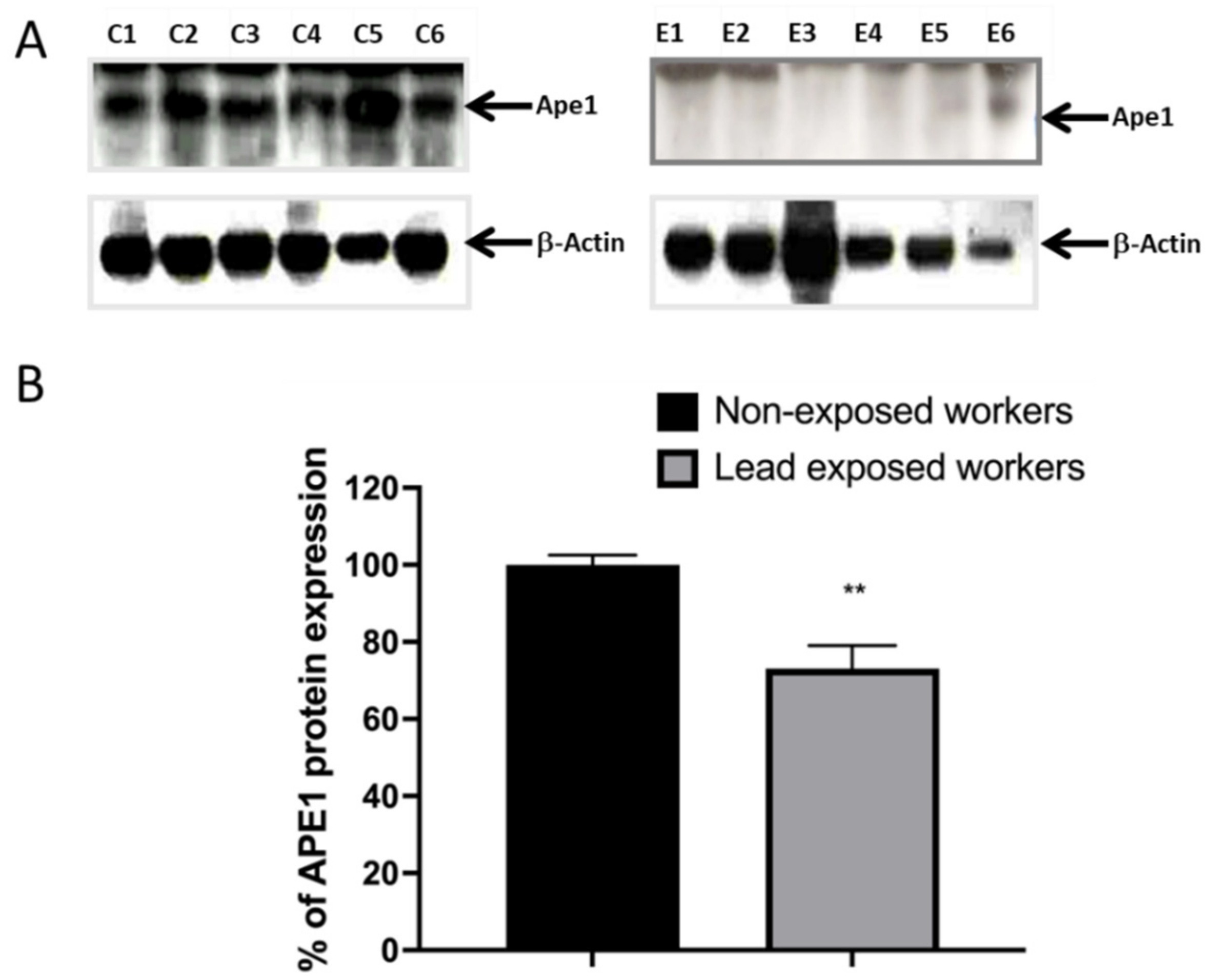
3.4. Impaired DNA Repair Capacity by APE1 in Lead Exposed Workers
3.5. In Silico Prediction of Transcription Factor Binding Sites
3.6. Interaction of Oxidative Markers of Lead Exposure and the Loss of Ape1 Activity in Exposed Workers
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abadin, H.; Ashizawa, A.; Stevens, Y.W.; Llados, F.; Diamond, G.; Sage, G.; Citra, M.; Quinones, A.; Bosch, S.J.; Swarts, S.G. Toxicological Profile for Lead; Agency for Toxic Substances and Disease Registry: Atlanta, GA, USA, 2007.
- Yadav, S.K.; Patil, G.P.; Virmagami1, A.; Bijalwan, V.; Devi, K.; Chauhan, A.; Gupta, S.K.; Fathima, S.; Naorem, C.D.; Yadav, S.; et al. Oxidative damage of DNA in subjects occupationally exposed to lead. Toxicol. Ind. Health 2022, 38, 139–150. [Google Scholar] [CrossRef]
- Firoozichahak, A.; Rahimnejad, S.; Rahmani, A.; Parvizimehr, A.; Aghaei, A.; Rahimpoor, R. Effect of occupational exposure to lead on serum levels of lipid profile and liver enzymes: An occupational cohort study. Toxicol. Rep. 2022, 9, 269–275. [Google Scholar] [CrossRef]
- Flora, G.; Gupta, D.; Tiwari, A. Toxicity of lead: A review with recent updates. Interdiscip. Toxicol. 2012, 5, 47–58. [Google Scholar] [CrossRef]
- Balali-Mood, M.; Naseri, K.; Tahergorabi, Z.; Khazdair, M.R.; Sadeghi, M. Toxic Mechanisms of Five Heavy Metals: Mercury, Lead, Chromium, Cadmium, and Arsenic. Front. Pharmacol. 2021, 12, 643972–643982. [Google Scholar] [CrossRef]
- Takeuchi, H.; Taki, Y.; Nouchi, R.; Yokoyama, R.; Kotozaki, Y.; Nakagawa, S.; Sekiguchi, A.; Iizuka, K.; Hanawa, S.; Araki, T.; et al. Lead exposure is associated with functional and microstructural changes in the healthy human brain. Commun. Biol. 2021, 4, 912. [Google Scholar] [CrossRef] [PubMed]
- Rendón-Ramirez, A.; Cerbón-Solórzano, J.; Maldonado-Vega, M.; Quintanar-Escorza, M.A.; Calderón-Salinas, J.V. Vitamin-E reduces the oxidative damage on delta-aminolevulinic dehydratase induced by lead intoxication in rat erythrocytes. Toxicol. In Vitro 2007, 21, 1121–1126. [Google Scholar] [CrossRef] [PubMed]
- Rendón-Ramírez, A.L.; Maldonado-Vega, M.; Quintanar-Escorza, M.A.; Hernández, G.; Arévalo-Rivas, B.I.; Zentella-Dehesa, A.; Calderón-Salinas, J.V. Effect of vitamin E and C supplementation on oxidative damage and total antioxidant capacity in lead-exposed workers. Environ. Toxicol. Pharmacol. 2014, 37, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Dorado, I.C.; Hernández, G.; Quintanar-Escorza, M.A.; Maldonado-Vega, M.; Rosas-Flores, M.; Calderón-Salinas, J.V. Eryptosis in lead-exposed workers. Toxicol. Appl. Pharmacol. 2014, 281, 195–202. [Google Scholar] [CrossRef]
- Hartwig, A.; Schwerdtle, T. Interactions by carcinogenic metal compounds with DNA repair processes: Toxicological implications. Toxicol. Lett. 2002, 127, 47–54. [Google Scholar] [CrossRef]
- Viau, M.; Sonzogni, L.; Ferlazzo, M.L.; Berthel, E.; Pereira, S.; Bodgi, L.; Granzotto, A.; Devic, C.; Fervers, B.; Charlet, L.; et al. DNA Double-Strand Breaks Induced in Human Cells by Twelve Metallic Species: Quantitative Inter-Comparisons and Influence of the ATM Protein. Biomolecules 2021, 11, 1462. [Google Scholar] [CrossRef]
- Hemmaphan, S.; Bordeerat, N.K. Genotoxic Effects of Lead and Their Impact on the Expression of DNA Repair Genes. Int. J. Environ. Res. Public Health 2022, 19, 4307. [Google Scholar] [CrossRef]
- Dizdaroglu, M. Oxidatively induced DNA damage and its repair in cancer. Mutat. Res. Rev. Mutat. Res. 2015, 763, 212–245. [Google Scholar] [CrossRef]
- Langie, S.A.; Koppen, G.; Desaulniers, D.; Al-Mulla, F.; Al-Temaimi, R.; Amedei, A.; Azqueta, A.; Bisson, W.H.; Brown, D.G.; Brunborg, G.; et al. Causes of genome instability: The effect of low dose chemical exposuRes. In modern society. Carcinogenesis 2015, 36 (Suppl. S1), S61–S88. [Google Scholar] [CrossRef]
- Koedrith, P.; Kim, H.; Weon, J.I.; Seo, Y.R. Toxicogenomic approaches for understanding molecular mechanisms of heavy metal mutagenicity and carcinogenicity. Int. J. Hyg. Environ. Health 2013, 216, 587–598. [Google Scholar] [CrossRef]
- Kim, H.S.; Kim, Y.J.; Seo, Y.R. An Overview of Carcinogenic Heavy Metal: Molecular Toxicity Mechanism and Prevention. J. Cancer Prev. 2015, 20, 232–240. [Google Scholar] [CrossRef]
- Gastaldo, J.; Viau, M.; Bouchot, M.; Joubert, A.; Charvet, A.M.; Foray, N. Induction and repair rate of DNA damage: A unified model for describing effects of external and internal irradiation and contamination with heavy metals. J. Theor. Biol. 2008, 251, 68–81. [Google Scholar] [CrossRef]
- Beyersmann, D.; Hartwig, A. Carcinogenic metal compounds: Recent insight into molecular and cellular mechanisms. Arch. Toxicol. 2008, 82, 493–512. [Google Scholar] [CrossRef]
- Al Bakheet, S.A.; Attafi, I.M.; Maayah, Z.H.; Abd-Allah, A.R.; Asiri, Y.A.; Korashy, H.M. Effect of long-term human exposure to environmental heavy metals on the expression of detoxification and DNA repair genes. Environ. Pollut. 2013, 181, 226–232. [Google Scholar] [CrossRef]
- Gastaldo, J.; Viau, M.; Bencokova, Z.; Joubert, A.; Charvet, A.M.; Balosso, J.; Foray, N. Lead contamination results in late and slowly repairable DNA double-strand breaks and impacts upon the ATM-dependent signaling pathways. Toxicol. Lett. 2007, 173, 201–214. [Google Scholar] [CrossRef]
- McNeill, D.R.; Narayana, A.; Wong, H.K.; Wilson, D.M. Inhibition of Ape1 nuclease activity by lead, iron, and cadmium. Environ. Health Perspect. 2004, 112, 799–804. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Franco, P.; Silva, M.; Franco, R.; Valverde, M.; Rojas, E. Lead facilitates foci formation in a Balb/c-3T3 two-step cell transformation model: Role of Ape1 function. Environ. Sci. Pollut. Res. Int. 2018, 25, 12150–12158. [Google Scholar] [CrossRef] [PubMed]
- McNeill, D.R.; Wong, H.K.; Narayana, A.; Wilson, D.M. Lead promotes abasic site accumulation and co-mutagenesis in mammalian cells by inhibiting the major abasic endonuclease Ape1. Mol. Carcinog. 2007, 46, 91–99. [Google Scholar] [CrossRef] [PubMed]
- McNeill, D.R.; Whitaker, A.M.; Stark, W.J.; Illuzzi, J.L.; McKinnon, P.J.; Freudenthal, B.D.; Wilson, D.M., III. Functions of the major abasic endonuclease (APE1) in cell viability and genotoxin resistance. Mutagenesis 2020, 35, 27–38. [Google Scholar] [CrossRef] [PubMed]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Smokeless tobacco and some tobacco-specific N-nitrosamines. IARC Monogr. Eval. Carcinog. Risks Hum. 2007, 89, 1–592. [Google Scholar]
- Valverde, M.; Trejo, C.; Rojas, E. Is the capacity of lead acetate and cadmium chloride to induce genotoxic damage due to direct DNA-metal interaction? Mutagenesis 2001, 16, 265–270. [Google Scholar] [CrossRef]
- Soto-Reyes, E.; Del Razo, L.M.; Valverde, M.; Rojas, E. Role of the alkali labile sites, reactive oxygen species and antioxidants in DNA damage induced by methylated trivalent metabolites of inorganic arsenic. BioMetals 2005, 18, 493–506. [Google Scholar] [CrossRef]
- Taylor, L.; Ashley, K.; Jones, R.L.; Deddens, J.A. Field evaluation of a portable blood lead analyzer in workers living at a high altitude: A follow-up investigation. Am. J. Ind. Med. 2004, 46, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Agency for Toxic Substances and Disease Registry (ATSDR). Toxicological Profile for Lead; Department of Health and Human Services, Public Health Service: Atlanta, GA, USA, 2020.
- Berlin, A.; Schaller, K.H. European standardized method for the determination of delta-aminolevulinic acid dehydratase activity in blood. Z. Klin. Chem. Klin. Biochem. 1974, 12, 389–390. [Google Scholar]
- Jain, S.K.; Ross, J.D.; Levy, G.J.; Duett, J. The effect of malonyldialdehyde on viscosity of normal and sickle red blood cells. Biochem. Med. Metab. Biol. 1990, 44, 37–41. [Google Scholar] [CrossRef]
- Singh, N.P.; McCoy, M.T.; Tice, R.R.; Schneider, E.L. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp. Cell Res. 1988, 175, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Maksimenko, A.; Ishchenko, A.A.; Sanz, G.; Laval, J.; Elder, R.H.; Saparbaev, M.K. A molecular beacon assay for measuring base excision repair activities. Biochem. Biophys. Res. Commun. 2004, 319, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Khan, D.A.; Qayyum, S.; Saleem, S.; Khan, F.A. Lead-induced oxidative stress adversely affects health of the occupational workers. Toxicol. Ind. Health 2008, 24, 611–618. [Google Scholar] [CrossRef]
- Restrepo, H.G.; Sicard, D.; Torres, M.M. DNA damage and repair in cells of lead exposed people. Am. J. Ind. Med. 2000, 38, 330–334. [Google Scholar] [CrossRef]
- Singh, P.; Mitra, P.; Goyal, T.; Sharma, S.; Sharma, P. Blood lead and cadmium levels in occupationally exposed workers and their effect on markers of DNA damage and repair. Environ. Geochem. Health 2021, 43, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, A. Metal interaction with redox regulation: An integrating concept in metal carcinogenesis? Free Radic. Biol. Med. 2013, 55, 63–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.-C.; Guo, K.-W.; Chu, J.-W.; Hsiao, Y.-Y. Understanding APE1 cellular functions by the structural preference of exonuclease activities. Compu. Struct. Biotech. J. 2021, 19, 3682–3691. [Google Scholar] [CrossRef]
- Miroshnokova, A.D.; Kuznetsova, A.A.; Vorobjev, Y.N.; Kuznetsov, N.A.; Fedorova, O. Effects of mono and divalent metal ions on DNA binding and catalysis of human apurinic/apyrimidinic endonuclease 1. Mol. Biosyst. 2016, 12, 1527–1539. [Google Scholar] [CrossRef] [Green Version]
- Tell, G.; Quadrifoglio, F.; Tiribelli, C.; Kelley, M.R. The many functions of APE1/Ref-1: Not only a DNA repair enzyme. Antioxid. Redox Signal. 2009, 11, 601–620. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Cobbina, S.J.; Mao, G.; Xu, H.; Zhang, Z.; Yang, L. A review of toxicity and mechanisms of individual and mixtures of heavy metals in the environment. Environ. Sci. Pollut. Res. Int. 2016, 23, 8244–8259. [Google Scholar] [CrossRef]
- Bae, D.S.; Hanneman, W.H.; Yang, R.S.; Campain, J.A. Characterization of gene expression changes associated with MNNG, arsenic, or metal mixture treatment in human keratinocytes: Application of cDNA microarray technology. Environ. Health Perspect. 2002, 110 (Suppl. S6), 931–941. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Lian, L.J.; Wu, C.; Wang, X.F.; Fu, W.Y.; Xu, L.H. Lead induces oxidative stress, DNA damage and alteration of p53, Bax and Bcl-2 expressions in mice. Food Chem. Toxicol. 2008, 46, 1488–1494. [Google Scholar] [CrossRef] [PubMed]
- Durham, T.R.; Snow, E.T. Metal ions and carcinogenesis. In Cancer: Cell Structures, Carcinogens and Genomic Instability; Part of the Experientia Supplementum Book Series; Springer: Berlin/Heidelberg, Germany, 2006; Volume 96, pp. 97–130. [Google Scholar] [CrossRef]
- Atkins, D.S.; Basha, M.R.; Zawia, N.H. Intracellular signaling pathways involved in mediating the effects of lead on the transcription factor Sp1. Int. J. Dev. Neurosci. 2003, 21, 235–244. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Non-Exposed (16) | Lead Exposed (37) |
|---|---|---|
| Age (years) | 37.15 ± 7.09 | 31.65 ± 8.56 |
| Duration of exposed (years) | - | 4.53 ± 3.29 |
| Smoking (%) | 18.70 | 16.00 |
| Blood lead concentration (µg/dL) | 1.42 ± 0.87 | 69.25 ± 24.95 *** |
| ALAD activity (nmol/mL/h) | 567.70 ± 46.20 | 312.86 ± 27.99 *** |
| MDA (nmol/mL) | 0.87 ± 0.03 | 1.52 ± 0.08 |
| # | Gene | Exposed/NonExposed | FC Log2 | t Test (p Value) | # | Gene | Exposed/Non Exposed | FC Log2 | t Test (p Value) | # | Gene | Exposed/Non Exposed | FC Long2 | t Test (p Value) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | ANXAS | 1.361 | 0.445 | 0.180 | 38 | GSR | 0.583 | −0.778 | 0.001 | 75 | PTGS2 | 1.347 | 0.429 | 0.128 |
| 2 | ATM | 0.850 | −0.234 | 0.224 | 39 | GSTM3 | 0.649 | −0.623 | 0.005 | 76 | RAD23A | 1.307 | 0.386 | 0.146 |
| 3 | BAX | 1.374 | 0.459 | 0.132 | 40 | HMOX1 | 0.546 | −0.873 | 0.001 | 77 | RAD50 | 0.883 | −0.179 | 0.257 |
| 4 | BCL2L1 | 1.046 | 0.064 | 0.413 | 41 | HMOX2 | 1.807 | 0.853 | 0.072 | 78 | CCL21 | 0.759 | −0.397 | 0.073 |
| 5 | BCL2L2 | 0.868 | −0.204 | 0.254 | 42 | HSF1 | 1.689 | 0.756 | 0.050 | 79 | CCL3 | 0.799 | −0.324 | 0.161 |
| 6 | CASPS | 0.836 | −0.258 | 0.182 | 43 | HSPH1 | 1.162 | 0.216 | 0.306 | 80 | CCL4 | 0.609 | −0.716 | 0.013 |
| 7 | CASP10 | 0.862 | −0.215 | 0.257 | 44 | HSPA1A | 1.074 | 0.103 | 0.395 | 81 | CXCL10 | 1.728 | 0.789 | 0.012 |
| 8 | CASP8 | 0.660 | −0.599 | 0.036 | 45 | PTGS1 | 0.657 | −0.607 | 0.004 | 82 | SERPINE1 | 1.565 | 0.646 | 0.041 |
| 9 | CAT | 1.752 | 0.809 | 0.071 | 46 | HSPA1L | 0.601 | −0.734 | 0.000 | 83 | SOD1 | 1.373 | 0.457 | 0.067 |
| 10 | CCNC | 1.521 | 0.605 | 0.095 | 47 | HSPA2 | 0.684 | −0.548 | 0.017 | 84 | SOD2 | 0.910 | −0.135 | 0.246 |
| 11 | CCND1 | 1.280 | 0.356 | 0.210 | 48 | HSPA4 | 0.582 | −0.780 | 0.004 | 85 | TNF | 0.707 | −0.500 | 0.011 |
| 12 | CCNG1 | 0.902 | −0.149 | 0.346 | 49 | HSPA5 | 1.682 | 0.750 | 0.094 | 86 | TNFRSF1A | 0.807 | −0.309 | 0.156 |
| 13 | CDKN1A | 0.728 | −0.458 | 0.075 | 50 | HSPA6 | 1.699 | 0.764 | 0.065 | 87 | TNFSF10 | 0.718 | −0.478 | 0.067 |
| 14 | CHEK2 | 0.831 | −0.268 | 0.166 | 51 | HSPA8 | 1.150 | 0.202 | 0.297 | 88 | FASLG | 0.608 | −0.718 | 0.011 |
| 15 | CRYAB | 1.297 | 0.375 | 0.157 | 52 | HSPA9B | 0.937 | −0.093 | 0.411 | 89 | TP53 | 1.332 | 0.413 | 0.079 |
| 16 | CSF2 | 0.884 | −0.178 | 0.307 | 53 | HSPAB1 | 0.593 | −0.753 | 0.000 | 90 | TRADD | 1.439 | 0.525 | 0.017 |
| 17 | CYP1A1 | 1.495 | 0.580 | 0.120 | 54 | HSPCA | 0.577 | −0.792 | 0.000 | 91 | UGT1A4 | 0.841 | −0.250 | 0.167 |
| 18 | CYP1B1 | 1.638 | 0.712 | 0.086 | 55 | HSPCB | 0.709 | −0.495 | 0.013 | 92 | UNG | 0.676 | −0.565 | 0.007 |
| 19 | CYP2E1 | 1.142 | 0.191 | 0.349 | 56 | HSPD1 | 0.622 | −0.684 | 0.003 | 93 | XRCC1 | 0.638 | −0.648 | 0.000 |
| 20 | CYP7A1 | 0.837 | −0.256 | 0.283 | 57 | HSPE1 | 1.216 | 0.282 | 0.242 | 94 | XRCC2 | 0.745 | −0.424 | 0.073 |
| 21 | CYP7B1 | 0.714 | −0.486 | 0.052 | 58 | IGFBP6 | 1.774 | 0.827 | 0.044 | 95 | XRCC4 | 0.900 | −0.151 | 0.312 |
| 22 | DDB1 | 0.749 | −0.417 | 0.059 | 59 | IL1B | 0.976 | −0.036 | 0.462 | 96 | XRCC5 | 0.788 | −0.344 | 0.165 |
| 23 | DDIT3 | 0.959 | −0.061 | 0.430 | 60 | IL1A | 0.988 | −0.017 | 0.483 | 97 | PUC18 | 1.271 | 0.346 | 0.172 |
| 24 | DNA1A1 | 0.678 | −0.561 | 0.032 | 61 | 1L1B | 0.631 | −0.664 | 0.003 | 98 | PUC18 | 1.232 | 0.301 | 0.148 |
| 25 | DNAJB4 | 0.796 | −0.328 | 0.163 | 62 | 1L6 | 0.712 | −0.490 | 0.020 | 99 | PUC18 | 0.920 | −0.120 | 0.303 |
| 26 | E2F1 | 1.431 | 0.517 | 0.118 | 63 | LTA | 0.739 | −0.436 | 0.072 | 103 | GAPDH | 1.032 | 0.046 | 0.430 |
| 27 | EGR1 | 1.015 | 0.022 | 0.478 | 64 | MDM2 | 0.643 | −0.636 | 0.020 | 104 | GAPDH | 0.950 | −0.074 | 0.402 |
| 28 | EPHX2 | 0.749 | −0.417 | 0.175 | 65 | MIF | 1.607 | 0.684 | 0.072 | 109 | RPL13A | 0.837 | −0.256 | 0.117 |
| 29 | ERCC1 | 0.712 | −0.490 | 0.080 | 66 | PRDX1 | 1.928 | 0.947 | 0.020 | 110 | RPL13A | 0.875 | −0.193 | 0.289 |
| 30 | ERCC3 | 0.655 | −0.611 | 0.008 | 67 | PRDX2 | 1.314 | 0.394 | 0.078 | 111 | ACTB | 0.933 | −0.101 | 0.383 |
| 31 | ERCC4 | 0.640 | −0.643 | 0.007 | 68 | MT2A | 1.101 | 0.138 | 0.030 | 112 | ACTB | 0.868 | 0.204 | 0.280 |
| 32 | ERCC5 | 0.584 | −0.777 | 0.004 | 69 | NFKB1 | 0.770 | −0.377 | 0.060 | |||||
| 33 | FM01 | 1.274 | 0.350 | 0.210 | 70 | NFKB1A | 0.753 | −0.409 | 0.054 | |||||
| 34 | FM05 | 1.592 | 0.671 | 0.059 | 71 | NOS2A | 0.668 | −0.583 | 0.025 | |||||
| 35 | GADD45A | 1.013 | 0.019 | 0.482 | 72 | PCNA | 0.591 | −0.759 | 0.009 | |||||
| 36 | GADD5B | 1.053 | 0.074 | 0.427 | 73 | GDF15 | 1.915 | 0.937 | 0.028 | |||||
| 37 | GPX1 | 0.736 | −0.441 | 0.036 | 74 | POR | 1.924 | 0.944 | 0.010 |
| Gene | Lead Exposed/Non Exposed | Fold Change log2 | t-Test (p Value) | Up/Down |
|---|---|---|---|---|
| HSPCA | 0.577 | −0.792 | 6.8 × 10−5 | Down |
| HSPA4 | 0.582 | −0.780 | 0.004 | Down |
| GSR | 0.583 | −0.778 | 0.001 | Down |
| ERCC5 | 0.584 | −0.777 | 0.004 | Down |
| PCNA | 0.591 | −0.759 | 0.009 | Down |
| HSPB1 | 0.593 | −0.753 | 0.000 | Down |
| HSPA1L | 0.601 | −0.734 | 0.000 | Down |
| FASLG | 0.608 | −0.718 | 0.011 | Down |
| CCL4 | 0.609 | −0.716 | 0.013 | Down |
| HSPD1 | 0.622 | −0.684 | 0.003 | Down |
| IL1B | 0.631 | −0.664 | 0.003 | Down |
| XRCC1 | 0.638 | −0.648 | 0.000 | Down |
| ERCC4 | 0.640 | −0.643 | 0.007 | Down |
| MDM2 | 0.643 | −0.636 | 0.020 | Down |
| GSTM3 | 0.649 | −0.623 | 0.005 | Down |
| ERCC3 | 0.655 | −0.611 | 0.008 | Down |
| PTGS1 | 0.657 | −0.607 | 0.004 | Down |
| CASP8 | 0.660 | −0.599 | 0.036 | Down |
| NOS2A | 0.668 | −0.583 | 0.025 | Down |
| UNG | 0.676 | −0.565 | 0.007 | Down |
| DNAJA1 | 0.678 | −0.561 | 0.032 | Down |
| HSPA2 | 0.684 | −0.548 | 0.017 | Down |
| TNF | 0.707 | −0.500 | 0.011 | Down |
| HSPCB | 0.709 | −0.495 | 0.013 | Down |
| IL6 | 0.712 | −0.490 | 0.020 | Down |
| CYP7B1 | 0.714 | −0.486 | 0.050 | Down |
| GPX1 | 0.736 | −0.441 | 0.036 | Down |
| NFKBIA | 0.753 | −0.409 | 0.050 | Down |
| SERPINE1 | 1.565 | 0.646 | 0.041 | Up |
| HSF1 | 1.689 | 0.756 | 0.050 | Up |
| CXCL10 | 1.728 | 0.789 | 0.012 | Up |
| IGFBP6 | 1.774 | 0.827 | 0.044 | Up |
| GDF15 | 1.915 | 0.937 | 0.028 | Up |
| POR | 1.924 | 0.944 | 0.010 | Up |
| PRDX1 | 1.928 | 0.947 | 0.030 | UP |
| Transcription Factors Families | Predicted Number of Downregulated Target Genes | Predicted Number of DNA Repair Genes |
|---|---|---|
| CEBP (Basic leucine zipper factors (bZIP)) | 16 | 6 |
| SOX (High/mobility group (HMG) domain | 16 | 13 |
| HOX and POU (Home o domain factors) | 38 | 24 |
| FOS (Basic leucine zipper factors (bZIP)) | 20 | 17 |
| MYC (Basic helix-loop/helix factors (bHLH)) | 21 | 18 |
| E2F (Fork head/winged helix factors) | 24 | 24 |
| ELF/ELK (Tryptophan cluster factors) | 26 | 22 |
| Zn–TF (C2H2 zinc finger factors and nuclear receptors with C4 zinc finger) | 140 | 72 |
| TOTAL | 1014 | 764 |
| δ-ALAD Activity | [MDA] | Ape1 Activity | APE1 mRNA | Ape1 Protein | |
|---|---|---|---|---|---|
| [PbB] | −0.72 *** | 0.60 *** | −0.43 *** | −0.53 *** | −0.24 ** |
| δ-ALAD activity | −0.52 *** | 0.38 ** | 0.31 ** | NS | |
| [MDA] | −0.33 ** | 0.31 ** | NS | ||
| Ape1 activity | 0.37 ** | NS | |||
| APE1 mRNA | NS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernández-Franco, P.; Maldonado-Vega, M.; Calderón-Salinas, J.V.; Rojas, E.; Valverde, M. Role of Ape1 in Impaired DNA Repair Capacity in Battery Recycling Plant Workers Exposed to Lead. Int. J. Environ. Res. Public Health 2022, 19, 7961. https://doi.org/10.3390/ijerph19137961
Hernández-Franco P, Maldonado-Vega M, Calderón-Salinas JV, Rojas E, Valverde M. Role of Ape1 in Impaired DNA Repair Capacity in Battery Recycling Plant Workers Exposed to Lead. International Journal of Environmental Research and Public Health. 2022; 19(13):7961. https://doi.org/10.3390/ijerph19137961
Chicago/Turabian StyleHernández-Franco, Pablo, María Maldonado-Vega, José Víctor Calderón-Salinas, Emilio Rojas, and Mahara Valverde. 2022. "Role of Ape1 in Impaired DNA Repair Capacity in Battery Recycling Plant Workers Exposed to Lead" International Journal of Environmental Research and Public Health 19, no. 13: 7961. https://doi.org/10.3390/ijerph19137961