
Enhanced Molecular Networking Shows Microbacterium sp. V1 as a Factory of Antioxidant Proline-Rich Peptides
 ,
,  , , and
, , and
Abstract
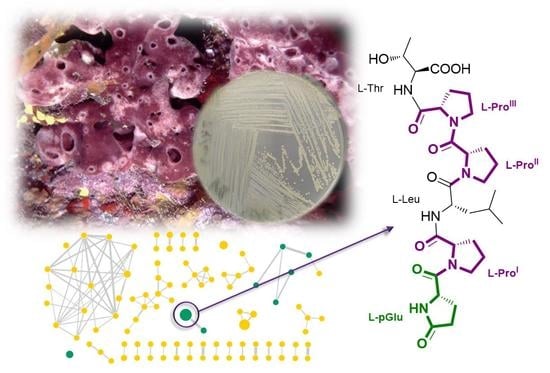
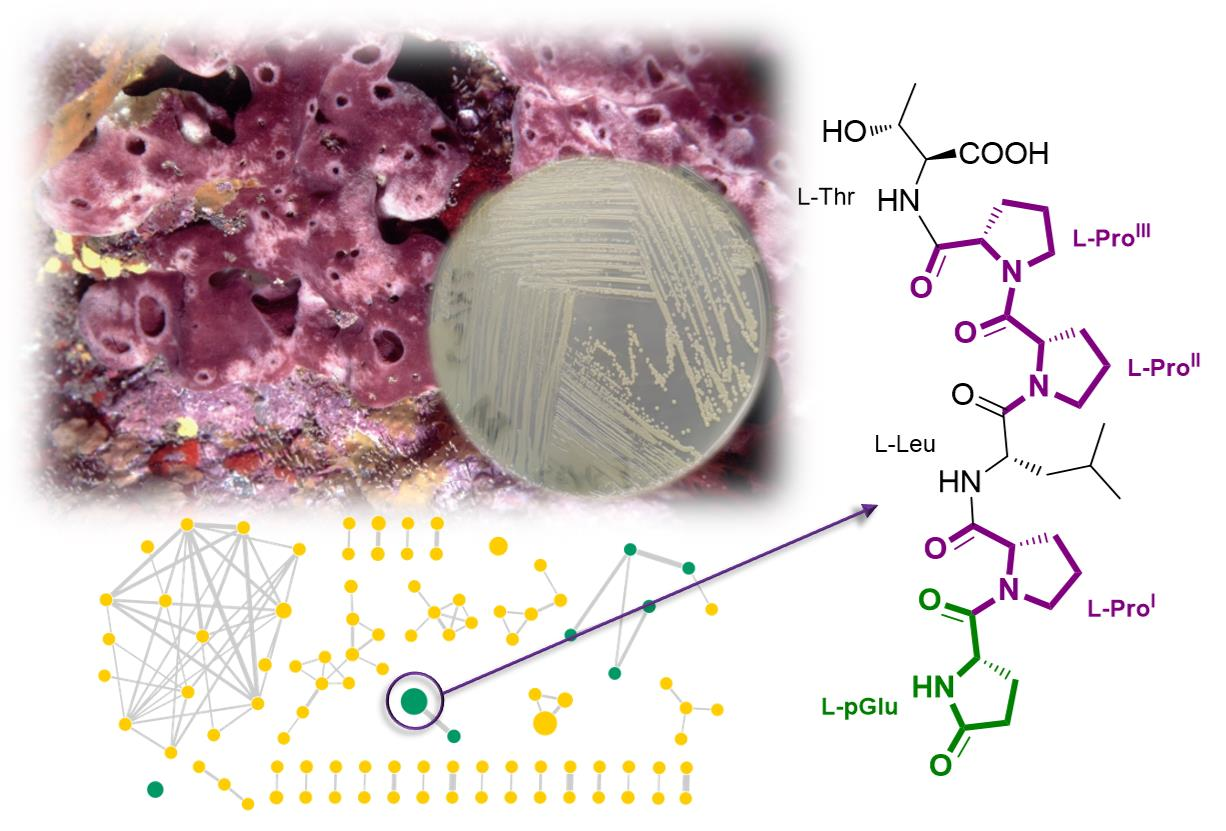
:
1. Introduction
2. Results and Discussion
2.1. Bacterial Isolation from the Sponge Petrosia Ficiformis
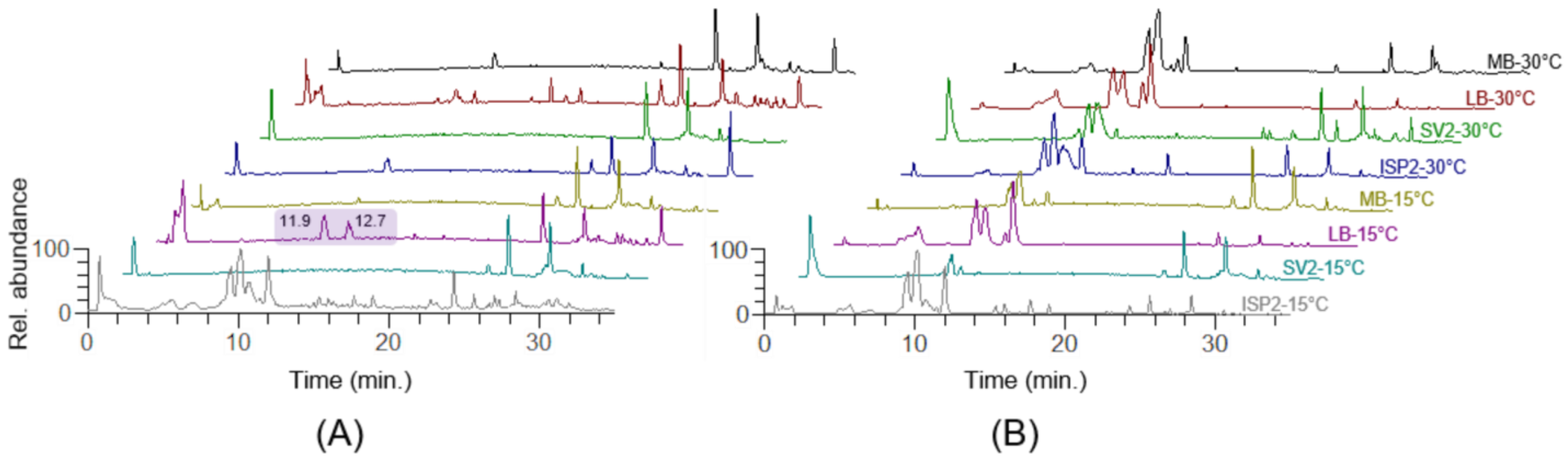
2.2. OSMAC-Based Cultivation and Comparative Metabolic Profiling
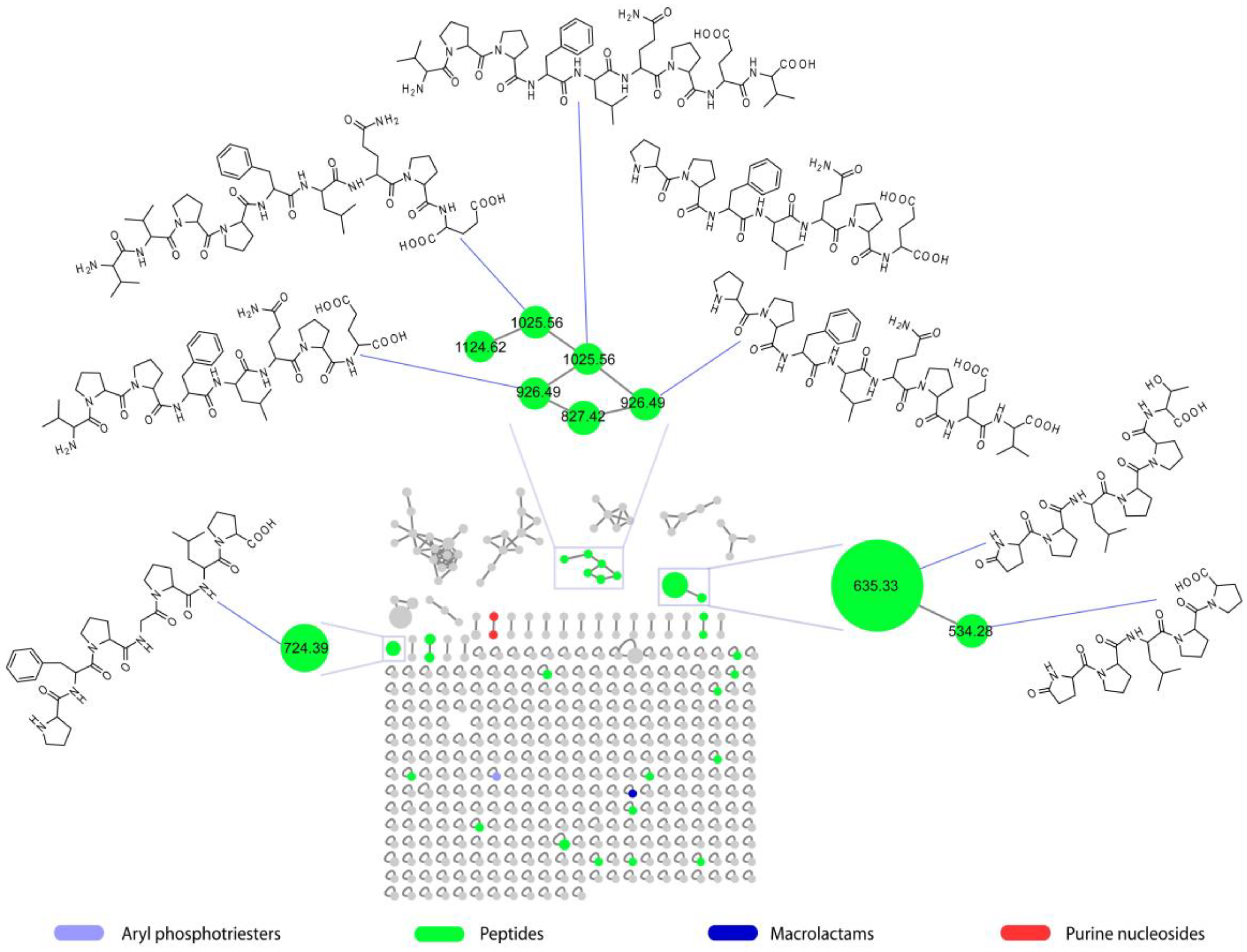
2.3. Molecular Network of Microbacterium sp. V1 Grown in LB Medium at 15 °C
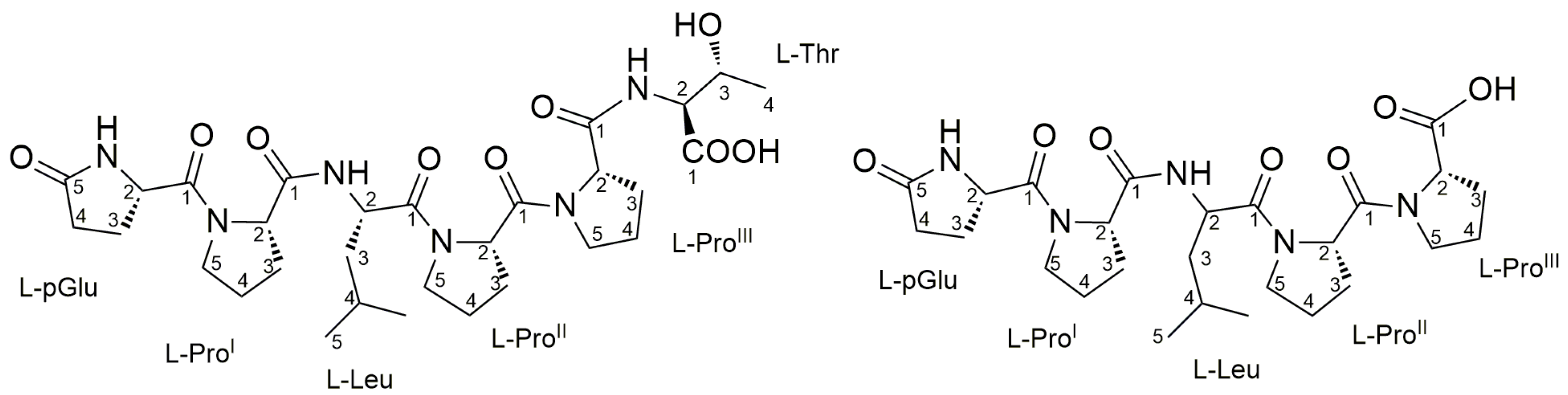
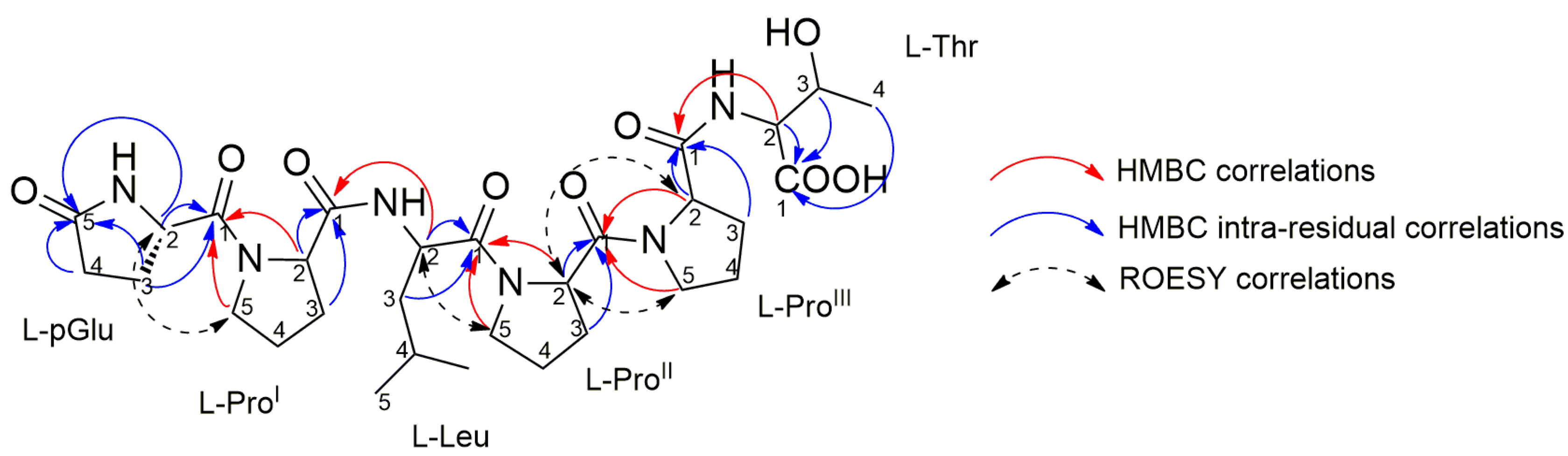
2.4. Purification and Structure Elucidation of Proline-Rich Peptides 1 and 2 from Microbacterium sp. V1
2.5. Structural Prediction of the Proline-Rich Peptides 3–8 from Microbacterium sp. V1
2.6. Assessment of Antioxidant Activity of Pure Metabolites
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Media and Buffers
3.3. OSMAC-Based Cultivation and Extraction Procedures
3.4. LC-HRMS and LC-HRMS2 Analysis of the Crude Extracts
3.5. Large-Scale Cultivation and Extract Fractionation and Purification
3.6. Metabolomic and Cheminformatic Analysis
3.7. Advanced Marfey’s Analysis
3.8. FRAP Assay of Pure Metabolites
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Genus Microbacterium. Available online: http://www.bacterio.net/microbacterium.html (accessed on 17 March 2023).
- Mondani, L.; Piette, L.; Christen, R.; Bachar, D.; Berthomieu, C.; Chapon, V. Microbacterium lemovicicum sp. nov., a bacterium isolated from a natural uranium-rich soil. Int. J. Syst. Evol. Microbiol. 2013, 63, 2600–2606. [Google Scholar] [CrossRef]
- Wang, H.; Xiang, T.; Wang, Y.; Song, J.; Zhai, Y.; Chen, X.; Li, Y.; Zhao, B.; Zhao, B.; Ruan, Z. Microbacterium petrolearium sp. nov., isolated from an oil-contaminated water sample. Int. J. Syst. Evol. Microbiol. 2014, 64, 4168–4172. [Google Scholar] [CrossRef]
- Madhaiyan, M.; Poonguzhali, S.; Lee, J.S.; Lee, K.C.; Saravanan, V.S.; Santhanakrishnan, P. Microbacterium azadirachtae sp. nov., a plant-growth-promoting actinobacterium isolated from the rhizoplane of neem seedlings. Int. J. Syst. Evol. Microbiol. 2010, 60, 1687–1692. [Google Scholar] [CrossRef]
- Subramani, R.; Sipkema, D. Marine Rare Actinomycetes: A Promising Source of Structurally Diverse and Unique Novel Natural Products. Mar. Drugs 2019, 17, 249. [Google Scholar] [CrossRef]
- Bai, Y.; Müller, D.B.; Srinivas, G.; Garrido-Oter, R.; Potthoff, E.; Rott, M.; Dombrowski, N.; Münch, P.C.; Spaepen, S.; Remus-Emsermann, M.; et al. Functional overlap of the Arabidopsis leaf and root microbiota. Nature 2015, 528, 364–369. [Google Scholar] [CrossRef]
- Corretto, E.; Antonielli, L.; Sessitsch, A.; Höfer, C.; Puschenreiter, M.; Widhalm, S.; Swarnalakshmi, K.; Brader, G. Comparative Genomics of Microbacterium Species to Reveal Diversity, Potential for Secondary Metabolites and Heavy Metal Resistance. Front. Microbiol. 2020, 11, 1869. [Google Scholar] [CrossRef]
- Udenigwe, C.C.; Aluko, R.E. Food protein-derived bioactive peptides: Production, processing, and potential health benefits. J. Food Sci. 2012, 77, R11-24. [Google Scholar] [CrossRef]
- Xu, Q.; Hong, H.; Wu, J.; Yan, X. Bioavailability of bioactive peptides derived from food proteins across the intestinal epithelial membrane: A review. Trends Food Sci. Technol. 2019, 86, 399–411. [Google Scholar] [CrossRef]
- Akbarian, M.; Khani, A.; Eghbalpour, S.; Uversky, V.N. Bioactive Peptides: Synthesis, Sources, Applications, and Proposed Mechanisms of Action. Int. J. Mol. Sci. 2022, 23, 1445. [Google Scholar] [CrossRef]
- Solieri, L.; De Vero, L.; Tagliazucchi, D. Peptidomic study of casein proteolysis in bovine milk by Lactobacillus casei PRA205 and Lactobacillus rhamnosus PRA331. Int. Dairy J. 2018, 85, 237–246. [Google Scholar] [CrossRef]
- Stressler, T.; Eisele, T.; Schlayer, M.; Lutz-Wahl, S.; Fischer, L. Characterization of the recombinant exopeptidases PepX and PepN from Lactobacillus helveticus ATCC 12046 important for food protein hydrolysis. PLoS ONE 2013, 8, e70055. [Google Scholar] [CrossRef] [PubMed]
- Raveschot, C.; Cudennec, B.; Coutte, F.; Flahaut, C.; Fremont, M.; Drider, D.; Dhulster, P. Production of Bioactive Peptides by Lactobacillus Species: From Gene to Application. Front. Microbiol. 2018, 9, 2354. [Google Scholar] [CrossRef] [PubMed]
- Tonolo, F.; Fiorese, F.; Moretto, L.; Folda, A.; Scalcon, V.; Grinzato, A.; Ferro, S.; Arrigoni, G.; Bindoli, A.; Feller, E.; et al. Identification of New Peptides from Fermented Milk Showing Antioxidant Properties: Mechanism of Action. Antioxidants 2020, 9, 117. [Google Scholar] [CrossRef]
- Hernández-Ledesma, B.; García-Nebot, M.J.; Fernández-Tomé, S.; Amigo, L.; Recio, I. Dairy protein hydrolysates: Peptides for health benefits. Int. Dairy J. 2014, 38, 82–100. [Google Scholar] [CrossRef]
- Hafeez, Z.; Cakir-Kiefer, C.; Roux, E.; Perrin, C.; Miclo, L.; Dary-Mourot, A. Strategies of producing bioactive peptides from milk proteins to functionalize fermented milk products. Food Res. Int. 2014, 63, 71–80. [Google Scholar] [CrossRef]
- Teschemacher, H. Opioid receptor ligands derived from food proteins. Curr. Pharm. Des. 2003, 9, 1331–1344. [Google Scholar] [CrossRef]
- Stefanovic, E.; Fitzgerald, G.; McAuliffe, O. Advances in the genomics and metabolomics of dairy lactobacilli: A review. Food Microbiol. 2017, 61, 33–49. [Google Scholar] [CrossRef]
- Vitale, G.A.; Sciarretta, M.; Palma Esposito, F.; January, G.G.; Giaccio, M.; Bunk, B.; Spröer, C.; Bajerski, F.; Power, D.; Festa, C.; et al. Genomics-Metabolomics Profiling Disclosed Marine Vibrio spartinae 3.6 as a Producer of a New Branched Side Chain Prodigiosin. J. Nat. Prod. 2020, 83, 1495–1504. [Google Scholar] [CrossRef]
- Teta, R.; Della Sala, G.; Mangoni, A.; Lega, M.; Costantino, V. Tracing cyanobacterial blooms to assess the impact of wastewaters discharges on coastal areas and lakes. Int. J. Sustain. Dev. Plan. 2016, 11, 804–811. [Google Scholar] [CrossRef]
- Della Sala, G.; Mangoni, A.; Costantino, V.; Teta, R. Identification of the Biosynthetic Gene Cluster of Thermoactinoamides and Discovery of New Congeners by Integrated Genome Mining and MS-Based Molecular Networking. Front. Chem. 2020, 8, 397. [Google Scholar] [CrossRef]
- Scarpato, S.; Teta, R.; Della Sala, G.; Pawlik, J.R.; Costantino, V.; Mangoni, A. New Tricks with an Old Sponge: Feature-Based Molecular Networking Led to Fast Identification of New Stylissamide L from Stylissa caribica. Mar. Drugs 2020, 18, 443. [Google Scholar] [CrossRef]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed]
- Stravs, M.A.; Dührkop, K.; Böcker, S.; Zamboni, N. MSNovelist: De novo structure generation from mass spectra. Nat. Methods 2022, 19, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Dührkop, K.; Fleischauer, M.; Ludwig, M.; Aksenov, A.A.; Melnik, A.V.; Meusel, M.; Dorrestein, P.C.; Rousu, J.; Böcker, S. SIRIUS 4: A rapid tool for turning tandem mass spectra into metabolite structure information. Nat. Methods 2019, 16, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Romano, S.; Jackson, S.A.; Patry, S.; Dobson, A.D.W. Extending the "One Strain Many Compounds" (OSMAC) Principle to Marine Microorganisms. Mar. Drugs 2018, 16, 244. [Google Scholar] [CrossRef]
- Goodwin, C.; Rodolfo-Metalpa, R.; Picton, B.; Hall-Spencer, J.M. Effects of ocean acidification on sponge communities. Mar. Ecol. 2014, 35, 41–49. [Google Scholar] [CrossRef]
- Vitale, G.A.; Sciarretta, M.; Cassiano, C.; Buonocore, C.; Festa, C.; Mazzella, V.; Núñez Pons, L.; D’Auria, M.V.; de Pascale, D. Molecular Network and Culture Media Variation Reveal a Complex Metabolic Profile in Pantoea cf. eucrina D2 Associated with an Acidified Marine Sponge. Int. J. Mol. Sci. 2020, 21, 6307. [Google Scholar] [CrossRef]
- Palma Esposito, F.; Giugliano, R.; Della Sala, G.; Vitale, G.A.; Buonocore, C.; Ausuri, J.; Galasso, C.; Coppola, D.; Franci, G.; Galdiero, M.; et al. Combining OSMAC Approach and Untargeted Metabolomics for the Identification of New Glycolipids with Potent Antiviral Activity Produced by a Marine Rhodococcus. Int. J. Mol. Sci. 2021, 22, 9055. [Google Scholar] [CrossRef]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Oresic, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinform. 2010, 11, 395. [Google Scholar] [CrossRef]
- Djoumbou Feunang, Y.; Eisner, R.; Knox, C.; Chepelev, L.; Hastings, J.; Owen, G.; Fahy, E.; Steinbeck, C.; Subramanian, S.; Bolton, E.; et al. ClassyFire: Automated chemical classification with a comprehensive, computable taxonomy. J. Cheminformatics 2016, 8, 61. [Google Scholar] [CrossRef]
- Siemion, I.Z.; Wieland, T.; Pook, K.H. Influence of the distance of the proline carbonyl from the beta and gamma carbon on the 13C chemical shifts. Angew. Chem. (Int. Ed. Engl. ) 1975, 14, 702–703. [Google Scholar] [CrossRef] [PubMed]
- Minkiewicz, P.; Iwaniak, A.; Darewicz, M. BIOPEP-UWM Database of Bioactive Peptides: Current Opportunities. Int. J. Mol. Sci. 2019, 20, 5978. [Google Scholar] [CrossRef] [PubMed]
- BIOPEP-UWM. Available online: https://biochemia.uwm.edu.pl/en/biopep-uwm-2 (accessed on 15 February 2023).
- Uenishi, H.; Kabuki, T.; Seto, Y.; Serizawa, A.; Nakajima, H. Isolation and identification of casein-derived dipeptidyl-peptidase 4 (DPP-4)-inhibitory peptide LPQNIPPL from gouda-type cheese and its effect on plasma glucose in rats. Int. Dairy J. 2012, 22, 24–30. [Google Scholar] [CrossRef]
- Vitale, G.A.; Coppola, D.; Palma Esposito, F.; Buonocore, C.; Ausuri, J.; Tortorella, E.; de Pascale, D. Antioxidant Molecules from Marine Fungi: Methodologies and Perspectives. Antioxidants 2020, 9, 1183. [Google Scholar] [CrossRef]
- Birasuren, B.; Kim, N.Y.; Jeon, H.L.; Kim, M.R. Evaluation of the Antioxidant Capacity and Phenolic Content of Agriophyllum pungens Seed Extracts from Mongolia. Prev. Nutr. Food Sci. 2013, 18, 188–195. [Google Scholar] [CrossRef]
- Jacobsen, C.; Paiva-Martins, F.; Schwarz, K.; Bochkov, V. Lipid Oxidation and Antioxidants in Food and Nutrition. Eur. J. Lipid Sci. Technol. 2019, 121, 1900298. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef]
- Benzie, I.F.; Strain, J.J. The ferric reducing ability of plasma (FRAP) as a measure of "antioxidant power": The FRAP assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef]
- Amarowicz, R.; Pegg, R.B. Chapter One—Natural antioxidants of plant origin. In Advances in Food and Nutrition Research; Ferreira, I.C.F.R., Barros, L., Eds.; Academic Press: Cambridge, MA, USA, 2019; Volume 90, pp. 1–81. [Google Scholar]
- Nothias, L.F.; Petras, D.; Schmid, R.; Dührkop, K.; Rainer, J.; Sarvepalli, A.; Protsyuk, I.; Ernst, M.; Tsugawa, H.; Fleischauer, M.; et al. Feature-based molecular networking in the GNPS analysis environment. Nat. Methods 2020, 17, 905–908. [Google Scholar] [CrossRef]
- Ernst, M.; Kang, K.B.; Caraballo-Rodríguez, A.M.; Nothias, L.F.; Wandy, J.; Chen, C.; Wang, M.; Rogers, S.; Medema, M.H.; Dorrestein, P.C.; et al. MolNetEnhancer: Enhanced Molecular Networks by Integrating Metabolome Mining and Annotation Tools. Metabolites 2019, 9, 144. [Google Scholar] [CrossRef]
- Liaqat, H.; Kim, K.J.; Park, S.Y.; Jung, S.K.; Park, S.H.; Lim, S.; Kim, J.Y. Antioxidant Effect of Wheat Germ Extracts and Their Antilipidemic Effect in Palmitic Acid-Induced Steatosis in HepG2 and 3T3-L1 Cells. Foods 2021, 10, 1061. [Google Scholar] [CrossRef] [PubMed]
- Ritt, J.F.; Remize, F.; Grandvalet, C.; Guzzo, J.; Atlan, D.; Alexandre, H. Peptidases specific for proline-containing peptides and their unusual peptide-dependent regulation in Oenococcus oeni. J. Appl. Microbiol. 2009, 106, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Busk, P.K. Accurate, automatic annotation of peptidases with hotpep-protease. Green Chem. Eng. 2020, 1, 124–130. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AA | Pos. | δC, Type | δH, Mult (J in Hz) | ROESY | HMBC a | |
|---|---|---|---|---|---|---|
| pGlu | NH | |||||
| 1 | 172.95, C | |||||
| 2 | 56.25, CH | 4.56, dd (4.2, 8.9) | ProI-5a, ProI-5b | pGlu-1, pGlu-5 | ||
| 3 | 25.51, CH2 | a | 2.50, m | pGlu-1, pGlu-5 | ||
| b | 2.19, m | pGlu-1, pGlu-5 | ||||
| 4 | 30.31, CH2 | a | 2.40, m | pGlu-5 | ||
| b | 2.31, m | pGlu-5 | ||||
| 5 | 181.55, C | |||||
| ProI | 1 | 174.23, C | ||||
| 2 | 61.60, CH | 4.47, dd (4.3, 8.5) | pGlu-1, ProI-1 | |||
| 3 | 30.33, CH2 | a | 2.18, m | ProI-1 | ||
| b | 1.97, m | ProI-1 | ||||
| 4 | 25.94, CH2 | a | 2.05, m | |||
| b | 1.98, m | |||||
| 5 | 48.06, CH2 | a | 3.68, m | pGlu-2 | pGlu-1 | |
| b | 3.61, m | pGlu-2 | pGlu-1 | |||
| Leu | NH | |||||
| 1 | 172.91, C | |||||
| 2 | 51.09, CH | 4.62, t (7.2) | ProII-5a, ProII-5b | Leu-1, ProI-1, ProI-4 | ||
| 3 | 41.04, CH2 | 1.56, t (7.2) | Leu-1 | |||
| 4 | 25.76, CH | 1.81, nonet (6.6) | ||||
| 5 | 21.86, CH3 | 0.97, d (6.6) | ||||
| ProII | 1 | 172.75, C | ||||
| 2 | 59.62, CH | 4.69, dd (4.5, 8.3) | ProIII-2,ProIII-5a, ProIII-5b | Leu-1, ProII-1 | ||
| 3 | 29.41, CH2 | a | 2.27, m | ProII-1 | ||
| b | 2.04, m | ProII-1 | ||||
| 4 | 25.87, CH2 | a | 2.13, m | |||
| b | 2.01, m | |||||
| 5 | 48.56, CH2 | a | 3.84, m | Leu-2 | Leu-1 | |
| b | 3.65, m | Leu-2 | Leu-1 | |||
| ProIII | 1 | 174.66, C | ||||
| 2 | 61.47, CH | 4.55, m | ProII-2 | ProII-1, ProIII-1 | ||
| 3 | 30.29, CH2 | a | 2.21, m | ProIII-1 | ||
| b | 2.08, m | ProIII-1 | ||||
| 4 | 25.90, CH2 | a | 2.07, m | |||
| b | 2.04, m | |||||
| 5 | 48.50, CH2 | a | 3.80, m | ProII-2 | ProII-1 | |
| b | 3.67, m | ProII-2 | ProII-1 | |||
| Thr | NH | |||||
| 1 | 173.48, C | |||||
| 2 | 59.13, CH | 4.38, d (2.9) | ProIII-1, Thr-1 | |||
| 3 | 68.66, CH | 4.29, dd (2.9, 6.4) | Thr-1 | |||
| 4 | 20.51, CH3 | 1.20, d (6.4) | Thr-1 |
| Peptide | Sequence | Rt (min) | m/z | [M + H]+ | β-Casein Fragment |
|---|---|---|---|---|---|
| 1 | pGlu-Pro-Leu-Pro-Pro-Thr | 12.7 | 635.3392 | C30H47N6O9 | 164–169 |
| 2 | pGlu-Pro-Leu-Pro-Pro | 12.0 | 534.2915 | C26H40N5O7 | 164–168 |
| 3 | Val-Val-Pro-Pro-Phe-Leu-Gln-Pro-Glu | 17.3 | 1025.5657 | C50H77N10O13 | 98–106 |
| 4 | Val-Pro-Pro-Phe-Leu-Gln-Pro-Glu-Val | 18.4 | 1025.5657 | C50H77N10O13 | 99–107 |
| 5 | Pro-Phe-Pro-Gly-Pro-Ile-Pro | 17.4 | 724.4020 | C37H54N7O8 | 76–82 |
| 6 | Val-Pro-Pro-Phe-Leu-Gln-Pro-Glu | 16.0 | 926.4953 | C45H68N9O12 | 99–106 |
| 7 | Pro-Pro-Phe-Leu-Gln-Pro-Glu-Val | 17.0 | 926.4953 | C45H68N9O12 | 100–107 |
| 8 | Pro-Pro-Phe-Leu-Gln-Pro-Glu | 14.2 | 827.4273 | C40H59N8O11 | 100–106 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vitale, G.A.; Scarpato, S.; Mangoni, A.; D’Auria, M.V.; Della Sala, G.; de Pascale, D. Enhanced Molecular Networking Shows Microbacterium sp. V1 as a Factory of Antioxidant Proline-Rich Peptides. Mar. Drugs 2023, 21, 256. https://doi.org/10.3390/md21040256
Vitale GA, Scarpato S, Mangoni A, D’Auria MV, Della Sala G, de Pascale D. Enhanced Molecular Networking Shows Microbacterium sp. V1 as a Factory of Antioxidant Proline-Rich Peptides. Marine Drugs. 2023; 21(4):256. https://doi.org/10.3390/md21040256
Chicago/Turabian StyleVitale, Giovanni Andrea, Silvia Scarpato, Alfonso Mangoni, Maria Valeria D’Auria, Gerardo Della Sala, and Donatella de Pascale. 2023. "Enhanced Molecular Networking Shows Microbacterium sp. V1 as a Factory of Antioxidant Proline-Rich Peptides" Marine Drugs 21, no. 4: 256. https://doi.org/10.3390/md21040256