
Thiolactones and Δ8,9-Pregnene Steroids from the Marine-Derived Fungus Meira sp. 1210CH-42 and Their α-Glucosidase Inhibitory Activity

Abstract
:1. Introduction
2. Results and Discussion
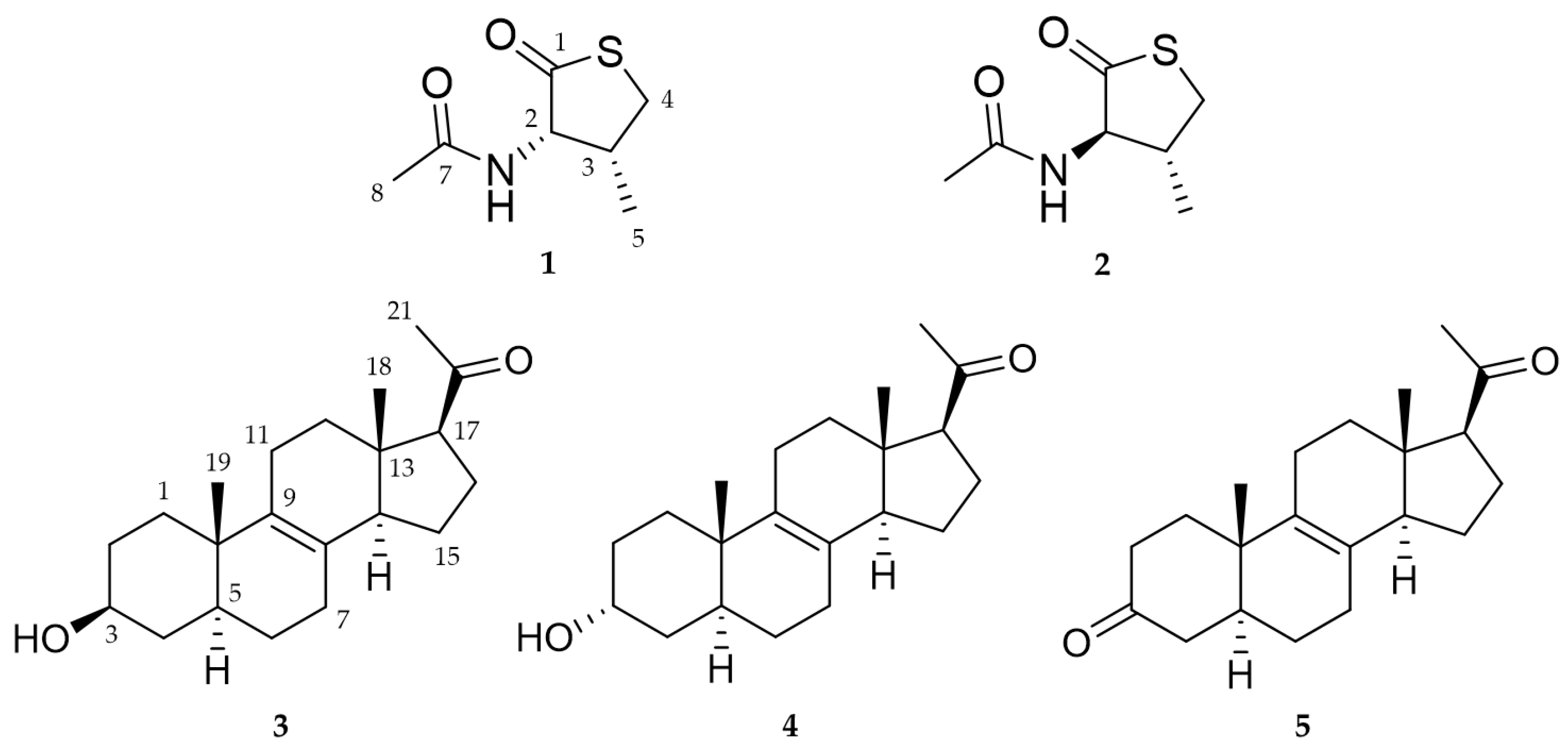
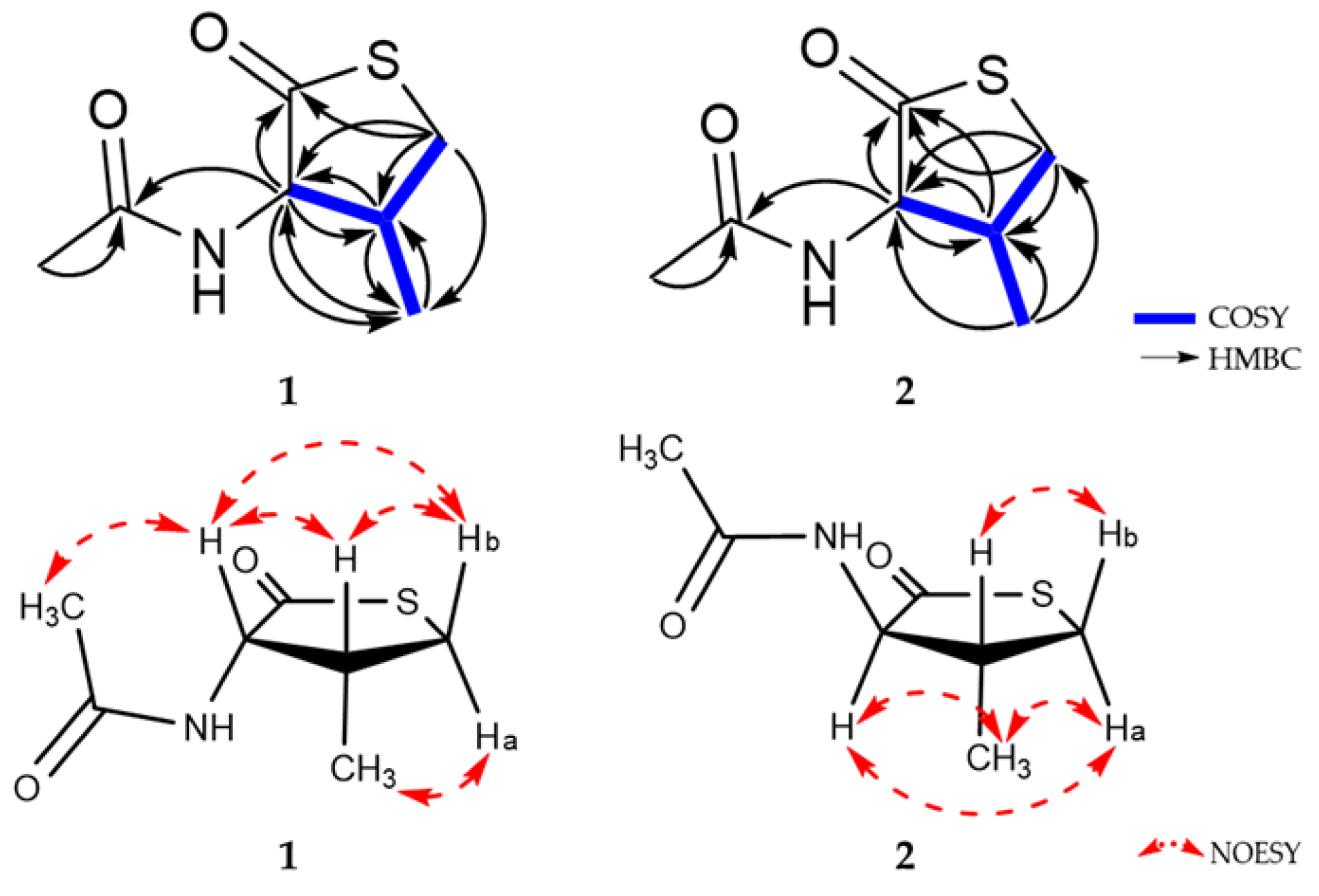
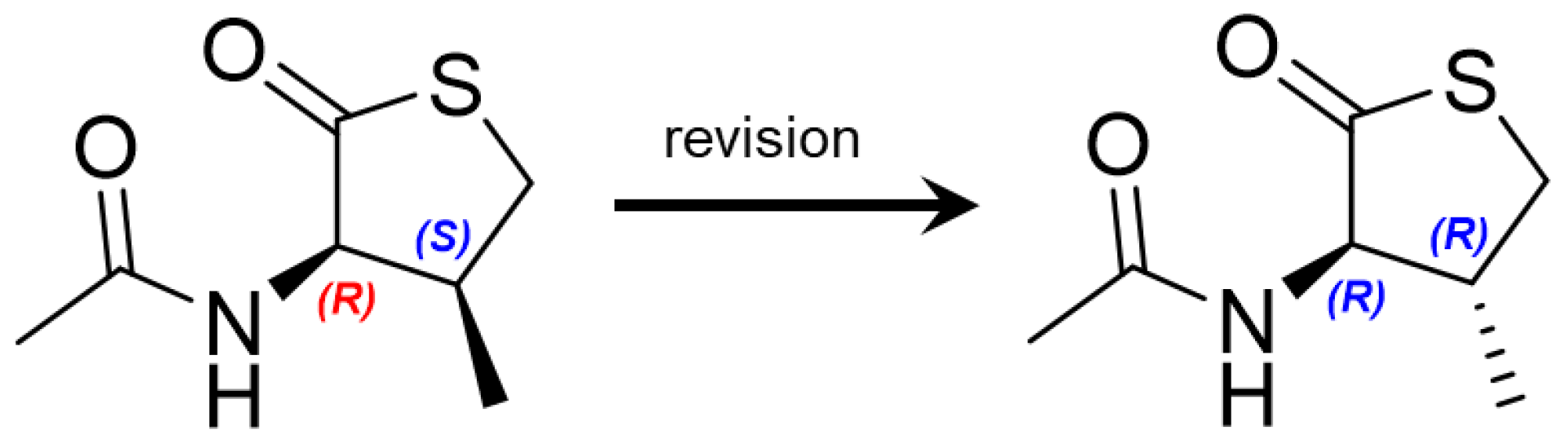
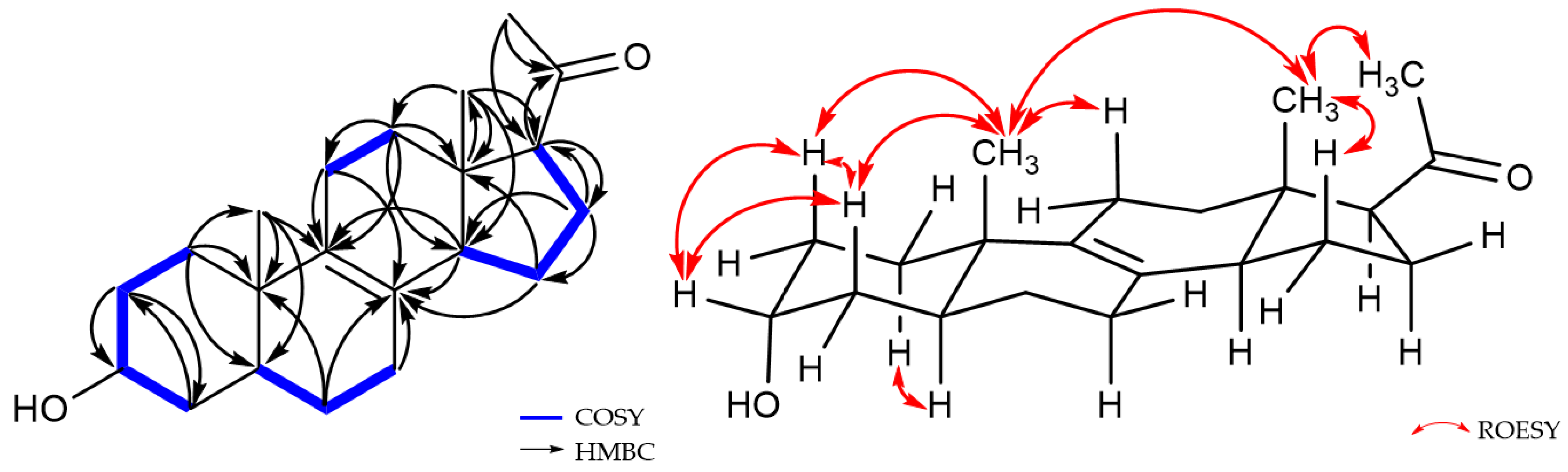
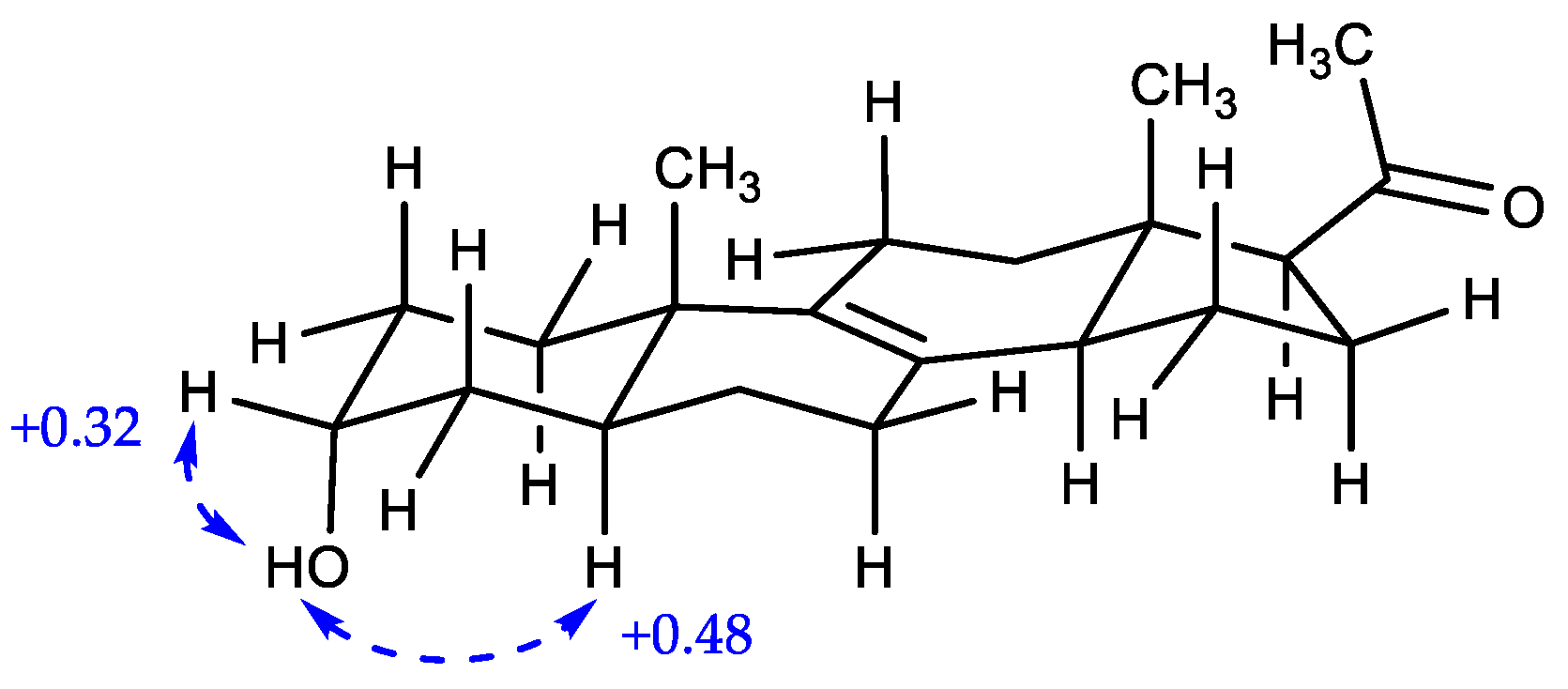
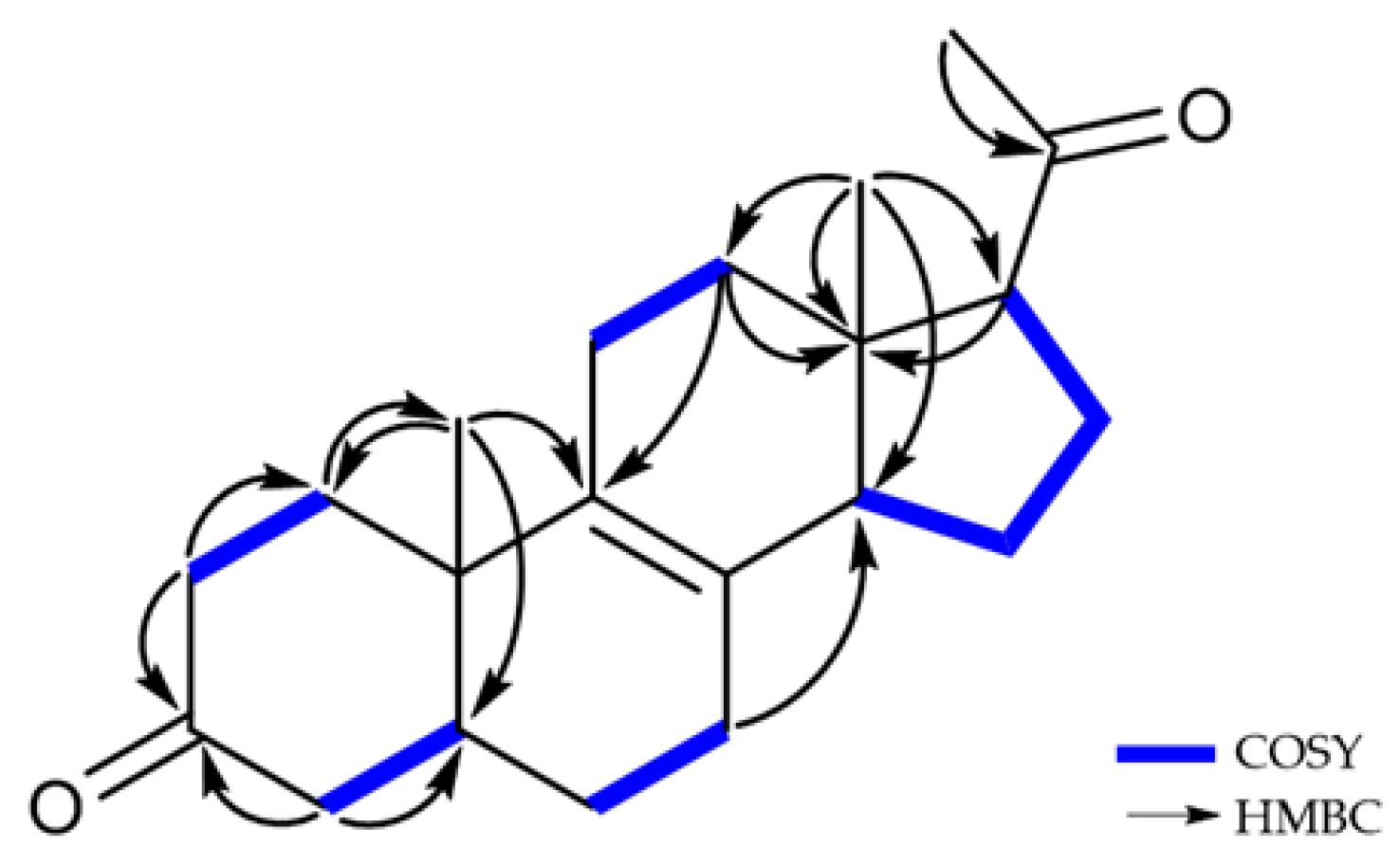
2.1. Structure Elucidation of New Compounds
2.2. α-Glucosidase Inhibitory Activities of Compounds
3. Materials and Methods
3.1. General Experimental Procedures and Reagents
3.2. Fungal Strain and Fermentation
3.3. Extraction and Isolation of Compounds 1–5
3.4. Computational Analysis
3.5. Measurement of α-Glucosidase Inhibitory Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schueffler, A.; Anke, T. Fungal natural products in research and development. Nat. Prod. Rep. 2014, 31, 1425–1448. [Google Scholar] [CrossRef] [PubMed]
- Duraes, F.; Szemeredi, N.; Kumla, D.; Pinto, M.; Kijjoa, A.; Spengler, G.; Sousa, E. Metabolites from Marine-Derived Fungi as Potential Antimicrobial Adjuvants. Mar. Drugs 2021, 19, 475. [Google Scholar] [CrossRef] [PubMed]
- Arrieche, D.; Cabrera-Pardo, J.R.; San-Martin, A.; Carrasco, H.; Taborga, L. Natural Products from Chilean and Antarctic Marine Fungi and Their Biomedical Relevance. Mar. Drugs 2023, 21, 98. [Google Scholar] [CrossRef] [PubMed]
- El-Demerdash, A.; Kumla, D.; Kijjoa, A. Chemical Diversity and Biological Activities of Meroterpenoids from Marine Derived-Fungi: A Comprehensive Update. Mar. Drugs 2020, 18, 317. [Google Scholar] [CrossRef]
- Hafez Ghoran, S.; Kijjoa, A. Marine-Derived Compounds with Anti-Alzheimer’s Disease Activities. Mar. Drugs 2021, 19, 410. [Google Scholar] [CrossRef]
- Jiang, M.; Wu, Z.; Guo, H.; Liu, L.; Chen, S. A Review of Terpenes from Marine-Derived Fungi: 2015–2019. Mar. Drugs 2020, 18, 321. [Google Scholar] [CrossRef] [PubMed]
- Rateb, M.E.; Ebel, R. Secondary metabolites of fungi from marine habitats. Nat. Prod. Rep. 2011, 28, 290–344. [Google Scholar] [CrossRef]
- Imhoff, J.F. Natural Products from Marine Fungi-Still an Underrepresented Resource. Mar. Drugs 2016, 14, 19. [Google Scholar] [CrossRef]
- Shin, H.J. Natural Products from Marine Fungi. Mar. Drugs 2020, 18, 230. [Google Scholar] [CrossRef]
- El-Bondkly, E.A.M.; El-Bondkly, A.A.M.; El-Bondkly, A.A.M. Marine endophytic fungal metabolites: A whole new world of pharmaceutical therapy exploration. Heliyon 2021, 7, e06362. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, M.F.M.; Esteves, A.C.; Alves, A. Marine Fungi: Opportunities and Challenges. Encyclopedia 2022, 2, 559–577. [Google Scholar] [CrossRef]
- Julianti, E.; Abrian, I.A.; Wibowo, M.S.; Azhari, M.; Tsurayya, N.; Izzati, F.; Juanssilfero, A.B.; Bayu, A.; Rahmawati, S.I.; Putra, M.Y. Secondary Metabolites from Marine-Derived Fungi and Actinobacteria as Potential Sources of Novel Colorectal Cancer Drugs. Mar. Drugs 2022, 20, 67. [Google Scholar] [CrossRef]
- Boekhout, T.; Theelen, B.; Houbraken, J.; Robert, V.; Scorzetti, G.; Gafni, A.; Gerson, U.; Sztejnberg, A. Novel anamorphic mite-associated fungi belonging to the Ustilaginomycetes: Meira geulakonigii gen. nov., sp nov., Meira argovae sp nov and Acaromyces ingoldii gen. nov., sp nov. Int. J. Syst. Evol. Micr. 2003, 53, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Rush, T.A.; Aime, M.C. The genus Meira: Phylogenetic placement and description of a new species. Anton. Leeuw. Int. J. G. 2013, 103, 1097–1106. [Google Scholar] [CrossRef]
- Gerson, U.; Gafni, A.; Paz, Z.; Sztejnberg, A. A tale of three acaropathogenic fungi in Israel: Hirsutella, Meira and Acaromyces. Exp. Appl. Acarol. 2008, 46, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Paz, Z.; Burdman, S.; Gerson, U.; Sztejnberg, A. Antagonistic effects of the endophytic fungus Meira geulakonigii on the citrus rust mite Phyllocoptruta oleivora. J. Appl. Microbiol. 2007, 103, 2570–2579. [Google Scholar] [CrossRef]
- Sztejnberg, A.; Paz, Z.; Boekhout, T.; Gafni, A.; Gerson, U. A new fungus with dual biocontrol capabilities: Reducing the numbers of phytophagous mites and powdery mildew disease damage. Crop. Prot. 2004, 23, 1125–1129. [Google Scholar] [CrossRef]
- Paz, Z.; Gerson, U.; Sztejnberg, A. Assaying three new fungi against citrus mites in the laboratory, and a field trial. Biocontrol 2007, 52, 855–862. [Google Scholar] [CrossRef]
- Cao, Y.; Li, P.-D.; Zhao, J.; Wang, H.-K.; Jeewon, R.; Bhoyroo, V.; Aruna, B.; Lin, F.-C.; Wang, Q. Morph-molecular characterization of Meira nicotianae sp. nov., a novel basidiomycetous, anamorphic yeast-like fungus associated with growth improvement in tobacco plant. Phytotaxa 2018, 365, 169–181. [Google Scholar] [CrossRef]
- Han, X.; Li, P.; Luo, X.; Qiao, D.; Tang, X.; Li, G. Two new compounds from the marine sponge derived fungus Penicillium chrysogenum. Nat. Prod. Res. 2020, 34, 2926–2930. [Google Scholar] [CrossRef]
- Dos Santos, A.; Rodrigues-Filho, E. NewΔ8,9-pregnene steroids isolated from the extremophile fungus Exophiala oligosperma. Nat. Prod. Res. 2021, 35, 2598–2601. [Google Scholar] [CrossRef]
- Shalit, Z.A.; Valdes, L.C.; Kim, W.S.; Micalizio, G.C. From an ent-Estrane, through a nat-Androstane, to the Total Synthesis of the Marine-Derived Δ8,9-Pregnene (+)-03219A. Org. Lett. 2021, 23, 2248–2252. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, X.; Huang, H.; Tian, X.; Song, Y.; Zhang, S.; Ju, J. 03219A, a new Δ8,9-pregnene isolated from Streptomyces sp. SCSIO 03219 obtained from a South China Sea sediment. J. Antibiot. 2013, 66, 327–331. [Google Scholar] [CrossRef]
- Qiu, S.X.; van Hung, N.; Xuan, L.T.; Gu, J.Q.; Lobkovsky, E.; Khanh, T.C.; Soejarto, D.D.; Clardy, J.; Pezzuto, J.M.; Dong, Y.M.; et al. A pregnane steroid from Aglaia lawii and structure confirmation of cabraleadiol monoacetate by X-ray crystallography. Phytochemistry 2001, 56, 775–780. [Google Scholar] [CrossRef]
- Cao, V.A.; Kwon, J.H.; Kang, J.S.; Lee, H.S.; Heo, C.S.; Shin, H.J. Aspersterols A-D, Ergostane-Type Sterols with an Unusual Unsaturated Side Chain from the Deep-Sea-Derived Fungus Aspergillus unguis. J. Nat. Prod. 2022, 85, 2177–2183. [Google Scholar] [CrossRef] [PubMed]
- Demarco, P.V.; Farkas, E.; Doddrell, D.; Mylari, B.L.; Wenkert, E. Pyridine-induced solvent shifts in the nuclear magnetic resonance spectra of hydroxylic compounds. J. Am. Chem. Soc. 1968, 90, 5480–5486. [Google Scholar] [CrossRef]
- Luo, X.; Li, F.; Shinde, P.B.; Hong, J.; Lee, C.O.; Im, K.S.; Jung, J.H. 26,27-cyclosterols and other polyoxygenated sterols from a marine sponge Topsentia sp. J. Nat. Prod. 2006, 69, 1760–1768. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Huang, Y.; Su, H.; Gao, Y.; Peng, X.; Zhou, L.; Li, X.; Qiu, M. C28 steroids from the fruiting bodies of Ganoderma resinaceum with potential anti-inflammatory activity. Phytochemistry 2019, 168, 112109. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Cheng, Y.; Tian, L.W. Semisynthesis of 22,25-Epoxylanostane Triterpenoids: Structure Revision and Protective Effects against Oxygen-Glucose Deprivation/Reoxygenation Injury in H9c2 Cells. J. Nat. Prod. 2023, 86, 406–415. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Kang, J.S.; Cho, D.Y.; Choi, D.K.; Shin, H.J. Isolation, Structure Determination, and Semisynthesis of Diphenazine Compounds from a Deep-Sea-Derived Strain of the Fungus Cystobasidium laryngis and Their Biological Activities. J. Nat. Prod. 2022, 85, 857–865. [Google Scholar] [CrossRef]
- Li, M.; Li, S.; Hu, J.; Gao, X.; Wang, Y.; Liu, Z.; Zhang, W. Thioester-Containing Benzoate Derivatives with alpha-Glucosidase Inhibitory Activity from the Deep-Sea-Derived Fungus Talaromyces indigoticus FS688. Mar. Drugs 2021, 20, 33. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Zeng, Y.; Chang, W.; Chen, H.; Wang, H.; Dai, H.; Lv, F. Potential α-Glucosidase Inhibitors from the Deep-Sea Sediment-Derived Fungus Aspergillus insulicola. Mar. Drugs 2023, 21, 157. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | ||
|---|---|---|---|---|
| δC, Type | δH, Mult. (J in Hz) | δC, Type | δH, Mult. (J in Hz) | |
| 1 | 206.5, C | 206.5, C | ||
| 2 | 63.9, CH | 4.79, d (6.6) | 65.7, CH | 4.29, d (12.5) |
| 3 | 36.0, CH | 2.86, m | 40.2, CH | 2.37, m |
| 4a | 35.9, CH2 | 3.10, dd (11.4, 2.2) | 34.7, CH2 | 3.08, t (11.2) |
| 4b | 3.64, dd (11.4, 5.4) | 3.34, d (11.2) | ||
| 5 | 13.0, CH3 | 1.04, d (6.9) | 17.5, CH3 | 1.20, d (6.5) |
| 6-NH | ||||
| 7 | 173.8, C | 174.0, C | ||
| 8 | 22.4, CH3 | 2.03, s | 22.6, CH3 | 2.02, s |
| Position | 3 | 4 | 5 | |||
|---|---|---|---|---|---|---|
| δC, Type | δH, Mult. (J in Hz) | δC, Type | δH, Mult. (J in Hz) | δC, Type | δH, Mult. (J in Hz) | |
| 1a | 36.6, CH2 | 1.22, td (16.2, 5.2) | 32.0, CH2 | 1.55, m | 38.0, CH2 | 1.55, m |
| 1b | 1.80, o.l 1 | |||||
| 2a | 32.4, CH2 | 1.42, o.l | 37.1, CH2 | 1.48, o.l | 39.1, CH2 | 2.31, o.l |
| 2b | 1.80, o.l | 1.54, o.l | 2.53, m | |||
| 3 | 71.8, CH | 3.53, m | 67.2, CH | 3.97, t (2.8) | 214.6, C | |
| 4a | 39.2, CH2 | 1.31, o.l | 30.0, CH2 | 1.68, m | 45.7, CH2 | 2.11, o.l |
| 4b | 1.61, m | 2.40, t (14.6) | ||||
| 5 | 42.3, CH | 1.40, o.l | 36.4, CH | 1.86, m | 44.4, CH | 1.80, m |
| 6a | 26.8, CH2 | 1.39, o.l | 26.7, CH2 | 1.32, m | 26.7, CH2 | 1.47, o.l |
| 6b | 1.52, m | 1.46, o.l | 1.60, m | |||
| 7 | 28.5, CH2 | 2.02, m | 28.4, CH2 | 2.02, m | 28.4, CH2 | 2.04, m |
| 8 | 129.2, C | 129.0, C | 130.1, C | |||
| 9 | 136.3, C | 137.2, C | 135.6, C | |||
| 10 | 37.1, CH | 37.6, CH | 37.3, CH | |||
| 11a | 24.0, CH2 | 2.15, m | 23.6, CH2 | 2.13, o.l | 24.1, CH2 | 2.20, m |
| 11b | 2.27, o.l | 2.28, o.l | 2.25, o.l | |||
| 12a | 37.3, CH2 | 1.69, m | 37.3, CH2 | 1.70, o.l | 37.2, CH2 | 1.70, o.l |
| 12b | 2.07, m | 2.07, o.l | 2.08, o.l | |||
| 13 | 45.0, C | 45.1, C | 45.0, C | |||
| 14 | 53.3, CH | 2.27, o.l | 53.4, CH | 2.30, o.l | 53.2, CH | 2.29, m |
| 15a | 25.3, CH2 | 1.42, o.l | 25.3, CH2 | 1.42, o.l | 25.3, CH2 | 1.45, o.l |
| 15b | 1.72, m | 1.72, o.l | ||||
| 16a | 24.3, CH2 | 1.72, o.l | 24.2, CH2 | 1.72, o.l | 24.3, CH2 | 1.72, o.l |
| 16b | 2.21, m | 2.21, m | 2.21, m | |||
| 17 | 63.5, CH | 2.69, t (8.7) | 63.5, CH | 2.70, t (8.6) | 63.5, CH | 2.70, t (8.7) |
| 18 | 13.2, CH3 | 0.57, s | 13.2, CH3 | 0.57, s | 13.2, CH3 | 0.60, s |
| 19 | 18.3, CH3 | 0.97, s | 17.3, CH3 | 0.94, s | 17.3, CH3 | 1.18, s |
| 20 | 212.4, C | 212.5, C | 212.3, C | |||
| 21 | 31.7, CH3 | 2.13, s | 31.7, CH3 | 2.13, s | 31.7, CH3 | 2.14, s |
| Compounds | IC50 (μM) 1 |
|---|---|
| 1 | >400 |
| 2 | 148.4 |
| 3 | 279.7 |
| 4 | 86.0 |
| Acarbose 2 | 418.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shin, H.J.; Lee, M.A.; Lee, H.-S.; Heo, C.-S. Thiolactones and Δ8,9-Pregnene Steroids from the Marine-Derived Fungus Meira sp. 1210CH-42 and Their α-Glucosidase Inhibitory Activity. Mar. Drugs 2023, 21, 246. https://doi.org/10.3390/md21040246
Shin HJ, Lee MA, Lee H-S, Heo C-S. Thiolactones and Δ8,9-Pregnene Steroids from the Marine-Derived Fungus Meira sp. 1210CH-42 and Their α-Glucosidase Inhibitory Activity. Marine Drugs. 2023; 21(4):246. https://doi.org/10.3390/md21040246
Chicago/Turabian StyleShin, Hee Jae, Min Ah Lee, Hwa-Sun Lee, and Chang-Su Heo. 2023. "Thiolactones and Δ8,9-Pregnene Steroids from the Marine-Derived Fungus Meira sp. 1210CH-42 and Their α-Glucosidase Inhibitory Activity" Marine Drugs 21, no. 4: 246. https://doi.org/10.3390/md21040246