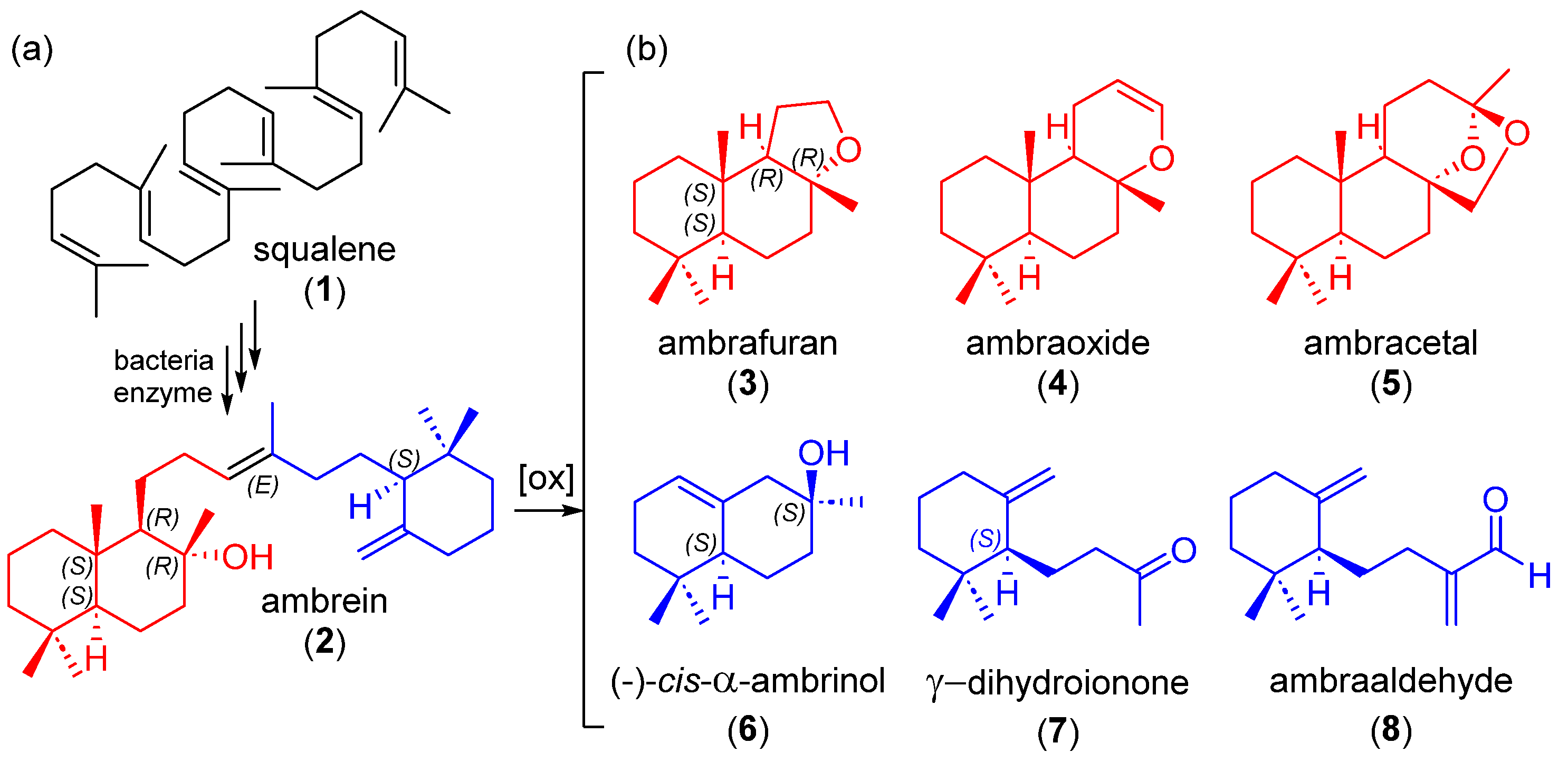
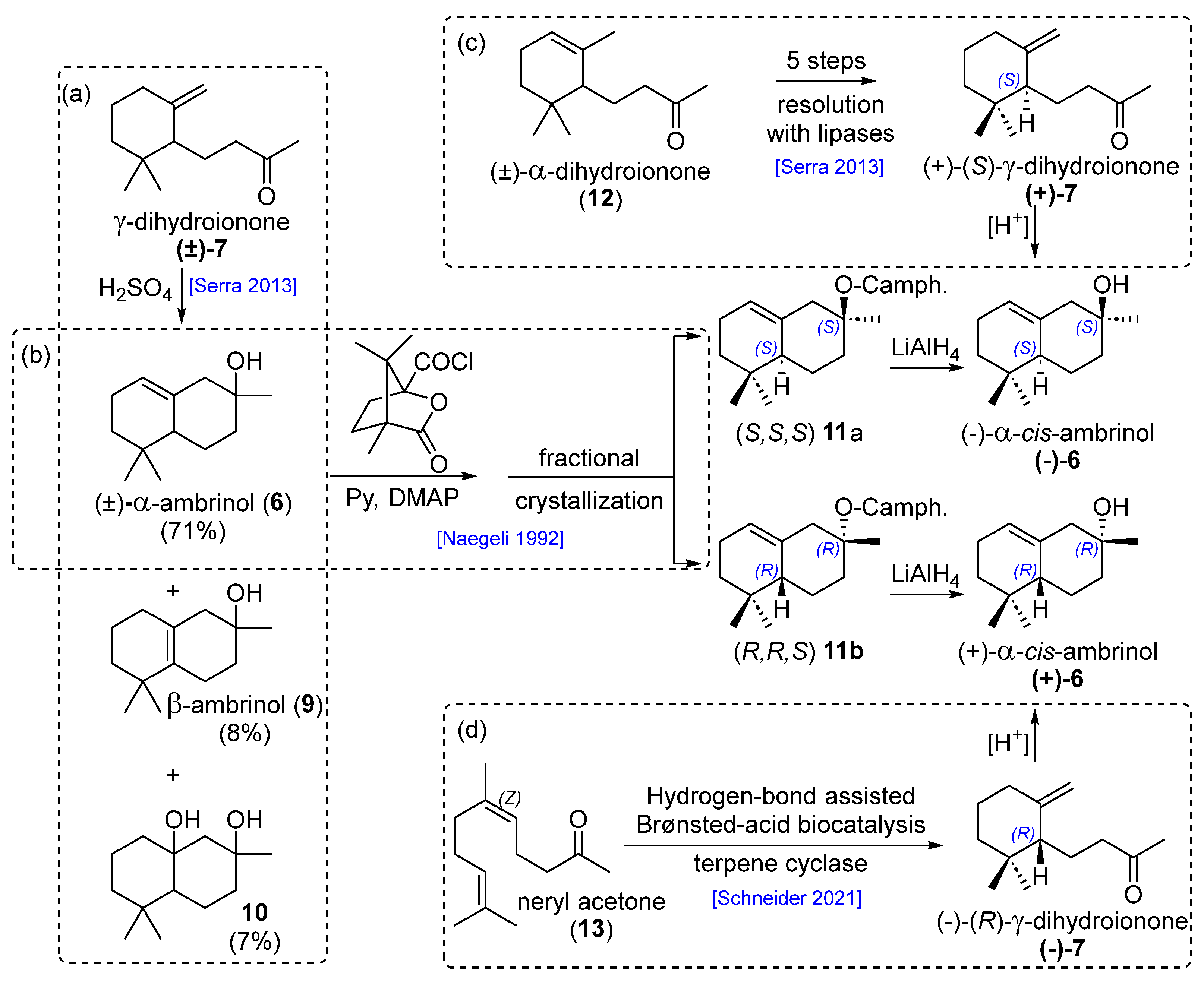
2. Results and Discussion
As part of our continued effort in the preparation of marine natural products [
14,
15,
16], and due to the fact that all the previously reported synthesis of
cis-α-ambrinol (
6) rely on an acid cyclization step that affords a mixture of regioisomers which is not easily purified by conventional chromatographic technics, we were interested in developing a concise diastereoselective total synthesis of
cis-α-ambrinol (
6).
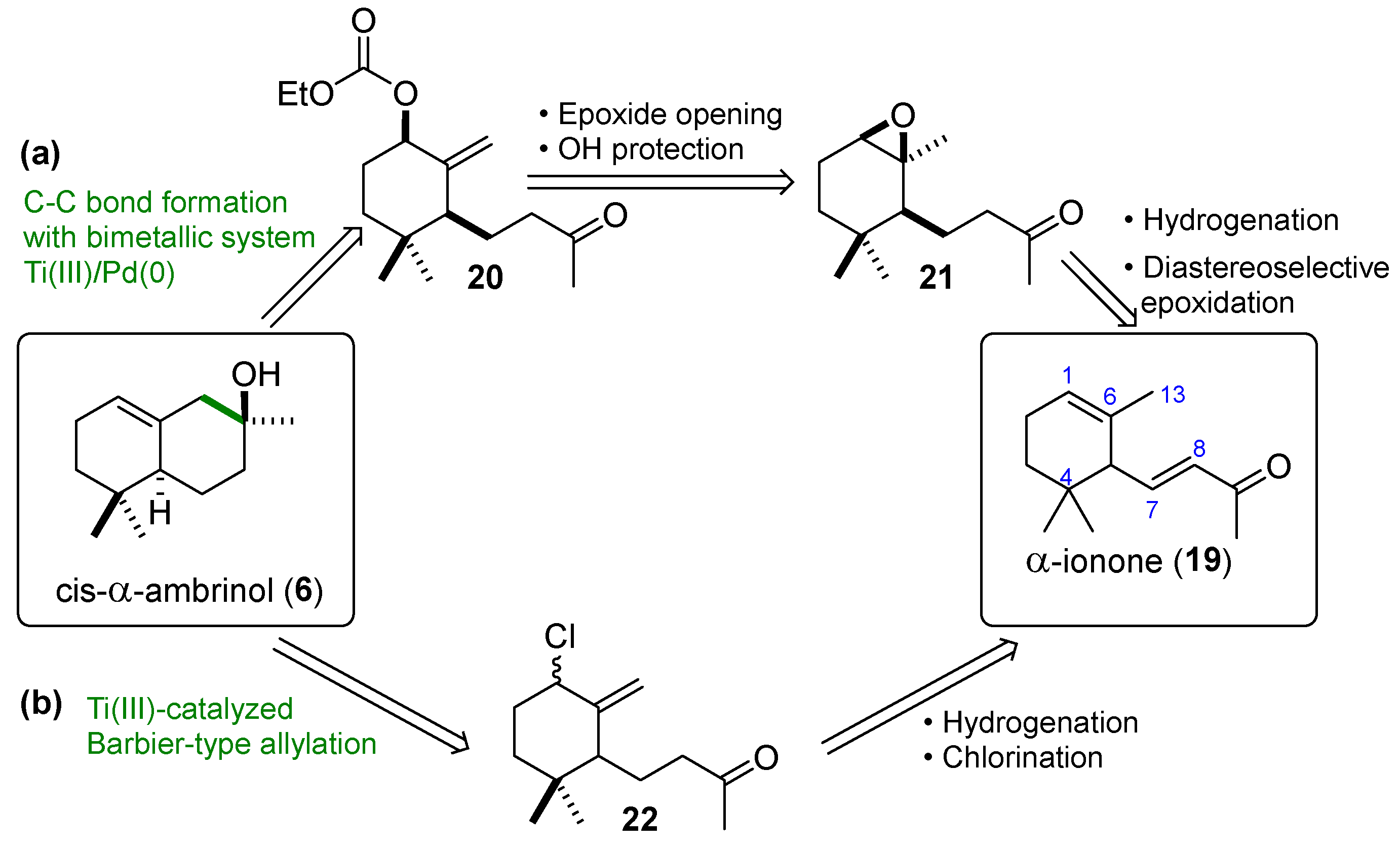
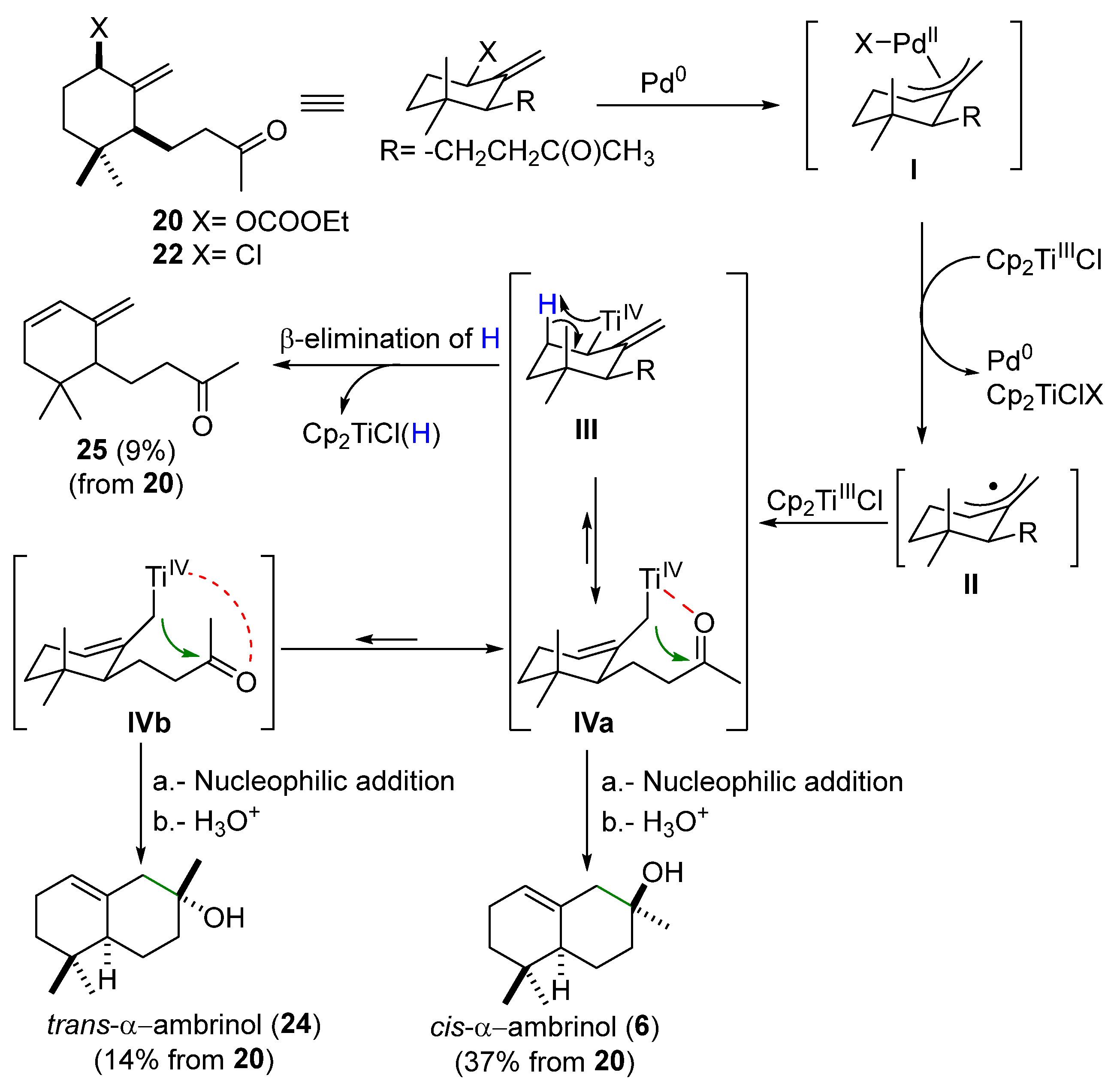
The synthesis of compound
6 was planned according to the retrosynthesis depicted in
Scheme 4 through two alternative pathways, both of them using commercially available α-ionone (
19) as a starting material. The first approach has as a key step a diastereoselective cyclization of the allylic carbonate (
20) using the bimetallic system Cp
2Ti
IIICl/Pd
0 (
Scheme 4a). The second one (
Scheme 4b), is based on a Barbier-type CpTi
IIICl
2-catalyzed intramolecular allylation of the chlorinated derivative (
22).
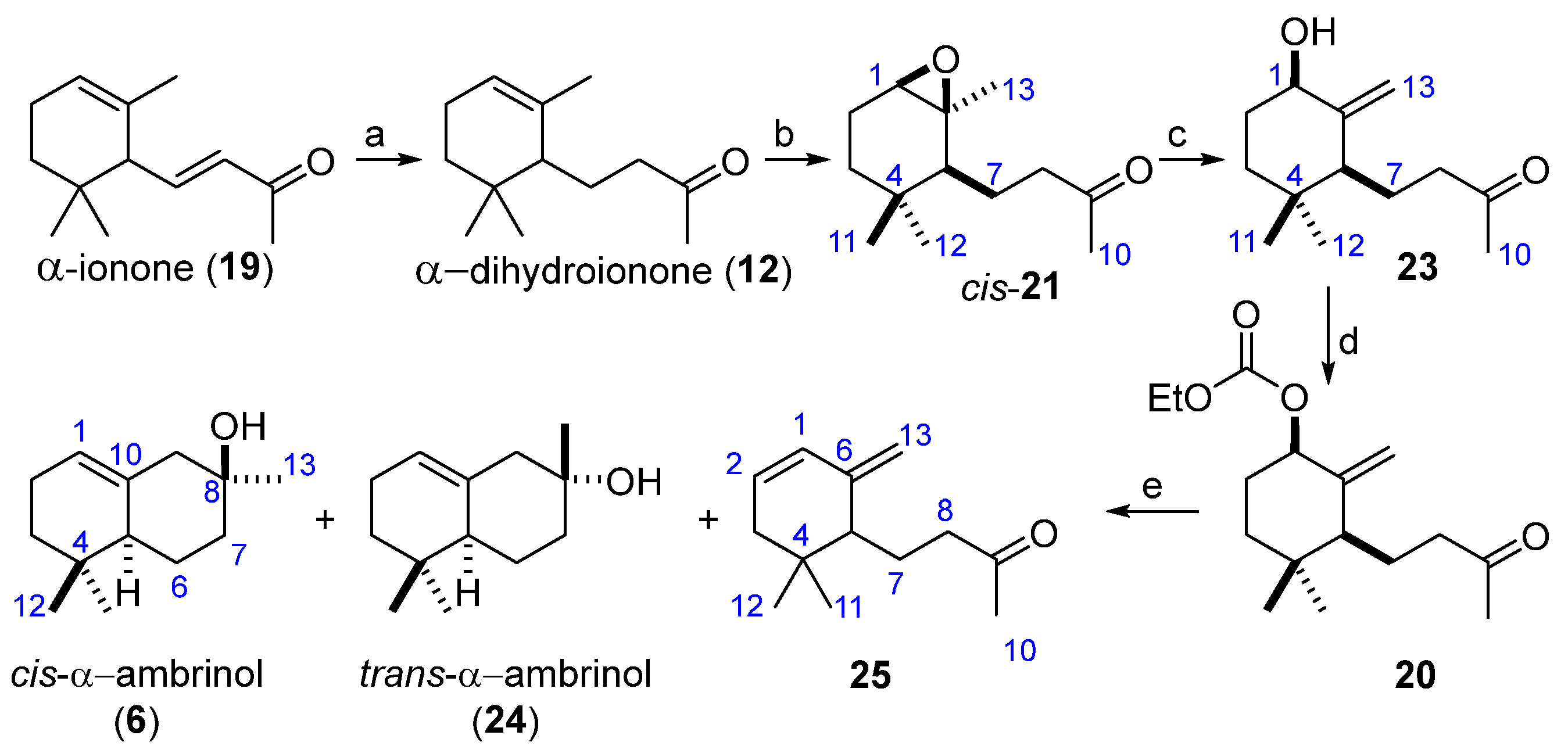
The synthetic route based on the bimetallic system Ti(III)/Pd(0) is depicted in
Scheme 5. The first step is the regioselective reduction of the conjugated double in α-ionone (
19), following a modification of a previously described methodology [
17], to give α-dihydroinone (
12). Epoxidation of (
12) with
m-CPBA afforded the epoxide (
21) as a diastereoselective mixture
cis:
trans (86:12) in an 98% yield. Acid treatment of the
cis-epoxide (
cis-21) gave 1-hydroxy-γ-dihydroionone (
23) in a 62% yield. The formation of carbonate (
20) was carried out using ethyl chloroformate under basic conditions, yielding the desired compound (
20) in an 80% yield. The key step of this synthetic approach relies on a cooperative catalytic method [
18,
19]. We first tried the reaction using the combination Cp
2TiCl/Ni(PPh
3)
2Cl
2, a bimetallic system which has been described as an efficient promoter for the allylation of carbonyl compounds [
19], although with little success in our case, as the process proved to be unproductive. However, treatment of allylic carbonate (
20) with a source of Ti(III)/Pd(0) [
18] (see
Section 3 for details) gave a mixture of the desired natural
cis-α-ambrinol (
6) (37% yield),
trans-α-ambrinol (
24) (14% yield), and the monocyclic compound (
25) (9% yield).
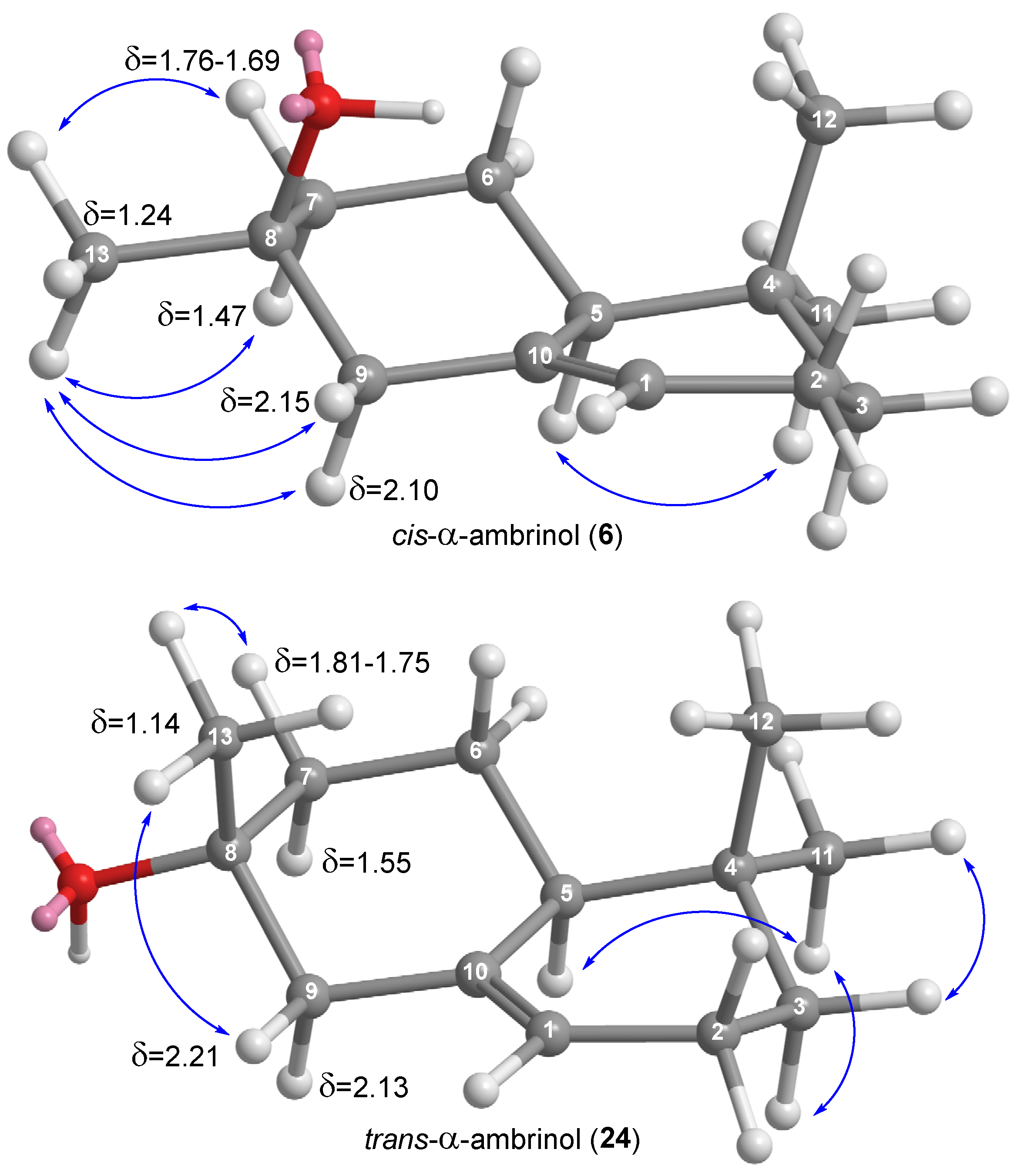
The relative stereochemistry of both isomers
cis-α-ambrinol (
6) and
trans-α-ambrinol (
24) was determined with the aid of NOE experiments (
Figure 1). Especially relevant is the presence of correlations in (
6) between the equatorial CH
3-13 (δ = 1.24 ppm) and all four hydrogens in CH
2-7 and CH
2-9 while in (
24), the observed correlations between the axial methyl CH
3-13 (δ = 1.14 ppm) are only those with the β H (equatorial) of CH
2-7 and CH
2-9 in each case.
Although the yield of (
6) was not completely satisfactory, the C-C bond formation reaction proceeds with high diastereoselectivity, and with simultaneous formation of the desired trisubstituted double bond. Spectroscopic data for synthetic
cis-α-ambrinol (
cis-6) were identical to those of the natural compound [
20,
21,
22].
The experimental results obtained in the cyclization of (
20) with the Ti(III)/Pd(0) bimetallic system can be fully explained by the mechanism tentatively proposed in
Scheme 6. Initially, an oxidative addition of the allylic electrophile carbonate (
20) to Pd
0 would give the corresponding ƞ
3-allyl-palladium intermediate
I. Monoelectronic reduction of intermediate
I by Cp
2TiCl generates a ƞ
3-allyl-palladium(II) intermediate, which could give the carbon-centered radical intermediate
II, while the Pd
0 complex is regenerated. This radical intermediate
II could be trapped by a second molecule of Cp
2TiCl to form two alkyl-Ti
IV species in metallotropic equilibrium (
III and
IV). β-Elimination of hydrogen in species
III would account for the formation of monocyclic diene (
25) and Cp
2TiCl(H). It has been previously reported that this titanium hydride spontaneously decomposes to regenerate Cp
2TiCl and molecular hydrogen [
23]. On the other hand, the least sterically hindered alkyl-Ti
IV intermediate
IV can evolve through two different rotational conformers,
IVa and
IVb. In one of them,
IVa, the carbonyl group oxygen is arranged spatially close to the titanium atom (axial-like orientation), and therefore the intramolecular nucleophilic addition of the alkyl-Ti
IV to the ketone favors the diastereoselective formation of
cis-α-ambrinol (
6) as the main product. However, if the ketone oxygen is located at a greater distance from the titanium atom (equatorial-like orientation,
IVb), the interaction between both atoms should be slightly weaker, resulting in a slower nucleophilic addition of the alkyl-Ti
IV intermediate (
IVb) to the carbonyl group, and further leading to the formation of the diastereoisomer
trans-α-ambrinol (
24) as a minor product.
The synthetic advantage of this synthetic route is that it could lead to the enantiopure
(−)-6 using an enantiopure alcohol (−)-
23. This was previously prepared by Serra [
10] using a lipase-mediated racemic resolution.
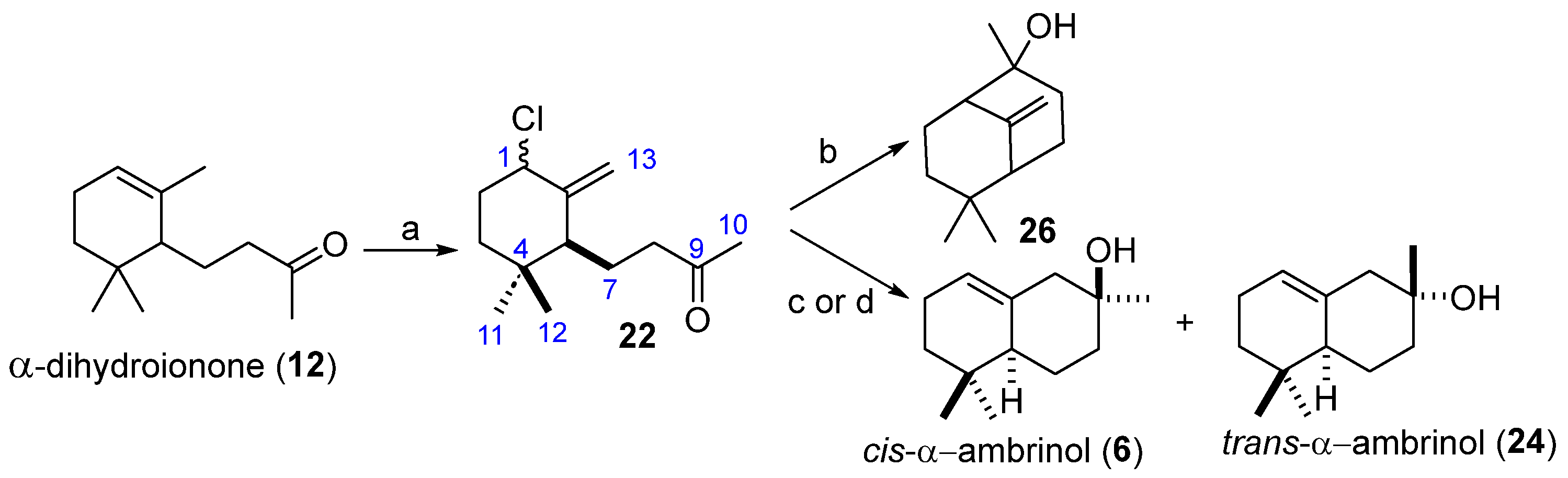
Our second synthetic approach is based on a Barbier-type Ti(III)-catalyzed intramolecular allylation of an allyl chloride, which is summarized in
Scheme 7. In this case, we used a starting material α-dihydroionone (
12), previously prepared in the other route.
Chlorination of 12 with NaClO afforded a diastereomeric mixture of allylic chlorides (22) in a 96% yield. With this substrate in hand, we first tried to induce the intramolecular allylation using an excess of Zn dust, which is known to react with allyl chlorides to form organometallic systems which can react with carbonyl groups. However, the reaction led to the formation of the bicyclic product (26), which originated as a result of the formation of a C-C bond between C1 and the carbonyl (C9). We next tried the allylation with two Ti(III) systems, the well-established single electron transfer reagent Cp2TiCl, and the half-sandwich titanocene CpTiCl2, both prepared by reduction with Mn of the appropriate Ti(IV) species.
The allylation was tested under catalytic and stoichiometric conditions for both systems. The results, summarized in
Table 1, show a similar behavior in all cases, although CpTiCl
3 seems to be superior both in terms of global yields and diastereoselectivity, particularly under stoichiometric conditions (
Table 1, entry 3). In the light of these results, we decided to check whether the Ti(III)/Pd(0) combination strategy could be performed with the half-sandwich titanocene reagent CpTiCl
2 using the ethylcarbonate (
20) as a substrate. Indeed, the reaction proved to be successful (
Table 1, entry 5), leading to a 73% global yield of the cyclic product, and with a 73:27 diastereoselectivity ratio using stoichiometric amounts of the Ti(III) source and catalytic of the Pd(0).
The diastereoselectivity observed in the cyclization of (
22) to give (
6) and (
24) mediated or catalyzed by CpTiCl
2 can be easily explained by a mechanism similar to the one discussed in
Scheme 6, although in this case the radical intermediate
II would be formed by the homolytic cleavage of the activated C-Cl bond present in (
22).
In conclusion, we have proved that
cis-α-ambrinol (
6) can be prepared from commercial α-ionone (
19) with an overall yield of 46% in only three steps using a stoichiometric amount of CpTiCl
2 for the intramolecular Barbier-type allylation of the chloro-derivative (
22).
cis-α-Ambrinol (
6) can also be prepared in five steps from the same starting material through a Ti(III)/Pd(0) cyclization of the carbonate intermediate (
20) with a 35% global yield. Finally, it should be mentioned that this synthesis of α-ambrinol (
6) constitutes a new application of the usefulness of CpTiCl
2 as a new monoelectronic transfer reagent, as we [
24,
25,
26] and others [
27,
28] have previously reported.
3. Materials and Methods
3.1. General Details
THF was distilled from Na/benzophenone under argon, and in all experiments involving titanocene (III) was deoxygenated prior to use, and oven-dried glassware was used in all cases. NMR spectra were recorded on Bruker Nanobay Avance III HD 300 MHz, and Avance III HD 600 MHz spectrometers. Proton-decoupled
13C{
1H} NMR and DEPT-135 were measured in all cases. When required, NOE 1D, COSY, HSQC and HMBC experiments were used for signal assignation. Chemical shifts (δ) are expressed in ppm and coupling constants (
J) in hertzs (Hz). Chemical shifts are reported using CDCl
3 as internal reference. IR Spectra were recorded with a Bruker Alpha spectrometer. Mass spectra were recorded in a Waters Xevo by LC-QTof-MS by electrospray ionization. All reactions were monitored by thin-layer chromatography (TLC) carried out on 0.2 mm DC-Fertigfolien Alugram
® XtraSil G/UV254 silica gel plates. The TLC plates were visualized with UV light and 7% phosphomolybdic acid or KMnO
4 in water/heat. Flash chromatography was performed on silicagel 60 (0.04–0.06 mm). Hard copies of NMR and IR spectra can be found as
Supplementary Materials.
3.2. Synthesis of α-Dihydroionone (12)
To a solution of α-ionone (
19) (864 mg, 4.5 mmol) in THF (10 mL) was added Ni-Raney (0.4 g). The mixture was stirred under H
2 (1 atm) for 30 min at room temperature in a hydrogenation apparatus. The mixture was filtered through celite, and the solvent evaporated, yielding (
12) (873 mg, 4.5 mmol, 100%) as a colorless oil. Spectroscopic data are in agreement with literature values [
29].
IR (ATR) v (cm−1): 2954, 2915, 2870, 1714, 1449, 1361, 1252, 1218, 1159, 961, 944, 811, 555.
1H NMR (300 MHz, CDCl3) δ (ppm): 5.36 (1H, bs, H1), 2.50 (1H, dd, J = 3.1, 7.0 Hz H8a), 2.47 (1H, dd, J = 2.0, 6.7 Hz H8b), 2.16 (3H, s, H9), 1.98 (2H, m), 1.85–1.73 (1H, m), 1.69 (3H, q, J = 1.7 Hz H13), 1.67–1.59 (1H, m), 1.51–1.48 (1H, m), 1.45–1.38 (1H, m), 1.19–1.12 (1H, m), 0.94 (3H, s), 0.89 (3H, s).
13C NMR (75 MHz, CDCl3) δ (ppm): 209.2 (C, C9), 135.6 (C, C6), 121.1 (CH, C1), 48.5 (CH), 43.8 (CH2), 32.6 (C, C4), 31.5 (CH2), 30.0 (CH3), 27.7 (CH3), 27.6 (CH3), 24.4 (CH2), 23.5 (CH3), 23.0 (CH2).
3.3. Preparation of Epoxide 21
To a solution of α-dihydroionone (12) (3.05 g, 15.67 mmol) in anhydrous CH
2Cl
2 (60 mL) at 0 °C, MCPBA (4.25 g, 17.24 mmol) was added. The mixture was stirred under N
2 and allowed to reach room temperature for 3 h. The reaction was quenched by stirring for 15 min. with saturated NaHCO
3 (30 mL) and another 30 mL of Na
2S
2O
3 (10% in water). The two phases were separated and the organic layer was washed with brine, dried over anhydrous MgSO
4 and the solvent was removed in a vacuum to give (
21) as a mixture 86:12
cis:trans (3.21 g, 98%). Compound (
cis-21) was purified by column chromatography (hexane:EtOAc 9:1) (83% yield from (
12)). Colorless oil. Spectroscopic data are in agreement with literature values [
17].
IR (ATR) v (cm−1): 2962, 2932, 2871, 1713, 1449, 1363, 1233, 1181, 1160, 1097, 1040, 995, 899, 864, 747, 557, 526.
1H NMR (600 MHz, CDCl3) δ (ppm): 2.96 (1H, s, H1), 2.76 (1H, ddd, J = 16.2, 10.1, 5.3 Hz, H8a), 2.52 (1H, ddd, J = 16.2, 9.9, 5.8 Hz, H8b), 2.18 (3H, s, H10), 1.94 (1H, dd, J = 15.5, 6.0 Hz, H2a), 1,87–1.81 (1H, m, H2b), 1.75–1.70 (1H, m, H7a), 1.61–1.54 (1H, m, H7b), 1.40 (1H, dd, J = 10.7, 6.0 Hz, H5), 1.34 (3H, s, H13), 1.32–1.27 (1H, m, H3a), 0.90 (3H, s, H11), 0.85 (3H, s, H12), 0.83 (1H, m, H3b).
13C NMR (75 MHz, CDCl3) δ (ppm): 209.4 (C, C9), 60.1 (CH, C1), 59.3 (C, C6), 46.2 (CH, C5), 43.1 (CH2, C8), 31.6 (C, C4), 30.1 (CH3, C10), 27.9 (CH3, C12), 27.3 (CH3, C11), 27.2 (CH3, C13), 26.7 (CH2, C3), 22.1 (CH2, C2), 21.6 (CH2, C7).
3.4. Synthesis of 1-Hydroxy-γ-dihydroionone (23)
To a solution of compound (
cis-21) (934 mg, 4.44 mmol) in CH
2Cl
2 (30 mL) at 0 °C,
p-TSA (76 mg, 0.44 mmol) was added. The mixture was stirred for 12 h (0 °C), and then
p-TSA (76 mg, 0.44 mmol) was again added and stirred for 4 h at room temperature. The mixture was washed with saturated NaHCO
3 (10 mL × 3) and dried over anhydrous MgSO
4. The solvent was removed in a vacuum and the residue purified by silica gel flash column chromatography (hexane/EtOAc, 8:2) to afford alcohol (
23) (575 mg, 62%) as a colorless oil. Spectroscopic data are in agreement with literature values [
30].
IR (ATR) v (cm−1): 3412, 2934, 2867, 1707, 1648, 1456, 1410, 1363, 1159, 1062, 1038, 898, 639, 584, 502.
1H NMR (600 MHz, CDCl3) δ (ppm): 5.18 (1H, s, H13a), 4.65 (1H, s, H13b), 3.96 (1H, dd, J = 4.8, 9.0 Hz, H1), 2.58 (1H, ddd, J = 5.4, 9.0, 18.0 Hz, H8a), 2.34 (1H, ddd, J = 7.8, 8.4, 18.0 Hz, H8b), 2.11 (3H, s, H10), 1.91 (1H, m, H2a), 1.85 (1H, m, H7a), 1.74–1.62 (3H, m, H7b, H5, OH), 1.50 (1H, m, H3a), 1.46 (1H, m, H2b), 1.38 (1H, m, 3b), 0.98 (3H, s, H11), 0.74 (3H, s, H12).
13C NMR (75 MHz, CDCl3) δ (ppm): 209.5 (C, C9), 150.3 (C, C6), 105.5 (CH2, C13), 73.6 (CH, C1), 51.3 (CH, C5), 42.5 (CH2, C8), 37.9 (CH2, C3), 35.8 (C, C4), 33.2 (CH2, C2), 30.0 (CH3, C10), 29.3 (CH3, C11), 21.0 (CH3, C12), 19.5 (CH2, C7).
3.5. Synthesis of Ethyl Carbonate (20)
In an N2 atmosphere at 0 °C, ethyl chloroformate (0.74 mL, 7.59 mmol), pyridine (1.54 mL, 18.98) and DMAP (64 mg. 0.51 mmol) were added to a solution of (23) (532 mg, 2.53 mmol) in CH2Cl2 (40 mL). After 10 min, the cooling bath was removed, and the mixture was stirred for 20 h. TIt was then diluted with Et2O and washed with HCl (3%) and water. The organic layer was dried over anhydrous MgSO4, the solvent was removed in a vacuum and the residue purified by silica gel flash column chromatography (gradient hexane/EtOAc) to afford carbonate (20) (571 mg, 80%) as a colorless oil.
IR (ATR) v (cm−1): 2949, 2871, 1741, 1715, 1650, 1456, 1367, 1251, 1162, 1007, 903, 856, 790. HREIMS (m/z) calcd. for C16H26O4 282.1831 [M]+, found 282.1833.
1H NMR (300 MHz, CDCl3) δ (ppm): 5.17 (1H, s, H13a), 4.96 (1H, dd, J = 4.5, 8.2 Hz, H1), 4.73 (1H, s, H13b), 4.22 (1H, q, J = 7.1 Hz, OCH2), 2.54 (1H, ddd, J = 4.5, 9.7, 17.6 Hz, H8a), 2.31 (1H, m, H8b), 2.13 (3H, s, H10), 1.98–1.83 (2H, m, H2a, H7a), 1.75–1.57 (4H, m, H2b, H7b, H5, H3a), 1.41 (1H, dd, J = 4.5, 10.3 Hz, H3b), 1.34 (3H, t, J = 7.1 Hz), 1.00 (3H, s, H11), 0.82 (3H, s, H12).
13C NMR (75 MHz, CDCl3) δ (ppm): 209.2 (C, C9), 154.5 (C, OCOO), 144.8 (C, C6), 109.2 (CH2, C13), 78.5 (CH, C1), 63.8 (CH2, OCH2), 51.4 (CH, C5), 42.3 (CH2, C8), 35.3 (C, C4), 30.0 (CH3, C10), 29.4 (CH2), 28.8 (CH3), 22.6 (CH3), 20.0 (CH2), 14.3 (CH3, OCH2CH3).
3.6. Synthesis of Ambrinol Using Cp2TiCl2
Deoxygenated, dry THF (6.5 mL) was added to a mixture of Cp2TiCl2 (195 mg, 0.76 mmol), Pd(PPh3)2Cl2 (54 mg, 0.076 mmol), and Mn dust (169 mg, 3.04 mmol) in an Ar atmosphere and the red suspension was stirred at room temperature until it turned lime green (after about 15 min). Then, a solution of (20) (107 mg, 0.38 mmol) in THF (2 mL) was added dropwise and the mixture was stirred for four days. The reaction was diluted in EtOAc, washed with HCl 12% and brine, dried (anhydrous Na2SO4) and the solvent was removed. The residue was purified by flash chromatography (hexane/EtOAc 9:1) yielding cis-α-ambrinol (6) (27 mg, 37%), trans-α-ambrinol (24) (10 mg, 14%) and compound (25) (7 mg, 9%).
cis-α-ambrinol (
6): Colorless oil; spectroscopic data are in agreement with literature values [
12]. IR (ATR)
v (cm
−1): 3449, 2957, 2913, 2868, 2843, 1451, 1383, 1363, 1321, 1263, 1237, 1186, 1134, 1111, 1097, 1019, 995, 926, 912, 878, 800, 727, 501. HREIMS (
m/
z) calcd. for C
13H
22O 194.1671 [M]
+, found 194.1670.
1H NMR (600 MHz, CDCl3) δ (ppm): 5.48 (1H, s, H1), 2.15 (1H, bd, J = 13.2 Hz, H9a), 2.10 (1H, dd, J = 13.2, 2.5 Hz, H9b), 2.04–2.00 (2H, m, H2), 1.87 (1H, s, OH), 1.76–1.69 (2H, m, H6a, H7eq), 1.52 (1H, bd, J = 12.6 Hz, H5), 1.47 (1H, td, J = 13.4, 4.2 Hz, H7ax), 1.39 (1H, dt, J = 13.0, 7.3 Hz, H3eq), 1.29 (1H, td, J = 13.0, 3.4 Hz, H6b), 1.24 (3H, s, H13), 1.21 (1H, m, H3ax), 0.94 (3H, s, H11), 0.89 (3H, s, H12).
13C NMR (151 MHz, CDCl3) δ (ppm): 137.5 (C, C10), 122.2 (CH2, C1), 70.4 (C, C8), 50.0 (CH2, C9), 47.4 (CH, C5), 39.1 (CH2, C7), 33.4 (CH2, C3), 31.2 (C, C4), 29.4 (CH3, C13), 28.2 (CH3, C11), 26.1 (CH3, C12), 25.2 (CH2, C6), 22.9 (CH2, C2).
trans-α-ambrinol (24): Colorless oil; IR (ATR) v (cm−1): 3368, 2923, 2869, 1710, 1664, 1451, 1364, 1260, 1198, 1119, 1062, 1020, 1010, 947, 919, 831, 820, 727, 682, 571. HREIMS (m/z) calcd. for C13H22O 194.1671 [M]+, found 194.167.
1H NMR (600 MHz, CDCl3) δ (ppm): 5.37 (1H, s, H1), 2.21 (1H, dd, J = 12.5, 2.6 Hz, H9eq), 2.13 (1H, bd, J = 12.5 Hz, H9ax), 1.97 (2H, m, H2), 1.81–1.75 (2H, m, H7eq, H6eq), 1.60 (1H, bd, J = 13.2 Hz, H5), 1.55 (1H, td, J = 13.3, 4.3 Hz, H7ax), 1.34 (1H, dt, J = 13.0, 6.1 Hz, H3eq), 1.25 (2H, m, H3ax. OH), 1.14 (3H, s, H13), 1.10 (1H, td, J = 6.5, 1.9 Hz, H6eq), 0.94 (3H, s, H11), 0.84 (3H, s, H12).
13C NMR (151 MHz, CDCl3) δ (ppm): 137.4 (C, C10), 120.9 (CH2, C1), 72.1 (C, C8), 50.9 (CH2, C9), 47.0 (CH, C5), 40.8 (CH2, C7), 35.2 (CH2, C3), 31.3 (C, C4), 28.7 (CH3, C11), 25.6 (CH3, C13), 25.3 (CH2, C6), 24.6 (CH3, C12), 22.8 (CH2, C2).
Compound (
25): Colorless oil [
31];
1H NMR (300 MHz, CDCl
3) δ 6.01 (1H, m, H1), 5.67 (1H, m, H2), 4.91 (1H, s, H13a), 4.70 (1H, s, H13b), 2.55–2.32 (2H, m), 2.14 (3H, s, H10), 1.84–1.70 (3H, m), 1.35–1.25 (2H, m), 1.02 (3H, s, H11), 0.88 (3H, s, H12).
3.7. Synthesis of Ambrinol Using CpTiCl3
Deoxygenated, dry THF (6 mL) was added to a mixture of CpTiCl3 (137 mg, 0.62 mmol), Pd(PPh3)2Cl2 (44 mg, 0.062 mmol), and Mn dust (138 mg, 2.48 mmol) in an Ar atmosphere red suspension was stirred at room temperature until it turned lime green (after about 15 min). Then, a solution of (20) (88 mg, 0.31 mmol) in THF (0.5 mL) was added dropwise and the mixture was stirred for 19 h. The reaction was quenched and purified as described above for Cp2TiCl2. Compound (6) (32 mg, 0.16 mmol, 53%) and compound (24) (12 mg, 0.06 mmol, 20%) were obtained.
3.8. Synthesis of 1-Chloro-γ-dihydroionone (22)
To a solution of α-dihydroionone (
12) (455 mg, 2.32 mmol) in hexane (1.5 mL), NaClO (aqueous solution 15%) (3 mL, 12.9 mmol) was added. The mixture was cooled at 0 °C and H
3PO
4 (aqueous solution 40%) (0.4 mL, 1.72 mmol) was added. After stirring for 2 h at 0 °C, water (15 mL) was added, and the mixture extracted with Et
2O (15 mL × 3). The combined organic layer was washed with brine and dried over anhydrous MgSO
4. The solvent was removed in a vacuum to give 1-chloro-γ-dihydroionone (
22) as a mixture of
cis (
R*S*):
trans (
R*R*) isomers (10:0.8 ratio); colourless oil (0.51 g, 96%). Spectroscopic data are in agreement with literature values [
32].
IR (ATR) v (cm−1): 2950, 2869, 1714, 1649, 1454, 1419, 1388, 1364, 1234, 1158, 989, 909, 886, 760, 705, 602, 537.
1H NMR (300 MHz, CDCl3) δ (ppm) (R*S*) isomer signals: 5.34 (1H, s, H13a), 4.78 (1H, s, H13b), 4.48 (1H, dd, J = 4.8, 6.6 Hz, H1), 0.94 (3H, s), 0.86 (3H, s); (R*R*) isomer signals: 5.22 (1H, s, H13a), 4.72 (1H, s, H13b), 4.62 (1H, t, J = 5.0 Hz, H1), 1.01 (3H, s), 0.79 (3H, s); both isomer signals: 2.64–2.53 (1H, m), 2.38–2.27 (1H, m), 2.16–2.06 (1H, m), 2.12 (3H, s, H10), 1.92–1.69 (5H, m), 1.30 (1H, ddd, J = 4.3, 8.3, 13.0 Hz).
13C NMR (75 MHz, CDCl3) δ (ppm): 209.3 (C, C9), 145.8 (C, C6), 111.2 (CH2, C13), 63.0 (CH, C1), 52.2 (CH, C5), 42.2 (CH2), 35.2 (CH2), 35.0 (C, C1), 33.9 (CH2), 30.1 (CH3), 28.5 (CH3), 21.3 (CH2).
3.9. Zinc Cyclization of 1-Chloro-γ-dihydroionone (22)
Zn (161 mg, 2.4 mmol) and HCOONa (169 mg, 2.4 mmol) were added to a solution of (22) (138 mg, 0.6 mmol) in THF (1 mL) and EtOH (3 mL). The mixture was heated at 80 °C and stirred overnight. The reaction was cooled, diluted with Et2O (10 mL) and filtered through celite. The celite was then washed with H2O (10 mL), the two phases were separated, and the aqueous layer was extracted with Et2O (10 mL × 3). The combined organic layer was washed with saturated NaHCO3 (30 mL) and brine (30 mL), dried over anhydrous MgSO4 and the solvent was removed in a vacuum. The residue was purified by flash chromatography (hexane/Et2O 9:1) afforded compound (26) (30 mg, 26%) as a colorless oil.
IR (ATR) v (cm−1): 3473, 3065, 2969, 2932, 2867, 1717, 1653, 1479, 1452, 1384, 1364, 1246, 1211, 1168, 1106, 983, 916, 882, 693, 541. HREIMS (m/z) calcd. for C13H22O 194.1671 [M]+, found 194.1675.
1H NMR (300 MHz, CDCl3) δ (ppm): 4.84 (2H, m), 2.12 (1H, m), 2.00–1.93 (1H, m), 1.89 (2H, m), 1.81–1.75 (2H, m), 1.73–1.64 (2H, m), 1.54 (1H, dd, J = 14.0, 7.1 Hz), 1.25 (3H, s), 1.21–1.19 (1H, m), 1.00 (3H, s), 0.93 (3H, s).
13C NMR (75 MHz, CDCl3) δ (ppm): 152.1 (C), 108.5 (CH2), 73.8 (C), 51.2 (CH), 49.3 (CH), 35.3 (C), 35.3 (CH2), 33.9 (CH2), 29.0 (CH3), 28.9 (CH3), 26.8 (CH3), 26.0 (CH2), 25.1 (CH2).
3.10. CpTiCl3-Catalyzed Cyclization of 1-Chloro-γ-dihydroionone (22)
Deoxygenated, dry THF (3 mL) was added to a mixture of CpTiCl3 (11 mg, 0.052 mmol) and Mn dust (57 mg, 1.04 mmol) in an Ar atmosphere resulting in a green suspension. Me3SiBr (0.22 mL, 0.52 mmol) was then added, and the mixture turned turquoise. Subsequently, a solution of (22) (119 mg, 0.52 mmol) in THF (1 mL) was added dropwise and the mixture was stirred for 4 h. The reaction was filtered, diluted in Et2O, washed with HCl 3% and brine, dried (anhydrous MgSO4), and the solvent was removed. The residue was purified by flash chromatography (hexane/EtOAc 8:2) afforded cis-α-ambrinol (6) (42 mg, 42%) and trans-α-ambrinol (24) (18 mg, 18%).
3.11. CpTiCl3-Induced Cyclization of 1-Chloro-γ-dihydroionone (22)
Deoxygenated, dry THF (3 mL) was added to a mixture of CpTiCl3 (105 mg, 0.48 mmol) and Mn dust (53 mg, 0.96 mmol) in an Ar atmosphere resulting in a green suspension. Subsequently, a solution of (22) (110 mg, 0.48 mmol) in THF (1 mL) was added dropwise and the mixture was stirred for 5 h. The reaction was quenched and purified as indicated for the catalytic version to give cis-α-ambrinol (6) (45 mg, 48%) and trans-α-ambrinol (24) (18 mg, 19%).
3.12. Cp2TiCl2 Cyclization of 1-Chloro-γ-dihydroionone (22)
For the catalytic cyclization, deoxygenated and dry THF (3 mL) was added to a mixture of Cp2TiCl2 (25 mg, 0.093 mmol) and Mn dust (160 mg, 2.85 mmol) in an Ar atmosphere and the red suspension was stirred at room temperature until it turned lime green (after about 15 min). Then, a mixture of 2,4,6-collidine (0.36 mL, 2.69 mmol) and Me3SiCl (0.2 mL, 1.54 mmol) in THF (1 mL) was added. Subsequently, a solution of (22) (110 mg, 0.48 mmol) in THF (1 mL) was added dropwise and the mixture was stirred for 1 h 45 min. The reaction was filtered, diluted in Et2O, washed with HCl 3% and brine, dried (anhydrous MgSO4), and the solvent was removed. The residue was purified by flash chromatography (hexane/EtOAc 8:2) affording cis-α-ambrinol (6) (31 mg, 33%) and trans-α-ambrinol (24) (16 mg, 17%).
With stoichiometric amounts of Cp2TiCl2, the reaction was carried out under the same conditions as above, employing Cp2TiCl2 (247 mg, 0.93 mmol), Mn dust (162 mg, 2.85 mmol) and (22) (100 mg, 0.44 mmol). The mixture was stirred for 2 h, quenched and purified as indicated above, and cis-α-ambrinol (6) (32 mg, 37%) and trans-α-ambrinol (24) (15 mg, 18%) were obtained.

 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}