
New Nostocyclophanes from Nostoc linckia
 ,
,
Abstract
:1. Introduction
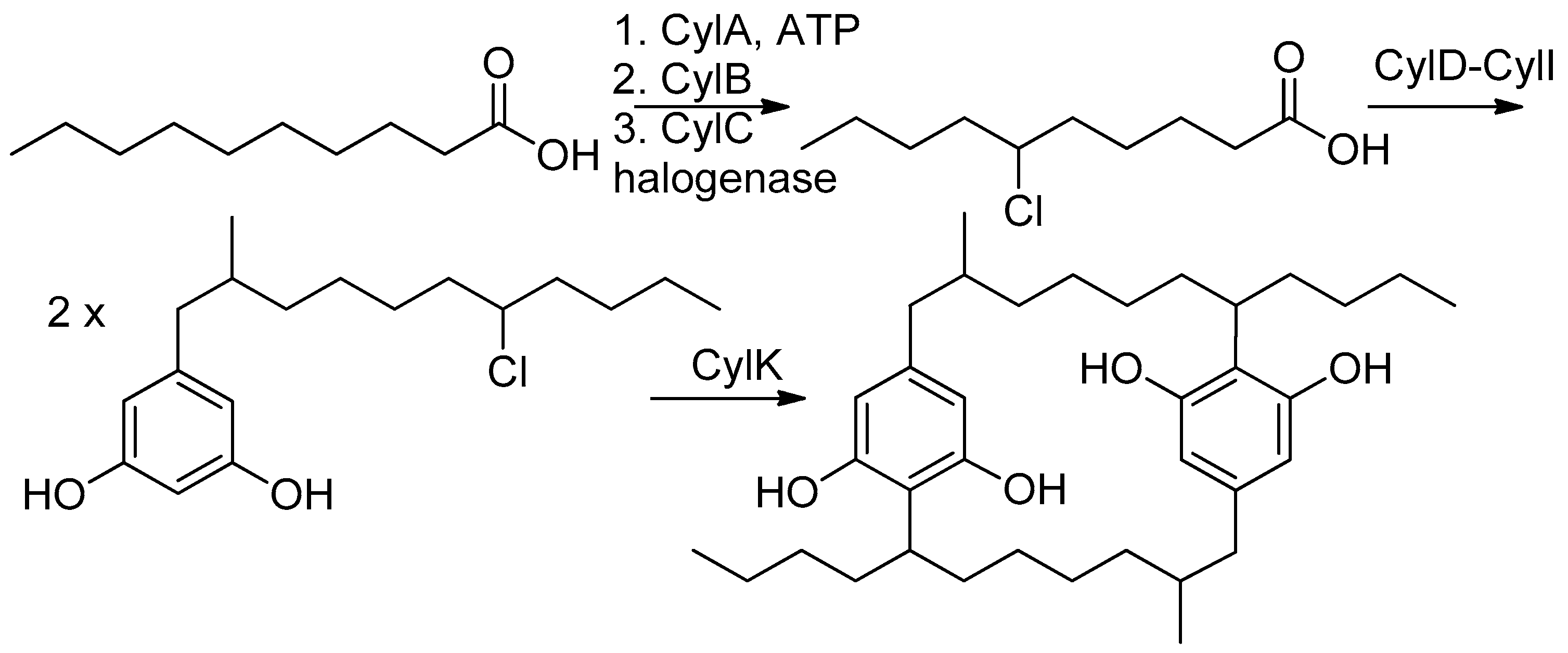
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Chemicals and Reagents
3.3. Biological Material
3.4. Extraction and Isolation of Metabolites
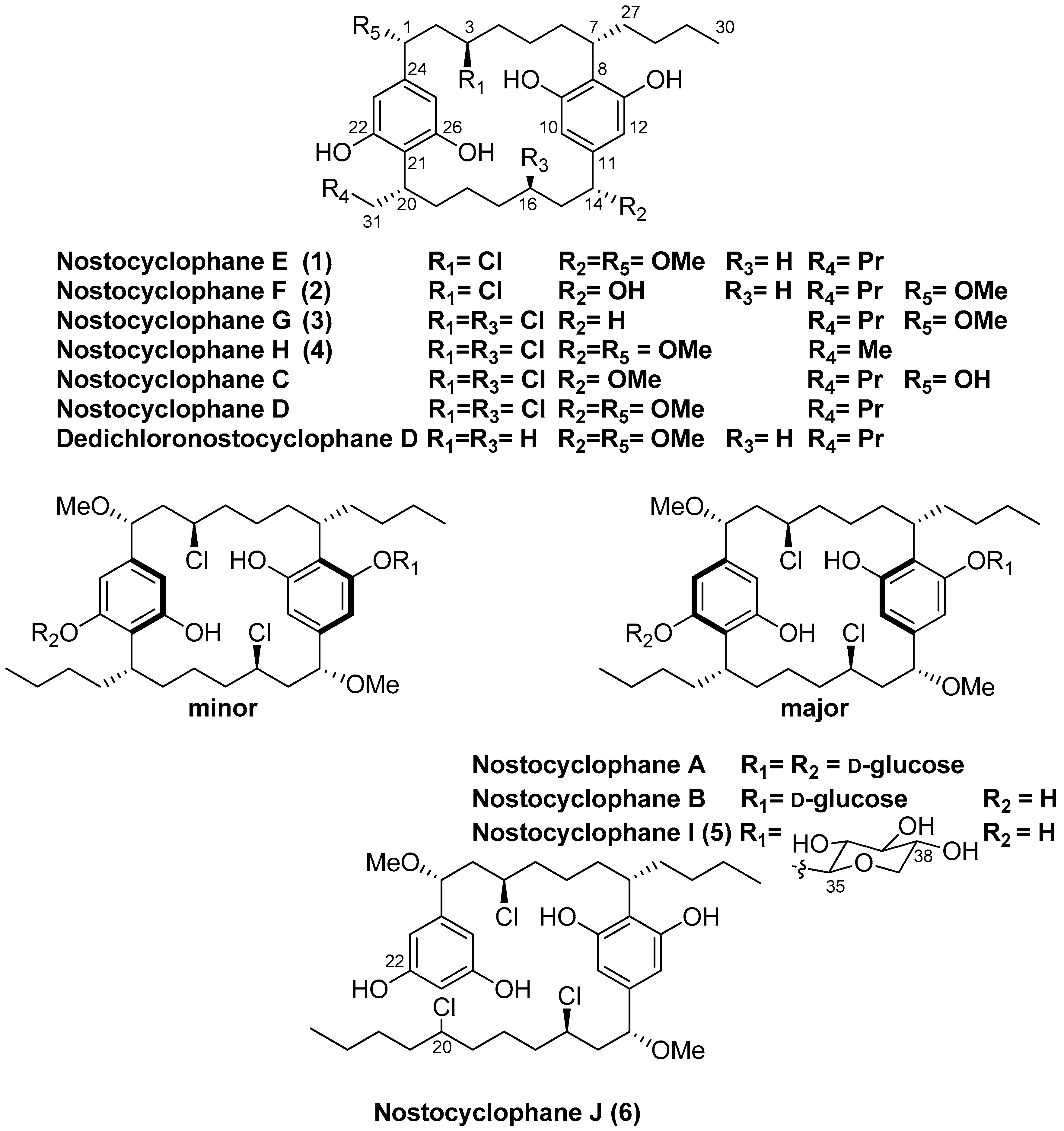
- Nostocyclophane E (1): amorphous white power; [α]D22 +5.7 (c 2.0, MeOH); UV (MeOH) λmax (log ε) 209 (2.5), 214 (2.5), 227 (1.3), 277 (0.2), 283 (0.2) nm; ECD (c 0.2 MeOH) λmax (Δ ε) 206 (0.80), 218 (−0.71), 240 (−0.20), 261 (−0.80), 284 (−0.090) nm; IR (film) νmax 3325, 2926, 2854, 1646, 1595, 1431, 1083, 1021 cm−1; see Table 1 and Table 2 for tabulated NMR spectroscopic data; HRESIMS m/z 617.3599 [M − H]− (calcd for C36H5435ClO6, 617.3609; Δ = −1.6 ppm).
- Nostocyclophane F (2): amorphous white power; [α]D22 −6.5 (c 0.2, MeOH); UV (MeOH) λmax (log ε) 210 (2.5), 214 (2.5), 227 (1.4), 277 (0.3), 283 (0.2) nm; ECD (c 0.2 MeOH) λmax (Δ ε) 205 (1.10), 217 (−1.80), 235 (−0.67), 272 (−0.26), 280 (−0.25) nm; IR (film) νmax 3345, 2958, 2864, 1650, 1431, 1020 cm−1; see Table 1 and Table 2 for tabulated NMR spectroscopic data; HRESIMS m/z 603.3454 [M − H]− (calcd for C35H5235ClO6−, 603.3453; Δ = 0.3 ppm).
- Nostocyclophane G (3): amorphous white power; [α]D22 −25.6 (c 2.0, MeOH); UV (MeOH) λmax (log ε) 208 (0.9), 229 (0.3), 274 (0.1) nm; ECD (c 0.2 MeOH) λmax (Δ ε) 202 (0.9), 217 (−0.20), 237 (−0.89), 285 (−0.02) nm; IR (film) νmax 3390, 2932, 2859, 1588, 1429, 1085 cm−1; see Table 1 and Table 2 for tabulated NMR spectroscopic data; HRESIMS m/z 621.3102 [M − H]− (calcd for C35H5135Cl2O5−, 621.3114; Δ = −1.9 ppm).
- Nostocyclophane H (4): amorphous white power; [α]D22 −0.2 (c 2.0, MeOH); UV (MeOH) λmax (log ε) 209 (2.4), 227 (0.9), 274 (0.2), 283 (0.2) nm; ECD (c 0.2 MeOH) λmax (Δ ε) 207 (0.22), 218 (−0.25), 233 (−0.09), 273 (−0.35), 282 (−0.37) nm; IR (film) νmax 3399, 2932, 2873, 1589, 1428, 1089 cm−1; see Table 1 and Table 2 for tabulated NMR spectroscopic data; HRESIMS m/z 623.2901 [M − H]− (calcd for C34H4935Cl2O6−, 623.2906; Δ = −0.8 ppm).
- Nostocyclophane I (5): amorphous white power; [α]D22 +2.0 (c 1.0, MeOH); UV (MeOH) λmax (log ε) 209 (2.0), 227 (0.6), 275 (0.1), 283 (0.1) nm; ECD (c 0.2 MeOH) λmax (Δ ε) 201 (4.9), 215 (−5.0), 233 (−1.45), 271 (−0.35), 283 (−0.34) nm; IR (film) νmax 3341, 2927, 2863, 1614, 1595, 1429, 1093 cm−1; see Table 3 for tabulated NMR spectroscopic data; HRESIMS m/z 783.3642 [M − H]− (calcd for C41H6135Cl2O10−, 783.3642; Δ = −2.1 ppm).
- Nostocyclophane J (6): amorphous white power; [α]D22 +5.0 (c 1.0, MeOH); UV (MeOH) λmax (log ε) 214 (2.6), 228 (1.7), 278 (0.4), 282 (0.4) nm; ECD (c 0.2 MeOH) λmax (Δ ε) 207 (2.6), 217 (−2.2), 228 (−0.8), 273 (−0.44), 293 (−0.37) nm. IR (film) νmax 3374, 2950, 2864, 1604, 1081 cm−1; see Table 5 for tabulated NMR spectroscopic data; HRESIMS m/z 687.2973 [M − H]− (calcd for C36H5435Cl3O6−, 687.2986; Δ = −1.9 ppm).
3.5. Growth Inhibition Assays
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Salvador-Reyes, L.A.; Luesch, H. Biological targets and mechanisms of action of natural products from marine cyanobacteria. Nat. Prod. Rep. 2015, 32, 478–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prinsep, M.R.; Caplan, F.R.; Moore, R.E.; Patterson, G.M.L.; Smith, C.D. Tolyporphin, A Novel Multidrug Resistance Reversing Agent from the Blue-green Alga Tolypothrix nodosa. J. Am. Chem. Soc. 1992, 114, 385–387. [Google Scholar] [CrossRef]
- Moore, B.S.; Chen, J.L.; Patterson, G.M.L.; Moore, R.E.; Brinen, L.S.; Kato, Y.; Clardy, J. [7.7]Paracyclophanes from blue-green algae. J. Am. Chem. Soc. 1990, 112, 4061–4063. [Google Scholar] [CrossRef]
- Chen, J.L.; Moore, R.E.; Patterson, G.M.L. Structures of Nostocyclophanes A–D. J. Org. Chem. 1991, 56, 4360–4364. [Google Scholar] [CrossRef]
- May, D.S.; Chen, W.-L.; Lantvit, D.D.; Zhang, X.; Krunic, A.; Burdette, J.E.; Eustaquio, A.; Orjala, J. Merocyclophanes C and D from the Cultured Freshwater Cyanobacterium Nostoc sp. (UIC 10110). J. Nat. Prod. 2017, 80, 1073–1080. [Google Scholar] [CrossRef] [Green Version]
- Preisitsch, M.; Heiden, S.E.; Beerbaum, M.; Niedermeyer, T.H.J.; Schneefeld, M.; Herrmann, J.; Kumpfmüller, J.; Thürmer, A.; Neidhardt, I.; Wiesner, C.; et al. Effects of Halide Ions on the Carbamidocyclophane Biosynthesis in Nostoc sp. CAVN2. Mar. Drugs 2016, 14, 21. [Google Scholar] [CrossRef]
- Preisitsch, M.; Niedermeyer, T.H.J.; Heiden, S.E.; Neidhardt, I.; Kumpfmüller, J.; Wurster, M.; Harmrolfs, K.; Wiesner, C.; Enke, H.; Müller, R.; et al. Cylindrofridins A–C, Linear Cylindrocyclophane-Related Alkylresorcinols from the Cyanobacterium Cylindrospermum stagnale. J. Nat. Prod. 2016, 79, 106–115. [Google Scholar] [CrossRef]
- Bobzin, S.C.; Moore, R.E. Biosynthetic Origin of [7.7]paracyclophanes from Cyanobacteria. Tetrahedron 1993, 49, 7615–7626. [Google Scholar] [CrossRef]
- Nakamura, H.; Hamer, H.A.; Sirasani, G.; Balskus, E.P. Cylindrocyclophane Biosynthesis Involves Functionalization of an Unactivated Carbon Center. J. Am. Chem. Soc. 2012, 134, 18518–18521. [Google Scholar] [CrossRef] [Green Version]
- Moore, B.S.; Chen, J.L.; Patterson, G.M.L.; Moore, R.E. Structures of Cylindrocyclophanes A-F. Tetrahedron 1992, 48, 3001–3006. [Google Scholar] [CrossRef]
- Schultz, E.E.; Braffman, N.R.; Luescher, M.U.; Hager, H.H.; Balskus, E.P. Biocatalytic Friedel–Crafts Alkylation Using a Promiscuous Biosynthetic Enzyme. Angew. Chem. Int. Ed. 2019, 58, 3151–3155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braffman, N.R.; Ruskoski, T.B.; Davis, K.M.; Glasser, N.R.; Johnson, C.; Okafor, C.D.; Boal, A.K.; Balskus, E.P. Structural Basis for an Unprecedented Enzymatic Alkylation in Cylindrocyclophane Biosynthesis. eLife 2022, 11, e75761. [Google Scholar] [CrossRef]
- Wang, H.-Q.; Mou, S.-B.; Xiao, W.; Zhou, H.; Hou, X.-D.; Wang, S.-J.; Wang, Q.; Gao, J.; Wei, Z.; Liu, L.; et al. Structural Basis for the Friedel–Crafts Alkylation in Cylindrocyclophane Biosynthesis. ACS Catal. 2022, 12, 2108–2117. [Google Scholar] [CrossRef]
- Thanh, N.V.; Thao, N.P.; Phong, N.V.; Cuong, N.X.; Nam, N.H.; Minh, C.V. A New [7.7]paracyclophane from Vietnamese Marine Snail Planaxis sulcatus (Born, 1780). Nat. Prod. Res. 2020, 34, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Van Santen, J.A.; Poynton, E.F.; Iskakova, D.; McMann, E.; Alsup, T.A.; Clark, T.N.; Fergusson, C.H.; Fewer, D.P.; Hughes, A.H.; McCadden, C.A.; et al. The Natural Products Atlas 2.0: A Database of Microbially-derived Natural Products. Nucleic Acids Res. 2021, 50, D1317–D1323. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Schultz, E.E.; Balskus, E.P. A New Strategy for Aromatic Ring Alkylation in Cylindrocyclophane Biosynthesis. Nat. Chem. Biol. 2017, 13, 916–921. [Google Scholar] [CrossRef]
- Sekine, M.; Kimura, T.; Katayama, Y.; Takahashi, D.; Toshima, K. The direct and one-pot transformation of xylan into the biodegradable surfactants, alkyl xylosides, is aided by an ionic liquid. RSC Adv. 2013, 3, 19756–19759. [Google Scholar] [CrossRef]
- Lee, J.; Kobayashi, Y.; Tezuka, K.; Kishi, Y. Toward Creation of a Universal NMR Database for the Stereochemical Assignment of Acyclic Compounds: Proof of Concept. Org. Lett. 1999, 1, 2181–2184. [Google Scholar] [CrossRef] [PubMed]
- Luesch, H.; Moore, R.E.; Paul, V.J.; Mooberry, S.L.; Corbett, T.H. Isolation of Dolastatin 10 From The Marine Cyanobacterium Symploca Species VP642 And Total Stereochemistry And Biological Evaluation Of Its Analogue Symplostatin 1. J. Nat. Prod. 2001, 64, 907–910. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Kamano, Y.; Herald, C.L.; Tuinman, A.A.; Boettner, F.E.; Kizu, H.; Schmidt, J.M.; Baczynskyj, L.; Tomer, K.B.; Bontems, R.J. The Isolation and Structure Of A Remarkable Marine Animal Antineoplastic Constituent: Dolastatin 10. J. Am. Chem. Soc. 1987, 109, 6883–6885. [Google Scholar] [CrossRef]
- Moore, R.E.; Cheuk, C.; Yang, X.Q.G.; Patterson, G.M.L.; Bonjouklian, R.; Smitka, T.A.; Mynderse, J.S.; Foster, R.S.; Jones, N.D. Hapalindoles, Antibacterial and Antimycotic Alkaloids from the Cyanophyte Hapalosiphon fontinalis. J. Org. Chem. 1987, 52, 1036–1043. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Position | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| δH (J in Hz) | δH (J in Hz) | δH (J in Hz) | δH (J in Hz) | |
| 1 | 4.04, dd (10.7, 3.9) | 4.03, dd (10.5, 3.9) | 4.04, dd (10.7, 4.1) | 4.03, dd (10.8, 3.9) |
| 2 | 2.07, ddd (11.0, 10.7, 3.9) | 2.07, ddd (11.1, 10.1, 3.8) | 2.08, ddd (11.2, 10.2, 3.3) | 2.07, ddd (13.2, 10.8, 3.9) |
| 1.82, m | 1.83, m | 1.82, m | 1.83, m | |
| 3 | 2.82, t (10.7) | 2.82, t (10.1) | 2.83, t (10.2) | 2.80, t (10.8) |
| 4 | 1.59, m | 1.59, m | 1.59, m | 1.61, m |
| 1.46, m | 1.45, m | 1.46, m | 1.46, m | |
| 5 | 1.36, m | 1.36, m | 1.36, m | 1.35, m |
| 0.52, m | 0.53, m | 0.58, m | 0.54, m | |
| 6 | 1.82, m | 1.82, m | 1.82, m | 1.83, m |
| 1.52, m | 1.50, m | 1.55, m | 1.52, m | |
| 7 | 3.07, m | 3.07, m | 3.09, m | 3.07, m |
| 10 | 6.09, s | 6.09, s | 6.06, s | 6.10, s |
| 12 | 6.13, s | 6.19, s | 6.15, s | 6.15, s |
| 14 | 3.69, dd (10.3, 4.4) | 4.05, dd (9.7, 4.0) | 2.40, m | 4.03, dd (10.8, 3.9) |
| 15 | 1.66, m | 1.60, m | 1.60, m | 2.07, ddd (13.2, 10.8, 3.9) |
| 1.30, m | 1.30, m | 1.83, m | ||
| 16 | 1.11, m | 1.11, m | 2.54, m | 2.80, t (10.8) |
| 1.00, m | 1.00, m | |||
| 17 | 0.98, m | 0.98, m | 1.59, m | 1.61, m |
| 0.51, m | 0.52, m | 1.46, m | 1.46, m | |
| 18 | 0.88, m | 0.89, m | 1.36, m | 1.35, m |
| 0.32, m | 0.32, m | 0.58, m | 0.54, m | |
| 19 | 1.82, m | 1.82, m | 1.82, m | 1.83, m |
| 1.52, m | 1.52, m | 1.55, m | 1.52, m | |
| 20 | 3.07, m | 3.07, m | 3.09, m | 3.07, m |
| 23 | 6.08, s | 6.13, s | 6.10, s | 6.10, s |
| 25 | 5.99, s | 5.95, s | 5.93, s | 6.15, s |
| 27 | 1.80, m | 1.80, m | 1.82, m | 1.80, m |
| 1.38, m | 1.30, m | 1.38, m | 1.35, m | |
| 28 | 1.18, m | 1.18, m | 1.18, m | 1.18, m |
| 29 | 1.25, m | 1.25, m | 1.25, m | 1.23, m |
| 30 | 0.77, t (7.2) | 0.76, t (7.2) | 0.77, t (7.2) | 0.76, t (7.2) |
| 31 | 1.80, m | 1.80, m | 1.82, m | 1.80, m |
| 1.38, m | 1.30, m | 1.38, m | 1.54, m | |
| 32 | 1.18, m | 1.18, m | 1.18, m | 0.69, t (7.4) |
| 33 | 1.25, m | 1.25, m | 1.25, m | |
| 34 | 0.77, t (7.2) | 0.76, t (7.2) | 0.77, t (7.2) | |
| 1-OMe | 3.03, s | 3.03, s | 3.03, s | 3.07, s |
| 14-OMe | 3.01, s | 3.07, s |
| Position | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| δC, Mult. | δC, Mult. | δC, Mult. | δC, Mult. | |
| 1 | 81.0, CH | 81.0, CH | 81.0, CH | 80.9, CH |
| 2 | 45.5, CH2 | 45.5, CH2 | 45.5, CH2 | 45.5, CH2 |
| 3 | 62.9, CH | 62.9, CH | 62.9, CH | 62.8, CH |
| 4 | 40.3, CH2 | 40.0, CH2 | 40.0, CH2 | 40.2, CH2 |
| 5 | 26.8, CH2 | 26.6, CH2 | 26.7, CH2 | 26.7, CH2 |
| 6 | 33.3, CH2 | 33.4, CH2 | 33.2, CH2 | 32.7, CH2 |
| 7 | 34.8, CH | 34.9, CH | 34.7, CH | 34.9, CH |
| 8 | 115.5, qC | 114.7, qC | 116.2, qC | 115.8, qC |
| 9 | 155.6, qC | 155.4, qC | 155.7, qC | 155.7, qC |
| 10 | 102.8, CH | 103.5, CH | 102.8, CH | 102.8, CH |
| 11 | 139.6, qC | 143.6, qC | 137.7, qC | 138.1, qC |
| 12 | 107.5, CH | 107.3, CH | 107.3, CH | 107.3, CH |
| 13 | 157.6, qC | 157.1, qC | 157.7, qC | 157.5, qC |
| 14 | 83.6, CH | 73.5, CH | 31.2, CH2 | 80.9, CH |
| 15 | 37.0, CH2 | 39.0, CH2 | 40.0, CH2 | 45.5, CH2 |
| 16 | 30.1, CH2 | 30.2, CH2 | 64.2, CH | 62.8, CH |
| 17 | 30.2, CH2 | 30.7, CH2 | 40.0, CH2 | 40.2, CH2 |
| 18 | 26.6, CH2 | 27.0, CH2 | 27.0, CH2 | 26.7, CH2 |
| 19 | 32.7, CH2 | 32.8, CH2 | 33.2, CH2 | 32.5, CH2 |
| 20 | 34.9, CH | 34.9, CH | 34.8, CH | 37.0, CH |
| 21 | 116.3, qC | 116.4, qC | 116.2, qC | 115.8, qC |
| 22 | 155.5, qC | 155.7, qC | 155.7, qC | 155.7, qC |
| 23 | 102.8, CH | 102.8, CH | 102.8, CH | 102.8, CH |
| 24 | 137.9, qC | 138.0, qC | 137.7, qC | 138.1, qC |
| 25 | 107.2, CH | 106.1, CH | 107.3, CH | 107.3, CH |
| 26 | 157.3, qC | 157.6, qC | 157.7, qC | 157.5, qC |
| 27 | 32.7, CH2 | 33.0, CH2 | 32.8, CH2 | 32.8, CH2 |
| 28 | 29.5, CH2 | 29.6, CH2 | 30.2, CH2 | 30.2, CH2 |
| 29 | 22.3, CH2 | 22.2, CH2 | 22.2, CH2 | 22.2, CH2 |
| 30 | 14.1, CH3 | 14.1, CH3 | 14.1, CH3 | 14.0, CH3 |
| 31 | 32.9, CH2 | 33.3, CH2 | 32.8, CH2 | 25.8, CH2 |
| 32 | 30.5, CH2 | 30.2, CH2 | 30.2, CH2 | 13.0, CH3 |
| 33 | 22.2, CH2 | 22.3, CH2 | 22.2, CH2 | |
| 34 | 14.1, CH3 | 14.1, CH3 | 14.1, CH3 | |
| 1-OMe | 55.7, CH3 | 55.7, CH3 | 55.8, CH3 | 55.7, CH3 |
| 14-OMe | 55.5, CH3 |
| Position | 5, Major | 5, Major | 5, Minor | 5, Minor |
|---|---|---|---|---|
| δH (J in Hz) | δc, Mult. | δH (J in Hz) | δc, Mult. | |
| 1 | 4.05, dd (10.7, 3.4) | 81.0, CH | 4.04, dd (10.7, 3.9) | 80.9, CH |
| 2 | 2.08, m | 45.6, CH2 | 2.06, m | 45.5, CH2 |
| 1.68, m | 1.80, m | |||
| 3 | 2.79, m | 62.7, CH | 2.82, m | 62.6, CH |
| 4 | 1.58, m | 39.6, CH2 | 1.47, m | 39.6, CH2 |
| 1.42, m | 1.36, m | |||
| 5 | 1.46, m | 26.7, CH2 | 1.34, m | 27.0, CH2 |
| 0.53, m | 0.45, m | |||
| 6 | 1.84, m | 32.4, CH2 | 2.12, m | 32.4, CH2 |
| 1.55, m | 1.58, m | |||
| 7 | 3.10, m | 35.1, CH | 3.08, m | 34.9, CH |
| 8 | 119.7, qC | 118.2, qC | ||
| 9 | 157.8, qC | 157.5, qC | ||
| 10 | 6.32, s | 100.8, CH | 6.42, s | 102.6, CH |
| 11 | 138.4, qC | 138.5, qC | ||
| 12 | 6.31, s | 107.4, CH | 6.30, s | 109.3, CH |
| 13 | 155.8, qC | 156.2, qC | ||
| 14 | 4.10, dd (10.7, 3.8) | 81.2, CH | 4.15, dd (10.7, 3.9) | 81.0, CH |
| 15 | 2.08, m | 45.9, CH2 | 2.08, m | 45.6, CH2 |
| 1.85, m | 1.80, m | |||
| 16 | 2.91, m | 63.1, CH | 2.79, m | 62.9, CH |
| 17 | 1.57, m | 40.2, CH2 | 1.47, m | 40.2, CH2 |
| 1.42, m | 1.36, m | |||
| 18 | 1.23, m | 26.2, CH2 | 1.27, m | 26.8, CH2 |
| 0.51, m | 0.54, m | |||
| 19 | 1.84, m | 33.1, CH2 | 1.85, m | 33.0, CH2 |
| 1.55, m | 1.50, m | |||
| 20 | 3.11, m | 35.0, CH | 3.11, m | 34.8, CH |
| 21 | 116.2, qC | 116.2, qC | ||
| 22 | 155.6, qC | 155.2, qC | ||
| 23 | 6.10, s | 102.7, CH | 6.09, s | 102.9, CH |
| 24 | 138.2, qC | 138.3, qC | ||
| 25 | 6.15, s | 107.0, CH | 6.14, s | 107.5, CH |
| 26 | 157.6, qC | 157.3, qC | ||
| 27 | 2.09, m | 32.7, CH2 | 1.72, m | 32.8, CH2 |
| 1.50, m | 1.35, m | |||
| 28 | 1.18, m | 30.2, CH2 | 1.18, m | 30.0, CH2 |
| 1.02, m | 1.02, m | |||
| 29 | 1.22, m | 22.3, CH2 | 1.22, m | 22.3, CH2 |
| 1.13, m | 1.13, m | |||
| 30 | 0.77, t (6.5) | 14.1, CH3 | 0.77, t (6.5) | 14.1, CH3 |
| 31 | 1.82, m | 32.5, CH2 | 1.72, m | 32.6, CH2 |
| 1.46, m | 1.35, m | |||
| 32 | 1.18, m | 30.2, CH2 | 1.18, m | 30.2, CH2 |
| 1.02, m | 1.02, m | |||
| 33 | 1.22, m | 22.3, CH2 | 1.22, m | 22.6, CH2 |
| 1.13, m | 1.12, m | |||
| 34 | 0.76, t (6.8) | 14.1, CH3 | 0.76, t (6.8) | 14.1, CH3 |
| 35 | 4.93, d (7.0) | 99.6, CH | 4.62, d (7.0) | 105.6, CH |
| 36 | 3.19, dd (8.8, 7.0) | 73.3, CH | 3.19, dd (8.8, 7.0) | 73.5, CH |
| 37 | 3.23, t (8.8) | 76.6, CH | 3.23, t (8.8) | 76.8, CH |
| 38 | 3.29, ddd (10.9, 8.8, 5.0) | 69.3, CH | 3.29, ddd (10.9, 8.8, 5.0) | 69.6, CH |
| 39 | 3.67, dd (10.9, 5.0) | 65.4, CH2 | 3.58, dd (10.9, 5.0) | 65.7, CH2 |
| 3.12, t (10.9) | 3.12, t (10.9) | |||
| 1-OMe | 3.04, s | 55.9, CH3 | 3.04, s | 55.9, CH3 |
| 14-OMe | 3.04, s | 55.8, CH3 | 3.04, s | 55.8, CH3 |
| Compound | GI50 (μM) |
|---|---|
| 1 | 0.72 |
| 2 | 0.94 |
| 3 | 5.1 |
| 4 | 1.2 |
| 5 | 1.7 |
| 6 | 8.2 |
| Nostocyclophane C | 0.95 |
| Position | 6 (DMSO-d6) | 6 (CDCl3) | ||
|---|---|---|---|---|
| δH (J in Hz) | δC, Mult. | δH (J in Hz) | δC, Mult. | |
| 1 | 4.03, m | 80.5, CH | 4.22, dd (8.7, 5.5) | 81.2, CH |
| 2 | 2.00, m | 45.6, CH2 | 2.14, ddd (15.3, 10.5, 5.3) | 45.3, CH2 |
| 1.83, m | 1.87, ddd (15.3, 8.7, 3.6) | |||
| 3 | 3.65, m | 61.0, CH | 3.52, m | 59.89, CH |
| 4 | 1.68, m | 37.5, CH2 | 1.74, m | 38.0, CH2 |
| 5 | 1.30, m | 24.2, CH2 | 1.30, m | 24.2, CH2 |
| 1.11, m | 1.20, m | |||
| 6 | 1.85, m | 32.7, CH2 | 1.84, m | 33.3, CH2 |
| 1.55, m | 1.62, m | |||
| 7 | 3.10, m | 33.9, CH | 3.09, m | 35.4, CH |
| 8 | 116.0, qC | 117.5, qC | ||
| 9 | 155.5, qC | 155.6, qC | ||
| 10 | 6.18, s | 104.5, CH | 6.43, s | 106.4, CH |
| 11 | 138.5, qC | 139.1, qC | ||
| 12 | 6.09, s | 104.5, CH | 6.43, s | 106.4, CH |
| 13 | 155.5, qC | 155.6, qC | ||
| 14 | 4.05, m | 80.5, CH | 4.27, dd (8.7, 5.3) | 81.3, CH |
| 15 | 2.14, m | 45.7, CH2 | 2.28, ddd (14.6, 9.7, 5.5) | 44.8, CH2 |
| 1.80, m | 1.96, ddd (14.6, 8.7, 3.5) | |||
| 16 | 3.35, m | 61.1, CH | 3.59, m | 59.88, CH |
| 17 | 1.68, m | 37.1, CH2 | 1.68, m | 37.8, CH2 |
| 18 | 1.64, m | 23.0, CH2 | 1.72, m | 23.2, CH2 |
| 1.33, m | 1.46, m | |||
| 19 | 1.64, m | 36.9, CH2 | 1.60, m | 37.9, CH2 |
| 20 | 3.97, m | 64.5, CH | 3.86, m | 64.0, CH |
| 21 | 6.09, s | 102.0, CH | 6.30, s | 102.6, CH |
| 22 | 158.5, qC | 157.0, qC | ||
| 23 | 6.11, s | 104.5, CH | 6.43, s | 106.4, CH |
| 24 | 142.9, qC | 143.5, qC | ||
| 25 | 6.18, s | 104.5, CH | 6.43, s | 106.4, CH |
| 26 | 158.5, qC | 157.0, qC | ||
| 27 | 1.74, m | 31.8, CH2 | 1.68, m | 38.2, CH2 |
| 1.48, m | 1.61, m | |||
| 28 | 1.00, m | 30.3, CH2 | 1.28, m | 30.6, CH2 |
| 1.11, m | 1.17, m | |||
| 29 | 1.24, m | 22.3, CH2 | 1.23, m | 22.0, CH2 |
| 1.17, m | ||||
| 30 | 0.77, t (7.1) | 14.1, CH3 | 0.83, t (7.1) | 14.1, CH3 |
| 31 | 1.64, m | 37.0, CH2 | 1.80, m 1.55, m | 32.3, CH2 |
| 32 | 1.40, m | 28.0, CH2 | 1.30, m | 28.7, CH2 |
| 33 | 1.25, m | 21.7, CH2 | 1.35, m 1.30, m | 22.2, CH2 |
| 34 | 0.85, t (7.2) | 13.9, CH3 | 0.90, t (7.2) | 13.9, CH3 |
| 1-OMe | 3.04, s | 55.8, CH3 | 3.20, s | 56.5, CH3 |
| 14-OMe | 3.05, s | 55.8, CH3 | 3.26, s | 56.6, CH3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, J.; Philbin, C.S.; Wakano, C.; Yoshida, W.Y.; Williams, P.G. New Nostocyclophanes from Nostoc linckia. Mar. Drugs 2023, 21, 101. https://doi.org/10.3390/md21020101
Dai J, Philbin CS, Wakano C, Yoshida WY, Williams PG. New Nostocyclophanes from Nostoc linckia. Marine Drugs. 2023; 21(2):101. https://doi.org/10.3390/md21020101
Chicago/Turabian StyleDai, Jingqiu, Casey S. Philbin, Clay Wakano, Wesley Y. Yoshida, and Philip G. Williams. 2023. "New Nostocyclophanes from Nostoc linckia" Marine Drugs 21, no. 2: 101. https://doi.org/10.3390/md21020101