
Marine Sponge and Octocoral-Associated Bacteria Show Versatile Secondary Metabolite Biosynthesis Potential and Antimicrobial Activities against Human Pathogens
 , , , , , , , , and
, , , , , , , , and 
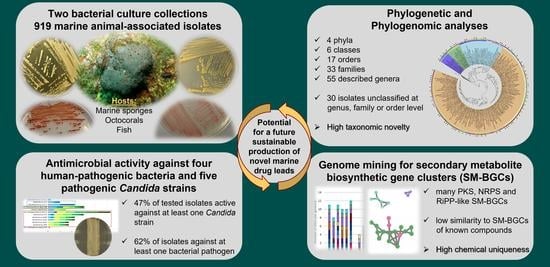
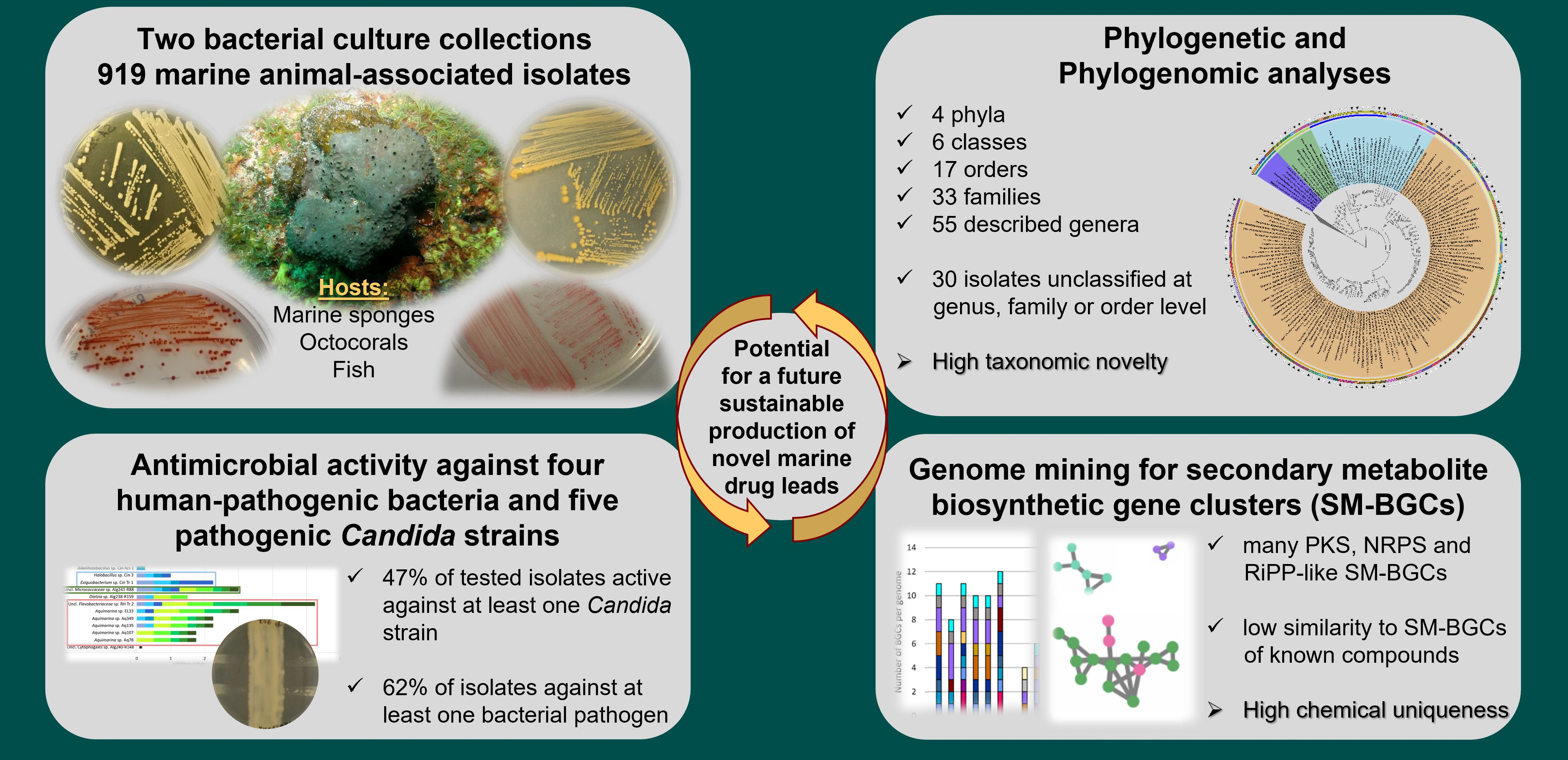
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
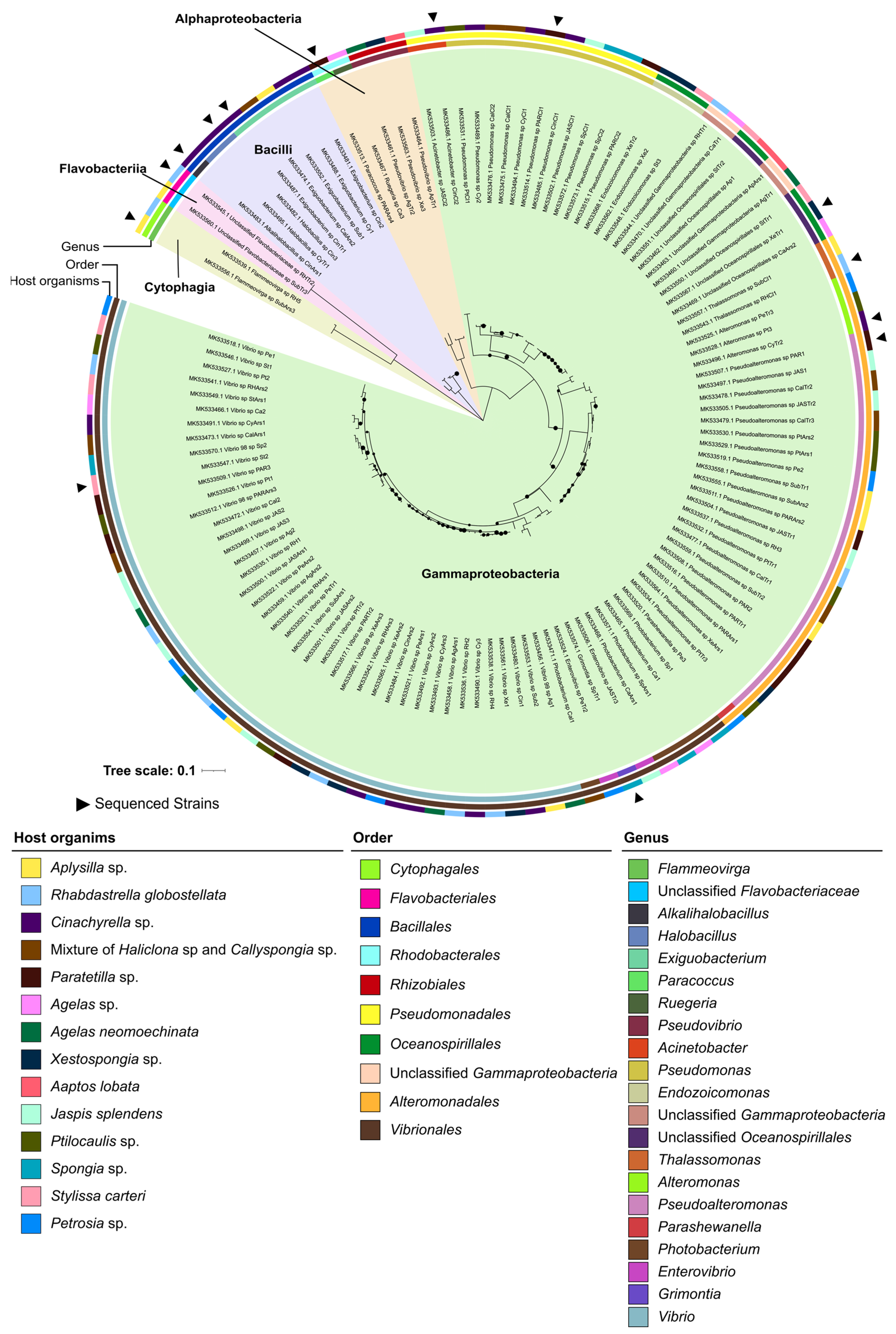
2.1. Taxonomic and Phylogenetic Diversity of the Two Marine Culture Collections


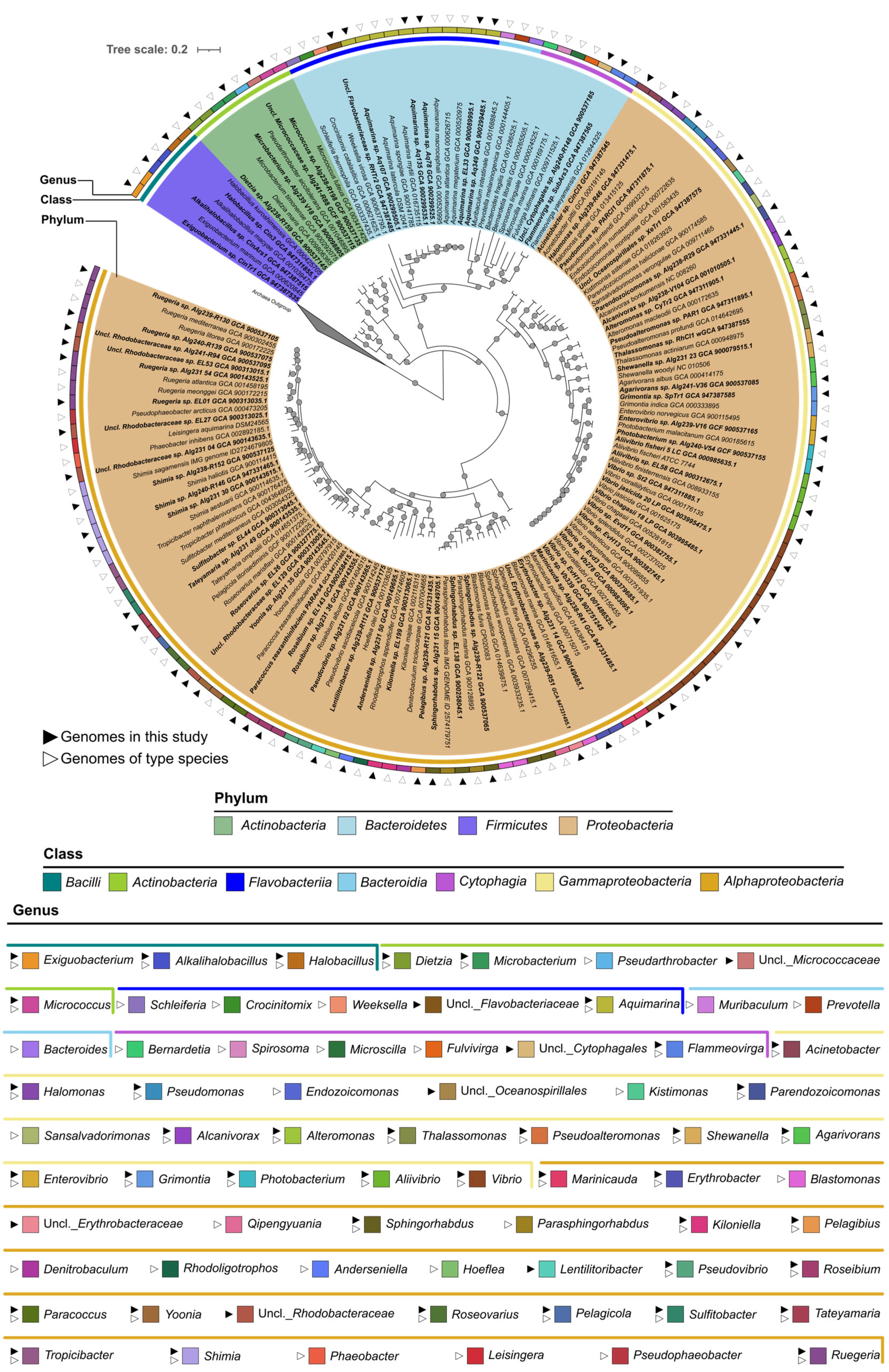
2.2. Phylogenomic Inference of Marine Host-Associated Bacteria

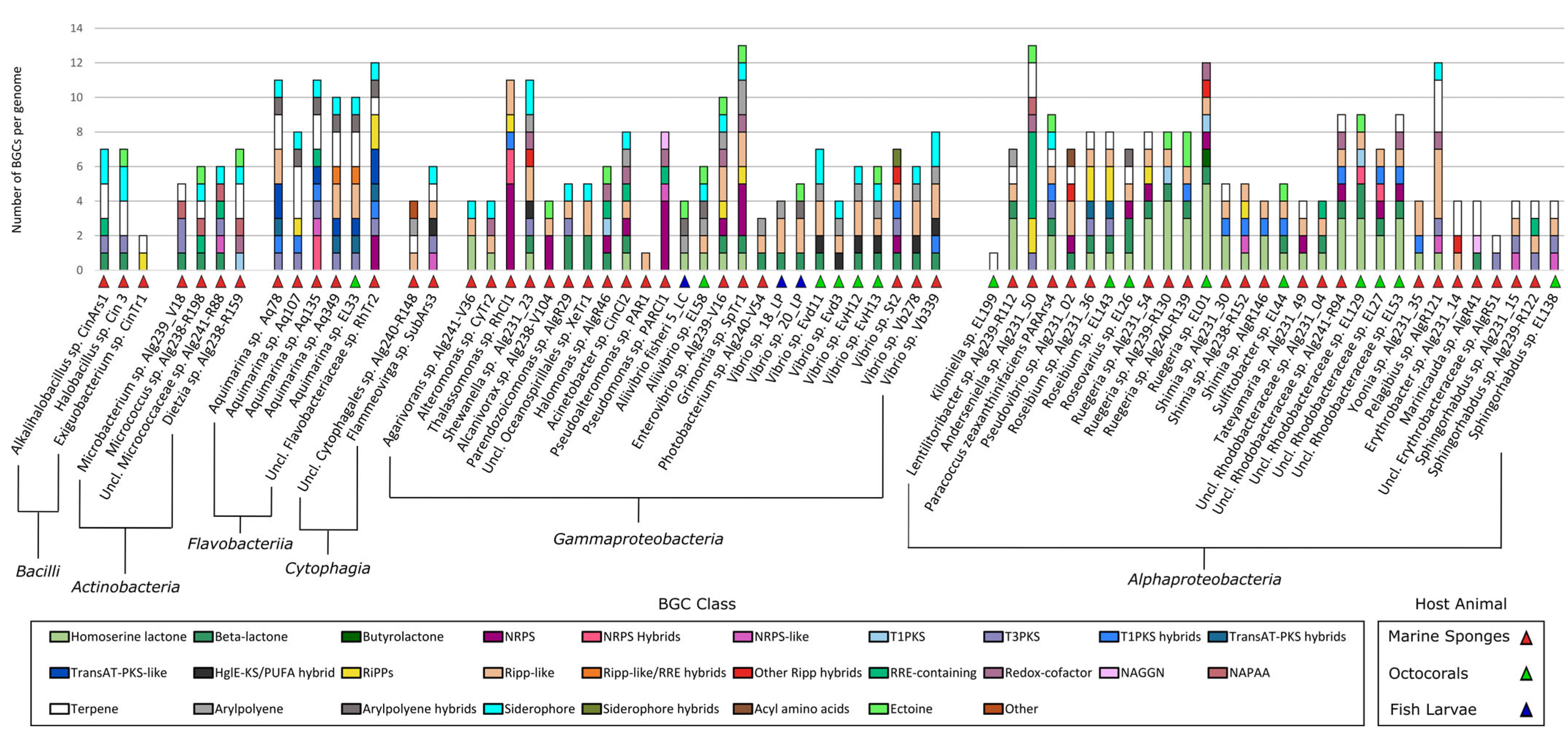
2.3. Identification of Secondary Metabolite Biosynthetic Gene Clusters (SM-BGCS) in Marine Bacterial Genomes
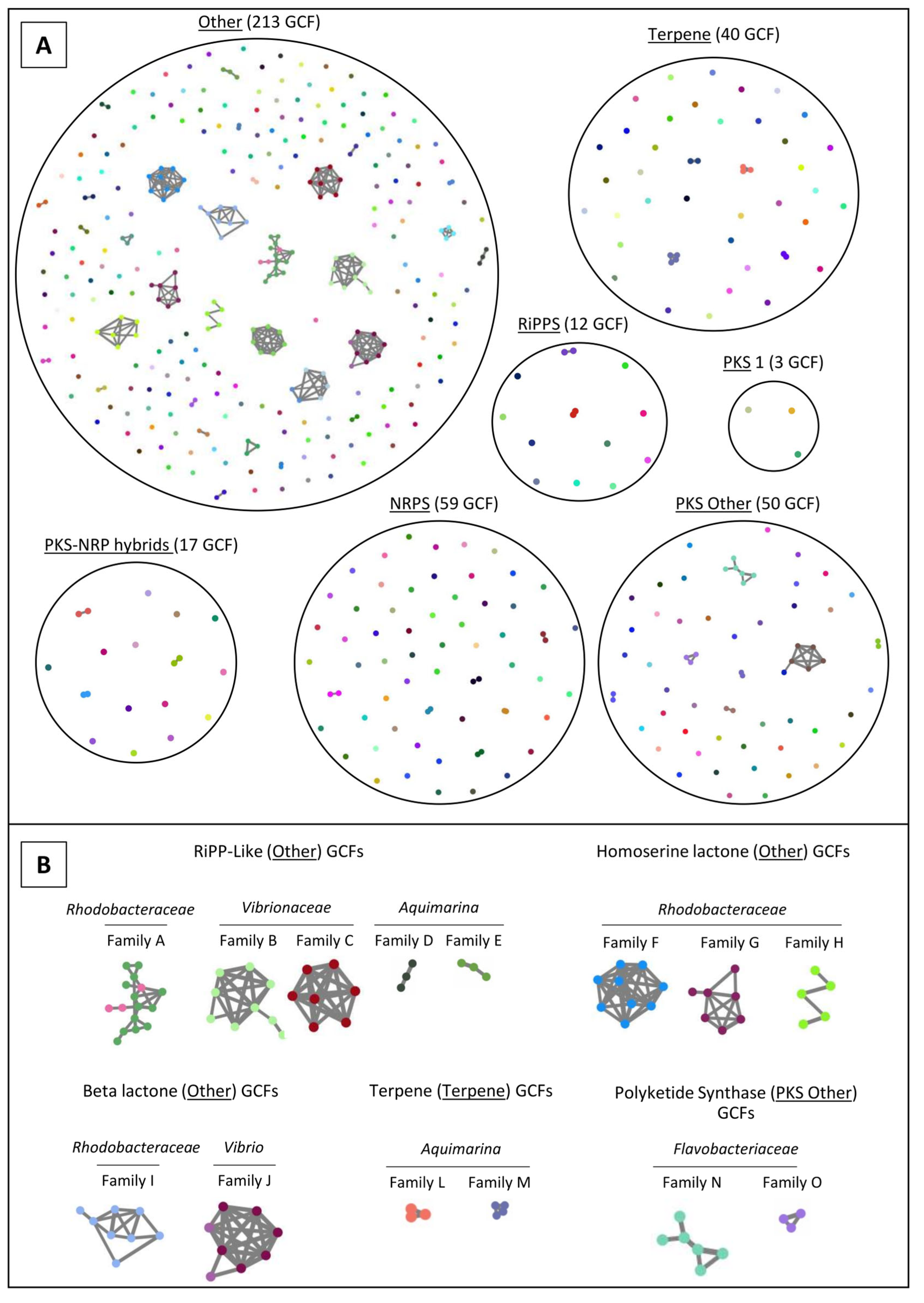
2.4. Network Analysis of SM-BGCs

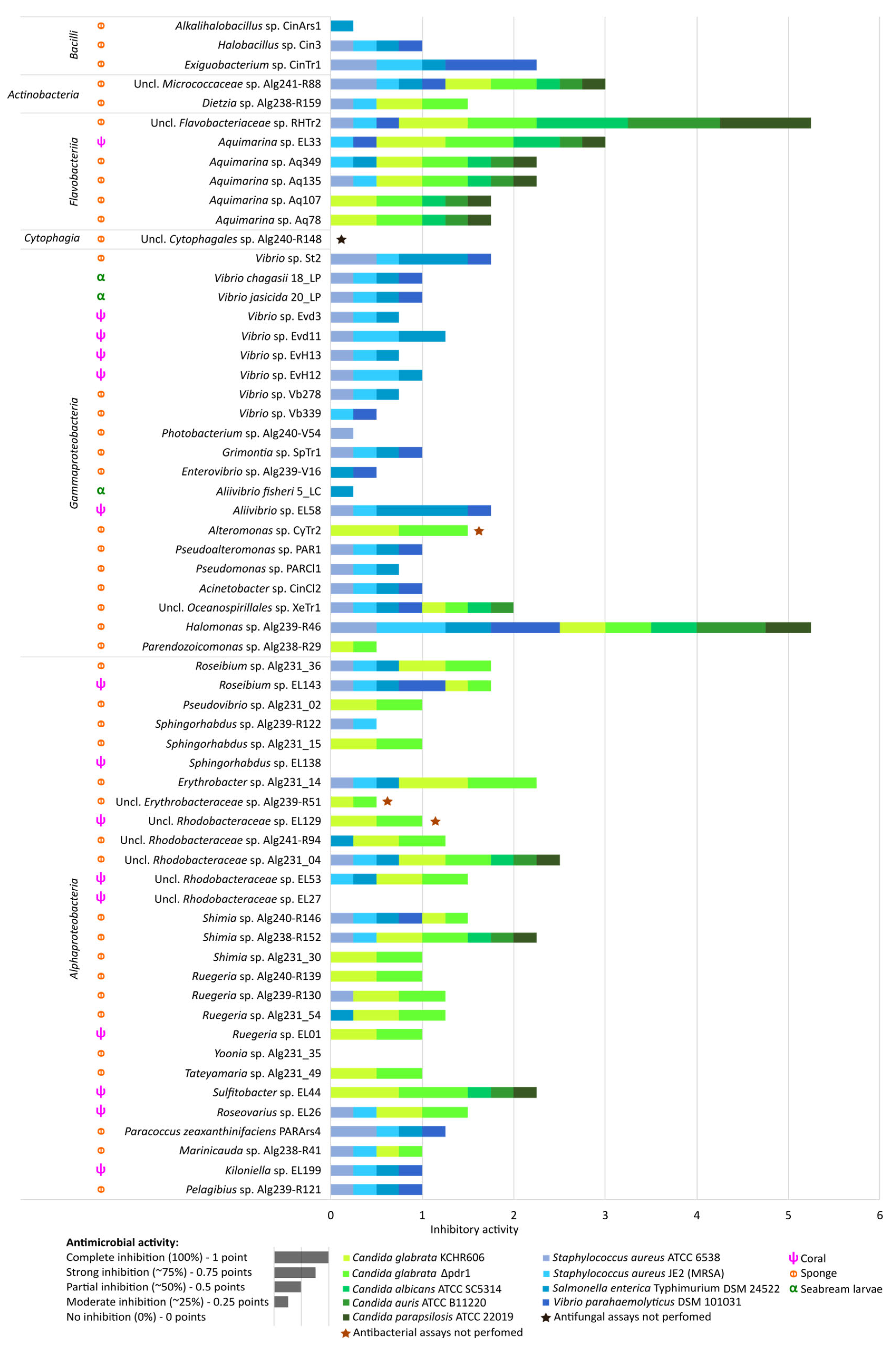
2.5. Antimicrobial Activities of Marine Bacterial Isolates

3. Discussion
3.1. The “MicroEcoEvo” and “EcoTech-SPONGE” Culture Collections Feature a Series of Underexplored and Taxonomically Novel Marine Bacteria
3.2. Both Culture Collections Comprise Taxa Typically Known for Their Symbiotic Lifestyles
3.3. Reduction of Carbon-Content and Prolonged Incubation Periods Promote an Increased Diversity of Culturable Marine Bacteria
3.4. The “MicroEcoEvo” and “EcoTech-SPONGE” Collections Are a Reservoir of Chemical Novelty with Most SM-BGCs Showing Little Homology to Those of Known Compounds
3.5. Taxon-Specific Differences in Antimicrobial Activity of Marine Bacteria Suggest Distinct Mechanisms Involved in Antagonism against Human-Pathogenic Bacteria versus Candida
4. Materials and Methods
4.1. The “MicroEcoEvo” and “EcoTech-SPONGE” Culture Collections
4.2. 16S rRNA Gene-Based Phylogenetic Analyses
4.3. Genome Sequencing and Assembly
4.4. Comparative Genomics and Phylogenomics Analysis
4.5. SM-BGC Identification and Network Analysis with antiSMASH and BiG-SCAPE
4.6. Marine Bacterial Isolates and Test Strains Used in Antimicrobial Assays
4.7. Cross-Streak Assays
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- O’Neil, J. Review on Antimicrobial Resistance. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations; Amr-Review: London, UK, 2014. [Google Scholar]
- Algammal, A.M.; Hetta, H.F.; Elkelish, A.; Alkhalifah, D.H.H.; Hozzein, W.N.; Batiha, G.E.-S.; El Nahhas, N.; Mabrok, M.A. Methicillin-Resistant Staphylococcus aureus (MRSA): One Health Perspective Approach to the Bacterium Epidemiology, Virulence Factors, Antibiotic-Resistance, and Zoonotic Impact. Infect. Drug Resist. 2020, 13, 3255–3265. [Google Scholar] [CrossRef]
- Köck, R.; Becker, K.; Cookson, B.; van Gemert-Pijnen, J.E.; Harbarth, S.; Kluytmans, J.; Mielke, M.; Peters, G.; Skov, R.L.; Struelens, M.J.; et al. Methicillin-Resistant Staphylococcus aureus (MRSA): Burden of Disease and Control Challenges in Europe. Eurosurveillance 2010, 15, 19688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patra, S.D.; Mohakud, N.K.; Panda, R.K.; Sahu, B.R.; Suar, M. Prevalence and Multidrug Resistance in Salmonella enterica Typhimurium: An Overview in South East Asia. World J. Microbiol. Biotechnol. 2021, 37, 185. [Google Scholar] [CrossRef]
- Ghenem, L.; Elhadi, N.; Alzahrani, F.; Nishibuchi, M. Vibrio parahaemolyticus: A Review on Distribution, Pathogenesis, Virulence Determinants and Epidemiology. Saudi J. Med. Med. Sci. 2017, 5, 93–103. [Google Scholar] [CrossRef]
- Balasubramanian, R.; Im, J.; Lee, J.-S.; Jeon, H.J.; Mogeni, O.D.; Kim, J.H.; Rakotozandrindrainy, R.; Baker, S.; Marks, F. The Global Burden and Epidemiology of Invasive Non-Typhoidal Salmonella Infections. Hum. Vaccines Immunother. 2019, 15, 1421–1426. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Roy, S.; Behera, B.K.; Bossier, P.; Das, B.K. Acute Hepatopancreatic Necrosis Disease (AHPND): Virulence, Pathogenesis and Mitigation Strategies in Shrimp Aquaculture. Toxins 2021, 13, 524. [Google Scholar] [CrossRef]
- Nelapati, S.; Nelapati, K.; Chinnam, B.K. Vibrio parahaemolyticus—An Emerging Foodborne Pathogen—A Review. Vet. World 2011, 5, 48–62. [Google Scholar] [CrossRef]
- Turner, S.A.; Butler, G. The Candida Pathogenic Species Complex. Cold Spring Harb. Perspect. Med. 2014, 4, a019778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geddes-McAlister, J.; Shapiro, R.S. New Pathogens, New Tricks: Emerging, Drug-Resistant Fungal Pathogens and Future Prospects for Antifungal Therapeutics: Drug-Resistant Fungal Pathogens. Ann. N. Y. Acad. Sci. 2019, 1435, 57–78. [Google Scholar] [CrossRef]
- Lockhart, S.R. Candida auris and Multidrug Resistance: Defining the New Normal. Fungal Genet. Biol. 2019, 131, 103243. [Google Scholar] [CrossRef]
- Vivas, R.; Barbosa, A.A.T.; Dolabela, S.S.; Jain, S. Multidrug-Resistant Bacteria and Alternative Methods to Control Them: An Overview. Microb. Drug Resist. 2019, 25, 890–908. [Google Scholar] [CrossRef]
- Raimundo, I.; Silva, S.; Costa, R.; Keller-Costa, T. Bioactive Secondary Metabolites from Octocoral-Associated Microbes—New Chances for Blue Growth. Mar. Drugs 2018, 16, 485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anjum, K.; Abbas, S.Q.; Shah, S.A.A.; Akhter, N.; Batool, S.; Hassan, S.S. Marine Sponges as a Drug Treasure. Biomol. Ther. 2016, 24, 347–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shetty, N.; Gupta, S. Eribulin Drug Review. South Asian J. Cancer 2014, 03, 057–059. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, C.; Francesch, A. Development of Yondelis® (Trabectedin, ET-743). A Semisynthetic Process Solves the Supply Problem. Nat. Prod. Rep. 2009, 26, 322. [Google Scholar] [CrossRef]
- Pita, L.; Rix, L.; Slaby, B.M.; Franke, A.; Hentschel, U. The Sponge Holobiont in a Changing Ocean: From Microbes to Ecosystems. Microbiome 2018, 6, 46. [Google Scholar] [CrossRef]
- van de Water, J.A.J.M.; Allemand, D.; Ferrier-Pagès, C. Host-Microbe Interactions in Octocoral Holobionts—Recent Advances and Perspectives. Microbiome 2018, 6, 64. [Google Scholar] [CrossRef] [Green Version]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. AntiSMASH 6.0: Improving Cluster Detection and Comparison Capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef]
- Karimi, E.; Ramos, M.; Gonçalves, J.M.S.; Xavier, J.R.; Reis, M.P.; Costa, R. Comparative Metagenomics Reveals the Distinctive Adaptive Features of the Spongia Officinalis Endosymbiotic Consortium. Front. Microbiol. 2017, 8, 2499. [Google Scholar] [CrossRef] [Green Version]
- Keller-Costa, T.; Lago-Lestón, A.; Saraiva, J.P.; Toscan, R.; Silva, S.G.; Gonçalves, J.; Cox, C.J.; Kyrpides, N.; Nunes da Rocha, U.; Costa, R. Metagenomic Insights into the Taxonomy, Function, and Dysbiosis of Prokaryotic Communities in Octocorals. Microbiome 2021, 9, 72. [Google Scholar] [CrossRef]
- Rodriguez Jimenez, A.; Dechamps, E.; Giaux, A.; Goetghebuer, L.; Bauwens, M.; Willenz, P.; Flahaut, S.; Laport, M.S.; George, I.F. The Sponges Hymeniacidon perlevis and Halichondria panicea Are Reservoirs of Antibiotic-producing Bacteria against Multi-drug Resistant Staphylococcus aureus. J. Appl. Microbiol. 2021, 131, 706–718. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.; Modolon, F.; Rosado, A.S.; Voolstra, C.R.; Sweet, M.; Peixoto, R.S. Methods and Strategies to Uncover Coral-Associated Microbial Dark Matter. mSystems 2022, 7, e00367-22. [Google Scholar] [CrossRef] [PubMed]
- Esteves, A.I.S.; Hardoim, C.C.P.; Xavier, J.R.; Gonçalves, J.M.S.; Costa, R. Molecular Richness and Biotechnological Potential of Bacteria Cultured from Irciniidae Sponges in the North-East Atlantic. FEMS Microbiol. Ecol. 2013, 85, 519–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karimi, E.; Keller-Costa, T.; Slaby, B.M.; Cox, C.J.; da Rocha, U.N.; Hentschel, U.; Costa, R. Genomic Blueprints of Sponge-Prokaryote Symbiosis Are Shared by Low Abundant and Cultivatable Alphaproteobacteria. Sci. Rep. 2019, 9, 1999. [Google Scholar] [CrossRef] [Green Version]
- Karimi, E.; Costa, R. Isolation and Genome Sequencing of 14 Spongia sp. Bacterial Associates Expands the Taxonomic and Functional Breadth of the Cultivatable Marine Sponge Microbiome. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Keller-Costa, T.; Eriksson, D.; Gonçalves, J.M.S.; Gomes, N.C.M.; Lago-Lestón, A.; Costa, R. The Gorgonian Coral Eunicella labiata Hosts a Distinct Prokaryotic Consortium Amenable to Cultivation. FEMS Microbiol. Ecol. 2017, 93. [Google Scholar] [CrossRef] [PubMed]
- Sanches-Fernandes, G.M.M.; Califano, G.; Keller-Costa, T.; Castanho, S.; Soares, F.; Ribeiro, L.; Pousão-Ferreira, P.; Mata, L.; Costa, R. Draft Genome Sequence of Vibrio chagasii 18LP, Isolated from Gilthead Seabream (Sparus aurata) Larvae Reared in Aquaculture. Microbiol. Resour. Announc. 2021, 10, e00658-21. [Google Scholar] [CrossRef]
- Sanches-Fernandes, G.M.M.; Califano, G.; Keller-Costa, T.; Castanho, S.; Soares, F.; Ribeiro, L.; Pousão-Ferreira, P.; Mata, L.; Costa, R. Draft Genome Sequence of Vibrio jasicida 20LP, an Opportunistic Bacterium Isolated from Fish Larvae. Microbiol. Resour. Announc. 2021, 10, e00813-21. [Google Scholar] [CrossRef]
- Califano, G.; Franco, T.; Gonçalves, A.C.S.; Castanho, S.; Soares, F.; Ribeiro, L.; Mata, L.; Costa, R. Draft Genome Sequence of Aliivibrio fischeri Strain 5LC, a Bacterium Retrieved from Gilthead Sea Bream (Sparus aurata) Larvae Reared in Aquaculture. Genome Announc. 2015, 3, e00593-15. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, V.; Polónia, A.R.M.; Cleary, D.F.R.; Huang, Y.M.; Voogd, N.J.; Rocha, U.N.; Gomes, N.C.M. Characterization of Putative Circular Plasmids in Sponge-associated Bacterial Communities Using a Selective Multiply-primed Rolling Circle Amplification. Mol. Ecol. Resour. 2021, 21, 110–121. [Google Scholar] [CrossRef]
- Oliveira, V.; Polónia, A.R.M.; Cleary, D.F.R.; Huang, Y.M.; de Voogd, N.J.; Keller-Costa, T.; Costa, R.; Gomes, N.C.M. Assessing the Genomic Composition, Putative Ecological Relevance and Biotechnological Potential of Plasmids from Sponge Bacterial Symbionts. Microbiol. Res. 2022, 265, 127183. [Google Scholar] [CrossRef] [PubMed]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and Tools for High Throughput RRNA Analysis. Nucleic Acids Res. 2014, 42, D633–D642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reasoner, D.J.; Geldreich, E.E. A New Medium for the Enumeration and Subculture of Bacteria from Potable Water. Appl. Env. Microbiol. 1985, 49, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Sait, M.; Hugenholtz, P.; Janssen, P.H. Cultivation of Globally Distributed Soil Bacteria from Phylogenetic Lineages Previously Only Detected in Cultivation-Independent Surveys: Cultivation of Soil Bacteria. Environ. Microbiol. 2002, 4, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Cúcio, A.C. Molecular Exploration of Bacterial Communities Associated with Azooxanthellate Gorgonians in the Coast of Algarve, South Portugal. Master’s Thesis, UALG, Faro, Portugal, 2011. [Google Scholar]
- Rodriguez-R, L.M.; Gunturu, S.; Harvey, W.T.; Rosselló-Mora, R.; Tiedje, J.M.; Cole, J.R.; Konstantinidis, K.T. The Microbial Genomes Atlas (MiGA) Webserver: Taxonomic and Gene Diversity Analysis of Archaea and Bacteria at the Whole Genome Level. Nucleic Acids Res. 2018, 46, W282–W288. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Muñoz, J.C.; Selem-Mojica, N.; Mullowney, M.W.; Kautsar, S.A.; Tryon, J.H.; Parkinson, E.I.; De Los Santos, E.L.C.; Yeong, M.; Cruz-Morales, P.; Abubucker, S.; et al. A Computational Framework to Explore Large-Scale Biosynthetic Diversity. Nat. Chem. Biol. 2020, 16, 60–68. [Google Scholar] [CrossRef]
- Ueno, K.; Uno, J.; Nakayama, H.; Sasamoto, K.; Mikami, Y.; Chibana, H. Development of a Highly Efficient Gene Targeting System Induced by Transient Repression of YKU80 Expression in Candida glabrata. Eukaryot. Cell 2007, 6, 1239–1247. [Google Scholar] [CrossRef] [Green Version]
- Galkina, K.V.; Okamoto, M.; Chibana, H.; Knorre, D.A.; Kajiwara, S. Deletion of CDR1 Reveals Redox Regulation of Pleiotropic Drug Resistance in Candida glabrata. Biochimie 2020, 170, 49–56. [Google Scholar] [CrossRef]
- Balouiri, M.; Sadiki, M.; Ibnsouda, S.K. Methods for in Vitro Evaluating Antimicrobial Activity: A Review. J. Pharm. Anal. 2016, 6, 71–79. [Google Scholar] [CrossRef] [Green Version]
- Silva, S.G.; Blom, J.; Keller-Costa, T.; Costa, R. Comparative Genomics Reveals Complex Natural Product Biosynthesis Capacities and Carbon Metabolism across Host-associated and Free-living Aquimarina (Bacteroidetes, Flavobacteriaceae) Species. Env. Microbiol. 2019, 21, 4002–4019. [Google Scholar] [CrossRef]
- Silva, S.G.; Paula, P.; da Silva, J.P.; Mil-Homens, D.; Teixeira, M.C.; Fialho, A.M.; Costa, R.; Keller-Costa, T. Insights into the Antimicrobial Activities and Metabolomes of Aquimarina (Flavobacteriaceae, Bacteroidetes) Species from the Rare Marine Biosphere. Mar. Drugs 2022, 20, 423. [Google Scholar] [CrossRef]
- Helfrich, E.J.N.; Ueoka, R.; Dolev, A.; Rust, M.; Meoded, R.A.; Bhushan, A.; Califano, G.; Costa, R.; Gugger, M.; Steinbeck, C.; et al. Automated Structure Prediction of Trans-Acyltransferase Polyketide Synthase Products. Nat. Chem. Biol. 2019, 15, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Dieterich, C.L.; Probst, S.I.; Ueoka, R.; Sandu, I.; Schäfle, D.; Dal Molin, M.; Minas, H.A.; Costa, R.; Oxenius, A.; Sander, P.; et al. Aquimarins, Peptide Antibiotics with Amino-Modified C-Termini from a Sponge-Derived Aquimarina sp. Bacterium. Angew. Chem. Int. Ed. Engl. 2021, 61, e202115802. [Google Scholar] [CrossRef]
- Steinert, G.; Whitfield, S.; Taylor, M.W.; Thoms, C.; Schupp, P.J. Application of Diffusion Growth Chambers for the Cultivation of Marine Sponge-Associated Bacteria. Mar. Biotechnol. 2014, 16, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Munaganti, R.K.; Muvva, V.; Konda, S.; Naragani, K.; Mangamuri, U.K.; Dorigondla, K.R.; Akkewar, D. Antimicrobial Profile of Arthrobacter kerguelensis VL-RK_09 Isolated from Mango Orchards. Braz. J. Microbiol. 2016, 47, 1030–1038. [Google Scholar] [CrossRef] [Green Version]
- Alvarado, P.; Huang, Y.; Wang, J.; Garrido, I.; Leiva, S. Phylogeny and Bioactivity of Epiphytic Gram-Positive Bacteria Isolated from Three Co-Occurring Antarctic Macroalgae. Antonie Van Leeuwenhoek 2018, 111, 1543–1555. [Google Scholar] [CrossRef]
- Hewitt, O.H.; Díez-Vives, C.; Taboada, S. Microbial Insights from Antarctic and Mediterranean Shallow-Water Bone-Eating Worms. Polar Biol. 2020, 43, 1605–1621. [Google Scholar] [CrossRef]
- Baelum, J.; Borglin, S.; Chakraborty, R.; Fortney, J.L.; Lamendella, R.; Mason, O.U.; Auer, M.; Zemla, M.; Bill, M.; Conrad, M.E.; et al. Deep-Sea Bacteria Enriched by Oil and Dispersant from the Deepwater Horizon Spill: Enrichment of Oil Degraders from Gulf of Mexico. Environ. Microbiol. 2012, 14, 2405–2416. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.L.; Christenson, J.K.; Wackett, L.P. Biosynthesis and Chemical Diversity of β-Lactone Natural Products. Nat. Prod. Rep. 2019, 36, 458–475. [Google Scholar] [CrossRef]
- Bartz, J.-O.; Blom, J.; Busse, H.-J.; Mvie, J.B.; Hardt, M.; Schubert, P.; Wilke, T.; Goessmann, A.; Wilharm, G.; Bender, J.; et al. Parendozoicomonas haliclonae Gen. Nov. sp. Nov. Isolated from a Marine Sponge of the Genus Haliclona and Description of the Family Endozoicomonadaceae Fam. Nov. Comprising the Genera Endozoicomonas, Parendozoicomonas, and Kistimonas. Syst. Appl. Microbiol. 2018, 41, 73–84. [Google Scholar] [CrossRef]
- Neave, M.J.; Michell, C.T.; Apprill, A.; Voolstra, C.R. Endozoicomonas Genomes Reveal Functional Adaptation and Plasticity in Bacterial Strains Symbiotically Associated with Diverse Marine Hosts. Sci. Rep. 2017, 7, 40579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller-Costa, T.; Kozma, L.; Silva, S.G.; Toscan, R.; Gonçalves, J.; Lago-Lestón, A.; Kyrpides, N.C.; Nunes da Rocha, U.; Costa, R. Metagenomics-Resolved Genomics Provides Novel Insights into Chitin Turnover, Metabolic Specialization, and Niche Partitioning in the Octocoral Microbiome. Microbiome 2022, 10, 151. [Google Scholar] [CrossRef] [PubMed]
- Raimundo, I.; Silva, R.; Meunier, L.; Valente, S.M.; Lago-Lestón, A.; Keller-Costa, T.; Costa, R. Functional Metagenomics Reveals Differential Chitin Degradation and Utilization Features across Free-Living and Host-Associated Marine Microbiomes. Microbiome 2021, 9, 43. [Google Scholar] [CrossRef] [PubMed]
- Sweet, M.; Villela, H.; Keller-Costa, T.; Costa, R.; Romano, S.; Bourne, D.G.; Cárdenas, A.; Huggett, M.J.; Kerwin, A.H.; Kuek, F.; et al. Insights into the Cultured Bacterial Fraction of Corals. mSystems 2021, 6, e01249-20. [Google Scholar] [CrossRef] [PubMed]
- Suantika, G. The Use of Indigenous Probiotic Halomonas aquamarina and Shewanella algae for White Shrimp (Litopenaeus vannamei Boone) Hatchery Productivity in Zero Water Discharge System. J. Aquac. Res. Dev. 2013, 04, 1000194. [Google Scholar] [CrossRef] [Green Version]
- Velmurugan, S.; Raman, K.; Thanga Viji, V.; Donio, M.B.S.; Adlin Jenifer, J.; Babu, M.M.; Citarasu, T. Screening and Characterization of Antimicrobial Secondary Metabolites from Halomonas salifodinae MPM-TC and Its in Vivo Antiviral Influence on Indian White Shrimp Fenneropenaeus indicus against WSSV Challenge. J. King Saud Univ. Sci. 2013, 25, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Rosado, P.M.; Leite, D.C.A.; Duarte, G.A.S.; Chaloub, R.M.; Jospin, G.; Nunes da Rocha, U.; Saraiva, J.P.; Dini-Andreote, F.; Eisen, J.A.; Bourne, D.G.; et al. Marine Probiotics: Increasing Coral Resistance to Bleaching through Microbiome Manipulation. ISME J. 2019, 13, 921–936. [Google Scholar] [CrossRef] [Green Version]
- Dat, T.T.H.; Steinert, G.; Cuc, N.T.K.; Smidt, H.; Sipkema, D. Bacteria Cultivated From Sponges and Bacteria Not Yet Cultivated From Sponges—A Review. Front. Microbiol. 2021, 12, 737925. [Google Scholar] [CrossRef] [PubMed]
- Offret, C.; Desriac, F.; Le Chevalier, P.; Mounier, J.; Jégou, C.; Fleury, Y. Spotlight on Antimicrobial Metabolites from the Marine Bacteria Pseudoalteromonas: Chemodiversity and Ecological Significance. Mar. Drugs 2016, 14, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chau, R.; Pearson, L.A.; Cain, J.; Kalaitzis, J.A.; Neilan, B.A. A Pseudoalteromonas Clade with Remarkable Biosynthetic Potential. Appl. Env. Microbiol. 2021, 87, e02604-20. [Google Scholar] [CrossRef]
- Rodrigues, G.N.; Lago-Lestón, A.; Costa, R.; Keller-Costa, T. Draft Genome Sequence of Labrenzia sp. Strain EL143, a Coral-Associated Alphaproteobacterium with Versatile Symbiotic Living Capability and Strong Halogen Degradation Potential. Genome Announc. 2018, 6, e00132-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pujalte, M.J.; Carmen Macián, M.; Arahal, D.R.; Garay, E. Stappia alba sp. Nov., Isolated from Mediterranean Oysters. Syst. Appl. Microbiol. 2005, 28, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Raj Sharma, A.; Zhou, T.; Harunari, E.; Oku, N.; Trianto, A.; Igarashi, Y. Labrenzbactin from a Coral-Associated Bacterium Labrenzia sp. J. Antibiot. 2019, 72, 634–639. [Google Scholar] [CrossRef] [Green Version]
- Kačar, D.; Schleissner, C.; Cañedo, L.M.; Rodríguez, P.; de la Calle, F.; Galán, B.; García, J.L. Genome of Labrenzia sp. PHM005 Reveals a Complete and Active Trans-AT PKS Gene Cluster for the Biosynthesis of Labrenzin. Front. Microbiol. 2019, 10, 2561. [Google Scholar] [CrossRef] [Green Version]
- Couceiro, J.F.; Keller-Costa, T.; Marques, M.; Kyrpides, N.C.; Woyke, T.; Whitman, W.B.; Costa, R. The Roseibium album (Labrenzia alba) Genome Possesses Multiple Symbiosis Factors Possibly Underpinning Host-Microbe Relationships in the Marine Benthos. Microbiol. Resour. Announc. 2021, 10, e00320-21. [Google Scholar] [CrossRef] [PubMed]
- Parte, A.C.; Sardà Carbasse, J.; Meier-Kolthoff, J.P.; Reimer, L.C.; Göker, M. List of Prokaryotic Names with Standing in Nomenclature (LPSN) Moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 2020, 70, 5607–5612. [Google Scholar] [CrossRef] [PubMed]
- Bethesda (MD); National Library of Medicine (US); National Center for Biotechnology Information National Center for Biotechnology Information (NCBI)[Internet]. Available online: https://www.ncbi.nlm.nih.gov/ (accessed on 28 August 2022).
- Peixoto, R.S.; Rosado, P.M.; de Leite, D.C.A.; Rosado, A.S.; Bourne, D.G. Beneficial Microorganisms for Corals (BMC): Proposed Mechanisms for Coral Health and Resilience. Front. Microbiol. 2017, 8, 341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Sayed, A.K.; Hothersall, J.; Cooper, S.M.; Stephens, E.; Simpson, T.J.; Thomas, C.M. Characterization of the Mupirocin Biosynthesis Gene Cluster from Pseudomonas Fluorescens NCIMB 10586. Chem. Biol. 2003, 10, 419–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudek, S.; Lopanik, N.B.; Waggoner, L.E.; Hildebrand, M.; Anderson, C.; Liu, H.; Patel, A.; Sherman, D.H.; Haygood, M.G. Identification of the Putative Bryostatin Polyketide Synthase Gene Cluster from “Candidatus Endobugula Sertula”, the Uncultivated Microbial Symbiont of the Marine Bryozoan Bugula neritina. J. Nat. Prod. 2007, 70, 67–74. [Google Scholar] [CrossRef]
- Chakraborty, K.; Kizhakkekalam, V.K.; Joy, M. Chemical Mining of Heterotrophic Shewanella algae Reveals Anti-Infective Potential of Macrocyclic Polyketides against Multidrug-Resistant Pathogens. Bioorgan. Chem. 2021, 108, 104533. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, O.N.; Méjean, V.; Iobbi-Nivol, C. The Shewanella Genus: Ubiquitous Organisms Sustaining and Preserving Aquatic Ecosystems. FEMS Microbiol. Rev. 2020, 44, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Gachhui, R.; Mukherjee, J. Enhanced Biofilm Formation and Melanin Synthesis by the Oyster Settlement-Promoting Shewanella colwelliana Is Related to Hydrophobic Surface and Simulated Intertidal Environment. Biofouling 2015, 31, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, V.; Jasmin, C.; Anas, A.; Parakkaparambil Kuttan, S.; Vinothkumar, S.; Perunninakulath Subrayan, P.; Nair, S. Sponge-Associated Bacteria Produce Non-Cytotoxic Melanin Which Protects Animal Cells from Photo-Toxicity. Appl. Biochem. Biotechnol. 2017, 183, 396–411. [Google Scholar] [CrossRef]
- Cámara-Ruiz, M.; Balebona, M.C.; Moriñigo, M.Á.; Esteban, M.Á. Probiotic Shewanella putrefaciens (SpPdp11) as a Fish Health Modulator: A Review. Microorganisms 2020, 8, 1990. [Google Scholar] [CrossRef]
- Interaminense, J.A.; Vogeley, J.L.; Gouveia, C.K.; Portela, R.W.S.; Oliveira, J.P.; Andrade, H.A.; Peixoto, S.M.; Soares, R.B.; Buarque, D.S.; Bezerra, R.S. In Vitro and in Vivo Potential Probiotic Activity of Bacillus subtilis and Shewanella algae for Use in Litopenaeus vannamei Rearing. Aquaculture 2018, 488, 114–122. [Google Scholar] [CrossRef]
- Thetsana, C.; Ijichi, S.; Kaweewan, I.; Nakagawa, H.; Kodani, S. Heterologous Expression of a Cryptic Gene Cluster from a Marine Proteobacterium Thalassomonas actiniarum Affords New Lanthipeptides Thalassomonasins A and B. J. Appl. Microbiol. 2022, 132, 3629–3639. [Google Scholar] [CrossRef]
- Joerger, R. Alternatives to Antibiotics: Bacteriocins, Antimicrobial Peptides and Bacteriophages. Poult. Sci. 2003, 82, 640–647. [Google Scholar] [CrossRef]
- Li, Y.; Rebuffat, S. The Manifold Roles of Microbial Ribosomal Peptide–Based Natural Products in Physiology and Ecology. J. Biol. Chem. 2020, 295, 34–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martens, T.; Gram, L.; Grossart, H.-P.; Kessler, D.; Müller, R.; Simon, M.; Wenzel, S.C.; Brinkhoff, T. Bacteria of the Roseobacter Clade Show Potential for Secondary Metabolite Production. Microb. Ecol. 2007, 54, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Buchan, A.; Mitchell, A.; Cude, W.N.; Campagna, S. Acyl-Homoserine Lactone-Based Quorum Sensing in Members of the Marine Bacterial Roseobacter Clade: Complex Cell-to-Cell Communication Controls Multiple Physiologies. In Stress and Environmental Regulation of Gene Expression and Adaptation in Bacteria; de Bruijn, F.J., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; pp. 225–233. ISBN 978-1-119-00481-3. [Google Scholar]
- Pais, P.; Galocha, M.; Viana, R.; Cavalheiro, M.; Pereira, D.; Teixeira, M.C. Microevolution of the Pathogenic Yeasts Candida albicans and Candida glabrata during Antifungal Therapy and Host Infection. Microb. Cell 2019, 6, 142–159. [Google Scholar] [CrossRef] [PubMed]
- Pristov, K.E.; Ghannoum, M.A. Resistance of Candida to Azoles and Echinocandins Worldwide. Clin. Microbiol. Infect. 2019, 25, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Shanthakumar, S.P.; Duraisamy, P.; Vishwanath, G.; Selvanesan, B.C.; Ramaraj, V.; Vasantharaj David, B. Broad Spectrum Antimicrobial Compounds from the Bacterium Exiguobacterium mexicanum MSSRFS9. Microbiol. Res. 2015, 178, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Romano, S.; Jackson, S.; Patry, S.; Dobson, A. Extending the “One Strain Many Compounds” (OSMAC) Principle to Marine Microorganisms. Mar. Drugs 2018, 16, 244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Califano, G.; Castanho, S.; Soares, F.; Ribeiro, L.; Cox, C.J.; Mata, L.; Costa, R. Molecular Taxonomic Profiling of Bacterial Communities in a Gilthead Seabream (Sparus aurata) Hatchery. Front. Microbiol. 2017, 8, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (ITOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Inkscape Project Inkscape. Available online: https://inkscape.org (accessed on 26 August 2021).
- Keller-Costa, T.; Silva, R.; Lago-Lestón, A.; Costa, R. Genomic Insights into Aquimarina sp. Strain EL33, a Bacterial Symbiont of the Gorgonian Coral Eunicella labiata. Genome Announc. 2016, 4, e00855-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franco, T.; Califano, G.; Gonçalves, A.C.S.; Cúcio, C.; Costa, R. Draft Genome Sequence of Vibrio sp. Strain Evh12, a Bacterium Retrieved from the Gorgonian Coral Eunicella verrucosa. Genome Announc. 2016, 4, e01729-15. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Chen, I.-M.A.; Chu, K.; Palaniappan, K.; Ratner, A.; Huang, J.; Huntemann, M.; Hajek, P.; Ritter, S.; Varghese, N.; Seshadri, R.; et al. The IMG/M Data Management and Analysis System v.6.0: New Tools and Advanced Capabilities. Nucleic Acids Res. 2021, 49, D751–D763. [Google Scholar] [CrossRef]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2012, 41, D36–D42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arkin, A.P.; Cottingham, R.W.; Henry, C.S.; Harris, N.L.; Stevens, R.L.; Maslov, S.; Dehal, P.; Ware, D.; Perez, F.; Canon, S.; et al. KBase: The United States Department of Energy Systems Biology Knowledgebase. Nat. Biotechnol. 2018, 36, 566–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Kautsar, S.A.; Blin, K.; Shaw, S.; Navarro-Muñoz, J.C.; Terlouw, B.R.; van der Hooft, J.J.J.; van Santen, J.A.; Tracanna, V.; Suarez Duran, H.G.; Pascal Andreu, V.; et al. MIBiG 2.0: A Repository for Biosynthetic Gene Clusters of Known Function. Nucleic Acids Res. 2019, 48, D454–D458. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almeida, J.F.; Marques, M.; Oliveira, V.; Egas, C.; Mil-Homens, D.; Viana, R.; Cleary, D.F.R.; Huang, Y.M.; Fialho, A.M.; Teixeira, M.C.; et al. Marine Sponge and Octocoral-Associated Bacteria Show Versatile Secondary Metabolite Biosynthesis Potential and Antimicrobial Activities against Human Pathogens. Mar. Drugs 2023, 21, 34. https://doi.org/10.3390/md21010034
Almeida JF, Marques M, Oliveira V, Egas C, Mil-Homens D, Viana R, Cleary DFR, Huang YM, Fialho AM, Teixeira MC, et al. Marine Sponge and Octocoral-Associated Bacteria Show Versatile Secondary Metabolite Biosynthesis Potential and Antimicrobial Activities against Human Pathogens. Marine Drugs. 2023; 21(1):34. https://doi.org/10.3390/md21010034
Chicago/Turabian StyleAlmeida, João F., Matilde Marques, Vanessa Oliveira, Conceição Egas, Dalila Mil-Homens, Romeu Viana, Daniel F. R. Cleary, Yusheng M. Huang, Arsénio M. Fialho, Miguel C. Teixeira, and et al. 2023. "Marine Sponge and Octocoral-Associated Bacteria Show Versatile Secondary Metabolite Biosynthesis Potential and Antimicrobial Activities against Human Pathogens" Marine Drugs 21, no. 1: 34. https://doi.org/10.3390/md21010034