
Antibody-Drug Conjugates Containing Payloads from Marine Origin

 , , and
, , and
Abstract
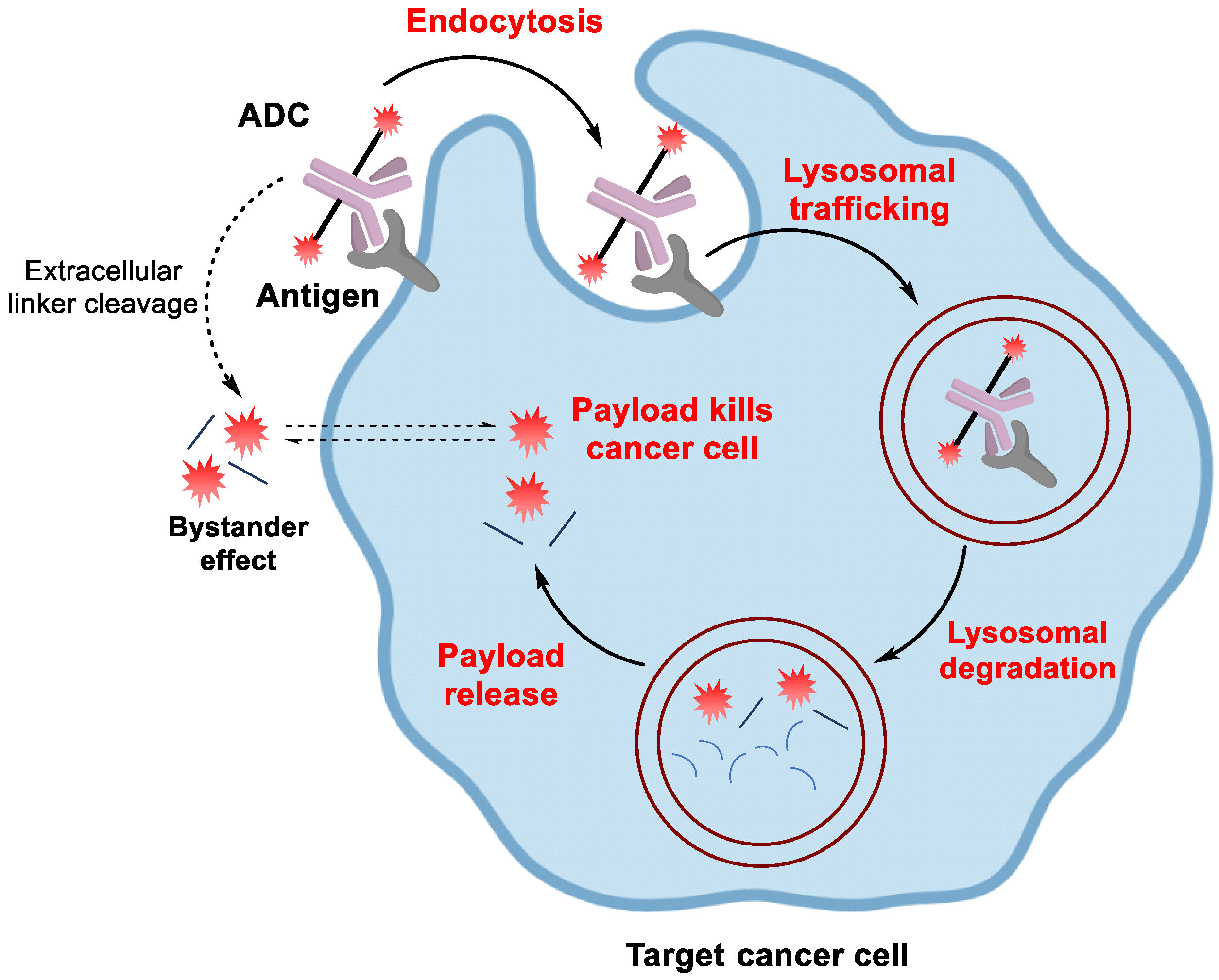
:1. Introduction
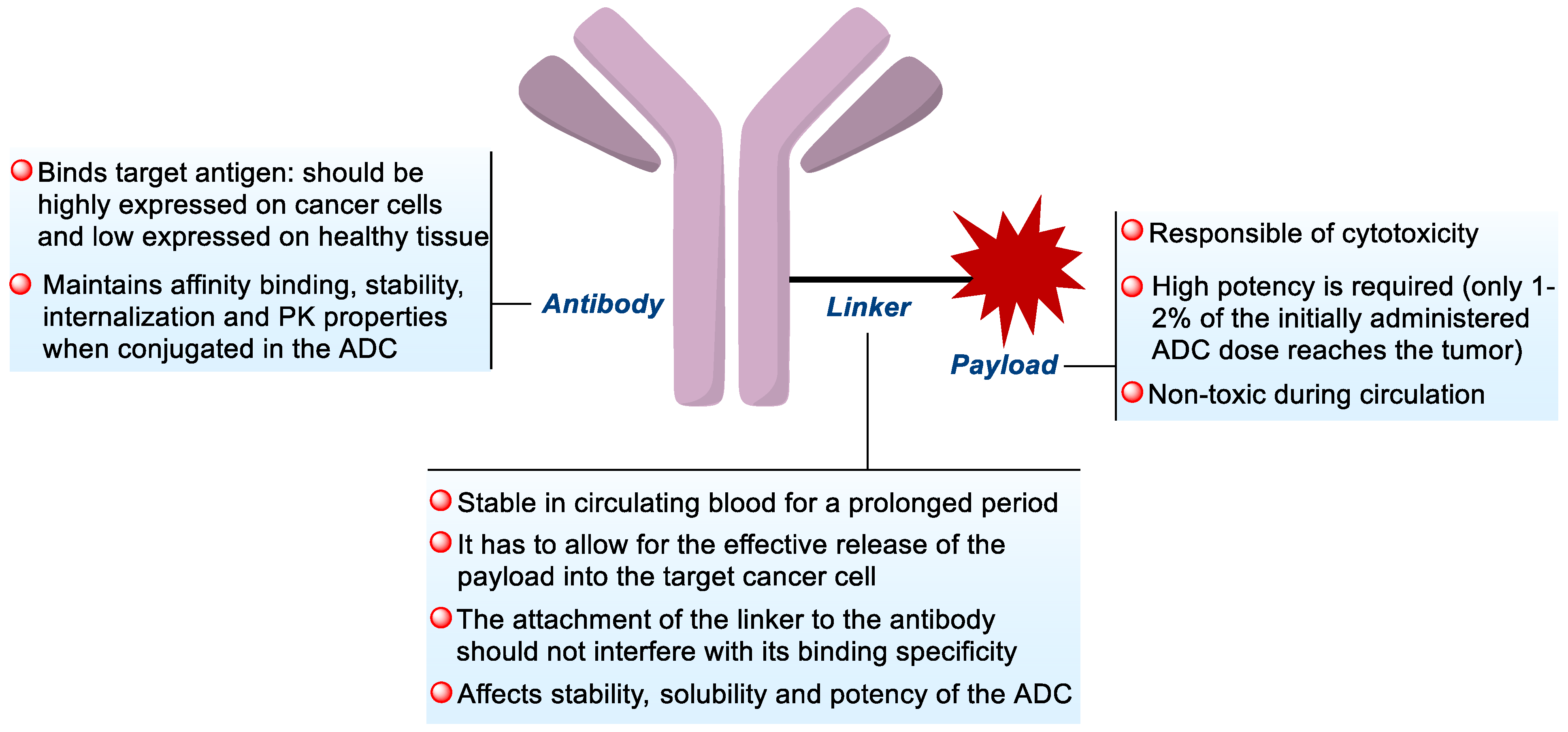
1.1. ADC Design
1.1.1. Antibody
1.1.2. Linker
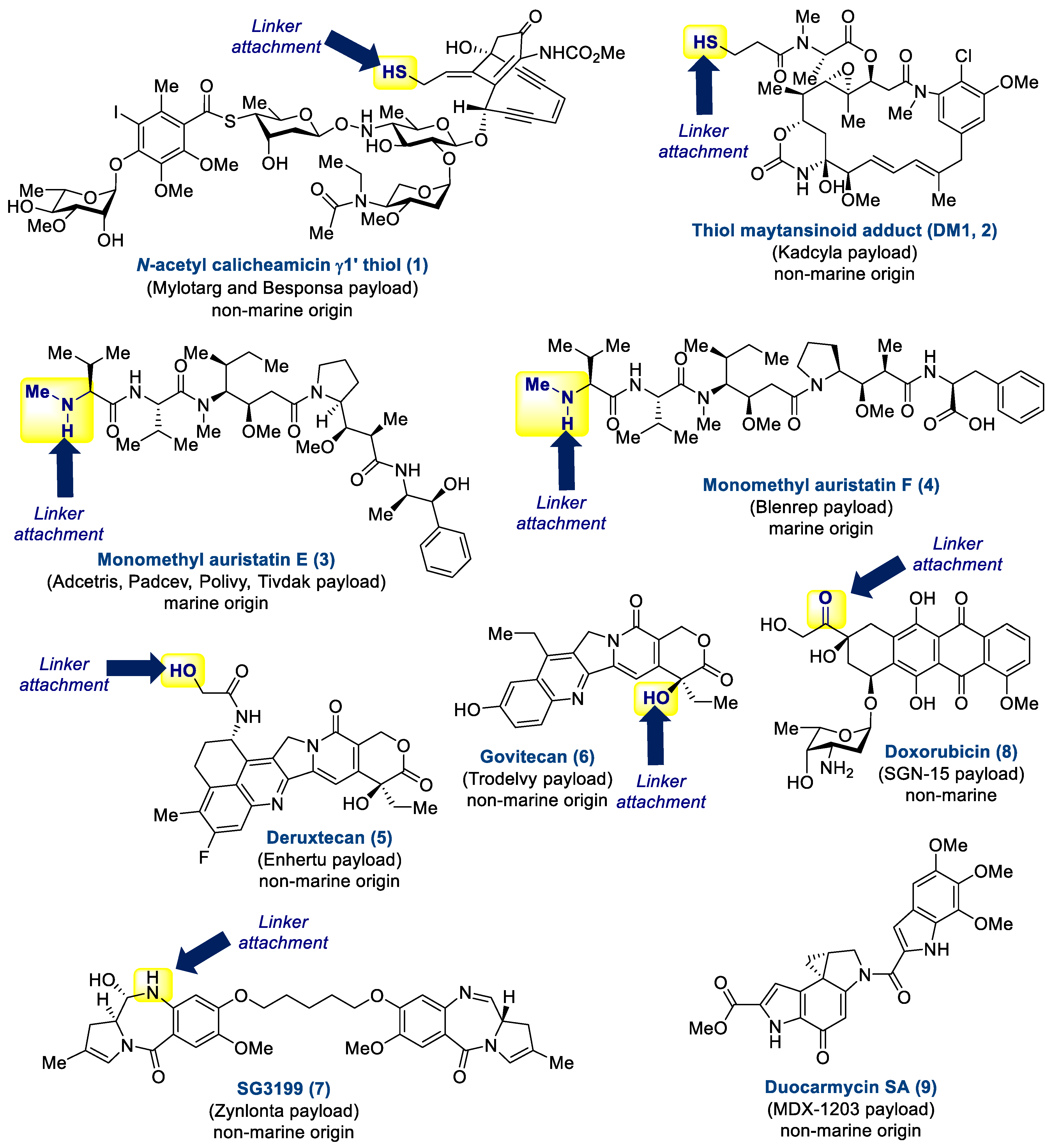
1.1.3. Payload
2. Chemistry and Biology of Marine Antibody-Drug Conjugates
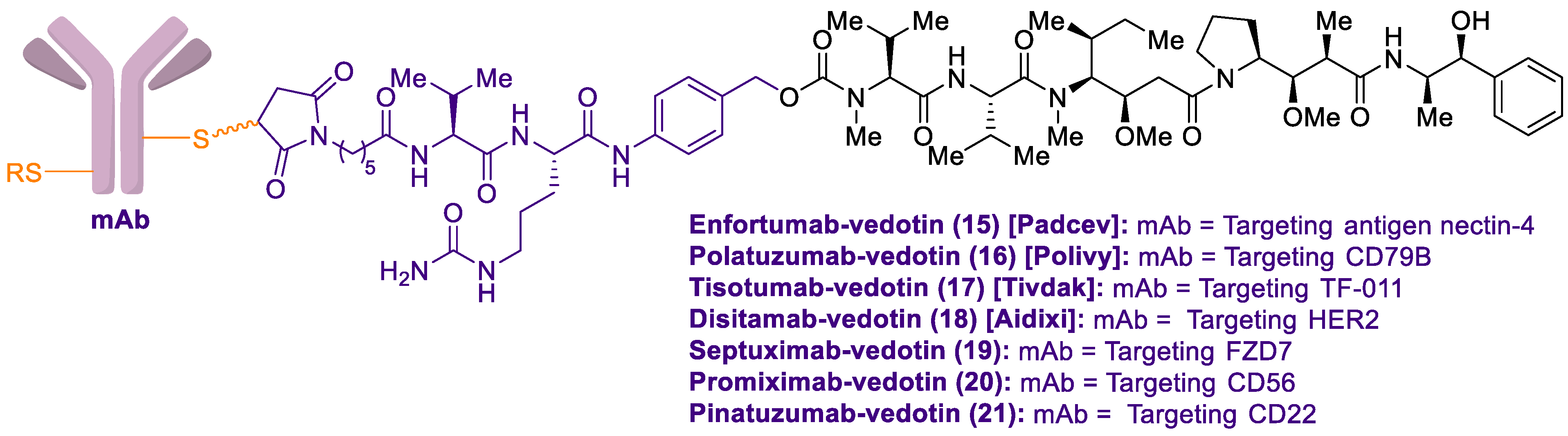
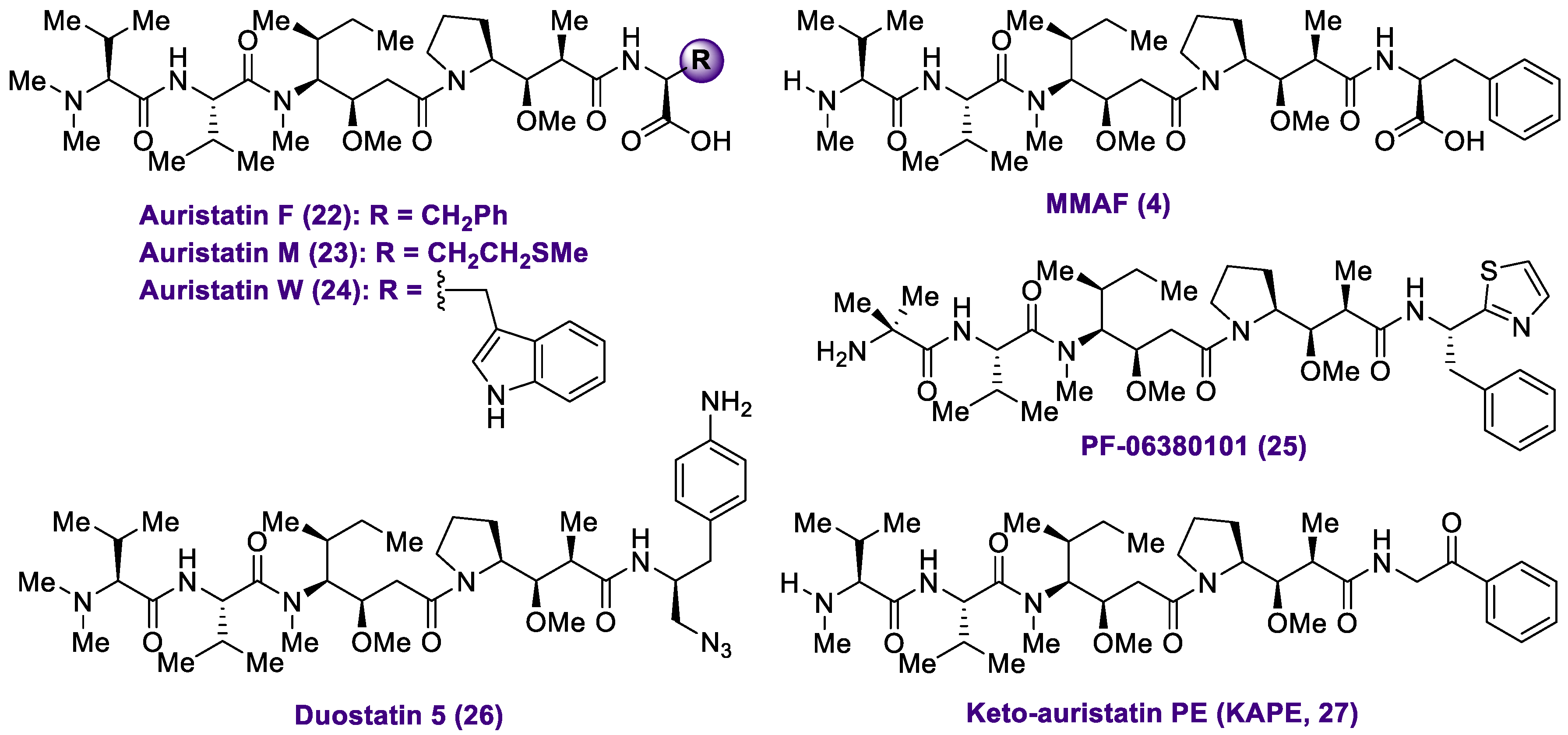
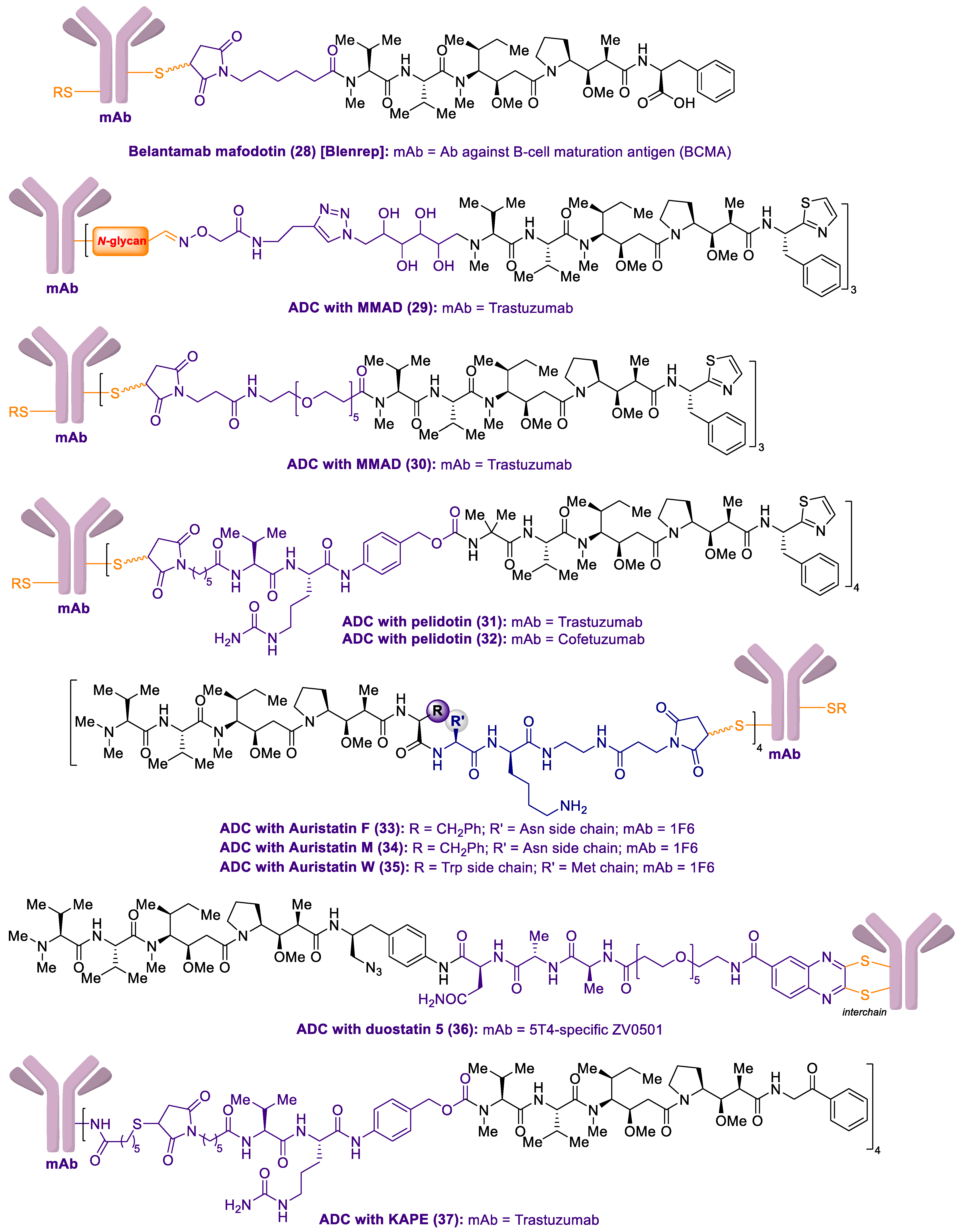
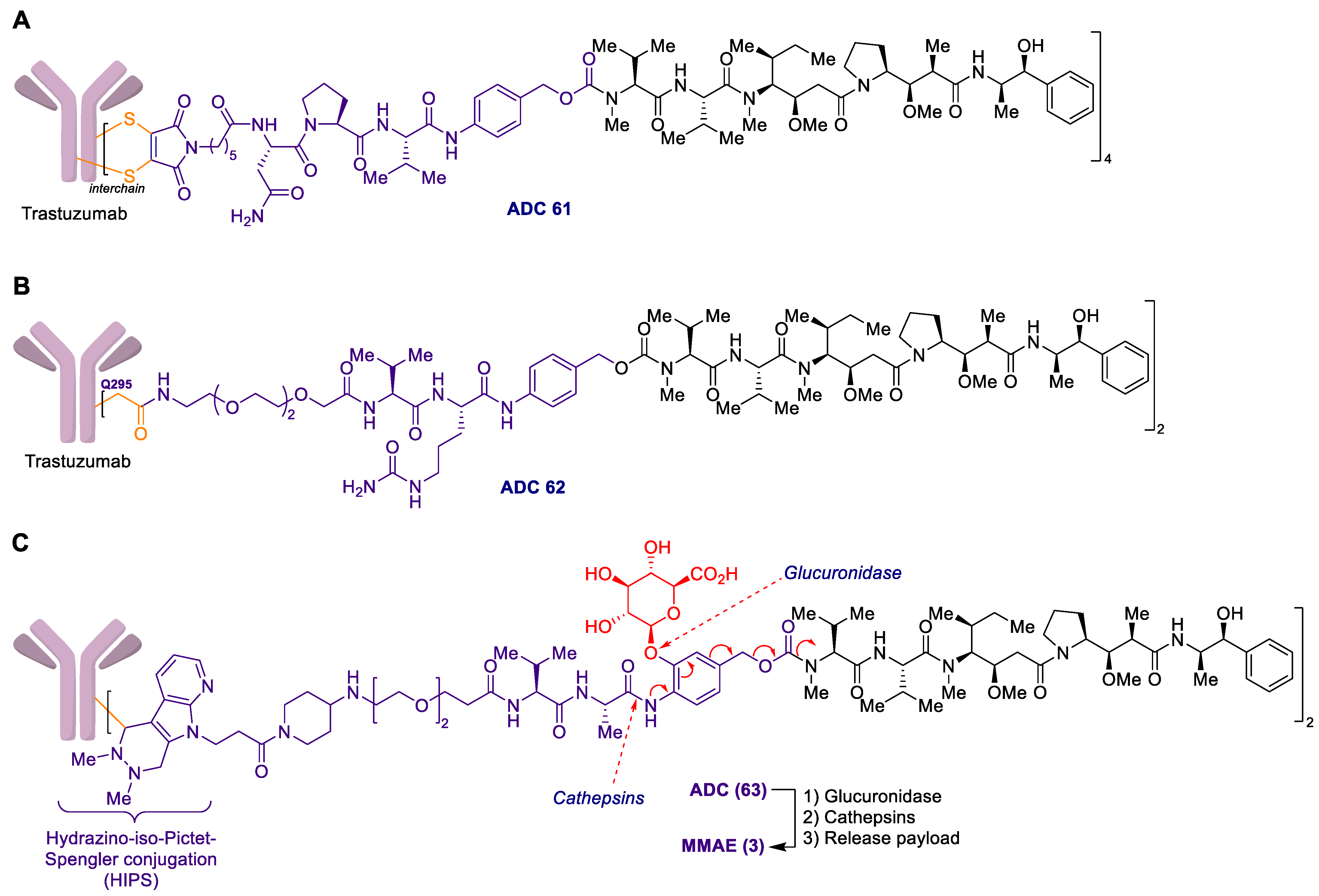
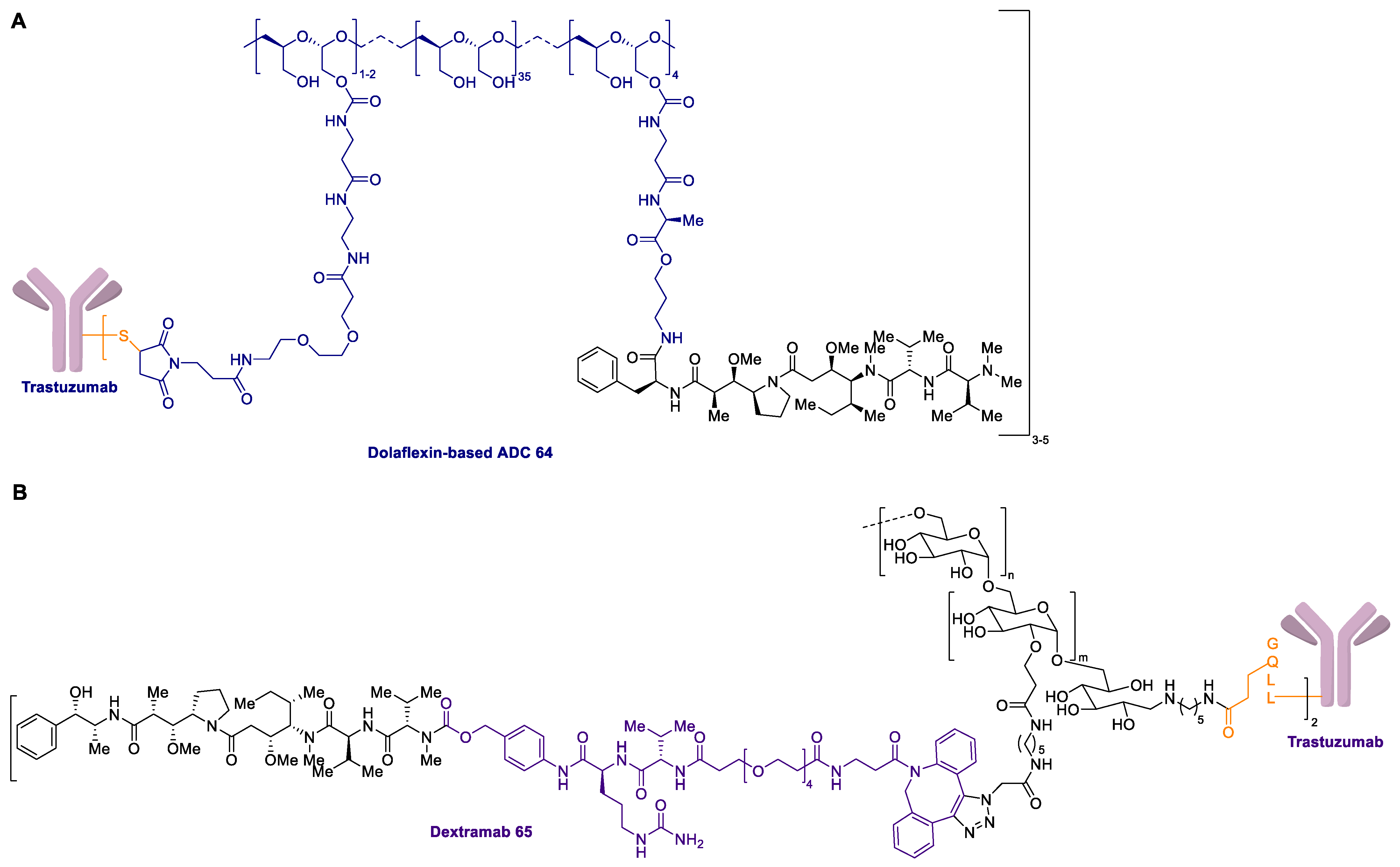
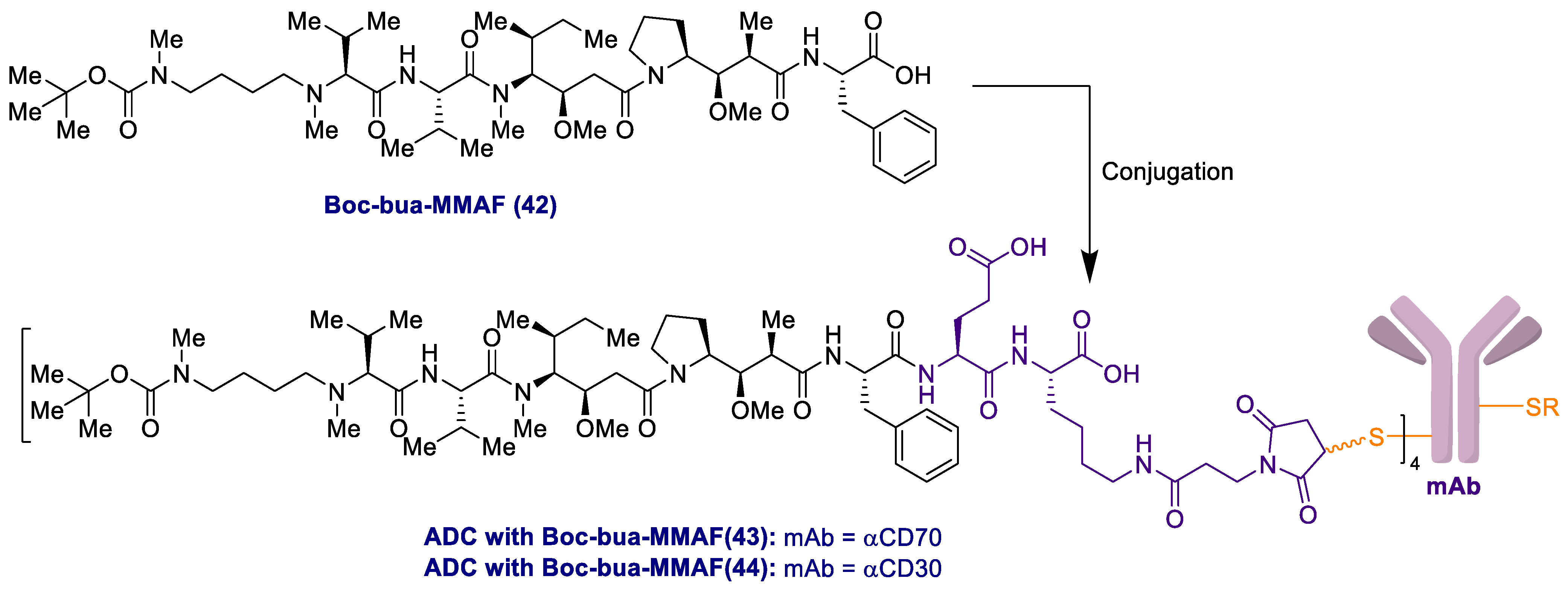
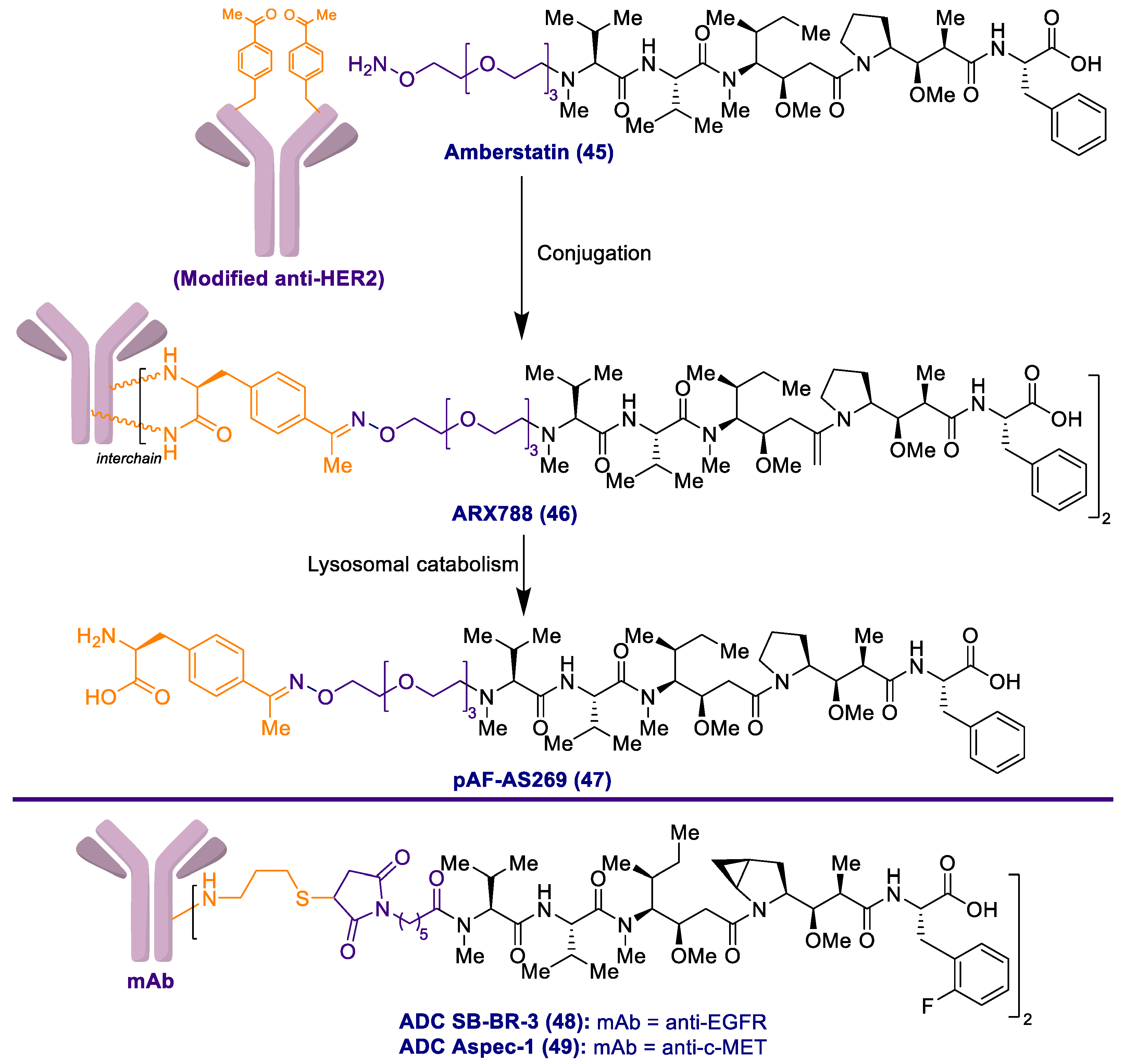
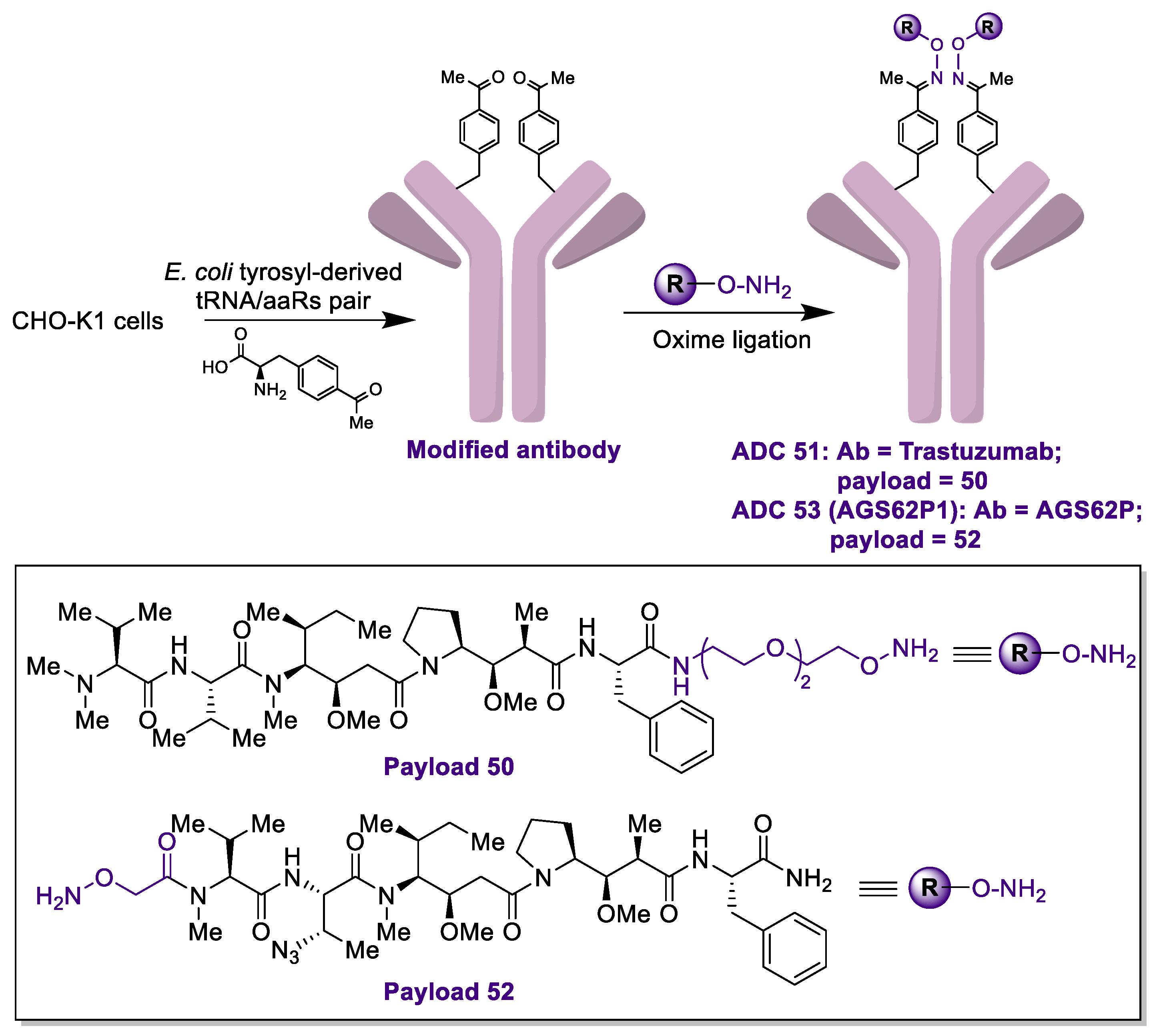
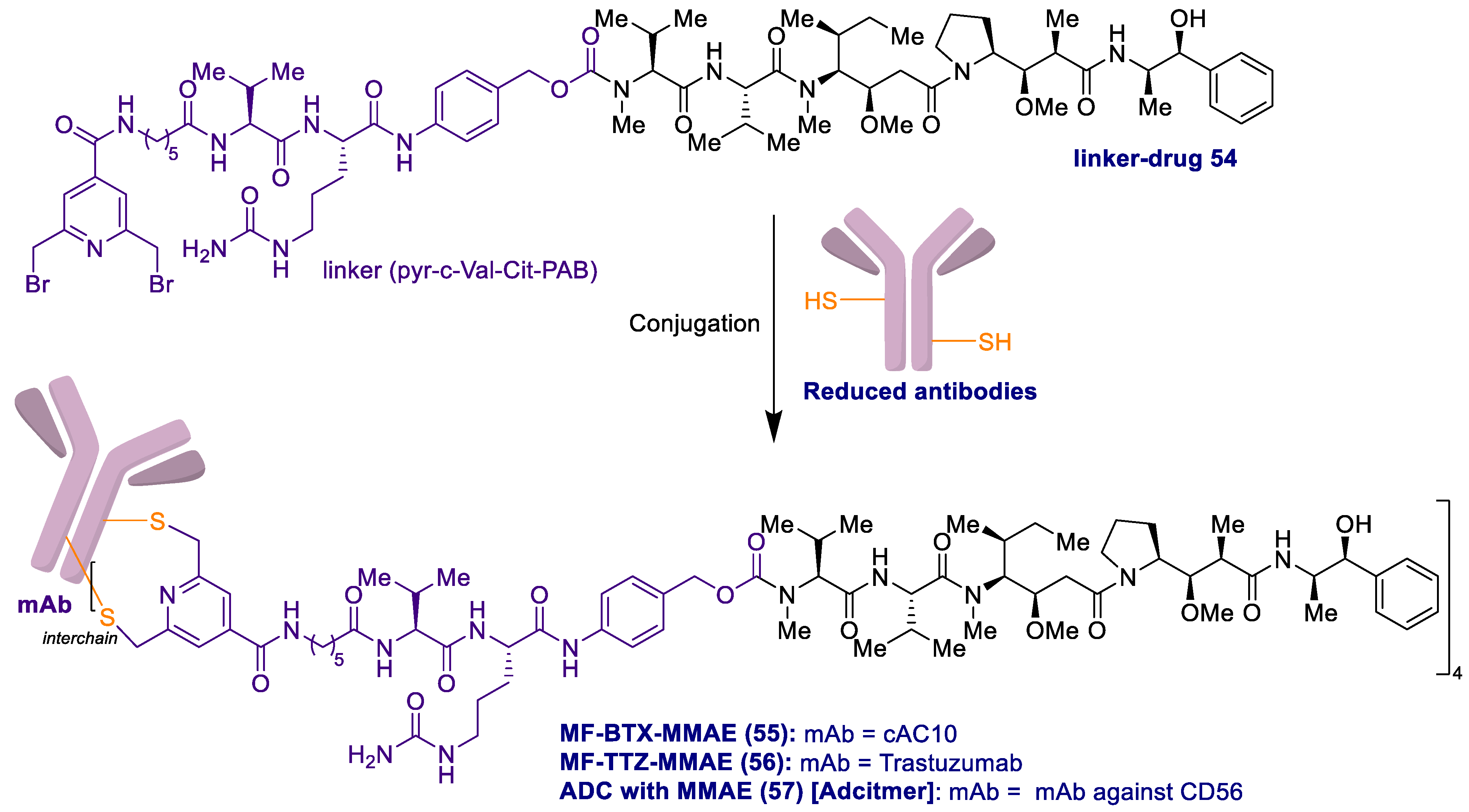
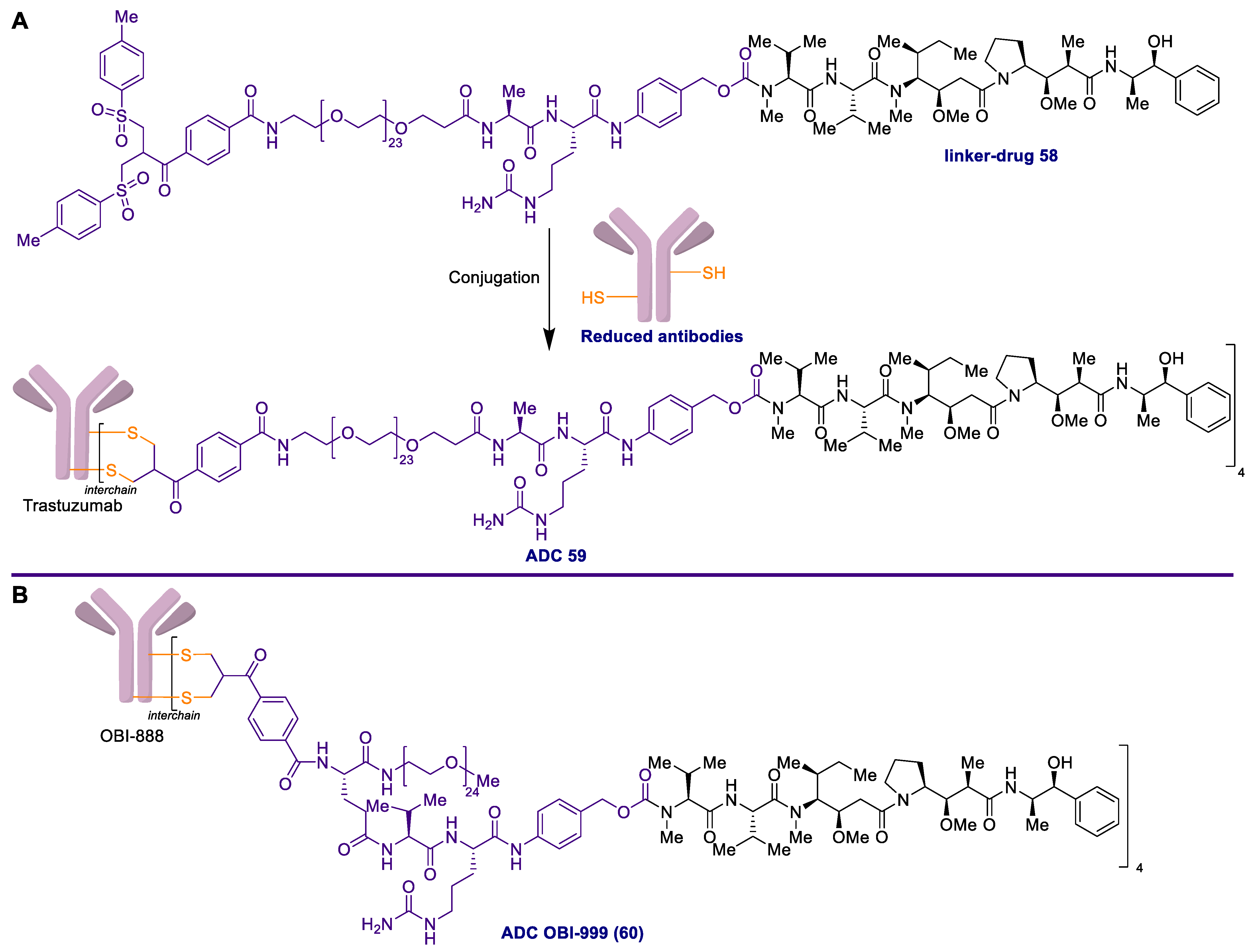
2.1. Antibody-Drug Conjugates Based on the Auristatins
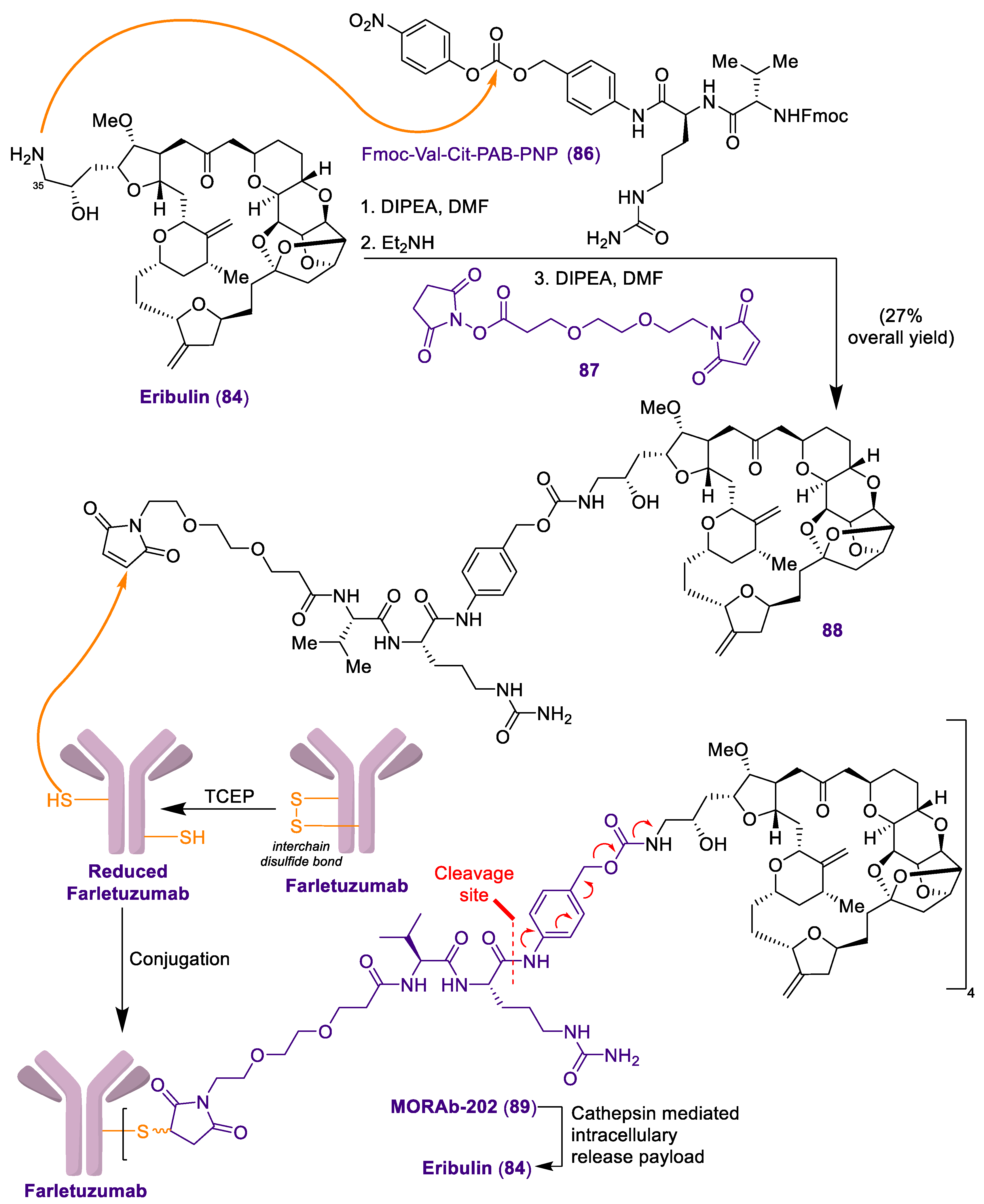
2.2. Antibody-Drug Conjugate Based on Halichondrin B
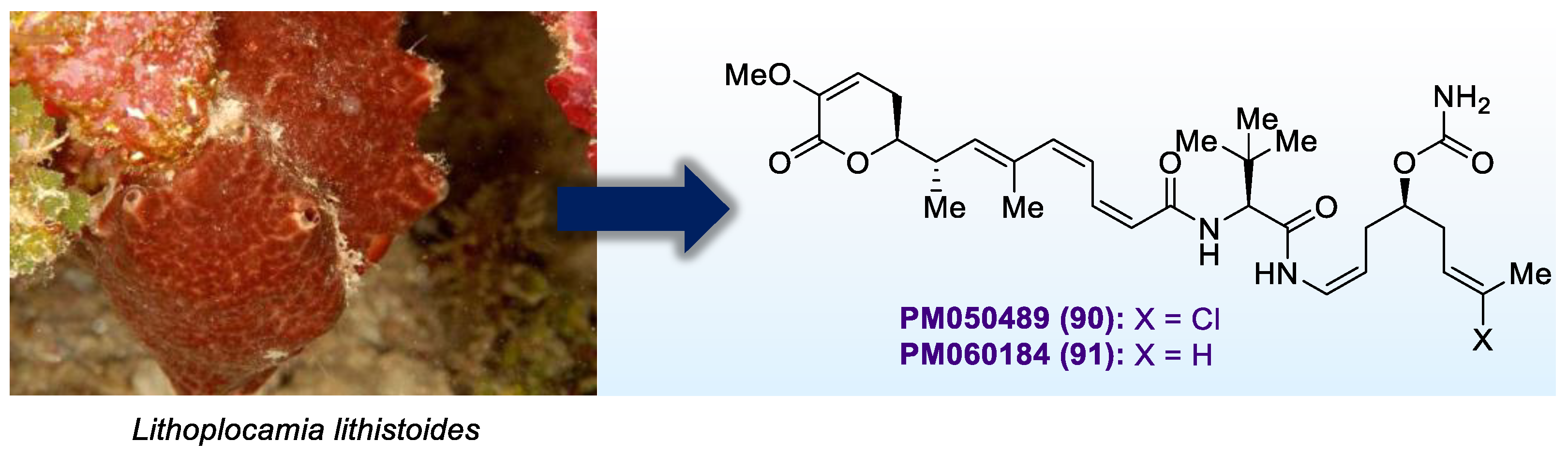
2.3. Antibody-Drug Conjugates Based on PM050489
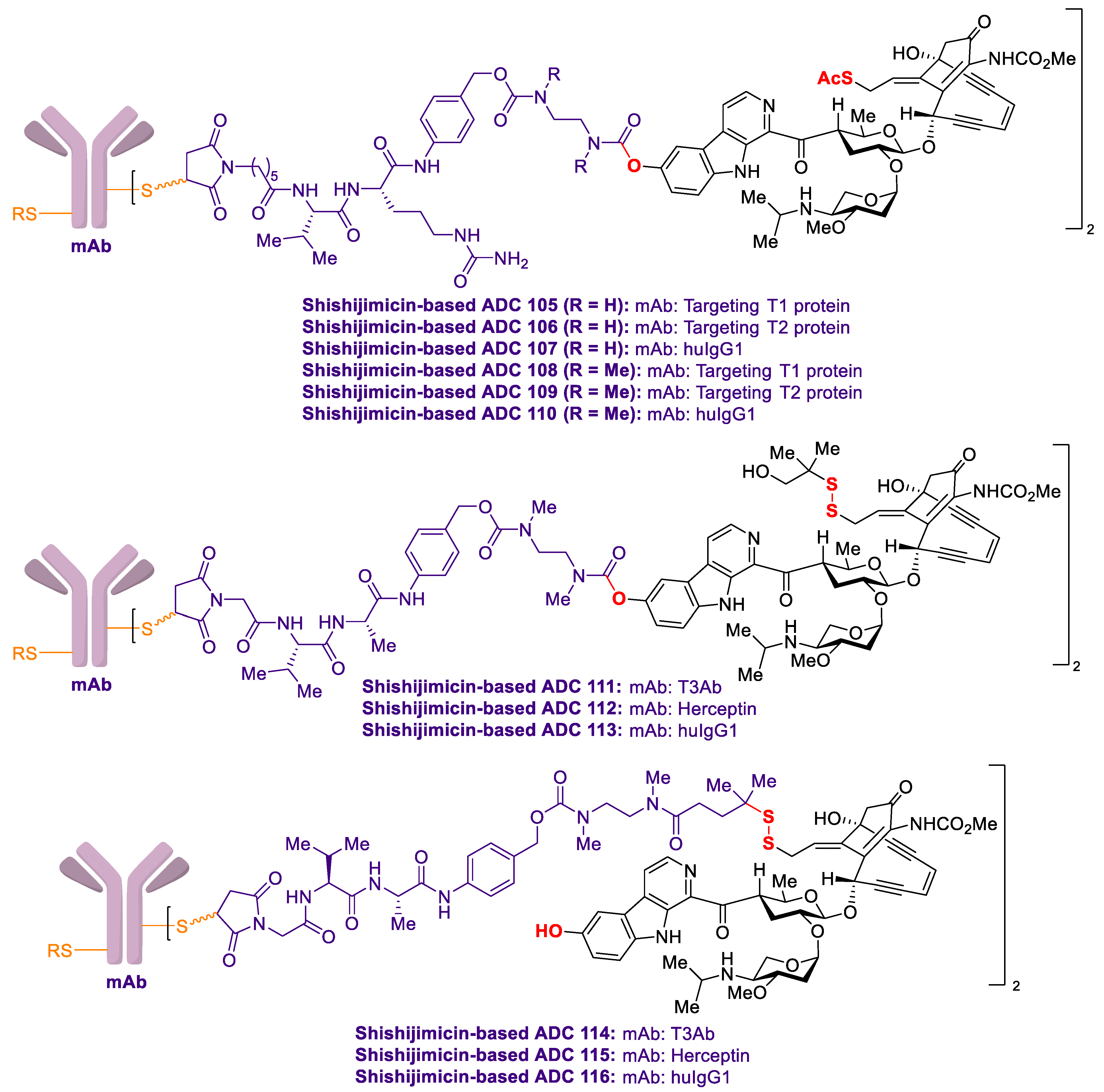
2.4. Antibody-Drug Conjugates Based on Shishijimicin A
2.5. Antibody-Drug Conjugates Based on Aplyronines
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ehrlich, P. Address in pathology, on chemotherapy: Delivered before the seventeenth international congress of medicine. Br. Med. J. 1913, 2, 353–359. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.-C. Antibody-drug conjugates: A historical review. In Antibody Drug Conjugates—The 21st Century Magic Bullets for Cancer; Springer: Cham, Switzerland, 2015; Volume 17, ISBN 978-3-319-13080-4. [Google Scholar]
- Pietersz, G.A.; Krauer, K. Antibody-targeted drugs for the therapy of cancer. J. Drug Target. 1994, 2, 183–215. [Google Scholar] [CrossRef]
- Petersen, B.H.; DeHerdt, S.V.; Schneck, D.W.; Bumol, T.F. The human immune response to KS1/4-desacetylvinblastine (LY256787) and KS1/4-desacetylvinblastine hydrazide (LY203728) in single and multiple dose clinical studies. Cancer Res. 1991, 51, 2286–2290. [Google Scholar]
- Sievers, E.L.; Larson, R.A.; Stadtmauer, E.A.; Estey, E.; Löwenberg, B.; Dombret, H.; Karanes, C.; Theobald, M.; Bennett, J.M.; Sherman, M.L.; et al. Efficacy and safety of gemtuzumab ozogamicin in patients with with CD33-positive acute myeloid leukemia in first relapse. J. Clin. Oncol. 2001, 19, 3244–3254. [Google Scholar] [CrossRef]
- Linenberger, M.L.; Flowers, T.H.D.; Sievers, E.L.; Gooley, T.A.; Bennett, J.M.; Berger, M.S.; Leopold, L.H.; Appelbaum, F.R.; Bernstein, I.D. Multidrug-resistance phenotype and clinical responses to gemtuzumab ozogamicin. Blood 2001, 98, 988–994. [Google Scholar] [CrossRef] [Green Version]
- Younes, A.; Bartlett, N.L.; Leonard, J.P.; Kennedy, D.A.; Lynch, C.M.; Sievers, E.L.; Forero-Torres, A. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N. Engl. J. Med. 2010, 363, 1812–1821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senter, P.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapse Hodgkin lymphoma and systematic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef]
- LoRusso, P.M.; Weiss, D.; Guardino, E.; Girish, S.; Sliwkowski, M.X. Trastuzumab emtansine: A unique antibody-drug conjugate in development for human epidermal growth factor receptor 2-positive cancer. Clin. Cancer Res. 2011, 17, 6437–6447. [Google Scholar] [CrossRef] [Green Version]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.-Y.; Diéras, V.; Guardino, E.; et al. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef] [Green Version]
- Lamb, Y.N. Inotuzumab ozogamicin: First global approval. Drugs 2017, 77, 1603–1610. [Google Scholar] [CrossRef]
- Dhillon, S. Moxetumomab pasudotox: First global approval. Drugs 2018, 78, 1763–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deeks, E.D. Polatuzumab vedotin: First global approval. Drugs 2019, 79, 1467–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, E.; Weinstock, C.; Zhang, L.; Charlab, R.; Dorff, S.E.; Gong, Y.; Hsu, V.; Li, F.; Ricks, T.K.; Song, P.; et al. FDA approval summary: Enfortumab vedotin for locally advanced or metastatic urothelial carcinoma. Clin. Cancer Res. 2021, 15, 922–927. [Google Scholar] [CrossRef] [PubMed]
- Keam, S. Trastuzumab deruxtecan: First approval. Drugs 2020, 80, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Syed, Y.Y. Sacituzumab govitecan: First approval. Drugs 2020, 80, 1019–1025. [Google Scholar] [CrossRef]
- Markham, A. Belantamab mafodotin: First approval. Drugs 2020, 80, 1607–1613. [Google Scholar] [CrossRef] [PubMed]
- Lee, A. Loncastuximab tesirine: First approval. Drugs 2021, 81, 1229–1233. [Google Scholar] [CrossRef]
- Markham, A. Tisotumab vedotin: First approval. Drugs 2021, 81, 2141–2147. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Li, S.; Han, S.; Shi, C.; Zhang, Y. Antibody drug conjugate: The “biological missile” for targeted cancer therapy. Signal Transduct. Target. Ther. 2022, 7, 93. [Google Scholar] [CrossRef] [PubMed]
- Damelin, M.; Zhong, W.; Myers, J.; Sapra, P. Evolving strategies for target selection for antibody-drug conjugates. Pharm. Res. 2015, 32, 3494–3507. [Google Scholar] [CrossRef]
- Ritchie, M.; Tchistiakova, L.; Scott, N. Implications of receptor-mediated endocytosis and intracellular trafficking dynamics in the development of antibody drug conjugates. mAbs 2013, 5, 13–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staudacher, A.H.; Brown, M.P. Antibody drug conjugates and bystander killing: Is antigen-dependent internalisation required? Br. J. Cancer 2017, 117, 1736–1742. [Google Scholar] [CrossRef] [PubMed]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef]
- Mahalingaiah, P.K.; Ciurlionis, R.; Durbin, K.R.; Yeager, R.L.; Philip, B.K.; Bawa, B.; Mantena, S.R.; Enright, B.P.; Liguori, M.J.; Van Vleet, T.R. Potential mechanisms of target-independent uptake and toxicity of antibody-drug conjugates. Pharmacol. Ther. 2019, 200, 110–125. [Google Scholar] [CrossRef] [PubMed]
- Chari, R.V.J.; Miller, M.L.; Widdison, W.C. Antibody-drug conjugates: An emerging concept in cancer therapy. Angew. Chem. Int. Ed. 2014, 53, 3796–3827. [Google Scholar] [CrossRef] [PubMed]
- Press, M.F.; Cordon-Cardo, C.; Slamon, D.J. Expression of the HER-2/neu proto-oncogene in normal human adult and fetal tissues. Oncogene 1990, 5, 953–962. [Google Scholar] [PubMed]
- Goldmacher, V.S.; Kovtun, Y.V. Antibody-drug conjugates: Using monoclonal antibodies for delivery of cytotoxic payloads to cancer cells. Ther. Deliv. 2011, 2, 397–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adair, J.R.; Howard, P.W.; Hartley, J.A.; Williams, D.G.; Chester, K.A. Antibody-drug conjugates—A perfect synergy. Exp. Opin. Biol. Ther. 2012, 12, 1191–1206. [Google Scholar] [CrossRef]
- Lu, J.; Jiang, F.; Lu, A.; Zhang, G. Linkers having a crucial role in antibody-drug conjugates. Int. J. Mol. Sci. 2016, 17, 561. [Google Scholar] [CrossRef]
- Lyon, R.P.; Bovee, T.D.; Doronina1, S.O.; Burke, P.J.; Hunter, J.H.; Neff-LaFord, H.D.; Jonas, M.; Anderson, M.E.; Setter, J.R.; Senter, P.D. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat. Biotech. 2015, 33, 733–736. [Google Scholar] [CrossRef]
- Su, D.; Zhang, D. Linker design impacts antibody-drug conjugate pharmacokinetics and efficacy via modulating the stability and payload release efficiency. Front. Pharmarcol. 2021, 12, 687926. [Google Scholar] [CrossRef] [PubMed]
- Bargh, J.D.; Isidro-Llobet, A.I.; Parker, J.S.; Spring, D.R. Cleavable linkers in antibody-drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef] [PubMed]
- Teicher, B.A.; Chari, R.V. Antibody-drug conjugate therapeutics: Challenges and potential. Clin. Cancer Res. 2011, 17, 6389–6397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, A.; Teicher, B.A.; Hassan, R. Antibody-drug conjugates for cancer therapy. Lancet Oncol. 2016, 17, e254–e262. [Google Scholar] [CrossRef]
- Tnag, H.; Liu, Y.; Yu, Z.; Sun, M.; Lin, L.; Liu, W.; Han, Q.; Wei, M.; Jin, Y. The analysis of key factors related to ADCs structural design. Front. Pharmacol. 2019, 10, 373. [Google Scholar] [CrossRef] [PubMed]
- Sapra, P.; Shor, B. Monoclonal antibody-based therapies in cancer: Advances and challenges. Pharmacol. Ther. 2013, 138, 452–469. [Google Scholar] [CrossRef]
- Fu, Y.; Ho, M. DNA damaging agent-based antibody-drug conjugates for cancer therapy. Antib. Ther. 2018, 1, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Owonikoko, T.K.; Hussain, A.; Stadler, W.M.; Smith, D.C.; Kluger, H.; Molina, A.M.; Gulati, P.; Shah, A.; Ahlers, C.M.; Cardarelli, P.M.; et al. First-in-human multicenter phase I study of BMS-936561 (MDX-1203), an antibody-drug conjugate targeting CD70. Cancer Chemother. Pharmacol. 2016, 77, 155–162. [Google Scholar] [CrossRef]
- Vollmar, B.S.; Frantz, C.; Schutten, M.M.; Zhong, F.; del Rosario, G.; Go, M.A.T.; Yu, S.-F.; Leipold, D.D.; Kamath, A.V.; Ng, C.; et al. Calicheamicin antibody-drug conjugates with improved properties. Mol. Cancer Ther. 2021, 20, 1112–1120. [Google Scholar] [CrossRef]
- Pahl, A.; Lutz, C.; Hechler, T. Amanitins and their development as payload for antibody-drug conjugates. Drug Discov. Today Technol. 2018, 30, 85–89. [Google Scholar] [CrossRef]
- Goldenberg, D.M.; Sharkey, R.M. Sacituzumab govitecan, a novel, third-generation, antibody-drug conjugate (ADC) for cancer therapy. Exp. Opin. Biol. Ther. 2020, 20, 871–885. [Google Scholar] [CrossRef] [PubMed]
- Mantaj, J.M.; Jackson, P.J.M.; Rahman, K.M.; Thurston, D.E. From anthramycin to pyrrolobenzodiazepine (PBD)-containing antibody–drug conjugates (ADCs). Angew. Chem. Int. Ed. 2016, 56, 462–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, S.J.; Bargh, J.D.; Dannheim, F.M.; Hanby, A.R.; Seki, H.; Counsell, A.J.; Ou, X.; Fowler, E.; Ashman, N.; Takada, Y.; et al. Site-selective modification strategies in antibody-drug conjugates. Chem. Soc. Rev. 2021, 50, 1305–1353. [Google Scholar] [CrossRef] [PubMed]
- Perez, H.L.; Cardarelli, P.M.; Deshpande, S.; Gangwar, S.; Schroeder, G.M.; Vite, G.D.; Borzilleri, R.M. Antibody-drug conjugates: Current status and future directions. Drug Discov. Today 2014, 19, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Mckertish, C.M.; Kayser, V. Advances and Limitations of Antibody Drug Conjugates for Cancer. Biomedicines 2021, 9, 872. [Google Scholar] [CrossRef]
- De Goeij, B.E.C.G.; Lambert, J.M. New developments for antibody-drug conjugate-based therapeutic approaches. Curr. Opin. Immun. 2016, 40, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Diamantis, N.; Banerji, U. Antibody-drug conjugates—An emerging class of cancer treatment. Br. J. Cancer 2016, 114, 362–367. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef]
- Yaghoubi, S.; Karimi, M.H.; Lotfinia, M.; Gharibi, T.; Mahi-Birjand, M.; Kavi, E.; Hosseini, F.; Sepehr, K.S.; Khatami, M.; Bagheri, N.; et al. Potential drugs used in the antibody-drug conjugate (ADC) architecture for cancer therapy. J. Cell. Physiol. 2020, 235, 31–64. [Google Scholar] [CrossRef] [PubMed]
- Hafeez, U.; Parakh, S.; Gan, H.K.; Scott, A.M. Antibody-drug conjugates for cancer therapy. Molecules 2020, 25, 4764. [Google Scholar] [CrossRef]
- Jin, S.; Sun, Y.; Liang, X.; Gu, X.; Ning, J.; Xu, Y.; Chen, S.; Pan, L. Emerging new therapeutic antibody derivatives for cancer treatment. Signal Transduct. Target. Ther. 2021, 7, 39. [Google Scholar] [CrossRef]
- Ceci, C.; Lacal, P.M.; Graziani, G. Antibody-drug conjugates: Resurgent anticancer agents with multi-targeted therapeutic potential. Pharmacol. Ther. 2022, 236, 108106. [Google Scholar] [CrossRef]
- Boghaert, E.R.; Cox, M.C.; Vaidya, K.S. Pathophysiologic and pharmacologic considerations to improve the design and application of antibody-drug conjugates. Cancer Res. 2022, 82, 1858–1869. [Google Scholar] [CrossRef] [PubMed]
- Baah, S.; Laws, M.; Rahman, K.M. Antibody-drug conjugates—A tutorial review. Molecules 2021, 26, 2943. [Google Scholar] [CrossRef]
- Dan, N.D.; Setua, S.; Kashyap, V.K.; Khan, S.; Jaggi, M.; Yallapu, M.M.; Chauhan, C. Antibody-drug conjugates for cancer therapy: Chemistry to clinical implications. Pharmaceuticals 2018, 11, 32. [Google Scholar] [CrossRef] [Green Version]
- Newman, D.J. Natural product based antibody drug conjugates: Clinical status as of November 9, 2020. J. Nat. Prod. 2021, 84, 917–931. [Google Scholar] [CrossRef]
- Chia, C.S.B. A patent review on FDA-Approved antibody-drug conjugates, their linkers and drug payloads. ChemMedChem 2022, 17, e202200032. [Google Scholar] [CrossRef]
- Murali, M.; Kumar, A.R.; Nair, B.; Pavithran, K.; Devan, A.R.; Pradeep, G.K.; Nath, L.R. Antibody-drug conjugate as targeted therapeutics against hepatocellular carcinoma: Preclinical studies and clinical relevance. Clin. Trans. Oncol. 2022, 24, 407–431. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Rigol, S. Total synthesis in search of potent antibody-drug conjugate payloads. From the fundamentals to the translational. Acc. Chem. Res. 2019, 52, 127–139. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Rigol, S. The role of organic synthesis in the emergence and development of antibody-drug conjugates as targeted cancer therapies. Angew. Chem. Int. Ed. 2019, 58, 11206–11241. [Google Scholar] [CrossRef]
- Wang, Y.-J.; Li, Y.-Y.; Liu, X.-Y.; Lu, X.-L.; Cao, X.; Jiao, B.-H. Marine antibody-drug conjugates: Design strategies and research progress. Mar. Drugs 2017, 15, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettit, G.R.; Kamano, Y.; Herald, C.L.; Tuinman, A.A.; Boettner, F.E.; Kizu, H.; Schmidt, J.M.; Baczynskyj, L.; Tomer, K.B.; Bontems, R.J. The isolation and structure of a remarkable marine animal antineoplastic constituent: Dolastatin 10. J. Am. Chem. Soc. 1987, 109, 6883–6885. [Google Scholar] [CrossRef]
- Pettit, G.R.; Singh, S.B.; Hogan, F.; Lloyd-Williams, P.; Herald, D.L.; Burkett, D.D.; Clewlow, P.J. Antineoplastic agents. Part 189. The absolute configuration and synthesis of natural (–)-dolastatin-10. J. Am. Chem. Soc. 1989, 111, 5463–5465. [Google Scholar] [CrossRef]
- Flahive, E.; Srirangam, J. The Dolastatins. In Anticancer Agents from Natural Products, 2nd ed.; Cragg, D.J., Kingston, G.M., Newman, D.G.I., Eds.; CRC Press: Boca Raton, FL, USA, 2011; pp. 263–290. [Google Scholar]
- Bai, R.; Pettit, G.R.; Hamel, E. Dolastatin 10, a powerful cytostatic peptide derived from a marine animal. Inhibition of tubulin polymerization mediated through the vinca alkaloid binding domain. Biochem. Pharmacol. 1990, 39, 1941–1949. [Google Scholar] [CrossRef]
- Maderna, A.; Doroski, M.; Subramanyam, C.; Porte, A.; Leverett, C.A.; Vetelino, B.C.; Chen, Z.; Risley, H.; Parris, K.; Pandit, J.; et al. Discovery of cytotoxic dolastatin-10 analogs with N-terminal modifications. J. Med. Chem. 2014, 57, 10527–10543. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.A.; Hillman, D.W.; Fishkin, P.A.; Krook, J.E.; Tan, W.W.; Kuriakose, P.A.; Alberts, S.R.; Dakhil, S.R. Phase II trial of dolastatin-10 in patients with advanced breast cancer. Investig. New Drugs 2005, 23, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Srirangam, J.K.; Barkoczy, J.; Williams, M.D.; Durkin, K.P.; Boyd, M.R.; Bai, R.; Hamel, E.; Schmidt, J.M.; Chapuis, J.C. Antineoplastic agents. 337. Synthesis of dolastatin 10 structural modifications. Anti-Cancer Drug Des. 1995, 10, 529–544. [Google Scholar]
- Miyazaki, K.; Kobayashi, M.; Natsume, T.; Gondo, M.; Mikami, T.; Sakakibara, K.; Tsukagoshi, S. Synthesis and antitumor activity of novel dolastatin 10 analogs. Chem. Pharm. Bull. 1995, 43, 1706–1718. [Google Scholar] [CrossRef] [Green Version]
- Pettit, G.R.; Srirangam, J.K.; Barkoczy, J.; Williams, M.D.; Boyd, M.R.; Hamel, E.; Pettit, R.K.; Hogan, F.; Bai, R.; Chapuis, J.C.; et al. Antineoplastic agents 365. Dolastatin 10 SAR probes. Anticancer Drug Des. 1998, 13, 243–277. [Google Scholar]
- Doronina, S.O.; Toki, B.E.; Torgov, M.Y.; Mendelsohn, B.A.; Cerveny, C.G.; Chace, D.F.; DeBlanc, R.L.; Gearing, R.P.; Bovee, T.D.; Siegall, C.B.; et al. Development of potent monoclonal antibody auristatin conjugates for cancer therapy. Nat. Biotechnol. 2003, 21, 778–784. [Google Scholar] [CrossRef]
- Maderna, A.; Leverett, C.A. Recent advances in the development of new auristatins: Structural modifications and application in antibody drug conjugates. Mol. Pharm. 2015, 12, 1798–1812. [Google Scholar] [CrossRef]
- Singh, S.B. Discovery and development of dolastatin 10-derived antibody drug conjugate anticancer drugs. J. Nat. Prod. 2022, 85, 666–687. [Google Scholar] [CrossRef]
- Newman, D.J. The “utility” of highly toxic marine-sourced compounds. Mar. Drugs 2019, 17, 324. [Google Scholar] [CrossRef] [Green Version]
- Dyshlovoy, S.A.; Honecker, F. Marine compounds and cancer: Updates 2020. Mar. Drugs 2020, 18, 643. [Google Scholar] [CrossRef]
- Song, X.; Li, R.; Wang, H.; Song, P.; Guo, W.; Chen, Z.-S. Tisotumab vedotin for the treatment of cervical carcinoma. Drugs Today 2022, 58, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Disitamab vedotin: A novel antibody-drug conjugates for cancer therapy. Drug Delivery 2022, 29, 1335–1344. [Google Scholar]
- Do, M.; Wu, C.C.N.; Sonavane, P.R.; Juarez, E.F.; Adams, S.R.; Ross, J.; Rodriguez y Baena, A.; Patel, C.; Mesirov, J.P.; Carson, D.A.; et al. A FZD7-specific antibody-drug conjugate induces ovarian tumor regression in preclinical models. Mol. Cancer Ther. 2022, 21, 113–124. [Google Scholar] [CrossRef]
- Yu, L.; Yao, Y.; Wang, Y.; Zhou, S.; Lai, Q.; Lu, Y.; Liu, Y.; Zhang, R.; Wang, R.; Liu, C.; et al. Preparation and anti-cancer evaluation of Promiximab-MMAE, an anti-CD56 antibody drug conjugate, in small cell lung cancer cell line xenograft models. J. Drug Target. 2018, 26, 905–912. [Google Scholar] [CrossRef]
- Advani, R.H.; Lebovic, D.; Chen, A.; Brunvand, M.; Goy, A.; Chang, J.E.; Hochberg, E.; Yalamanchili, S.; Kahn, R.; Lu, D.; et al. Phase I study of the anti-CD22 antibody-drug conjugate pinatuzumab vedotin with/without rituximab in patients with relapsed/refractory B-cell Non-Hodgkin lymphoma. Clin. Cancer Res. 2017, 23, 1167–1176. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, A.; Cusmai, A.; Acquafredda, S.; Rinaldi, L.; Palmiotti, G. Ladiratuzumab vedotin for metastatic triple negative cancer: Preliminary results, key challenges and clinical potential. Exp. Opin. Investig. Drugs 2022, 31, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Anderson, M.G.; Oleksijew, A.; Vaidya, K.S.; Boghaert, E.R.; Tucker, L.; Zhang, Q.; Han, E.K.; Palma, J.P.; Naumovski, L.; et al. ABBV-339, a c-Met antibody-drug conjugate that targets both MET-amplified and c-MET-overexpressing tumors, irrespective of MET pathway dependence. Clin. Cancer Res. 2017, 23, 992–1000. [Google Scholar] [CrossRef] [Green Version]
- Gerber, D.E.; Infante, J.R.; Gordon, M.S.; Goldberg, S.B.; Martín, M.; Felip, E.; Martínez García, M.; Schiller, J.H.; Spigel, D.R.; Cordova, J.; et al. Phase Ia study of anti-NaPi2b antibody-drug conjugate lifastuzumab vedotin DNIB0600A in patients with non-small cell lung cancer and platinum-resistant ovarian cancer. Clin. Cancer Res. 2020, 26, 364–372. [Google Scholar] [CrossRef] [Green Version]
- Petrylak, D.P.; Vogelzang, N.J.; Chatta, K.; Fleming, M.T.; Smith, D.C.; Appleman, L.J.; Hussain, A.; Modiano, M.; Singh, P.; Tagawa, S.T.; et al. PSMA ADC monotherapy in patients with progressive metastatic castration-resistant prostate cancer following abiraterone and/or enzalutamide: Efficacy and safety in open-label single-arm phase 2 study. Prostate 2020, 80, 99–108. [Google Scholar] [CrossRef]
- Gallery, M.; Zhang, J.; Bradley, D.P.; Brauer, P.; Cvet, D.; Estevam, J.; Danaee, H.; Greenfield, E.; Li, P.; Manfredi, M.; et al. A monomethyl auristatin E-conjugated antibody to guanylyl cyclase C is cytotoxic to target-expressing cells in vitro and in vivo. PLoS ONE 2018, 13, e0191046. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Zhang, X.; Xu, Z.; Li, L.; Liu, W.; Dai, Z.; Zhao, Z.; Xiao, L.; Li, H.; Hu, C. Preclinical evaluation of MRG002, a novel HER2-targeting antibody-drug conjugate with potent antitumor activity against HER2-positive solid tumors. Antibody Ther. 2021, 4, 175–184. [Google Scholar] [CrossRef]
- Hu, X.; Jiang, H.; Bai, W.; Liu, X.; Miao, Q.; Wang, L.; Jin, J.; Cui, A.; Liu, R.; Li, Z. Synthesis, characterization, and targeted chemotherapy of SCT200-linker-monomethyl auristatin E conjugates. Eur. J. Med. Chem. 2021, 216, 113297. [Google Scholar] [CrossRef]
- Li, D.; Poon, K.A.; Yu, S.-F.; Dere, R.; Go, M.; Lau, J.; Zheng, B.; Elkins, K.; Danilenko, D.; Kozak, K.R.; et al. DCDT2980S, an anti-CD22-monomethyl auristatin E antibody-drug conjugate, is a potential treatment for Non-Hodgkin lymphoma. Mol. Cancer Ther. 2013, 12, 1255–1265. [Google Scholar] [CrossRef] [Green Version]
- Von Achenbach, C.; Silginer, M.; Blot, V.; Weiss, W.A.; Weller, M. Depatuxizumab mafodotin (ABT-414)-induced glioblastoma cell death requires EGFR overexpression, but not EGFRY1068 phosporylation. Mol. Cancer Ther. 2020, 19, 1328–1339. [Google Scholar] [CrossRef]
- Satomaa, T.; Helin, J.; Ekholm, F.S. Glycoprotein Toxic Payload Conjugates. U.S. Patent 10973922B2, 13 April 2021. [Google Scholar]
- Yang, K.; Chen, B.; Gianolio, D.A.; Stefano, J.E.; Busch, M.; Manning, C.; Alving, K.; Gregory, R.C.; Brondyk, W.H.; Miller, R.J. Convergent synthesis of hydrophilic monomethyl dolastatin 10 based drug linkers for antibody-drug conjugation. Org. Biomol. Chem. 2019, 17, 8115–8124. [Google Scholar] [CrossRef]
- Graziani, E.I.; Sung, M.; Ma, D.; Narayanan, B.; Marquette, K.; Puthenveetil, S.; Tumey, L.N.; Bikker, J.; Casavant, J.; Bennett, E.M.; et al. PF-06804103, a site-specific anti-HER2 antibody-drug conjugate for the treatment of HER2-expressing breast, gastric, and lung cancers. Mol. Cancer Ther. 2020, 19, 2068–2078. [Google Scholar] [CrossRef]
- Maitland, M.L.; Sachdev, J.C.; Sharma, M.R.; Moreno, V.; Boni, V.; Kummar, S.; Stringer-Reasor, E.; Lakhani, N.; Moreau, A.R.; Xuan, D.; et al. First-in-human study of PF-06647020 (cofetuzumab pelidotin), an antibody-drug conjugate targeting protein tyrosine kinase 7, in advanced solid tumors. Clin. Cancer Res. 2021, 27, 4511–4520. [Google Scholar] [CrossRef] [PubMed]
- King, G.T.; Eaton, K.D.; Beagle, B.R.; Zopf, C.J.; Wong, G.Y.; Krupka, H.I.; Hua, S.Y.; Messersmith, W.A.; El-Khoueiry, A.B. A phase 1, dose-escalation study of PF-06664178, an anti-Trop-2/Aur0101 antibody-drug conjugate in patients with advanced or metastatic solid tumors. Investig. New Drugs 2018, 36, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Rosen, L.S.; Wesolowski, R.; Baffa, R.; Liao, K.-H.; Hua, S.Y.; Gibson, B.L.; Pirie-Shepherd, S.; Tolcher, A.W. A phase I, dose-escalation study of PF-06650808, an anti-Notch3 antibody-drug conjugate, in patients with breast cancer and other advanced solid tumors. Investig. New Drugs 2020, 38, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Doronina, S.O.; Bovee, T.D.; Meyer, D.W.; Miyamoto, J.B.; Anderson, M.E.; Morris-Tilden, C.A.; Senter, P.D. Novel peptide linkers for highly potent antibody-auristatin conjugate. Bioconjugate Chem. 2008, 19, 1960–1963. [Google Scholar] [CrossRef]
- Shi, B.; Wu, M.; Li, Z.; Xie, Z.; Wei, X.; Fan, J.; Xu, Y.; Ding, D.; Akash, S.H.; Chen, S.; et al. Antitumor activity of a 5T4 targeting antibody drug conjugate with a novel payload derived from MMAF via C-lock linker. Cancer Med. 2019, 8, 1793–1805. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.J.; Jeong, J.-k.; Choi, Y.M.; Minseob, L.; Kim, Y.J.; Kim, K.S.; Choi, J.H.; Lee, J.S.; Cho, E.J.; Song, H. Antibody-Linker-Drug Conjugate, Preparation Method Therof, and Anticancer Drug Composition Containing Same. U.S. Patent 9814784B2, 14 November 2017. [Google Scholar]
- Lerchen, H.-G.; Sheikh, S.E.; Stelte-Ludwig, B.; Golfier, S.; Schuhmacher, J.; Gnoth, M.; Krenz, U. N-Carboxyalkyl-Auristatin and the Use Thereof. U.S. Patent 8987209B2, 24 March 2015. [Google Scholar]
- Moquist, P.N.; Bovee, T.D.; Waight, A.B.; Mitchell, J.A.; Miyamoto, J.B.; Mason, M.L.; Emmerton, K.K.; Stevens, N.; Balasubramanian, C.; Simmons, J.K.; et al. Novel auristatins with high bystander and cytotoxic activities in drug efflux-positive tumor models. Mol. Cancer Ther. 2021, 20, 320–328. [Google Scholar] [CrossRef]
- Skidmore, L.; Sakamuri, S.; Knudsen, N.A.; Hewet, A.G.; Milutinovic, S.; Barkho, W.; Biroc, S.L.; Kirtley, J.; Marsden, R.; Storey, K.; et al. ARX788, a site-specific anti-HER2 antibody-drug conjugate, demonstrates potent and selective activity in HER2-low and T-DM1-resistant breast and gastric cancers. Mol. Cancer Ther. 2020, 19, 1833–1843. [Google Scholar] [CrossRef]
- Shastri, P.N.; Zhu, J.; Skidmore, L.; Liang, X.; Ji, Y.; Gu, Y.; Tian, F.; Yao, S.; Xia, G. Nonclinical development of next-generation site-specific HER2-targeting antibody-drug conjugate (ARX788) for breast cancer treatment. Mol. Cancer Ther. 2020, 19, 1822–1832. [Google Scholar] [CrossRef]
- He, K.; Xu, J.; Liang, J.; Jiang, J.; Tang, M.; Ye, X.; Zhang, Z.; Zhang, L.; Fu, B.; Li, Y.; et al. Discovery of a novel EGFR-targeting antibody-drug conjugate, SHR-A1307, for the treatment of solid tumors resistant or refractory to anti-EGFR therapies. Mol. Cancer Ther. 2019, 18, 1104–1114. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.-Y.; Wang, L.; Sun, X.; Tang, M.; Quan, H.-T.; Zhang, L.-S.; Lou, L.-G.; Gou, S.-H. SHR-A1403, a novel c-Met antibody-drug conjugate, exerts encouraging anti-tumor activity in c-Met-overexpressing models. Acta Pharmacol. Sin. 2019, 40, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Alley, S.C.; Benjamin, D.R.; Jeffrey, S.C.; Okeley, N.M.; Meyer, D.L.; Sanderson, R.J.; Senter, P.D. Contribution of linker stability to the activities of anticancer immunoconjugates. Bioconjugate Chem. 2008, 19, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Lyon, R.P.; Setter, J.R.; Bovee, T.D.; Doronina, S.O.; Hunter, J.H.; Anderson, M.E.; Balasubramanian, C.L.; Duniho, S.M.; Leiske, C.I.; Senter, P.D. Self-hydrolyzing maleimides improve the stability and pharmacological properties of antibody-drug conjugates. Nat. Biotechnol. 2014, 32, 1059–1062. [Google Scholar] [CrossRef]
- Axup, J.Y.; Bajjuri, K.M.; Ritland, M.; Hutchins, B.M.; Kim, C.H.; Kazane, S.A.; Halder, R.; Forsyth, J.S.; Santidrian, A.F.; Stafin, K.; et al. Synthesis of site-specific antibody-drug conjugates using unnatural amino acids. Proc. Natl. Acad. Sci. USA 2012, 109, 16101–16106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, J.T.; Malinao, M.-C.; Dugal-Tessier, J.; Atkinson, J.E.; Anand, B.S.; Okada, A.; Mendelsohn, B.A. Metabolism of an oxime-linked antibody drug conjugate, AGS62P1, and characterization of its identified metabolite. Mol. Pharmaceutics 2018, 15, 2384–2390. [Google Scholar] [CrossRef]
- Bryden, F.; Martin, C.; Letast, S.; Lles, E.; Viéitez-Villemin, I.; Rousseau, A.; Colas, C.; Brachet-Botineau, M.; Allard-Vannier, E.; Larbouret, C.; et al. Impact of cathepsin B-sensitive triggers and hydrophilic linkers on in vitro efficacy of novel site-specific antibody-drug conjugates. Org. Biomol. Chem. 2018, 16, 1882–1889. [Google Scholar] [CrossRef]
- Juen, L.; Baltus, C.B.; Gély, C.; Feuillatre, O.; Desgranges, A.; Viaud-Massuard, M.-C.; Martin, C. Innovative bioconjugation technology for antibody-drug conjugates: Proof of concept in a CD30-positive lymphoma mouse model. Bioconjugate Chem. 2021, 32, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Juen, L.; Baltus, C.B.; Gély, C.; Kervarrec, T.; Feuillatre, O.; Desgranges, A.; Viaud-Massuard, M.-C.; Martin, C. Therapeutic potential of MF-TTZ-MMAE, a site-specifically conjugated antibody-drug conjugate, for the treatment of HER2-overexpressing breast cancer. Bioconjugate Chem. 2022, 33, 418–426. [Google Scholar] [CrossRef]
- Esnault, C.; Leblond, V.; Martin, C.; Desgranges, A.; Baltus, C.B.; Aubrey, N.; Lakhrif, Z.; Lajoie, L.; Lantier, L.; Clémenceau, B.; et al. Adcitmer®, a new CD56-targeting monomethyl auristatin E-conjugated antibody, is a potential therapeutic approach in Merkel cell carcinoma. Br. J. Dermatol. 2022, 186, 295–306. [Google Scholar] [CrossRef]
- Badescu, G.; Bryant, P.; Bird, M.; Henseleit, K.; Swierkosz, J.; Parekh, V.; Tommasi, R.; Pawlisz, E.; Jurlewicz, K.; Farys, M.; et al. Bridging disulfides for stable and defined antibody drug conjugates. Bioconjugate Chem. 2014, 25, 1124–1136. [Google Scholar] [CrossRef]
- Bryant, P.; Pabst, M.; Badescu, G.; Bird, M.; McDowell, W.; Jamieson, E.; Swierkosz, J.; Jurlewicz, K.; Tommasi, R.; Henseleit, K.; et al. In vitro and in vivo evaluation of cysteine rebridged trastuzumab-MMAE antibody drug conjugates with defined drug-to-antibody ratios. Mol. Pharmaceutics 2015, 12, 1872–1879. [Google Scholar] [CrossRef]
- Yang, M.-C.; Shia, C.-S.; Li, W.-F.; Wang, C.-C.; Chen, I.-J.; Huang, T.-Y.; Chen, Y.-J.; Chang, H.-W.; Lu, C.-H.; Wu, Y.-C.; et al. Preclinical studies of OBI-999: A novel Globo H-targeting antibody-drug conjugate. Mol. Cancer Ther. 2021, 20, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Amar, I.A.M.; Huvelle, S.; Douez, E.; Letast, S.; Henrion, S.; Viaud-Massuard, M.-C.; Aubrey, N.; Allard-Vannier, E.; Joubert, N.; Denevault-Sabourin, C. Dual intra- and extracellular release of monomethyl auristatin E from a neutrophil elastase-sensitive antibody-drug conjugate. Eur. J. Med. Chem. 2022, 229, 114063. [Google Scholar] [CrossRef]
- Hui, X.; Yuan, C.; Cao, W.; Ge, W.; Zhang, D.; Dan, M.; Zhao, Q.; Liu, B.; Yao, B. An innovative site-specific anti-HER2 antibody-drug conjugate with high homogeneity and improved therapeutic index. Oncotargets Ther. 2022, 15, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Boschanski, M.; Krüger, T.; Karsten, L.; Falck, G.; Alam, S.; Gerlach, M.; Müller, B.; Müller, K.M.; Sewald, N.; Dierks, T. Site-specific conjugation strategy for dual antibody-drug conjugates using aerobic formylglycine-generating enzymes. Bioconjugate Chem. 2021, 32, 1167–1174. [Google Scholar] [CrossRef]
- Chuprakov, S.; Ogunkoya, A.O.; Barfield, R.M.; Bauzon, M.; Hickle, C.; Kim, Y.C.; Yeo, D.; Zhang, F.; Rabuka, D.; Drake, P.M. Tandem-cleavage linkers improve the in vivo stability and tolerability of antibody-drug conjugates. Bioconjugate Chem. 2021, 32, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Kudirka, R.; Albers, A.E.; Barfield, R.M.; de Hart, G.W.; Drake, P.M.; Jones, L.C.; Rabuka, D. Hydrazino-Pictet-Spengler ligation as a biocompatible method for the generation of stable protein conjugates. Bioconjugate Chem. 2013, 24, 846–851. [Google Scholar] [CrossRef]
- Yurkovetskiy, A.V.; Bodyak, N.D.; Yin, M.; Thomas, J.D.; Clardy, S.M.; Conlon, P.R.; Stevenson, C.A.; Uttard, A.; Qin, L.; Gumerov, D.R.; et al. Dolaflexin: A novel antibody-drug conjugate platform featuring high drug loading and a controlled bystander effect. Mol. Cancer Ther. 2021, 20, 885–895. [Google Scholar] [CrossRef]
- Bodyak, N.D.; Mosher, R.; Yurkovetskiy, A.V.; Yin, M.; Bu, C.; Conlon, P.R.; Demady, D.R.; DeVit, M.J.; Gumerov, D.R.; Gurijala, V.R.; et al. The dolaflexin-based antibody-drug conjugate XMT-1536 targets the solid tumor lineage antigen SLC34A2/NaPi2b. Mol. Cancer Ther. 2021, 20, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.; Deweid, L.; Pirzer, T.; Yanakieva, D.; Englert, S.; Becker, B.; Avrutina, O.; Kolmar, H. Dextramabs: A novel format of antibody-drug conjugates featuring a multivalent polysaccharide scaffold. ChemistryOpen 2019, 8, 354–357. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Zhang, X.-N.; Chen, J.; Cheng, Q.; Pei, H.; Louie, S.G.; Zhang, Y. A poly-ADP-ribose polymer-based antibody-drug conjugate. Chem. Sci. 2020, 11, 9303–9308. [Google Scholar] [CrossRef]
- Märcher, A.; Nijenhuis, M.A.D.; Gothelf, K.V. A wireframe DNA cube: Antibody conjugate for targeted delivery of multiple copies of monomethyl auristatin E. Angew. Chem. Int. Ed. 2021, 60, 21691–21696. [Google Scholar] [CrossRef] [PubMed]
- Serpell, C.J.; Edwardson, T.G.W.; Chidchob, P.; Carneiro, K.M.M.; Sleiman, H.F. Precision polymers and 3D DNA nanostructures: Emergent assemblies from new parameters space. J. Am. Chem. Soc. 2014, 136, 15767–15774. [Google Scholar] [CrossRef] [PubMed]
- Dovgan, I.; Ehkirch, A.; Lehot, V.; Kuhn, I.; Koniev, O.; Kolodych, S.; Hentz, A.; Ripoll, M.; Ursuegui, S.; Nothisen, M.; et al. On the use of DNA as a linker in antibody-drug conjugates: Synthesis, stability and in vivo potency. Sci. Rep. 2020, 10, 7691. [Google Scholar] [CrossRef]
- Machulkin, A.E.; Uspenskaya, A.A.; Zyk, N.U.; Nimenko, E.A.; Ber, A.P.; Petrov, S.A.; Polshakov, V.I.; Shafikov, R.R.; Skortsov, D.A.; Plotnikova, E.A.; et al. Synthesis, characterization, and preclinical evaluation of a small-molecule prostate-specific membrane antigen-targeted monomethyl auristatin E conjugate. J. Med. Chem. 2021, 64, 17123–17145. [Google Scholar] [CrossRef] [PubMed]
- Paulus, J.; Sewald, N. Synthesis and evaluation of a non-peptide small-molecule drug conjugate targeting integrin ανβ3. Front. Chem. 2022, 10, 869639. [Google Scholar] [CrossRef]
- Zhang, H.; Jin, C.; Zhang, L.; Peng, B.; Zhang, Y.; Liu, Y.; Li, L.; Ye, M.; Xiong, W.; Tan, W. CD71-specific aptamer conjugated with monomethyl auristatin E for the treatment of uveal melanoma. ACS Appl. Mater. Interfaces 2022, 14, 32–40. [Google Scholar] [CrossRef]
- Luo, D.; Wang, W.; Walker, E.; Springer, S.; Ramamurthy, G.; Burda, C.; Basilion, J.P. Targeted chemoradiotherapy of prostate cancer using gold nanoclusters with protease activatable monomethyl auristatin E. ACS Appl. Mater. Interfaces 2022, 14, 14916–14927. [Google Scholar] [CrossRef] [PubMed]
- Cahuzac, H.; Sallustrau, A.; Malgorn, C.; Beau, F.; Barbe, P.; Babin, V.; Dubois, S.; Palazzolo, A.; Thai, R.; Correia, I.; et al. Monitoring in vivo performances of protein-drug conjugates using site-selective dual radiolabelling and ex vivo digital imaging. J. Med. Chem. 2022, 65, 6953–6968. [Google Scholar] [CrossRef]
- Uemura, D.; Takahashi, K.; Yamamoto, T. Norhalichondrin A: An antitumor polyether macrolide from a marine sponge. J. Am. Chem. Soc. 1985, 107, 4796–4798. [Google Scholar] [CrossRef]
- Pettit, G.R.; Tan, R.; Gao, F.; Williams, M.D.; Doubek, D.L.; Boyd, M.R.; Schmidt, J.M.; Chapuis, J.-C.; Hamel, E.; Bai, R.; et al. Isolation and structure of halistatin 1 from the eastern Indian Ocean marine sponge Phakellia carteri. J. Org. Chem. 1993, 58, 2538–2543. [Google Scholar] [CrossRef]
- Litaudon, M.; Hart, J.B.; Blunt, J.W.; Lake, R.J.; Munro, M.H.G. Isohomohalichondrin B, a new antitumour polyether macrolide from the New Zealand deep-water sponge Lissodendoryx sp. Tetrahedron Lett. 1994, 35, 9435–9438. [Google Scholar] [CrossRef]
- Litaudon, M.; Hickford, S.J.H.; Lill, R.E.; Lake, R.J.; Blunt, J.W.; Munro, M.H.G. Antitumor Polyether Macrolides: New and Hemisynthetic Halichondrins from the New Zealand Deep-Water Sponge Lissodendoryx sp. J. Org. Chem. 1997, 62, 1868–1871. [Google Scholar] [CrossRef]
- Hickford, S.J.H.; Blunt, J.W.; Munro, M.H.G. Antitumour polyether macrolides: Four new halichondrins from the New Zealand deep-water marine sponge Lissodendoryx sp. Bioorg. Med. Chem. 2009, 17, 2199–2203. [Google Scholar] [CrossRef]
- Pettit, G.R.; Herald, C.L.; Boyd, M.R.; Leed, J.E.; Dufrense, C.; Doubek, D.L.; Schmidt, J.M.; Cerny, R.L.; Hooper, J.N.A.; Rützler, K.C. Antineoplastic agents. 219. Isolation and structure of the cell growth inhibitory constituents from the western Pacific marine sponge Axinella sp. J. Med. Chem. 1991, 34, 3339–3340. [Google Scholar] [CrossRef]
- Ueda, A.; Yamamoto, A.; Kato, D.; Kishi, Y. Total Synthesis of Halichondrin A, the Missing Member in the Halichondrin Class of Natural Products. J. Am. Chem. Soc. 2014, 136, 5171–5176. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Paull, K.D.; Heraldy, C.L.; Malspeis, L.; Pettit, G.R.; Hamel, E. Halichondrin B and homohalichondrin B, marine natural products binding in the vinca domain of tubulin. Discovery of tubulin-based mechanism of action by analysis of differential cytotoxicity data. J. Biol. Chem. 1991, 266, 15882–15889. [Google Scholar] [CrossRef]
- Hirata, Y.; Uemura, D. Halichondrins—Antitumor polyether macrolides from a marine sponge. Pure Appl. Chem. 1986, 58, 701–710. [Google Scholar] [CrossRef] [Green Version]
- Dabydeen, D.; Burnett, J.C.; Bai, R.; Verdier-Pinard, P.; Hickford, S.J.H.; Pettit, G.R.; Blunt, J.W.; Munro, M.H.G.; Gussio, R.; Hamel, E. Comparison of the Activities of the Truncated Halichondrin B Analog NSC 707389 (E7389) with Those of the Parent Compound and a Proposed Binding Site on Tubulin. Mol. Pharmacol. 2006, 70, 1866–1875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, M.A.; Kamath, K.; Manna, T.; Okouneva, T.; Miller, H.P.; Davis, C.; Littlefield, B.A.; Wilson, L. The primary antimitotic mechanism of action of the synthetic halichondrin E7389 is suppression of microtubule growth. Mol. Cancer Ther. 2005, 4, 1086–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overmoyer, B. Options for the Treatment of Patients with Taxane-Refractory Metastatic Breast Cancer. Clin. Breast Cancer 2008, 8, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Aicher, T.D.; Buszek, K.R.; Fang, F.G.; Forsyth, C.J.; Jung, S.H.; Kishi, Y.; Matelich, M.C.; Scola, P.M.; Spero, D.M.; Yoon, S.K. Total synthesis of halichondrin B and norhalichondrin B. J. Am. Chem. Soc. 1992, 114, 3162–3164. [Google Scholar] [CrossRef]
- Towle, M.J.; Salvato, K.A.; Budrow, J.; Wels, B.F.; Kuznetsov, G.; Aalfs, K.K.; Welsh, S.; Zheng, W.; Seletsk, B.M.; Palme, M.H.; et al. In vitro and in vivo anticancer activities of synthetic macrocyclic ketone analogues of Halichondrin B. Cancer Res. 2001, 61, 1013–1021. [Google Scholar] [PubMed]
- Towle, M.J.; Nomoto, K.; Asano, M.; Kishi, Y.; Yu, M.J.; Littlefield, B.A. Broad spectrum preclinical antitumor activity of eribulin (Halaven®): Optimal effectiveness under intermitent dosimg conditions. Anticancer Res. 2012, 32, 1611–1619. [Google Scholar]
- Funahashi, Y.; Okamoto, K.; Adachi, Y.; Semba, T.; Uesugi, M.; Ozawa, Y.; Tohyama, O.; Uehara, T.; Kimura, T.; Watanabe, H.; et al. Eribulin mesylate reduces tumor microenvironment abnormality by vascular remodeling in preclinical human breast cancer models. Cancer Sci. 2014, 105, 1334–1343. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Ozawa, Y.; Kimura, T. Eribulin mesilate suppresses experimental metastasis of breast cancer cells by reversing phenotype from epithelial-mesenchymal transition (EMT) to mesenchymal-epithelial transition (MET) states. Br. J. Cancer 2014, 110, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.; O’Shaughnessy, J.; Loesch, D.; Blum, J.L.; Vahdat, L.T.; Petrakova, K.; Chollet, P.; Manikas, A.; Diéras, V.; Delozier, T.; et al. Eribulin monotherapy versus treatment of physician’s choice in patients with metastatic breast cancer (EMBRACE): A phase 3 open-label randomised study. Lancet 2011, 377, 914–923. [Google Scholar] [CrossRef]
- Bauer, A. Story of Eribulin Mesylate: Development of the Longest Drug Synthesis. Synthesis of Heterocycles in Contemporary Medicinal Chemistry. In Topics in Heterocyclic Chemistry; Springer: Cham, Switzerland, 2016; Volume 44, ISBN 978-3-319-39917-1/978-3-319-39915-7. [Google Scholar]
- Cheng, X.; Li, J.; Tanaka, K.; Majumder, U.; Milinichik, A.Z.; Verdi, A.C.; Maddage, C.J.; Rybinski, K.A.; Fernando, S.; Fernando, D.; et al. MORAb-202, an antibody-drug conjugate utilizing humanized anti-human Frα farletuzumab and the microtubule-targeting agent eribulin, has potent antitumor activity. Mol. Cancer Ther. 2018, 17, 2665–2675. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, Y.; Yamaoka, T.; Ohba, M.; Miura, S.; Masuda, H.; Sangai, T.; Takimoto, M.; Nakamura, S.; Tsurutani, J. Novel anti-FOLR1 antibody-drug conjugate MORAb-202 in breast cancer and non-small cell lung cancer cells. Antibodies 2021, 10, 6. [Google Scholar] [CrossRef]
- Martín, M.J.; Coello, L.; Fernández, R.; Reyes, R.; Rodríguez, A.; Murcia, C.; Garranzo, M.; Mateo, C.; Sánchez-Sancho, F.; Bueno, S.; et al. Isolation and first total synthesis of PM050489 and PM060184, two new marine anticancer compounds. J. Am. Chem. Soc. 2013, 135, 10164–10171. [Google Scholar] [CrossRef]
- Pera, B.; Barasoain, I.; Pantazopoulou, A.; Canales, A.; Matesanz, R.; Rodriguez-Salarichs, J.; García-Fernandez, L.F.; Moneo, V.; Jiménez-Barbero, J.; Galmarini, C.M.; et al. New interfacial microtubule inhibitors of marine origin, PM050489/PM060184, with potent antitumor activity and a distinct mechanism. ACS Chem. Biol. 2013, 8, 2084–2094. [Google Scholar] [CrossRef]
- Avilés, P.; Domínguez, J.M.; Guillén, M.J.; Muñoz-Alonso, M.J.; Mateo, C.; Rodriguez-Acebes, R.; Molina-Guijarro, J.M.; Francesch, A.; Martínez-Leal, J.F.; Munt, S.; et al. MI130004, a novel antibody-drug conjugate combining trastuzumab with a molecule of marine origin, shows outstanding in vivo activity against HER2-expressing tumors. Mol. Cancer Ther. 2018, 17, 786–794. [Google Scholar] [CrossRef] [Green Version]
- Domínguez, J.M.; Pérez-Chacón, G.; Guillén, M.J.; Muñoz-Alonso, M.J.; Somovilla-Crespo, B.; Cibrián, D.; Acosta-Iborra, B.; Adrados, M.; Muñoz-Calleja, C.; Cuevas, C.; et al. CD13 as a new tumor target for antibody-drug conjugates: Validation with the conjugate MI130110. J. Hematol. Oncol. 2020, 13, 32. [Google Scholar] [CrossRef]
- Oku, N.; Matsunaga, S.; Fusetani, N. Shishijimicins A-C, novel enediyne antitumor antibiotics from the ascidian Didemnum proliferum. J. Am. Chem. Soc. 2003, 125, 2044–2045. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Lu, Z.; Li, R.; Woods, J.R.; Sohn, T.-I. Total synthesis of shishijimicin A. J. Am. Chem. Soc. 2015, 137, 8716–8719. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Li, R.; Lu, Z.; Pitsinos, E.N.; Alemany, L.B.; Aujay, M.; Lee, C.; Sandoval, J.; Gavrilyuk, J. Streamlined total synthesis of shishijimicin A and its application to the design, synthesis, and biological evaluation of analogues thereof and practical syntheses of PhthNSSMe and related sulfenylating reagents. J. Am. Chem. Soc. 2018, 140, 12120–12136. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, R.; Ba, S.; Lu, Z.; Pitsinos, E.N.; Li, T.; Nicolaou, K.C. DNA binding and cleavage modes of shishijimicin A. J. Am. Chem. Soc. 2019, 141, 7842–7852. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Li, R.; Chen, Q.; Lu, Z.; Pitsinos, E.N.; Schammel, A.; Lin, B.; Gu, C.; Sarvaiya, H.; Tchelepi, R.; et al. Synthesis and biological evaluation of shishijimicin A-type linker-drugs and antibody-drug conjugates. J. Am. Chem. Soc. 2020, 142, 12890–12899. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Ojika, M.; Ishigaki, T.; Yoshida, Y. Aplyronine A, a potent antitumor substance, and the congeners aplyronines B and C isolated from the sea hare Aplysia kurodai. J. Am. Chem. Soc. 1993, 115, 11020–11021. [Google Scholar] [CrossRef]
- Ojida, M.; Kigoshi, H.; Ishigaki, T.; Yamada, K. Further studies on aplyronine A, an antitumor substance isolated from the sea hare Aplysia kurodai. Tetrahedron Lett. 1993, 34, 8501–8504. [Google Scholar]
- Ojida, M.; Kigoshi, H.; Ishigaki, T.; Nisiwaki, M.; Tsukada, I.; Mizuta, K.; Yamada, K. Studies on the stereochemistry of aplyronine A: Determination of the stereochemistry of the C21-C34 fragment. Tetrahedron Lett. 1993, 34, 8505–8508. [Google Scholar]
- Ojida, M.; Kigoshi, H.; Ishigaki, T.; Tsukada, I.; Tsuboi, T.; Ogawa, T.; Yamada, K. Absolute stereochemistry of aplyronine A, a potent antitumor substance of marine origin. J. Am. Chem. Soc. 1994, 116, 7441–7442. [Google Scholar]
- Ojida, M.; Kigoshi, H.; Ishigaki, T.; Suenaga, K.; Mutou, T.; Sakakura, A.; Ogawa, T.; Yamada, K. Total Synthesis of Aplyronine A, a Potent Antitumor Substance of Marine Origin. J. Am. Chem. Soc. 1994, 116, 7443–7444. [Google Scholar]
- Yamada, K.; Ojika, M.; Kigoshi, H.; Suenaga, K. Aplyronine A, a potent antitumor macrolide of marine origin, and the congeners aplyronines B-H: Chemistry and biology. Nat. Prod. Rep. 2009, 26, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Ojida, M.; Kigoshi, H.; Suneaga, K.; Imamura, Y.; Yoshikawa, K.; Ishigaki, T.; Sakakura, A.; Mutou, T.; Yamada, K. Aplyronines D-H from the sea hare Aplysia kurodai: Isolation, structures and cytotoxicity. Tetrahedron 2012, 68, 982–987. [Google Scholar]
- Saito, S.; Karaki, H. A family of novel actin-inhibiting marine toxins. Clin. Exp. Pharm. Phys. 1996, 23, 743–746. [Google Scholar] [CrossRef]
- Saito, S.; Watabe, S.; Ozaki, H.; Ozaki, H.; Kigoshi, H.; Yamada, K.; Fusetani, N.; Karaki, H. Novel actin depolymerizing macrolide aplyronine A. J. Biochem. 1996, 120, 552–555. [Google Scholar] [CrossRef]
- Suneaga, K.; Kamei, N.; Okugawa, Y.; Takagi, M.; Akao, A.; Kigoshi, H.; Yamada, K. Cytotoxicity and actin depolymerizing activity of aplyronine A, a potent antitumor macrolide of marine origin, and the natural and artificial analogs. Bioorg. Med. Chem. Lett. 1997, 7, 269–274. [Google Scholar] [CrossRef]
- Kigoshi, H.; Suneaga, K.; Takagi, M.; Akao, A.; Kanematsu, K.; Kamei, N.; Okugawa, Y.; Yamada, K. Cytotoxicity and actin-depolymerizing activity of aplyronine A, a potent antitumor macrolide of marine origin, and its analogs. Tetrahedron 2002, 58, 1075–1102. [Google Scholar] [CrossRef]
- Kuroda, T.; Suneaga, K.; Sakakura, A.; Handa, T.; Okamoto, K.; Kigoshi, H. Study of the interaction between actin and antitumor substance aplyronine A with a novel fluorescent photoaffinity probe. Bioconjugate Chem. 2006, 17, 524–529. [Google Scholar] [CrossRef]
- Hirata, K.; Muraoka, S.; Suneaga, K.; Kuroda, T.; Kato, K.; Tanaka, H.; Yamamoto, M.; Takata, M.; Yamada, K.; Kigoshi, H. Structure basis for antitumor effect of aplyronine A. J. Mol. Biol. 2006, 356, 945–954. [Google Scholar] [CrossRef]
- Kita, M.; Hirayama, Y.; Yoneda, K.; Yamagishi, K.; Chinen, T.; Usui, T.; Sumiya, E.; Uesugi, M.; Kigoshi, H. Inhibition of microtubule assembly by a complex of actin and antitumor macrolide aplyronine A. J. Am. Chem. Soc. 2013, 135, 18089–18095. [Google Scholar] [CrossRef] [PubMed]
- Anzicek, N.; Williams, S.; Housden, M.P.; Paterson, I. Toward aplyronine payloads for antibody-drug conjugates: Total synthesis of aplyronines A and D. Org. Biomol. Chem. 2018, 16, 1343–1350. [Google Scholar] [CrossRef] [PubMed]


























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Payload | Origin | Mechanism of Action | ADC |
|---|---|---|---|
| Calicheamicin | Non-marine | DNA-damaging | Mylotarg, Besponsa |
| MMAE | Marine | Microtubule inhibitor | Adcetris, Polivy, Padcev, Tivdak |
| Maytansine DM1 | Non-marine | Microtubule inhibitor | Kadcyla |
| Pseudomonas exotoxin 38 | Non-marine | Elongation factor-2 inhibitor | Lumoxiti |
| Deruxtecan | Non-marine | DNA-damaging | Enhertu |
| Govitecan | Non-marine | DNA-damaging | Trodelvy |
| MMAF | Marine | Microtubule inhibitor | Blenrep |
| SG3199 | Non-marine | DNA-damaging | Zynlonta |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng-Sánchez, I.; Moya-Utrera, F.; Porras-Alcalá, C.; López-Romero, J.M.; Sarabia, F. Antibody-Drug Conjugates Containing Payloads from Marine Origin. Mar. Drugs 2022, 20, 494. https://doi.org/10.3390/md20080494
Cheng-Sánchez I, Moya-Utrera F, Porras-Alcalá C, López-Romero JM, Sarabia F. Antibody-Drug Conjugates Containing Payloads from Marine Origin. Marine Drugs. 2022; 20(8):494. https://doi.org/10.3390/md20080494
Chicago/Turabian StyleCheng-Sánchez, Iván, Federico Moya-Utrera, Cristina Porras-Alcalá, Juan M. López-Romero, and Francisco Sarabia. 2022. "Antibody-Drug Conjugates Containing Payloads from Marine Origin" Marine Drugs 20, no. 8: 494. https://doi.org/10.3390/md20080494