
The Development of the Bengamides as New Antibiotics against Drug-Resistant Bacteria

 ,
,  , , , , , , , , and
, , , , , , , , and
Abstract
:1. Introduction
2. Recent Progress in the Chemistry and Biology of the Bengamides
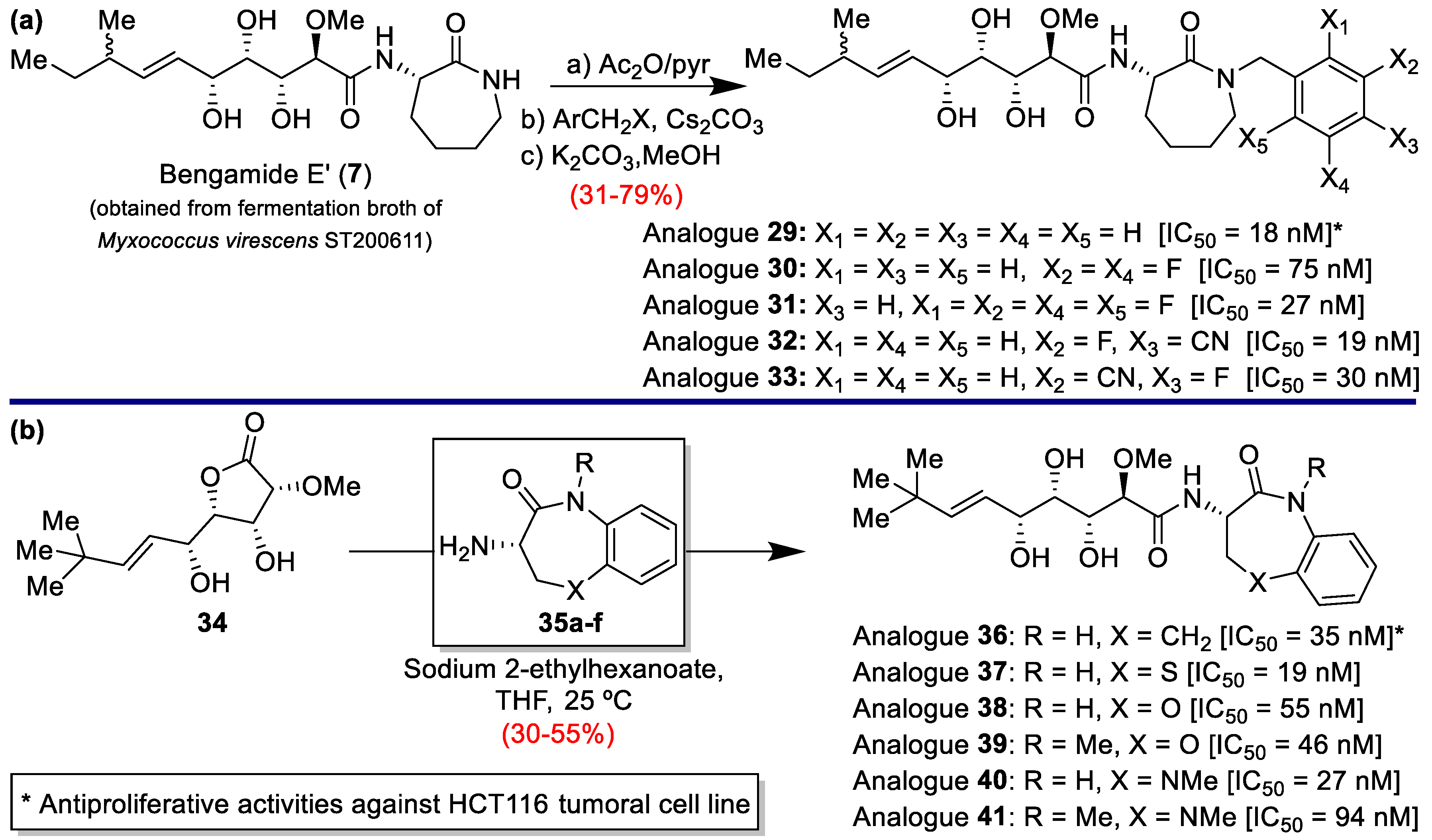
2.1. New Progress in Antitumor Properties of the Bengamides
2.2. Antifungal Activities of the Bengamides
2.3. Antiviral Activity of the Bengamides
3. Development of the Bengamides and Their Analogues as New Antibiotics
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- García-Ruiz, C.; Sarabia, F. Chemistry and biology of bengamides and bengazoles, bioactive natural products from Jaspis sponges. Mar. Drugs 2014, 12, 1580–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quiñoá, E.; Adamczeski, M.; Crews, P.; Bakus, G.J. Bengamides, heterocyclic anthelminthics from a Jaspidae marine sponge. J. Org. Chem. 1986, 51, 4494–4497. [Google Scholar] [CrossRef]
- Adamczeski, M.; Quiñoá, E.; Crews, P. Novel sponge-derived amino acids. 5. Structures, stereochemistry, and synthesis of several new heterocycles. J. Am. Chem. Soc. 1989, 111, 647–654. [Google Scholar]
- Thale, Z.; Kinder, F.R.; Bair, K.W.; Bontempo, J.; Czuchta, A.M.; Versace, R.W.; Phillips, P.E.; Sanders, M.L.; Wattanasin, S.; Crews, P. Bengamides revisited: New structures and antitumor studies. J. Org. Chem. 2001, 66, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- Rudi, A.; Kashman, Y.; Benayahu, Y.; Schleyer, M. Amino acid derivatives from the marine sponge Jaspis digonoxea. J. Nat. Prod. 1994, 57, 829–832. [Google Scholar] [CrossRef]
- DʼAuria, M.V.; Giannini, C.; Minale, L.; Zampella, A.; Debitus, C.; Frostin, M. Bengamides and related new amino acid derivatives from the new caledonian marine sponge Jaspis carteri. J. Nat. Prod. 1997, 60, 814–816. [Google Scholar] [CrossRef]
- Groweiss, A.; Newcomer, J.J.; OʼKeefe, B.R.; Blackman, A.; Boyd, M.R. Cytotoxic metabolites from an Australian collection of the sponge Jaspis species. J. Nat. Prod. 1999, 62, 1691–1693. [Google Scholar] [CrossRef]
- Fernández, R.; Dherbomez, M.; Letourneux, Y.; Nabil, M.; Verbist, J.F.; Biard, J.F. Antifungal metabolites from the marine sponge Pachastrissa sp.: New bengamide and bengazole derivatives. J. Nat. Prod. 1999, 62, 678–680. [Google Scholar] [CrossRef]
- Pettit, G.R.; Hogan, F.; Xu, J.-P.; Tan, R.; Nogawa, T.; Cichacz, Z.; Pettit, R.K.; Du, J.; Ye, Q.-H.; Cragg, G.M.; et al. Antineoplastic agents. 536. New sources of naturally occurring cancer cell growth inhibitors from marine organisms, terrestrial plants and microorganisms. J. Nat. Prod. 2008, 71, 438–444. [Google Scholar] [CrossRef]
- Ovenden, S.P.B.; Nielson, J.L.; Liptrot, C.H.; Willis, R.H.; Tapiolas, D.M.; Wright, A.D.; Motti, C.A. A new diketopiperazine, cyclo-(4-S-hydroxy-R-proline-R-isoleucine), from an australian specimen of the sponge Stelletta sp. Mar. Drugs 2011, 9, 2469–2478. [Google Scholar] [CrossRef]
- Susilowati, F.; Swasono, R.T.; Okino, T.; Haryadi, W. In vitro cytotoxic anticancer potential of bioactive fraction isolated from indonesian tidal sponge Calthropella sp. Asian J. Pharm. Clin. Res. 2019, 12, 380–383. [Google Scholar] [CrossRef] [Green Version]
- Johnson, T.A.; Sohn, J.; Vaske, Y.M.; White, K.N.; Cohen, T.L.; Vervoot, H.C.; Tenney, K.; Valeriote, F.A.; Bjeldanes, L.F.; Crews, P. Myxobacteria versus sponge-derived alkaloids: The bengamide family identified as potent immune modulating agents by scrutiny of LC-MS/ELSD libraries. Bioorg. Med. Chem. Lett. 2012, 20, 4348–4355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenzel, S.C.; Hoffmann, H.; Zhang, J.; Debussche, L.; Haag-Ritcher, S.; Kurz, M.; Nardi, F.; Lukat, P.; Kochems, I.; Tietgen, H.; et al. Production of the bengamide class of marine natural products in Myxobacteria: Biosynthesis and structure-activity relationships. Angew. Chem. Int. Ed. 2015, 54, 15560–15564. [Google Scholar] [CrossRef] [PubMed]
- McCauley, E.; Radjasa, O.K.; Trianto, A.; Crews, M.S.; Smith, A.; Smith, G.C.; Zerebinski, P.; Sabdono, A.; Crews, P. The UNDIP-UCSC campaign to culture chemically prolifix gram-negative bacteria from Jaspis sponges. Arkivoc 2018, 4, 123–131. [Google Scholar] [CrossRef] [Green Version]
- Carballeira, N.; Thompson, J.E.; Ayanoglu, E.; Djerassi, C. Biosynthetic studies of marine lipids. 5. The biosynthesis of long-chain branched fatty acids in marine sponges. J. Org. Chem. 1986, 51, 2751–2756. [Google Scholar] [CrossRef]
- Acquah, K.S.; Beukes, D.R.; Seldon, R.; Jordaan, A.; Sunassee, S.N.; Warner, D.F.; Gammon, D.W. Identification of antimycobacterial natural products from a library of marine invertebrate extracts. Medicines 2022, 9, 9. [Google Scholar] [CrossRef]
- Adamczeski, M.; Quiñoá, E.; Crews, P. Novel sponge-derived amino acids. 11. The entire absolute stereochemistry of the bengamides. J. Org. Chem. 1990, 55, 240–242. [Google Scholar] [CrossRef]
- Phillips, P.E.; Bair, K.W.; Bontempo, J.; Crews, P.; Czuchta, M.; Kinder, F.R.; Versace, R.W.; Wang, B.; Wang, J.; Wood, A.; et al. Bengamide E arrests cells at the G1/S restriction point and within the G2/M phase of the cell cycle. Proc. Am. Assoc. Cancer Res. 2000, 41, 59. [Google Scholar]
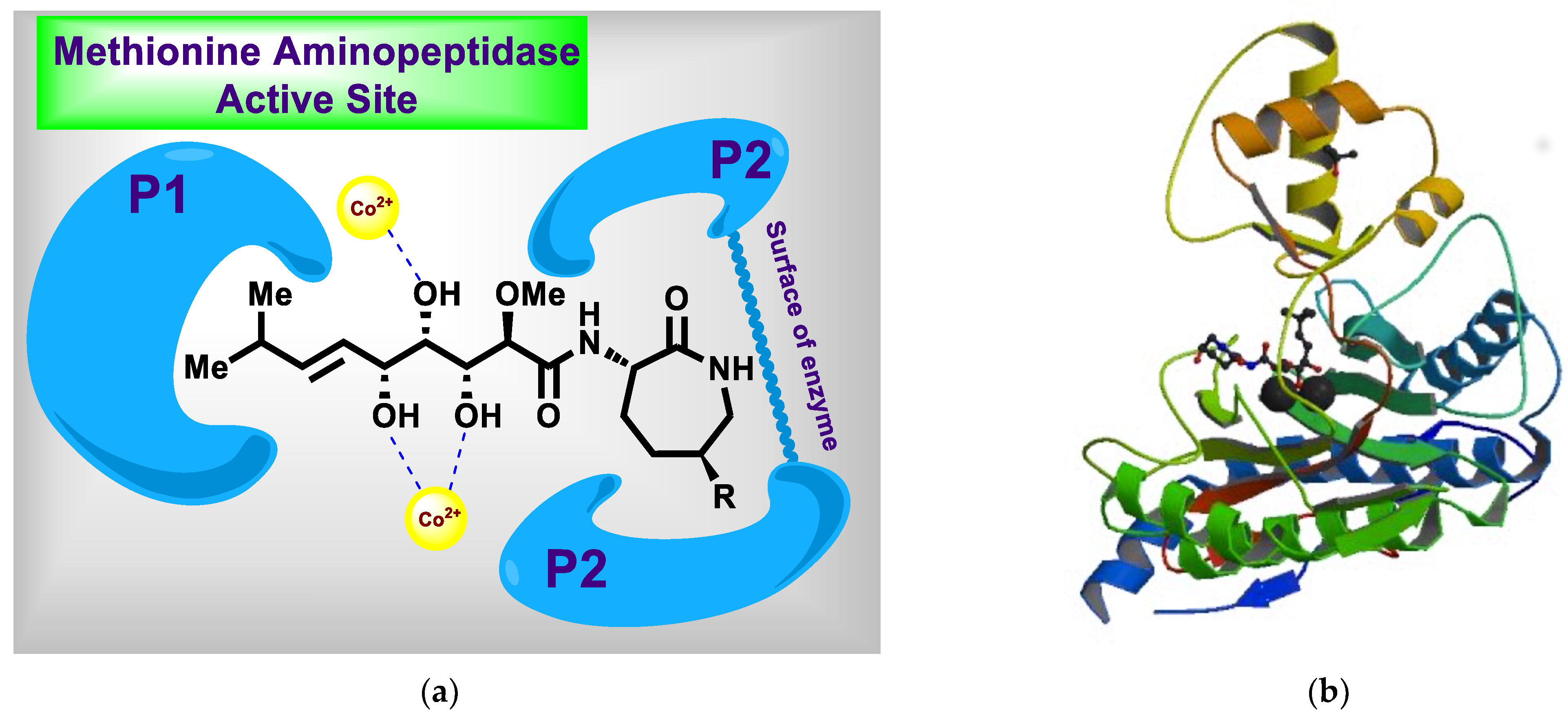
- Towbin, H.; Bair, K.W.; DeCaprio, J.A.; Eck, M.J.; Kim, S.; Kinder, F.R.; Morollo, A.; Mueller, D.R.; Schindler, P.; Song, H.K.; et al. Proteomics-based target identification: Bengamides as a new class of methionine aminopeptidase inhibitors. J. Biol. Chem. 2003, 278, 52964–52971. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Dang, Y.; Tenney, K.; Crews, P.; Tsai, C.W.; Sixt, K.M.; Cole, P.A.; Liu, J.O. Regulation of c-Src nonreceptor tyrosine kinase activity by bengamide A through inhibition of methionine aminopeptidases. Chem. Biol. 2007, 14, 764–774. [Google Scholar] [CrossRef] [Green Version]
- Griffith, E.C.; Su, Z.; Niwayama, S.; Ramsay, C.A.; Chang, Y.H.; Liu, J.O. Molecular recognition of angiogenesis inhibitors fumagillin and ovalicin by methionine aminopeptidase 2. Proc. Natl. Acad. Sci. USA 1998, 95, 15183–15188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffith, E.C.; Su, Z.; Turk, B.E.; Chen, S.; Chang, Y.H.; Wu, Z.; Biemann, K.; Liu, J.O. Methionine aminopeptidase (type 2) is the common target for angiogenesis inhibitors AGM-1470 and ovalicin. Chem. Biol. 1997, 4, 461–471. [Google Scholar] [CrossRef] [Green Version]
- Joharapurkar, A.A.; Dhanesha, N.A.; Jain, M.R. Inhibition of the methionine aminopeptidase 2 enzyme for the treatment of obesity. Diabetes Metab. Syndr. Obes. Targets Ther. 2014, 7, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Sashidhara, K.V.; White, K.N.; Crews, P. A selective account of effective paradigms and significant outcomes in the discovery of inspirational marine natural products. J. Nat. Prod. 2009, 72, 588–603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; LaMontagne, K.; Sabio, M.; Sharma, S.; Versace, R.W.; Yusuff, N.; Phillips, P.E. Depletion of methionine aminopeptidase 2 does not alter cell response to fumagillin and bengamides. Cancer Res. 2004, 64, 2984–2987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, S.-Q.; Wang, J.-J.; Zhang, C.-M.; Liu, Z.-P. The development of MetAp-2 inhibitors in cancer treatment. Curr. Med. Chem. 2012, 19, 1021–1035. [Google Scholar] [CrossRef] [PubMed]
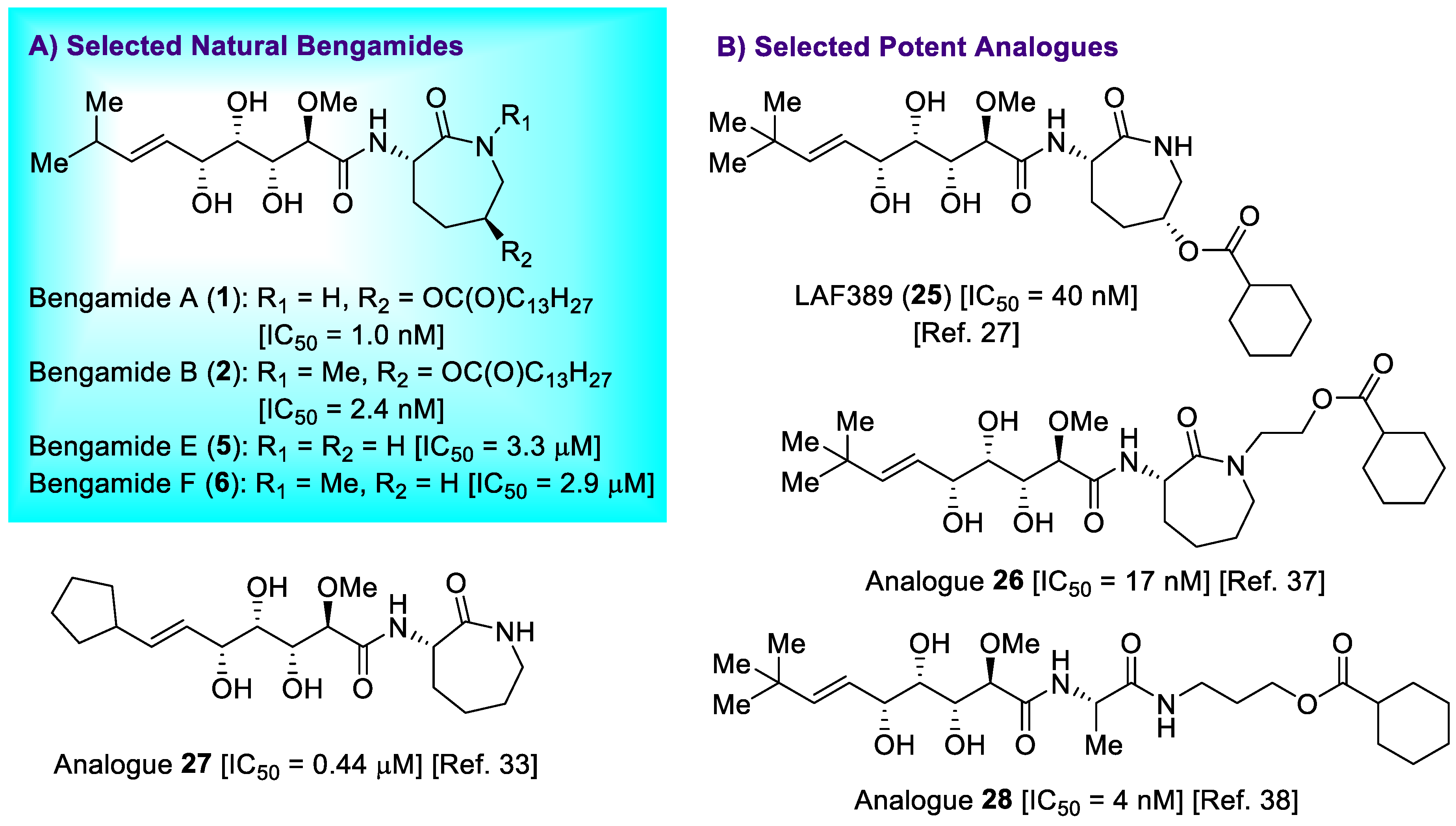
- Kinder, F.R., Jr.; Versace, R.W.; Bair, K.W.; Bontempo, J.M.; Cesarz, D.; Chen, S.; Crews, P.; Czuchta, A.M.; Jagoe, C.T.; Mou, Y.; et al. Synthesis and antitumor activity of ester-modified analogues of bengamide B. J. Med. Chem. 2001, 44, 3692–3699. [Google Scholar] [CrossRef]
- Xu, D.D.; Waykole, L.; Calienni, J.V.; Ciszewski, L.; Lee, G.T.; Liu, W.; Szewczyk, J.; Vargas, K.; Prasad, K.; Repi, O.; et al. An expedient synthesis of LAF389, a bengamide B analogue. Org. Process. Res. Dev. 2003, 7, 856–865. [Google Scholar] [CrossRef]
- Dumez, H.; Gall, H.; Capdeville, R.; Dutreix, C.; van Oosterom, A.T.; Giaccone, G. A phase I and pharmacokinetic study of LAF389 administered to patients with advanced cancer. Anti-Cancer Drugs 2007, 18, 219–225. [Google Scholar] [CrossRef]
- Sarabia, F.; Sánchez-Ruiz, A. A diversity-oriented synthetic approach to bengamides. Tetrahedron Lett. 2005, 46, 1131–1135. [Google Scholar] [CrossRef]
- Sarabia, F.; Sánchez-Ruiz, A. Total synthesis of bengamide E and analogues by modification at C-2 and at terminal olefinic positions. J. Org. Chem. 2005, 70, 9514–9520. [Google Scholar] [CrossRef] [PubMed]
- Sarabia, F.; Martín-Gálvez, F.; Chammaa, S.; Martín-Ortiz, L.; Sánchez-Ruiz, A. Chiral sulfur ylides for the synthesis of bengamide E and analogues. J. Org. Chem. 2010, 75, 5526–5532. [Google Scholar] [CrossRef] [PubMed]
- Martín-Gálvez, F.; García-Ruiz, C.; Sánchez-Ruiz, A.; Valeriote, F.A.; Sarabia, F. An array of bengamide E analogues modified at the terminal olefinic position: Synthesis and antitumor properties. ChemMedChem 2013, 8, 819–831. [Google Scholar] [CrossRef] [PubMed]
- Sarabia, F.; Martín-Gálvez, F.; García-Ruiz, C.; Sánchez-Ruiz, A.; Vivar-García, C. Epi-, epoxy-, and C2-modified bengamides: Synthesis and biological evaluation. J. Org. Chem. 2013, 78, 5239–5253. [Google Scholar] [CrossRef]
- García-Pinel, B.; Porras-Alcalá, C.; Cabeza, L.; Ortiz, R.; Prados, J.; Melguizo, C.; Cheng-Sánchez, I.; López-Romero, J.M.; Sarabia, F. Bengamide analogues show a potent antitumor activity against colon cancer cells: A preliminary study. Mar. Drugs 2020, 18, 240. [Google Scholar] [CrossRef]
- White, K.N.; Tenney, K.; Crews, P. The bengamides: A mini-review of natural sources, analogues, biological properties, biosynthetic origins and future prospects. J. Nat. Prod. 2017, 80, 740–755. [Google Scholar] [CrossRef]
- Liu, G.; Ma, Y.M.; Tai, W.Y.; Xie, C.M.; Li, Y.L.; Li, J.; Nan, F.J. Design, synthesis, and biological evaluation of caprolactam-modified bengamide analogues. ChemMedChem 2008, 3, 74–78. [Google Scholar] [CrossRef]
- Alam, S.; Dhimane, H. A concise synthesis of bengamide E and analogues via E-selective cross-metathesis olefination. Synlett 2010, 19, 2923–2927. [Google Scholar]
- Tai, W.Y.; Zhang, R.T.; Ma, Y.M.; Gu, M.; Liu, G.; Li, J.; Nan, F.J. Design, synthesis, and biological evaluation of ring-opened bengamide analogues. ChemMedChem 2011, 6, 1555–1558. [Google Scholar] [CrossRef]
- Liu, Q.J.; Li, H.; Chen, S.P.; Zhou, G.C. Synthesis of (3S,4R)-bengamide E. Chin. Chem. Lett. 2011, 22, 505–507. [Google Scholar] [CrossRef]
- Zhang, W.; Liang, Q.; Li, H.; Meng, X.; Li, Z. Concise synthesis and antitumor activity of bengamide E and its analogs. Tetrahedron 2013, 69, 664–672. [Google Scholar] [CrossRef]
- Kinder, F.R., Jr. Synthetic approaches toward the bengamide family of antitumor marine natural products. A review. Org. Prep. Proc. Int. 2002, 34, 559–583. [Google Scholar] [CrossRef]
- Liu, W.; Szewczyk, J.M.; Waykole, L.; Repic, O.; Blacklock, T.J. Total synthesis of bengamide E. Tetrahedron Lett. 2002, 43, 1373–1375. [Google Scholar] [CrossRef]
- Boeckman, R.K., Jr.; Clark, T.J.; Shook, B.C. A practical enantioselective total synthesis of the bengamides B, E, and Z. Org. Lett. 2002, 4, 2109–2112. [Google Scholar] [CrossRef]
- Boeckman, R.K., Jr.; Clark, T.J.; Shook, B.C. The development of a convergent and efficient enantioselective synthesis of the bengamides via a common polyol intermediate. Helv. Chim. Acta 2002, 85, 4532–4560. [Google Scholar] [CrossRef]
- Metri, P.K.; Schiess, R.; Prasad, K.R. Enantiospecific total synthesis of (−)-bengamide E. Chem. Asian J. 2013, 8, 488–493. [Google Scholar] [CrossRef]
- Phi, T.D.; Mai, H.D.T.; Tran, V.H.; Truong, B.N.; Tran, T.A.; Vu, V.L.; Chau, V.M.; Pham, V.C. Design, synthesis and cytotoxicity of bengamide analogues and their epimers. Med. Chem. Commun. 2017, 8, 445–451. [Google Scholar] [CrossRef]
- Banwell, M.G.; McRae, K.J. A chemoenzymatic total synthesis of ent-bengamide E. J. Org. Chem. 2001, 66, 6768–6774. [Google Scholar] [CrossRef]
- Phi, T.D.; Mai, H.D.T.; Tran, V.H.; Vu, V.L.; Truong, B.N.; Tran, T.A.; Chau, V.M.; Pham, V.C. Synthesis of bengamide E analogues and their cytotoxic activity. Tetrahedron Lett. 2017, 58, 1830–1833. [Google Scholar] [CrossRef]
- Mulder, R.J.; Shafer, C.M.; Molinski, T.F. First total synthesis of bengazole A. J. Org. Chem. 1999, 64, 4995–4998. [Google Scholar] [CrossRef]
- Mulder, R.J.; Shafer, C.M.; Dalisay, D.S.; Molinski, T.F. Synthesis and structure-activity relationships of bengazole A analogs. Bioorg. Med. Chem. Lett. 2009, 19, 2928–2930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molinski, T.F. Developments in marine natural products. Receptor-specific bioactive compounds. J. Nat. Prod. 1993, 56, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jamison, M.T.; Wang, X.; Cheng, T.; Molinski, T.F. Synergistic anti-Candida activity of bengazole A in the presence of bengamide A. Mar. Drugs 2019, 17, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tietjen, I.; Williams, D.E.; Read, S.; Kuang, X.T.; Mwimanzi, P.; Wilhelm, E.; Markle, T.; Kinloch, N.N.; Naphen, C.N.; Tenney, K.; et al. Inhibition of NF-κB-dependent HIV-1 replication by the marine natural product bengamide A. Antivir. Res. 2018, 152, 94–103. [Google Scholar] [CrossRef]
- Gogineni, V.; Schinazi, R.F.; Hamann, M.T. Role of marine natural products in the genesis of antiviral agents. Chem. Rev. 2015, 115, 9655–9706. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.P.; Yuan, X.H.; Yuan, H.; Wang, W.L.; Wan, B.; Franzblau, S.G.; Ye, Q.Z. Inhibition of Mycobacterium tuberculosis methionine aminopeptidases by bengamide derivatives. ChemMedChem 2011, 6, 1041–1048. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.Y.; McGary, E.C.; Chang, S. Methionine aminopeptidase gene of Escherichia coli is essential for cell growth. J. Bacteriol. 1989, 171, 4071–4072. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.-P.; Yuan, X.-H.; Ye, Q.-Z. Structural analysis of inhibition of Mycobacterium tuberculosis methionine aminopeptidase by bengamide derivatives. Eur. J. Med. Chem. 2012, 47, 479–484. [Google Scholar] [CrossRef] [Green Version]
- Luo, Q.-L.; Li, J.-Y.; Liu, Z.-Y.; Chen, L.-L.; Li, J.; Qian, Z.; Shen, Q.; Li, Y.; Lushington, G.H.; Ye, Q.-Z.; et al. Discovery and structural modification of inhibitors of methionine aminopeptidases from Escherichia coli and Saccharomyces Cerevisiae. J. Med. Chem. 2003, 46, 2631–2640. [Google Scholar] [CrossRef]
- Lu, J.-P.; Ye, Q.-Z. Expression and characterization of Mycobacterium tuberculosis methionine aminopeptidase type 1a. Bioorg. Med. Chem. Lett. 2010, 20, 2776–2779. [Google Scholar] [CrossRef] [Green Version]
- Chai, S.C.; Wang, W.-L.; Ye, Q.-Z. Fe(II) is the native cofactor for Escherichia coli methionine aminopeptidase. J. Biol. Chem. 2008, 283, 26879–26885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Q.-Z.; Xie, S.-X.; Huang, M.; Huang, W.-J.; Lu, J.-P.; Ma, Z.-Q. Metalloform-Selective inhibitors of Escherichia coli methionine aminopeptidase and X-ray structure of a Mn(II)-form enzyme complexed with an inhibitor. J. Am. Chem. Soc. 2004, 126, 13940–13941. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.-L.; Chai, S.C.; Huang, M.; He, H.-Z.; Hurley, T.D.; Ye, Q.-Z. Discovery of inhibitors of Escherichia coli methionine aminopeptidase with the Fe(II)-form. Selectivity and antibacterial activity. J. Med. Chem. 2008, 51, 6110–6120. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Lu, J.-P.; Ye, Q.-Z. Structural analysis of bengamide derivatives as inhibitors of methionine aminopeptidases. J. Med. Chem. 2012, 55, 8021–8027. [Google Scholar] [CrossRef] [Green Version]
- Quan, D.H.; Nagalingam, G.; Luck, I.; Proschogo, N.; Pillalamarri, V.; Addlagatta, A.; Martinez, E.; Sintchenko, V.; Rutledge, P.J.; Triccas, J.A. Bengamides display potent activity against drug-resistant Mycobacterium tuberculosis. Sci. Rep. 2019, 9, 14396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, X.-Q.; Wei, B.-Y.; Yu, C.-X.; Guan, X.-N.; Ma, W.-P.; Liu, G.; Yang, C.-G.; Nan, F.-J. Design, synthesis and biological evaluation of bengamide analogues as ClpP activators. Chin. J. Chem. 2020, 38, 1111–1115. [Google Scholar] [CrossRef]
- Moreno-Cinos, C.; Goossens, K.; Salado, I.G.; Van der Veken, P.; De Winter, H.; Augustyns, K. ClpP protease, a promising antimicrobial target. Int. J. Mol. Sci. 2019, 20, 2232. [Google Scholar] [CrossRef] [Green Version]
- Bhandari, V.; Wong, K.S.; Zhou, J.L.; Mabanglo, M.F.; Batey, R.A.; Houry, W.A. The role of ClpP protease in bacterial pathogenesis and human diseases. ACS Chem. Biol. 2018, 13, 1413–1425. [Google Scholar] [CrossRef]
- Ye, F.; Li, J.; Yang, C.-G. The development of small-molecule modulators for ClpP protease activity. Mol. BioSyst. 2017, 13, 23–31. [Google Scholar] [CrossRef]
- Leung, L.; Datti, A.; Cossette, M.; Goodreid, J.; McCaw, S.E.; Mah, M.; Nakhamchik, A.; Ogata, K.; El Bakkouri, M.; Cheng, Y.Q.; et al. Activators of cylindral proteases as antimicrobials: Identification and development of small molecule activators of ClpP protease. Chem. Biol. 2011, 18, 1167–1178. [Google Scholar] [CrossRef] [Green Version]
- Compton, C.L.; Schmitz, K.R.; Sauer, R.T.; Sello, J.K. Antibacterial activity of and resistance to small molecule inhibitors of the ClpP peptidase. ACS Chem. Biol. 2013, 8, 2669–2677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brotz-Oesterhelt, H.; Beyer, D.; Kroll, H.P.; Endermann, R.; Ladel, C.; Schroeder, W.; Hinzen, B.; Raddatz, S.; Paulsen, H.; Henninger, K.; et al. Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat. Med. 2005, 11, 1082–1087. [Google Scholar] [CrossRef] [PubMed]
- Malik, I.T.; Brotz-Oesterhelt, H. Conformational control of the bacterial Clp protease by natural product antibiotics. Nat. Prod. Rep. 2017, 34, 815–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatfield, M.J.; Umans, R.A.; Hyatt, J.L.; Edwards, C.C.; Wierdl, M.; Tsurkan, L.; Taylor, M.R.; Potter, P.M. Carboxylesterases: General detoxyfying enzymes. Chem. Biol. Interact. 2016, 259, 327–331. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ye, F.; Lan, L.; Jiang, H.; Luo, C.; Yang, C.-G. Switching of Staphylococcus aureus Clp protease. J. Biol. Chem. 2011, 286, 37590–37601. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.-X.; Wei, B.-Y.; Kong, X.-Q.; Yang, C.-G.; Nan, F.-J. Synthesis and structure-activity relationships of ring-opened bengamide analogues against methicillin-resistant Staphylococcus aureus. Chin. J. Chem. 2021, 39, 671–676. [Google Scholar] [CrossRef]
- Lakhundi, S.; Zhang, K. Methicillin-resistant Staphylococcus aureus: Molecular characterization, evolution and epidemiology. Clin. Microbiol. Rev. 2018, 31, e00020-18. [Google Scholar] [CrossRef] [Green Version]
- Nguta, J.M.; Appiah-Opong, R.; Nyarko, A.K.; Yeboah-Manu, D.; Addo, P.G.A. Current perspectives in drug discovery against tuberculosis from natural products. Int. J. Mycobacteriol. 2015, 4, 165–183. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.; Wang, C.; Gerwick, W.H.; Shao, C. Marine natural products as potential anti-tubercular agents. Eur. J. Med. Chem. 2019, 165, 273–292. [Google Scholar] [CrossRef]
- Kazlauskas, R.; Murphy, P.T.; Quinn, R.J.; Wells, R.J. Heteronemin, a new scalarin type sesterterpene from the sponge Heteronema erecta. Tetrahedron Lett. 1976, 17, 2631–2634. [Google Scholar] [CrossRef]
- Alahdal, A.M.; Asfour, H.Z.; Ahmed, S.A.; Noor, A.O.; Al-Abd, A.M.; Elfaky, M.A.; Elhady, S.S. Anti-Helicobacter, antitubercular and cytotoxic activities of scalaranes from the red sea sponge Hyrtios erectus. Molecules 2018, 23, 978. [Google Scholar] [CrossRef] [PubMed] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
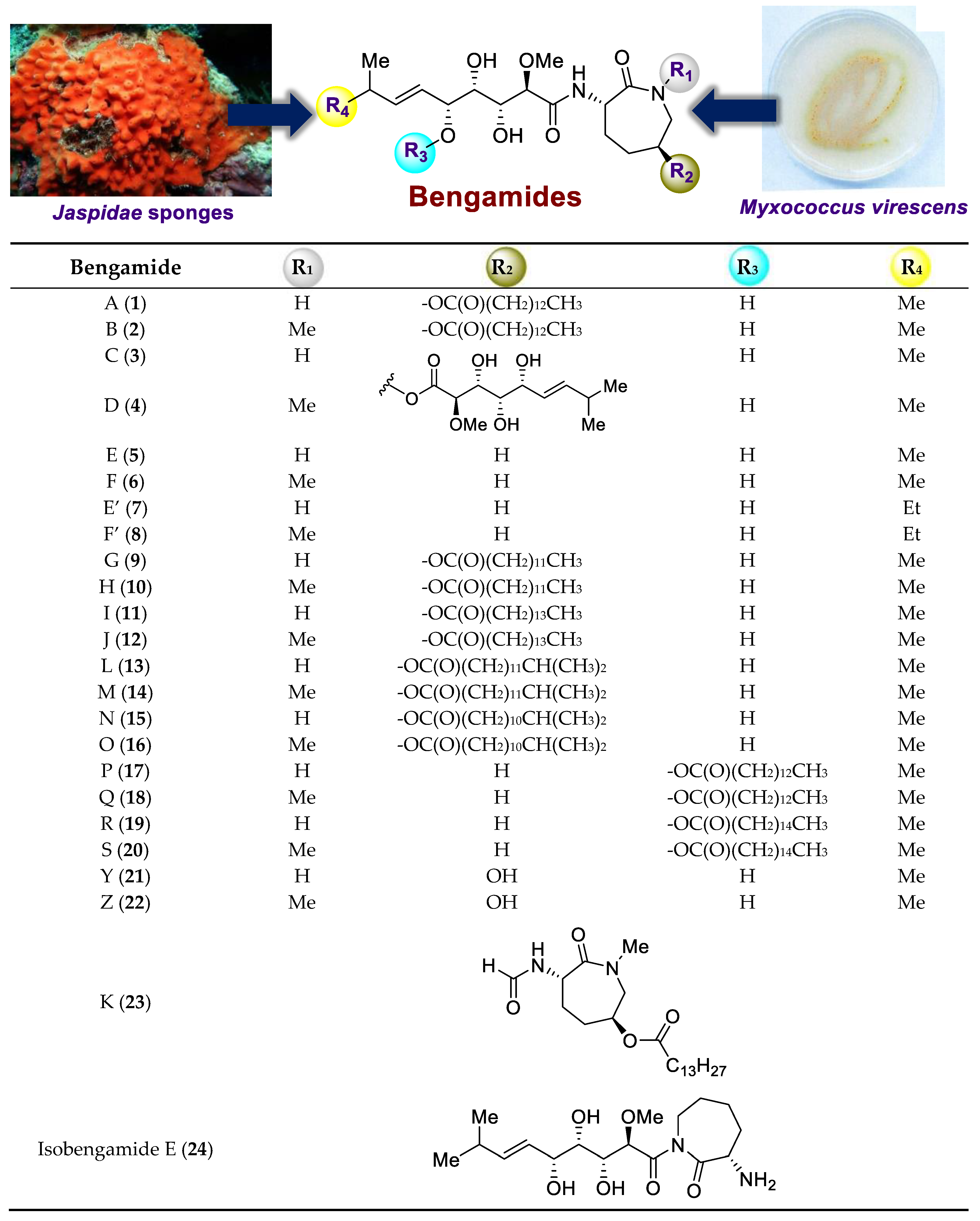
| Year | Natural Source (Collection Site) | Research Group | Isolated Compounds [Ref.] |
|---|---|---|---|
| 1986 | Jaspis cf. coriacea (Fiji Islands) | P. Crews | Bengamides A and B [2] |
| 1989 | Jaspis cf. coriacea (Fiji Islands) | P. Crews | Bengamides A–F [3] |
| 1994 | Jaspis digonoxea (South Africa) | Y. Kashman | Bengamides A and B [5] |
| 1997 | Jaspis carteri (New Caledonia) | M. V. D’Auria | Bengamides A, B, G–J, K [6] |
| 1999 | Jaspis splendens (Serrurion Island) | M. R. Boyd | Bengamides A, B, Y, Z [7] |
| 1999 | Pachasctrissa sp. (Musha Archipelago) | Y. Letourneux | Bengamides A, B, L [8] |
| 2001 | Jaspis cf. coriacea (Fiji Islands) | P. Crews | Bengamides M–R [4] |
| 2008 | Dorypleres splendens (Fiji Islands) | G. R. Pettit | Bengamide A [9] |
| 2011 | Stelleta sp. (Bonaparte Archipelago) | C. A. Motti | Bengamides A, F, N and Y [10] |
| 2012 | Myxococcus virescens (German Collection of Microorganisms and Cell Cultures) | P. Crews | Bengamides E, E’, F’ [12] |
| 2015 | Myxococcus virescens (Soil sample) | M. Brönstrup | Bengamides E, F, E’, F’ [13] |
| 2019 | Calthropella sp. (Indonesia) | R. T. Swasono | Bengamide Q [11] |
| 2022 | Jaspis splendens (Mauritius) | D. W. Gammon | Bengamides A, B, H, I, J, L, M, N, O, P, Q and R [16] |
| Cancer Cell Line [a] | Analogue 36 | Analogue 39 | LAF389 (25) |
|---|---|---|---|
| A549 | 9 | 39 | 13 |
| B16-F10 | 33 | 47 | 29 |
| H460 | 59 | 42 | 9 |
| HCT116 | 44 | 51 | 23 |
| HCT15 | 550 | 45 | 1300 |
| HT29 | 120 | 100 | 27 |
| MCF-7 | 110 | 19 | |
| MDA-A1 | 4800 | 1100 | >10,000 |
| MDA-MB-231 | 110 | 140 | 22 |
| PC3 | 270 | 270 | 20 |
| CCRF-CEM | 350 | 290 | 65 |
| HL60 | 280 | 370 | 58 |
| NHDF | 260 | 140 | |
| PBL | >5900 | >7900 |
| Cancer Cell Line [a] |  | Reference Compound [b] |
|---|---|---|
| CCD18 | 5.08 ± 0.39 | 7.35 ± 0.41 |
| T84 | 0.07 ± 0.02 | 2.68 ± 0.16 |
| SW480 | 0.08 ± 0.00 | 6.35 ± 0.54 |
| HCT15 | 2.44 ± 0.25 | 6.58 ± 0.35 |
| HT29 | 0.66 ± 0.18 | 6.14 ± 0.94 |
| MC38 | 6.51 ± 1.12 | 0.33 ± 0.01 |
| MCF-7 | 0.13 ± 0.01 | 0.04 ± 0.01 |
| Analogue | R | Tumor Cell Line | |||||
|---|---|---|---|---|---|---|---|
| Sconfiguration, n = 1 | KB [a] | LU1 [b] | HepG-2 [c] | MCF-7 [d] | HL-60 [e] | HeLa [f] | |
| 51a | Cyclohexyl | 8.2 | 12.7 | 11.8 | 20.1 | >50 | >50 |
| 52a | Phenyl | 2.9 | 7.5 | 11.3 | 17.8 | 55.8 | 39.7 |
| 53a | Cinnamyl | 2.3 | 5.8 | 6.6 | 4.3 | 36.8 | 36.8 |
| Rconfiguration, n = 1 | |||||||
| 51b | Cyclohexyl | 2.4 | 2.6 | 6.6 | 8.8 | >50 | >50 |
| 52b | Phenyl | 4.4 | 2.7 | 12.0 | 20.4 | 29.0 | 19.7 |
| 53b | Cinnamyl | 1.7 | 2.3 | 2.4 | 15.1 | 21.5 | 16.1 |
| Sconfiguration, n = 2 | |||||||
| 54a | Cyclohexyl | 11.8 | 17.1 | >50 | >50 | >50 | >50 |
| 55a | Phenyl | 5.7 | 7.7 | 4.8 | 32.5 | 45.6 | 41.5 |
| 56a | Cinnamyl | 1.9 | 23.6 | 1.5 | 4.9 | 13.4 | 26.9 |
| Rconfiguration, n = 2 | |||||||
| 54b | Cyclohexyl | 0.4 | 0.3 | 1.0 | 10.4 | 19.5 | 39.1 |
| 55b | Phenyl | 1.0 | 1.1 | 1.9 | 8.6 | 10.6 | 16.2 |
| 56b | Cinnamyl | 1.1 | 25.1 | 0.4 | 2.7 | 8.9 | 9.5 |
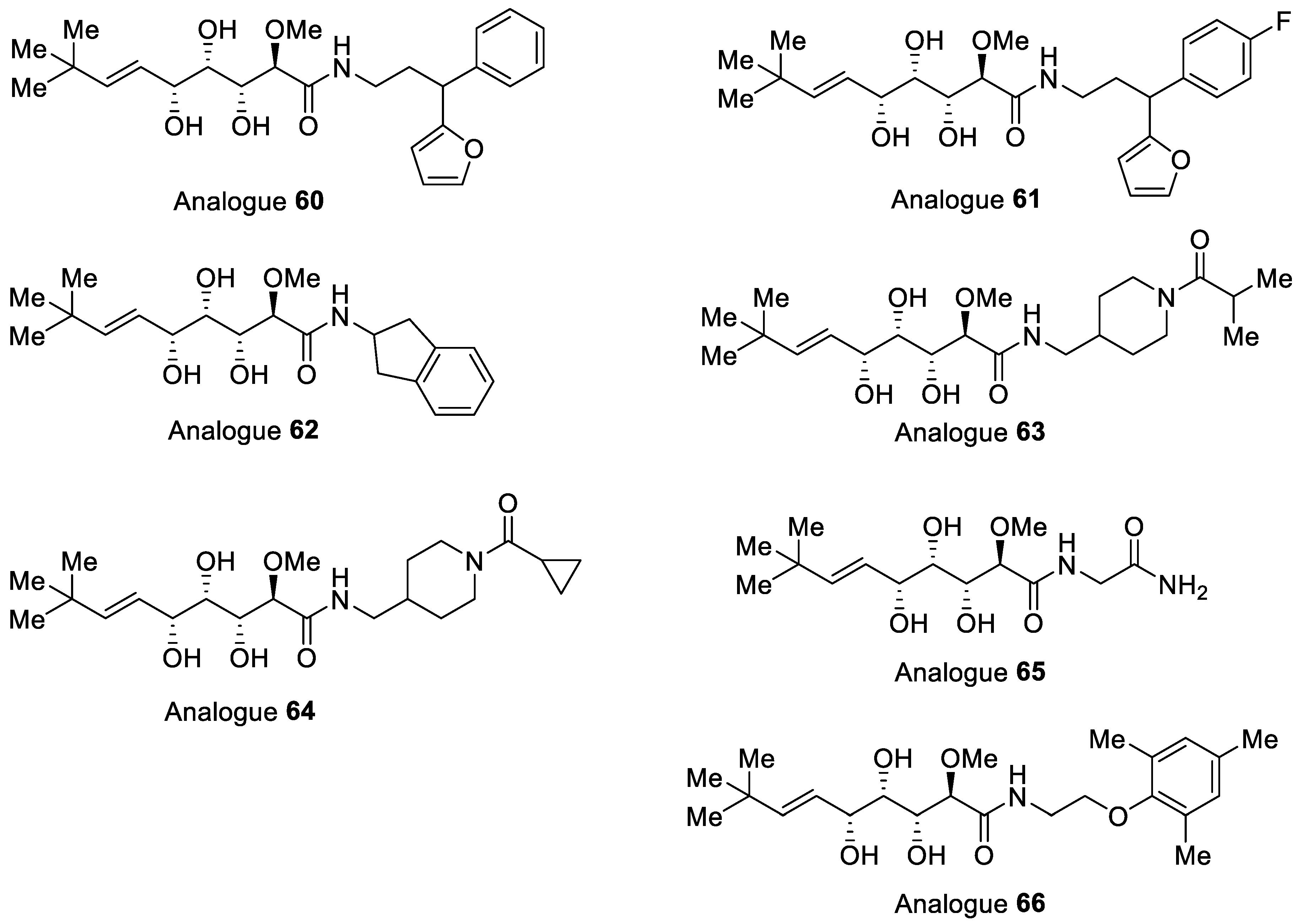
| Analogue | IC50 [μM] against MtMetAP1a [a] | M. tuberculosis MIC [μM] [b] | K562 [e] IC50 [μM] | |||
|---|---|---|---|---|---|---|
| CoII | MnII | FeII | MABA [c] | LORA [d] | ||
| 60 | 6.0 | 11 | 5.5 | 15% | 8% | 79.6 |
| 61 | 8.1 | 12 | 6.7 | 48% | 12% | 96.5 |
| 62 | 31 | 11 | 18 | 29% | 28% | >333 |
| 63 | 45 | 68 | 67 | 0% | 0% | >333 |
| 64 | 88 | 187 | 110 | 0% | 0% | >333 |
| 65 | 21 | 49 | 14 | 122 | 0% | >333 |
| 66 | 7.9 | 6.9 | 4.5 | 50.6 | 107.4 | 37.8 |
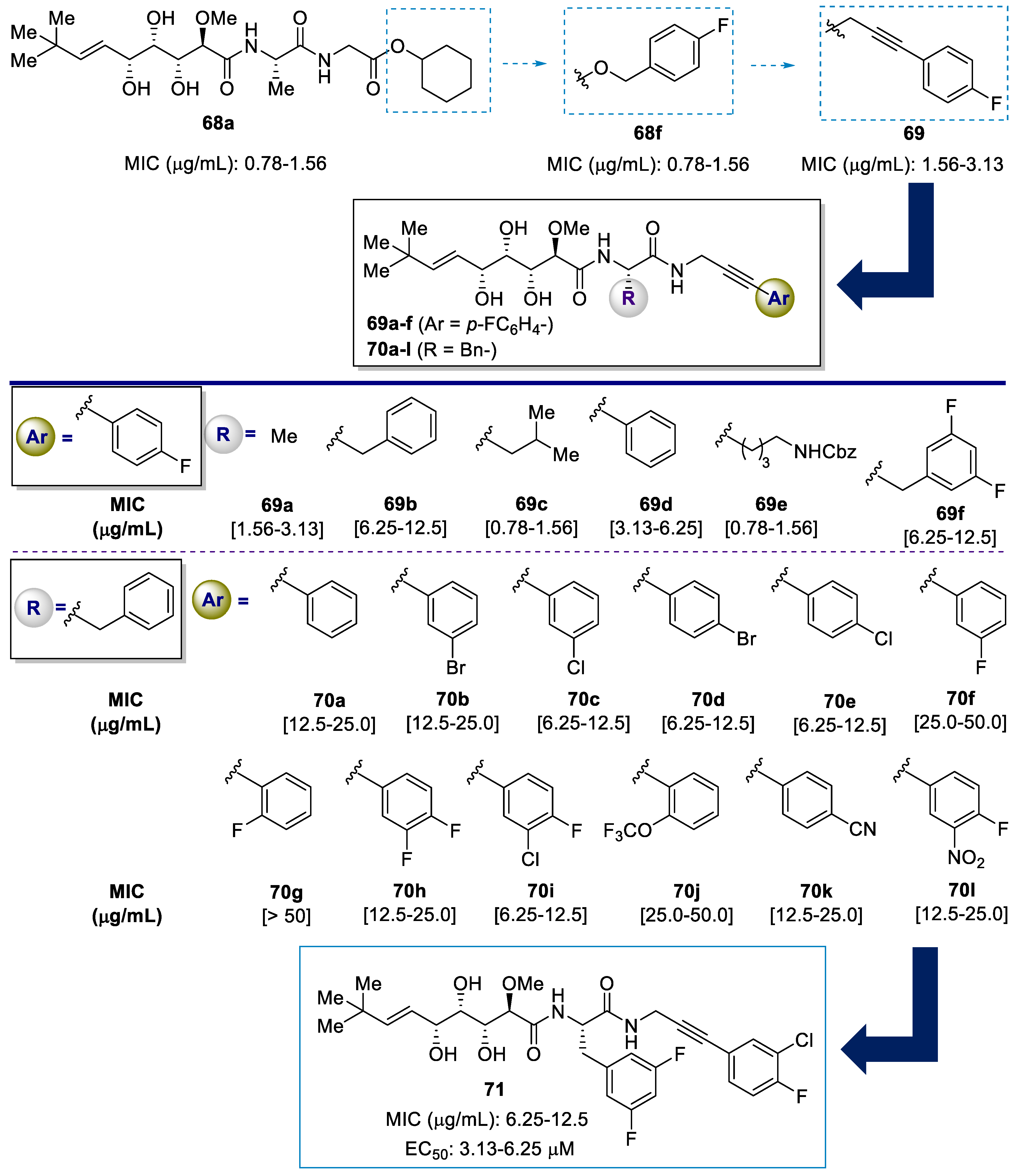
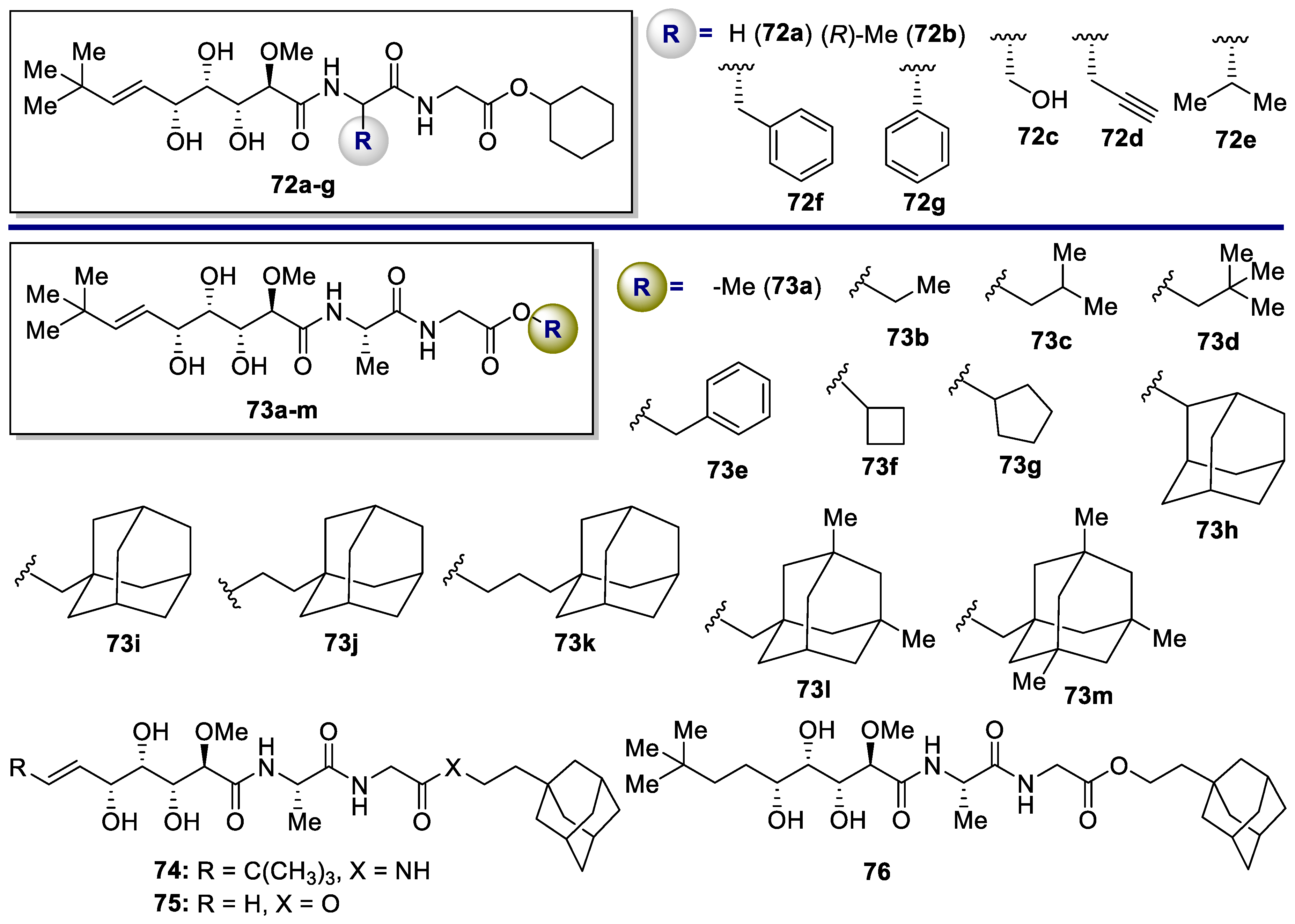
| Analogue | Methicillin-Resistant Staphylococcus Aureus (MRSA) MIC [μg/mL] [a] | ||||||
|---|---|---|---|---|---|---|---|
| Newman | USA300 | NRS-1 | NRS-70 | NRS-271 | NRS-108 | NRS-100 | |
| 68a | 4.00 | 2.00 | 4.00 | 1.00 | 4.00 | 8.00 | 2.00 |
| Van | 0.78–1.56 | 1.56–3.13 | 0.78–1.56 | 0.78–1.56 | 0.78–1.56 | 0.78–1.56 | 0.78–1.56 |
| Tetra | 0.10–0.20 | 12.5–25.0 | 25.0 | 0.39–0.78 | 0.20–0.39 | 0.10–0.20 | 0.10–0.20 |
| 72a | 12.5–25.0 | 12.5–25.0 | 6.25–12.5 | 0.78–1.56 | 25.0–50.0 | 6.25–12.5 | 6.25–12.5 |
| 72b | >50 | >50 | >50 | >50 | >50 | >50 | >50 |
| 72c | 25.0–50.0 | 50.0 | 50.0 | 50.0 | 50.0 | 50.0 | 50.0 |
| 72d | 6.25–12.5 | 12.5–25.0 | 6.25–12.5 | 6.25–12.5 | 12.5–25 | 6.25–12.5 | 6.25–12.5 |
| 72e | 3.13–6.25 | 1.56–3.13 | 3.13–6.25 | 1.56–3.13 | 6.25–12.5 | 0.78–1.56 | 0.78–1.56 |
| 72f | 12.5–25.0 | 25.0–50.0 | 25.0–50.0 | 50 | 12.5–25.0 | 25.0–50.0 | 12.5–25.0 |
| 72g | 25.0–50.0 | 25 | 25.0–50.0 | > 50 | 25.0–50.0 | 50 | 12.5–25.0 |
| 73a | 16.0 | 32.0 | 32.0 | 8.0 | 32.0 | 32.0 | 16.0 |
| 73b | 50 | 50 | 50 | 50 | 50 | 50 | 50 |
| 73c | 6.25–12.5 | 3.13–6.25 | 3.13–6.25 | 0.78–1.56 | 6.25–12.5 | 1.56–3.13 | 1.56–3.13 |
| 73d | 6.25–12.5 | 6.25–12.5 | 6.25–12.5 | 12.5–25.0 | 12.5–25.0 | 12.5–25.0 | 6.25–12.5 |
| 73e | 1.56–3.13 | 1.56–3.13 | 1.56–3.13 | 1.56–3.13 | 6.25–12.5 | 1.56–3.13 | 0.78–1.56 |
| 73f | 6.35–12.5 | 12.5–25.0 | 12.5–25.0 | 12.5–25.0 | 12.5–25.0 | 12.5–25.0 | 6.25–12.5 |
| 73g | 12.5–25.0 | 12.5–25.0 | 6.25–12.5 | 0.78–1.56 | 25–50 | 6.25–12.5 | 6.25–12.5 |
| 73h | 1.56–3.13 | 0.78–1.56 | 1.56–3.13 | 1.56–3.13 | 3.13–6.25 | 3.13–6.25 | 0.78–1.56 |
| 73i | 0.39–0.78 | 0.78–1.56 | 0.39–0.78 | 0.78–1.56 | 0.78–1.56 | 0.78–1.56 | <0.39 |
| 73j | 0.078–0.156 | 0.156–0.31 | 0.04–0.08 | 0.16–0.31 | 0.1–0.31 | 0.08–0.16 | 0.04–0.08 |
| 73k | 0.78–1.56 | 0.78–1.56 | 0.78–1.56 | 0.78–1.56 | 0.78–1.56 | 0.78–1.56 | 0.39–0.78 |
| 73l | 0.78–1.56 | 0.78–1.56 | 0.78–1.56 | 1.56–3.13 | 1.56–3.13 | 0.78–1.56 | 0.39–0.78 |
| 73m | 0.78–1.56 | 0.78–1.56 | 0.78–1.56 | 0.78–1.56 | 1.56–3.13 | 0.78–1.56 | 0.39–0.78 |
| 74 | 4.00 | 4.00 | 4.00 | 2.00 | 4.00 | 4.00 | 4.00 |
| 75 | >50 | >50 | >50 | >50 | >50 | >50 | >50 |
| 76 | 4.00 | 8.00 | 4.00 | 1.00 | 4.00 | 8.00 | 2.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porras-Alcalá, C.; Moya-Utrera, F.; García-Castro, M.; Sánchez-Ruiz, A.; López-Romero, J.M.; Pino-González, M.S.; Díaz-Morilla, A.; Kitamura, S.; Wolan, D.W.; Prados, J.; et al. The Development of the Bengamides as New Antibiotics against Drug-Resistant Bacteria. Mar. Drugs 2022, 20, 373. https://doi.org/10.3390/md20060373
Porras-Alcalá C, Moya-Utrera F, García-Castro M, Sánchez-Ruiz A, López-Romero JM, Pino-González MS, Díaz-Morilla A, Kitamura S, Wolan DW, Prados J, et al. The Development of the Bengamides as New Antibiotics against Drug-Resistant Bacteria. Marine Drugs. 2022; 20(6):373. https://doi.org/10.3390/md20060373
Chicago/Turabian StylePorras-Alcalá, Cristina, Federico Moya-Utrera, Miguel García-Castro, Antonio Sánchez-Ruiz, Juan Manuel López-Romero, María Soledad Pino-González, Amelia Díaz-Morilla, Seiya Kitamura, Dennis W. Wolan, José Prados, and et al. 2022. "The Development of the Bengamides as New Antibiotics against Drug-Resistant Bacteria" Marine Drugs 20, no. 6: 373. https://doi.org/10.3390/md20060373