
A Metabolomics-Based Toolbox to Assess and Compare the Metabolic Potential of Unexplored, Difficult-to-Grow Bacteria
 , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
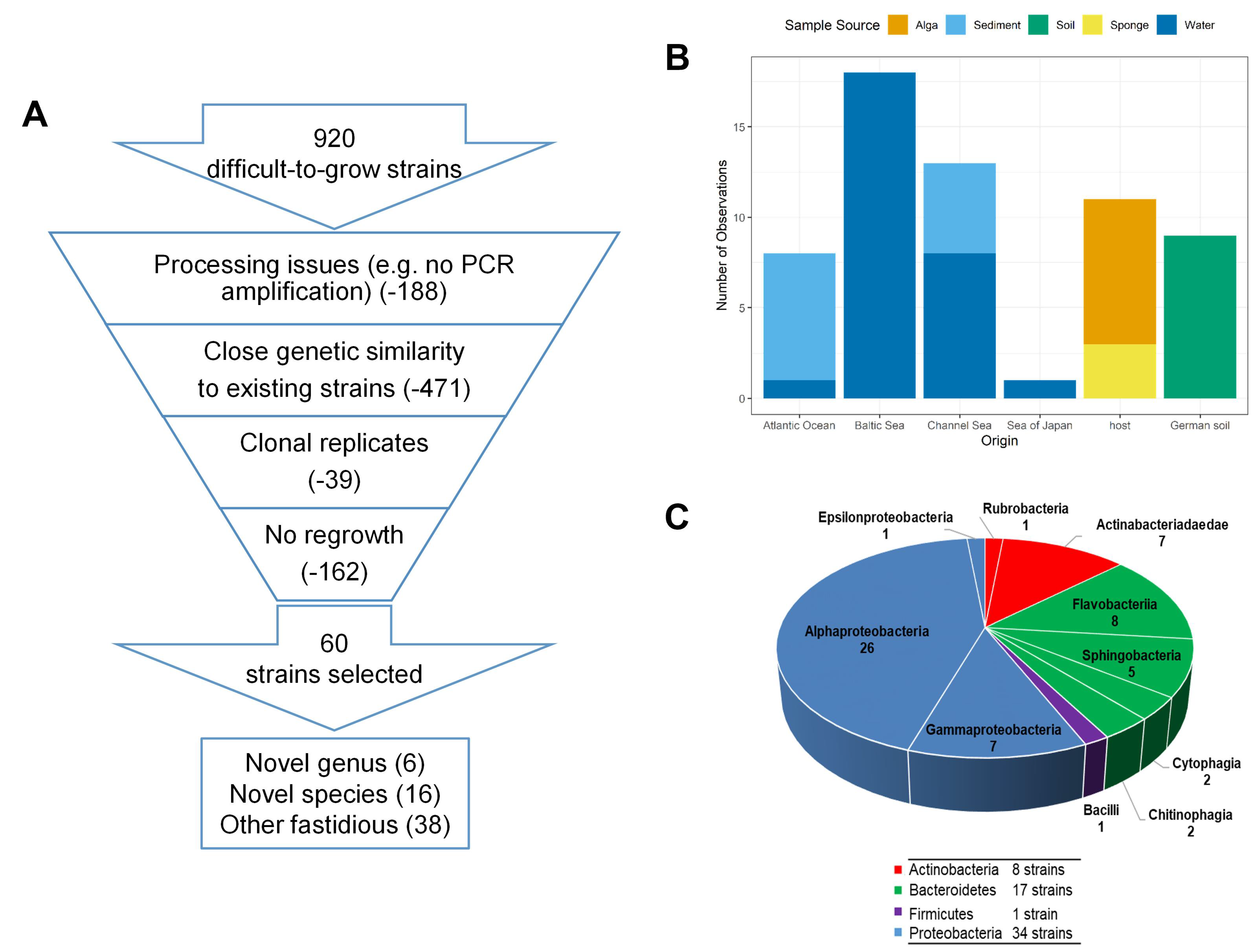
2.1. Samples Sources, Isolation and Cultivation
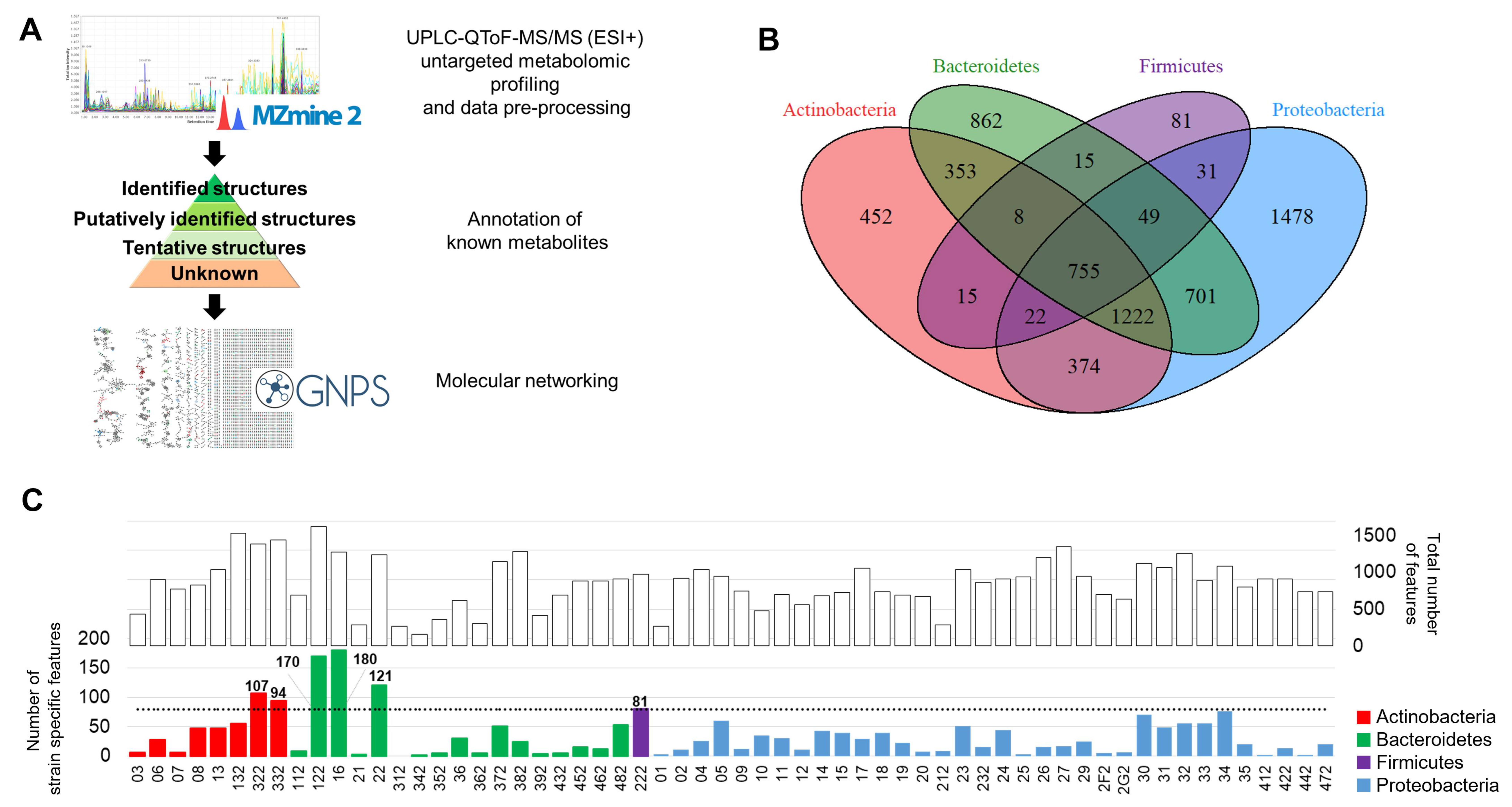
2.2. Untargeted Metabolomics Analysis
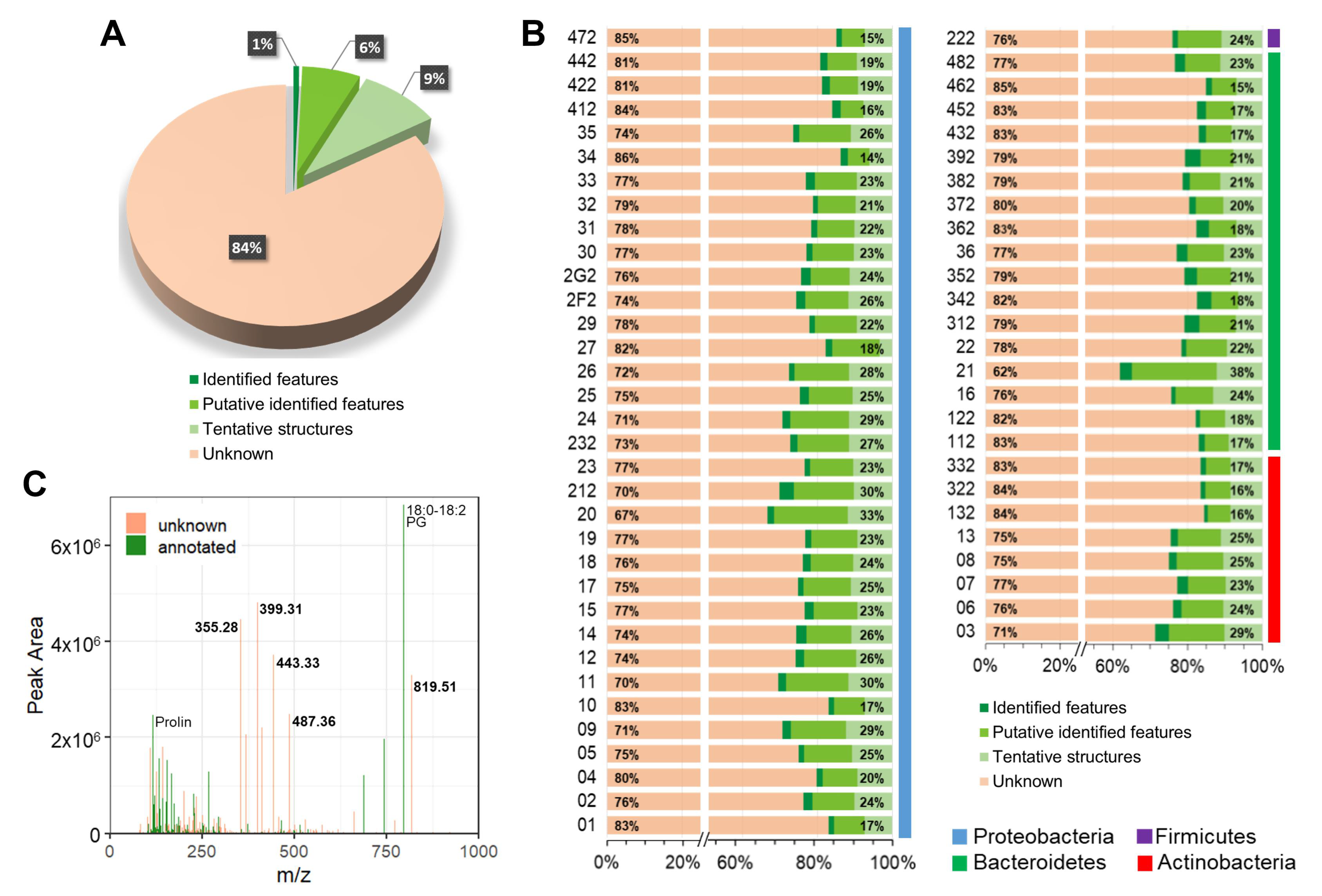
2.3. Metabolite Annotation
2.4. Metabolite Distribution and Chemical Richness
3. Conclusions
4. Materials and Methods
4.1. Samples Collection
4.2. Cultivation Strategies
4.2.1. Single Dilution High-Throughput Cultivation in Liquid Media
4.2.2. Direct Plating Method
4.2.3. Growth in Biofilms
4.2.4. Chemotaxis Chambers
4.3. Taxonomic Affiliation of Isolates
4.4. Fermentation of Bacteria for Natural Product Analysis
4.5. Untargeted Metabolomic Profiling
4.6. Data Processing and Metabolomics Analysis
4.6.1. Feature Detection
4.6.2. Metabolite Annotation
4.6.3. Molecular Networking
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; El-Hossary, E.M.; Oelschlaeger, T.A.; Donia, M.S.; Quinn, R.J.; Abdelmohsen, U.R. Potential of Marine Natural Products against Drug-Resistant Bacterial Infections. Lancet Infect. Dis. 2019, 19, e237–e245. [Google Scholar] [CrossRef]
- Wright, G.D. Opportunities for Natural Products in 21st Century Antibiotic Discovery. Nat. Prod. Rep. 2017, 34, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Orhan, I.E.; Banach, M.; Rollinger, J.M.; Barreca, D.; Weckwerth, W.; Bauer, R.; Bayer, E.A.; et al. Natural Products in Drug Discovery: Advances and Opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Goodfellow, M.; Fiedler, H.-P. A Guide to Successful Bioprospecting: Informed by Actinobacterial Systematics. Antonie Van Leeuwenhoek 2010, 98, 119–142. [Google Scholar] [CrossRef]
- Fenical, W.; Jensen, P.R. Developing a New Resource for Drug Discovery: Marine Actinomycete Bacteria. Nat. Chem. Biol. 2006, 2, 666–673. [Google Scholar] [CrossRef]
- Hoffmann, T.; Krug, D.; Bozkurt, N.; Duddela, S.; Jansen, R.; Garcia, R.; Gerth, K.; Steinmetz, H.; Müller, R. Correlating Chemical Diversity with Taxonomic Distance for Discovery of Natural Products in Myxobacteria. Nat. Commun. 2018, 9, 803. [Google Scholar] [CrossRef] [Green Version]
- Zdouc, M.M.; Iorio, M.; Maffioli, S.I.; Crüsemann, M.; Donadio, S.; Sosio, M. Planomonospora: A Metabolomics Perspective on an Underexplored Actinobacteria Genus. J. Nat. Prod. 2021, 84, 204–219. [Google Scholar] [CrossRef]
- Reimer, L.C.; Vetcininova, A.; Carbasse, J.S.; Söhngen, C.; Gleim, D.; Ebeling, C.; Overmann, J. BacDive in 2019: Bacterial Phenotypic Data for High-Throughput Biodiversity Analysis. Nucleic. Acids Res. 2019, 47, D631–D636. [Google Scholar] [CrossRef] [Green Version]
- Overmann, J. Principles of Enrichment, Isolation, Cultivation, and Preservation of Prokaryotes. In The Prokaryotes: Prokaryotic Biology and Symbiotic Associations; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 149–207. ISBN 978-3-642-30194-0. [Google Scholar]
- Kim, H.; Kim, S.; Kim, M.; Lee, C.; Yang, I.; Nam, S.J. Bioactive Natural Products from the Genus Salinospora: A Review. Arch. Pharm. Res. 2020, 43, 1230–1258. [Google Scholar] [CrossRef]
- Crüsemann, M.; O’Neill, E.C.; Larson, C.B.; Melnik, A.V.; Floros, D.J.; da Silva, R.R.; Jensen, P.R.; Dorrestein, P.C.; Moore, B.S. Prioritizing Natural Product Diversity in a Collection of 146 Bacterial Strains Based on Growth and Extraction Protocols. J. Nat. Prod. 2017, 80, 588–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfender, J.-L.; Nuzillard, J.-M.; van der Hooft, J.J.J.; Renault, J.-H.; Bertrand, S. Accelerating Metabolite Identification in Natural Product Research: Toward an Ideal Combination of Liquid Chromatography–High-Resolution Tandem Mass Spectrometry and NMR Profiling, in Silico Databases, and Chemometrics. Anal. Chem. 2019, 91, 704–742. [Google Scholar] [CrossRef] [PubMed]
- Hubert, J.; Nuzillard, J.-M.; Renault, J.-H. Dereplication Strategies in Natural Product Research: How Many Tools and Methodologies behind the Same Concept? Phytochem. Rev. 2017, 16, 55–95. [Google Scholar] [CrossRef]
- Medema, M.H. The Year 2020 in Natural Product Bioinformatics: An Overview of the Latest Tools and Databases. Nat. Prod. Rep. 2021, 38, 301–306. [Google Scholar] [CrossRef]
- van Santen, J.A.; Kautsar, S.A.; Medema, M.H.; Linington, R.G. Microbial Natural Product Databases: Moving Forward in the Multi-Omics Era. Nat. Prod. Rep. 2021, 38, 264–278. [Google Scholar] [CrossRef]
- Dührkop, K.; Fleischauer, M.; Ludwig, M.; Aksenov, A.A.; Melnik, A.V.; Meusel, M.; Dorrestein, P.C.; Rousu, J.; Böcker, S. SIRIUS 4: A Rapid Tool for Turning Tandem Mass Spectra into Metabolite Structure Information. Nat. Methods 2019, 16, 299–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and Community Curation of Mass Spectrometry Data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [Green Version]
- Maansson, M.; Vynne, N.G.; Klitgaard, A.; Nybo, J.L.; Melchiorsen, J.; Nguyen, D.D.; Sanchez, L.M.; Ziemert, N.; Dorrestein, P.C.; Andersen, M.R.; et al. An Integrated Metabolomic and Genomic Mining Workflow To Uncover the Biosynthetic Potential of Bacteria. mSystems 2016, 1, e00028-15. [Google Scholar] [CrossRef] [Green Version]
- Wolfender, J.-L.; Litaudon, M.; Touboul, D.; Queiroz, E.F. Innovative Omics-Based Approaches for Prioritisation and Targeted Isolation of Natural Products—New Strategies for Drug Discovery. Nat. Prod. Rep. 2019, 36, 855–868. [Google Scholar] [CrossRef] [Green Version]
- Yi, L.; Dong, N.; Yun, Y.; Deng, B.; Ren, D.; Liu, S.; Liang, Y. Chemometric Methods in Data Processing of Mass Spectrometry-Based Metabolomics: A Review. Anal. Chim. Acta 2016, 914, 17–34. [Google Scholar] [CrossRef]
- Misra, B.B. New Software Tools, Databases, and Resources in Metabolomics: Updates from 2020. Metabolomics 2021, 17, 49. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Zhou, G.; Ewald, J.; Chang, L.; Hacariz, O.; Basu, N.; Xia, J. Using MetaboAnalyst 5.0 for LC–HRMS Spectra Processing, Multi-Omics Integration and Covariate Adjustment of Global Metabolomics Data. Nat. Protoc. 2022, 17, 1735–1761. [Google Scholar] [CrossRef] [PubMed]
- Giacomoni, F.; le Corguillé, G.; Monsoor, M.; Landi, M.; Pericard, P.; Pétéra, M.; Duperier, C.; Tremblay-Franco, M.; Martin, J.-F.; Jacob, D.; et al. Workflow4Metabolomics: A Collaborative Research Infrastructure for Computational Metabolomics. Bioinformatics 2015, 31, 1493–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plyushchenko, I.V.; Fedorova, E.S.; Potoldykova, N.V.; Polyakovskiy, K.A.; Glukhov, A.I.; Rodin, I.A. Omics Untargeted Key Script: R-Based Software Toolbox for Untargeted Metabolomics with Bladder Cancer Biomarkers Discovery Case Study. J. Proteome Res. 2022, 21, 833–847. [Google Scholar] [CrossRef] [PubMed]
- Stackebrandt, E.; Goebel, B.M. Taxonomic Note: A Place for DNA-DNA Reassociation and 16S RRNA Sequence Analysis in the Present Species Definition in Bacteriology. Int. J. Syst. Evol. Microbiol. 1994, 44, 846–849. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Oh, H.-S.; Park, S.-C.; Chun, J. Towards a Taxonomic Coherence between Average Nucleotide Identity and 16S RRNA Gene Sequence Similarity for Species Demarcation of Prokaryotes. Int. J. Syst. Evol. Microbiol. 2014, 64, 346–351. [Google Scholar] [CrossRef]
- Amiri Moghaddam, J.; Crüsemann, M.; Alanjary, M.; Harms, H.; Dávila-Céspedes, A.; Blom, J.; Poehlein, A.; Ziemert, N.; König, G.M.; Schäberle, T.F. Analysis of the Genome and Metabolome of Marine Myxobacteria Reveals High Potential for Biosynthesis of Novel Specialized Metabolites. Sci. Rep. 2018, 8, 16600. [Google Scholar] [CrossRef] [Green Version]
- Krug, D.; Zurek, G.; Revermann, O.; Vos, M.; Velicer, G.J.; Müller, R. Discovering the Hidden Secondary Metabolome of Myxococcus Xanthus: A Study of Intraspecific Diversity. Appl. Environ. Microbiol. 2008, 74, 3058–3068. [Google Scholar] [CrossRef] [Green Version]
- Böcker, S.; Letzel, M.C.; Lipták, Z.; Pervukhin, A. SIRIUS: Decomposing Isotope Patterns for Metabolite Identification. Bioinformatics 2009, 25, 218–224. [Google Scholar] [CrossRef] [Green Version]
- Dührkop, K.; Shen, H.; Meusel, M.; Rousu, J.; Böcker, S. Searching Molecular Structure Databases with Tandem Mass Spectra Using CSI:FingerID. Proc. Natl. Acad. Sci. USA 2015, 112, 12580–12585. [Google Scholar] [CrossRef]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glöckner, F.O.; Ludwig, W.; Schleifer, K.-H.; Whitman, W.B.; Euzéby, J.; Amann, R.; Rosselló-Móra, R. Uniting the Classification of Cultured and Uncultured Bacteria and Archaea Using 16S RRNA Gene Sequences. Nat. Rev. Microbiol. 2014, 12, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Schrimpe-Rutledge, A.C.; Codreanu, S.G.; Sherrod, S.D.; McLean, J.A. Untargeted Metabolomics Strategies—Challenges and Emerging Directions. J. Am. Soc. Mass Spectrom. 2016, 27, 1897–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.-M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed Minimum Reporting Standards for Chemical Analysis. Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depke, T.; Franke, R.; Brönstrup, M. Clustering of MS2spectra Using Unsupervised Methods to Aid the Identification of Secondary Metabolites from Pseudomonas Aeruginosa. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1071, 19–28. [Google Scholar] [CrossRef]
- Djoumbou Feunang, Y.; Eisner, R.; Knox, C.; Chepelev, L.; Hastings, J.; Owen, G.; Fahy, E.; Steinbeck, C.; Subramanian, S.; Bolton, E.; et al. ClassyFire: Automated Chemical Classification with a Comprehensive, Computable Taxonomy. J. Cheminform. 2016, 8, 61–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.-M.; Rock, C.O. Membrane Lipid Homeostasis in Bacteria. Nat. Rev. Microbiol. 2008, 6, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.Y.; Salton, M.R.J. Fatty Acid Composition of Bacterial Membrane and Wall Lipids. Biochim. Biophys. Acta (BBA) Lipids Lipid Metab. 1966, 116, 73–79. [Google Scholar] [CrossRef]
- Bajerski, F.; Wagner, D.; Mangelsdorf, K. Cell Membrane Fatty Acid Composition of Chryseobacterium Frigidisoli PB4T, Isolated from Antarctic Glacier Forefield Soils, in Response to Changing Temperature and PH Conditions. Front. Microbiol. 2017, 8, 677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legendre, S.; Letellier, L.; Shechter, E. Influence of Lipids with Branched-Chain Fatty Acids on the Physical, Morphological and Functional Properties of Escherichia Coli Cytoplasmic Membrane. Biochim. Biophys. Acta (BBA) Biomembr. 1980, 602, 491–505. [Google Scholar] [CrossRef]
- Lim, J.; Lee, S.H.; Cho, S.; Lee, I.-S.; Kang, B.Y.; Choi, H.J. 4-Methoxychalcone Enhances Cisplatin-Induced Oxidative Stress and Cytotoxicity by Inhibiting the Nrf2/ARE-Mediated Defense Mechanism in A549 Lung Cancer Cells. Mol. Cells 2013, 36, 340–346. [Google Scholar] [CrossRef]
- Buso Bortolotto, L.F.; Azevedo, B.C.; Silva, G.; Marins, M.; Fachin, A.L. Cytotoxic Activity Evaluation of Chalcones on Human and Mouse Cell Lines. BMC Proc. 2014, 8, P52. [Google Scholar] [CrossRef] [Green Version]
- Sivakumar, P.M.; Iyer, G.; Natesan, L.; Doble, M. 3′-Hydroxy-4-Methoxychalcone as a Potential Antibacterial Coating on Polymeric Biomaterials. Appl. Surf. Sci. 2010, 256, 6018–6024. [Google Scholar] [CrossRef]
- Stompor, M.; Zarowska, B. Antimicrobial Activity of Xanthohumol and Its Selected Structural Analogues. Molecules 2016, 21, 608. [Google Scholar] [CrossRef] [PubMed]
- Martín, J.F.; Liras, P. Comparative Molecular Mechanisms of Biosynthesis of Naringenin and Related Chalcones in Actinobacteria and Plants: Relevance for the Obtention of Potent Bioactive Metabolites. Antibiotics 2022, 11, 82. [Google Scholar] [CrossRef] [PubMed]
- Bringmann, G.; Noll, T.F.; Gulder, T.A.M.; Grüne, M.; Dreyer, M.; Wilde, C.; Pankewitz, F.; Hilker, M.; Payne, G.D.; Jones, A.L.; et al. Different Polyketide Folding Modes Converge to an Identical Molecular Architecture. Nat. Chem. Biol. 2006, 2, 429–433. [Google Scholar] [CrossRef]
- Nothias, L.-F.; Petras, D.; Schmid, R.; Dührkop, K.; Rainer, J.; Sarvepalli, A.; Protsyuk, I.; Ernst, M.; Tsugawa, H.; Fleischauer, M.; et al. Feature-Based Molecular Networking in the GNPS Analysis Environment. Nat. Methods 2020, 17, 905–908. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Ernst, M.; Kang, K.B.; Caraballo-Rodríguez, A.M.; Nothias, L.-F.; Wandy, J.; Chen, C.; Wang, M.; Rogers, S.; Medema, M.H.; Dorrestein, P.C.; et al. MolNetEnhancer: Enhanced Molecular Networks by Integrating Metabolome Mining and Annotation Tools. Metabolites 2019, 9, 144. [Google Scholar] [CrossRef] [Green Version]
- Dührkop, K.; Nothias, L.-F.; Fleischauer, M.; Reher, R.; Ludwig, M.; Hoffmann, M.A.; Petras, D.; Gerwick, W.H.; Rousu, J.; Dorrestein, P.C.; et al. Systematic Classification of Unknown Metabolites Using High-Resolution Fragmentation Mass Spectra. Nat. Biotechnol. 2021, 39, 462–471. [Google Scholar] [CrossRef]
- Schmid, R.; Petras, D.; Nothias, L.-F.; Wang, M.; Aron, A.T.; Jagels, A.; Tsugawa, H.; Rainer, J.; Garcia-Aloy, M.; Dührkop, K.; et al. Ion Identity Molecular Networking for Mass Spectrometry-Based Metabolomics in the GNPS Environment. Nat. Commun. 2021, 12, 3832. [Google Scholar] [CrossRef]
- Afoullouss, S.; Balsam, A.; Allcock, A.L.; Thomas, O.P. Optimization of LC-MS2 Data Acquisition Parameters for Molecular Networking Applied to Marine Natural Products. Metabolites 2022, 12, 245. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Lu, H.; Lee, Y.H. Challenges and Emergent Solutions for LC-MS/MS Based Untargeted Metabolomics in Diseases. Mass Spectrom. Rev. 2018, 37, 772–792. [Google Scholar] [CrossRef]
- Nett, M.; Ikeda, H.; Moore, B.S. Genomic Basis for Natural Product Biosynthetic Diversity in the Actinomycetes. Nat. Prod. Rep. 2009, 26, 1362–1384. [Google Scholar] [CrossRef] [PubMed]
- Gulder, T.A.M.; Moore, B.S. Chasing the Treasures of the Sea—Bacterial Marine Natural Products. Curr. Opin. Microbiol. 2009, 12, 252–260. [Google Scholar] [CrossRef] [Green Version]
- Crüsemann, M. Coupling Mass Spectral and Genomic Information to Improve Bacterial Natural Product Discovery Workflows. Mar. Drugs 2021, 19, 142. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Bossdorf, O.; Gockel, S.; Hänsel, F.; Hemp, A.; Hessenmöller, D.; Korte, G.; Nieschulze, J.; Pfeiffer, S.; Prati, D.; et al. Implementing Large-Scale and Long-Term Functional Biodiversity Research: The Biodiversity Exploratories. Basic Appl. Ecol. 2010, 11, 473–485. [Google Scholar] [CrossRef]
- Connon, S.A.; Giovannoni, S.J. High-Throughput Methods for Culturing Microorganisms in Very-Low-Nutrient Media Yield Diverse New Marine Isolates. Appl. Environ. Microbiol. 2002, 68, 3878–3885. [Google Scholar] [CrossRef] [Green Version]
- Pascual, J.; Wüst, P.K.; Geppert, A.; Foesel, B.U.; Huber, K.J.; Overmann, J. Novel Isolates Double the Number of Chemotrophic Species and Allow the First Description of Higher Taxa in Acidobacteria Subdivision 4. Syst. Appl. Microbiol. 2015, 38, 534–544. [Google Scholar] [CrossRef]
- Overmann, J.; Abt, B.; Sikorski, J. Present and Future of Culturing Bacteria. Annu. Rev. Microbiol. 2017, 71, 711–730. [Google Scholar] [CrossRef]
- Bruns, A.; Hoffelner, H.; Overmann, J. A Novel Approach for High Throughput Cultivation Assays and the Isolation of Planktonic Bacteria. FEMS Microbiol. Ecol. 2003, 45, 161–171. [Google Scholar] [CrossRef]
- Camarinha-Silva, A.; Jáuregui, R.; Chaves-Moreno, D.; Oxley, A.P.A.; Schaumburg, F.; Becker, K.; Wos-Oxley, M.L.; Pieper, D.H. Comparing the Anterior Nare Bacterial Community of Two Discrete Human Populations Using Illumina Amplicon Sequencing. Environ. Microbiol. 2014, 16, 2939–2952. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic. Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and Clustering Orders of Magnitude Faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Gich, F.; Janys, M.A.; König, M.; Overmann, J. Enrichment of Previously Uncultured Bacteria from Natural Complex Communities by Adhesion to Solid Surfaces. Environ. Microbiol. 2012, 14, 2984–2997. [Google Scholar] [CrossRef] [PubMed]
- Kjelleberg, S.; Humphrey, B.A.; Marshall, K.C. Effect of Interfaces on Small, Starved Marine Bacteria. Appl. Environ. Microbiol. 1982, 43, 1166–1172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adler, J. A Method for Measuring Chemotaxis and Use of the Method to Determine Optimum Conditions for Chemotaxis by Escherichia Coli. Microbiology 1973, 74, 77–91. [Google Scholar] [CrossRef] [Green Version]
- Fröstl, J.M.; Overmann, J. Physiology and Tactic Response of the Phototrophic Consortium “Chlorochromatium Aggregatum”. Arch. Microbiol. 1998, 169, 129–135. [Google Scholar] [CrossRef]
- Overmann, J. Chemotaxis and Behavioral Physiology of Not-yet-Cultivated Microbes. Methods Enzym. 2005, 397, 133–147. [Google Scholar]
- Galkiewicz, J.P.; Kellogg, C.A. Cross-Kingdom Amplification Using Bacteria-Specific Primers: Complications for Studies of Coral Microbial Ecology. Appl. Environ. Microbiol. 2008, 74, 7828–7831. [Google Scholar] [CrossRef] [Green Version]
- Stackebrandt, E.; Goodfellow, M. Nucleic Acid Techniques in Bacterial Systematics; John Wiley and Sons: Hoboken, NJ, USA, 1991. [Google Scholar]
- Muyzer, G.; de Waal, E.C.; Uitterlinden, A.G. Profiling of Complex Microbial Populations by Denaturing Gradient Gel Electrophoresis Analysis of Polymerase Chain Reaction-Amplified Genes Coding for 16S RRNA. Appl. Environ. Microbiol. 1993, 59, 695–700. [Google Scholar] [CrossRef] [Green Version]
- Yoon, S.-H.; Ha, S.-M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A Taxonomically United Database of 16S RRNA Gene Sequences and Whole-Genome Assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Göker, M.; Spröer, C.; Klenk, H.-P. When Should a DDH Experiment Be Mandatory in Microbial Taxonomy? Arch. Microbiol. 2013, 195, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A Database Tandem for Fast and Reliable Genome-Based Classification and Nomenclature of Prokaryotes. Nucleic Acids Res. 2022, 50, D801–D807. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Hahnke, R.L.; Petersen, J.; Scheuner, C.; Michael, V.; Fiebig, A.; Rohde, C.; Rohde, M.; Fartmann, B.; Goodwin, L.A.; et al. Complete Genome Sequence of DSM 30083(T), the Type Strain (U5/41(T)) of Escherichia Coli, and a Proposal for Delineating Subspecies in Microbial Taxonomy. Stand. Genom. Sci. 2014, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome Sequence-Based Species Delimitation with Confidence Intervals and Improved Distance Functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goloboff, P.A.; Farris, J.S.; Nixon, K.C. TNT, a Free Program for Phylogenetic Analysis. Cladistics 2008, 24, 774–786. [Google Scholar] [CrossRef]
- Pattengale, N.D.; Alipour, M.; Bininda-Emonds, O.R.P.; Moret, B.M.E.; Stamatakis, A. How Many Bootstrap Replicates Are Necessary? J. Comput. Biol. 2010, 17, 337–354. [Google Scholar] [CrossRef]
- Swofford, D.L. PAUP*: Phylogenetic Analysis Using Parsimony (* and Other Methods); Version 4.0 B10; Sinauer Associates: Sunderland, MA, USA, 2002. [Google Scholar]
- Ivica Letunic, P.B. Interactive Tree of Life (ITOL): An Online Tool for Phylogenetic Tree Display and Annotation. Bioinformatics 2007, 23, 127–128. [Google Scholar]
- van der Hooft, J.J.J.; Wandy, J.; Barret, M.P.; Burgess, K.E.V.; Rogers, S. Topic Modeling for Untargeted Substructure Exploration in Metabolomics. Proc. Natl. Acad. Sci. USA 2016, 113, 13738–13743. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Extract ID | Strain ID | Accession Number | Source | Origin | Cultivation Strategy | Isolation Medium | Phylum | Genus | Closest Relative | Similarity (%) |
|---|---|---|---|---|---|---|---|---|---|---|
| 01 | HEG41_91 | OP776843 | Soil | German soil | Direct plating | SSE 1:10 HD | Proteobacteria | Bradyrhizobium | Bradyrhizobium uaiense UFLA03 164 KC879705 | 97.02 * |
| 02 | 4RS2_G4 | OP776844 | Sediment | Channel Sea | Biofilm | ASWsalts 1:10 HD | Proteobacteria | Sulfitobacter | Sulfitobacter dubius DQ915635 | 99.67 |
| 03 | JAB_HD_127b | OP776845 | Water | Baltic Sea | Multiwell plate | ABWsalts 1:10 HD | Actinobacteria | Rhodococcus | Rhodococcus qingshengii JCM 15477 DQ090961 | 100.00 |
| 04 | PCS2D_E11 | OP776846 | Sediment | Atlantic Ocean | Multiwell plate | ASWsalts 1:10 HD Polymer | Proteobacteria | Oceanisphaera | Oceanisphaera psychrotolerans KF418814 | 99.89 |
| 05 | JAB_HD_128b | OP776847 | Water | Baltic Sea | Multiwell plate | ABWsalts 1:10 HD | Proteobacteria | Devosia | Devosia psychrophila GU441678 | 98.83 |
| 06 | JAB_HD_2a | OP776848 | Water | Baltic Sea | Multiwell plate | ABWsalts 1:10 HD | Actinobacteria | Rhodococcus | Rhodococcus qingshengii JCM 15477DQ090961 | 100.00 |
| 07 | JAB_HD_137a | OP776849 | Water | Baltic Sea | Multiwell plate | ABWsalts 1:10 HD | Actinobacteria | Rhodococcus | Rhodococcus jostii KF410370 | 99.24 |
| 08 | JAB_HD_121a | OP776850 | Water | Baltic Sea | Multiwell plate | ABWsalts 1:10 HD | Actinobacteria | Microbacterium | Microbacterium marinum EF204420 | 100.00 |
| 09 | 4RS2_G3b | OP776852 | Sediment | Channel Sea | Biofilm | ASWsalts 1:10 HD Glass | Proteobacteria | Aliidiomarina | Aliidiomarina soli KX548074 | 97.10 |
| 10 | 4RW5_PS1 | OP776853 | Water | Channel Sea | Biofilm | ASWsalts 1:10 HD Polymer | Proteobacteria | Alteromonas | Alteromonas macleodii AB681740 | 99.35 |
| 11 | CS1_PP3 | OP776854 | Sediment | Atlantic Ocean | Multiwell plate | ASWsalts 1:10 HD Polymer | Proteobacteria | Pseudoalteromonas | Pseudoalteromonas shioyasakiensis AB720724 | 99.65 |
| 12 | 4CH2_twe | OP776855 | Sponge | host | Chemotaxis | ASWsalts 1:10 HD | Proteobacteria | Vibrio | Vibrio kanaloae CAIM 485 MT757984 | 99.85 |
| 13 | JAB_HD_4a2 | OP776856 | Water | Baltic Sea | Multiwell plate | ABWsalts 1:10 HD | Actinobacteria | Aeromicrobium | Aeromicrobium ginsengisoli AB245394 | 99.47 |
| 14 | 4RS2_G3a | OP776857 | Sediment | Channel Sea | Biofilm | ASWsalts 1:10 HD Glass | Proteobacteria | Halomonas | Halomonas alkaliphila AJ640133 | 99.93 |
| 15 | 4RW5_PS3 | OP776858 | Water | Channel Sea | Biofilm | ASWsalts 1:10 HD | Proteobacteria | Pseudovibrio | Pseudovibrio ascidiaceicola AB681198 | 98.51 |
| 16 | 3RW5_S4aa | OP776859 | Water | Channel Sea | Biofilm | ASWsalts 1:10 HD Steel | Bacteroidetes | Maribacter | Maribacter litoralis MG456900 | 99.93 |
| 17 | JAB_HD_102a2 | OP776860 | Water | Baltic Sea | Multiwell plate | ABWsalts 1:10 HD | Proteobacteria | Pseudomonas | Pseudomonas pelagia strain CL-AP6 EU888911 | 98.79 |
| 18 | 4d1_twe | OP776861 | Sponge | host | Chemotaxis | ASWsalts 1:10 HD | Proteobacteria | Pseudomonas | Pseudomonas knackmussii B13 AJ272544 | 99.67 |
| 19 | 4RS2_G7 | OP776862 | Sediment | Channel Sea | Biofilm | ASWsalts 1:10 HD Glass | Proteobacteria | Lutimaribacter | Lutimaribacter pacificus DQ659449 | 97.04 * |
| 20 | JAB_HD_109a | OP776863 | Water | Baltic Sea | Multiwell plate | ABWsalts 1:10 HD | Proteobacteria | Pseudorhodobacter | Pseudorhodobacter ponti KX771233 | 97.15 * |
| 21 | RW5_G2 | OP776864 | Water | Channel Sea | Biofilm | ASWsalts 1:10 HD Glass | Bacteroidetes | Altibacter-Rhodococcus | Rhodococcus yunnanensis AY602219 | 99.33 |
| 22 | CS1PS2a | OP776865 | Sediment | Atlantic Ocean | Biofilm | ASWsalts 1:10 HD Polymer | Proteobacteria | Paracoccus | Paracoccus indicus MG845150 | 99.77 |
| 23 | D100_Iso2 | OP776866 | Alga | host | Direct plating | MB | Proteobacteria | Aquicoccus | Aquicoccus porphyridii MF113254 | 96.82 * |
| 24 | MEBiC05055 | OP776870 | Sponge | host | Direct plating | MB | Proteobacteria | Tateyamaria | Tateyamaria armeniaca LC464518 | 98.34 |
| 25 | DSM_16472T | OP776867 | Water | Sea of Japan | Direct plating | MB | Proteobacteria | Sulfitobacter | Sulfitobacter dubius DQ915635 | 100.00 * |
| 26 | DSM_10251T | OP776871 | Alga | host | Direct plating | MB | Proteobacteria | Marinovum | Marinovum algicola DG898 DSM 27768 | 100.00 * |
| 27 | DSM_27768 | OP776872 | Alga | host | Direct plating | MB | Proteobacteria | Marinovum | Marinovum algicola FF3 DSM 10251T | 100.00 * |
| 29 | C05C_116 | OP776869 | Alga | host | Direct plating | L1ZM10 | Proteobacteria | Sulfitobacter | Sulfitobacter pseudonitzschiae KF006321 | 99.50 |
| 30 | A11D_105 | OP776868 | Alga | host | Direct plating | MB | Proteobacteria | Sulfitobacter | Sulfitobacter porphyrae AB758574 | 99.85 |
| 31 | A05D_005 | OP776873 | Alga | host | Direct plating | MB | Proteobacteria | Aquicoccus | Aquicoccus porphyridii MF113254 | 100.00 |
| 32 | C05C_110 | OP776875 | Alga | host | Direct plating | MB | Proteobacteria | Hoeflea | Hoeflea alexandrii MT760263 | 99.69 |
| 33 | H01Y_008A | OP776874 | Alga | host | Direct plating | MB | Proteobacteria | Fretibacter | Fretibacter rubidus FJ394547 | 97.12 * |
| 34 | RW5_G4 | OP776824 | Water | Channel Sea | Biofilm | ASWsalts 1:10 HD Glass | Proteobacteria | Amylibacter | Amylibacter cionae KX790330 | 99.19 |
| 35 | JAB_HD_121b | OP776851 | Water | Baltic Sea | Multiwell plate | ABWsalts 1:10 HD | Proteobacteria | Pseudorhodobacter | Pseudorhodobacter wandonensis JN247434 | 99.18 |
| 36 | JAB_HD_38 | OP776826 | Water | Baltic Sea | Multiwell plate | ASWsalts 1:10 HD | Bacteroidetes | Algoriphagus | Algoriphagus aquaemixtae KY661386 | 99.26 |
| 112 | M64 | OP776831 | Water | Baltic Sea | Biofilm | KM14 | Bacteroidetes | Flavobacterium | Flavobacterium circumlabens P5626 MH100898 | 98.80 |
| 122 | M66 | OP776832 | Water | Baltic Sea | Biofilm | KM14 | Bacteroidetes | Flavobacterium | Flavobacterium terriphilum CUG00004 KT592306 | 99.12 |
| 132 | M20 | OP776827 | Water | Baltic Sea | Biofilm | KM14 | Actinobacteria | Rubrobacter | Rubrobacter radiotolerans X87134 | 93.95 ** |
| 212 | M55 | OP776829 | Water | Baltic Sea | Biofilm | MB | Proteobacteria | Altererythrobacter | Altererythrobacter epoxidivorans DQ304436 | 97.94 * |
| 222 | M62 | OP776830 | Water | Baltic Sea | Biofilm | MB | Firmicutes | Bacillus | Bacillus mobilis MCCC 1A05942 KJ812449 | 99.93 |
| 232 | M09 | OP776828 | Water | Baltic Sea | Biofilm | MB | Proteobacteria | Altererythrobacter | Altererythrobacter aquiaggeris KX812543 | 98.73 |
| 312 | SEG27_38 | OP776841 | Soil | German soil | Direct plating | SSE 1:10 HD | Bacteroidetes | Chitinophaga | Chitinophaga flava MH553387 | 93.57 ** |
| 322 | AEG42_45 | OP776842 | Soil | German soil | Direct plating | SSE 1:10 HD | Actinobacteria | Sporichthya | Sporichthya brevicatena AB006164 | 96.46 * |
| 332 | AEG42_13 | OP776840 | Soil | German soil | Direct plating | SSE 1:10 HD | Actinobacteria | Nocardioides | Nocardioides humi EF623863 | 96.86 * |
| 342 | ACS3D_E6 | OP776819 | Sediment | Atlantic Ocean | Multiwell plate | SSE 1:10 HD | Bacteroidetes | Ulvibacter | Ulvibacter antarcticus AB681898 | 97.28 * |
| 352 | HEG41_64b | OP776836 | Soil | German soil | Direct plating | SSE 1:10 HD | Bacteroidetes | Niastella | Niastella populi EU877262 | 96.17 * |
| 362 | SEG27_44 | OP776837 | Soil | German soil | Direct plating | SSE 1:10 HD | Bacteroidetes | Pseudoflavitalea | Pseudoflavitalea rhizosphaerae KU379667 | 94.04 ** |
| 372 | AEG42_46 | OP776839 | Soil | German soil | Direct plating | SSE 1:10 HD | Bacteroidetes | Flavitalea | Flavitalea flava KX762320 | 99.80 |
| 382 | SEG27_28 | OP776838 | Soil | German soil | Direct plating | SSE 1:10 HD | Bacteroidetes | Niveitalea | Niveitalea solisilvae KX268597 | 92.80 ** |
| 392 | AEG42_23 | OP776835 | Soil | German soil | Direct plating | SSE 1:10 HD | Bacteroidetes | Ferruginibacter | Ferruginibacter yonginensis MT760289 | 93.85 ** |
| 412 | PCS2D_E7 | OP776816 | Sediment | Atlantic Ocean | Multiwell plate | ASWsalts 1:10 HD Polymer | Proteobacteria | Marinomonas | Marinomonas atlantica LN909522 | 99.86 |
| 422 | CS3_PS3b | OP776818 | Sediment | Atlantic Ocean | Biofilm | ASWsalts 1:10 HD Polymer | Proteobacteria | Amylibacter | Amylibacter lutimaris MF113253 | 99.85 |
| 432 | 3RW5_PP6 | OP776825 | Water | Channel Sea | Biofilm | ASWsalts 1:10 HD Polymer | Bacteroidetes | Ulvibacter | Ulvibacter antarcticus AB681898 | 96.77 * |
| 442 | ACS3C_E5 | OP776817 | Sediment | Atlantic Ocean | Multiwell plate | ASWsalts 1:10 HD | Proteobacteria | Pseudoalteromonas | Pseudoalteromonas shioyasakiensis SE3 AB720724 | 99.65 |
| 452 | 2CW3_G4 | OP776820 | Water | Atlantic Ocean | Biofilm | ASWsalts 1:10 HD Polymer | Bacteroidetes | Balneola | Balneola vulgaris AY576749 | 94.85 ** |
| 462 | M68 | OP776833 | Water | Baltic Sea | Biofilm | ABWsalts 1:10 HD | Bacteroidetes | Arenibacter | Arenibacter algicola FJ176555 | 99.91 |
| 472 | RS2_PS_4 | OP776823 | Sediment | Channel Sea | Biofilm | ASWsalts 1:10 HD Polymer | Proteobacteria | Pararhodobacter | Pararhodobacter oceanensis KY009733 | 99.85 |
| 482 | M72 | OP776834 | Water | Baltic Sea | Biofilm | ABWsalts 1:10 HD | Bacteroidetes | Algoriphagus | Algoriphagus jejuensis EF217418 | 98.79 |
| 2F2 | ARW1_2F2 | OP776821 | Water | Channel Sea | Multiwell plate | ASWsalts 1:10 HD | Proteobacteria | Arcobacter | Arcobacter lekithochrous LT629298 | 98.16 * |
| 2G2 | ARW1_2G2 | OP776822 | Water | Channel Sea | Multiwell plate | ASWsalts 1:10 HD | Proteobacteria | Arcobacter | Arcobacter lekithochrous LT629298 | 98.17 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fiorini, F.; Bajerski, F.; Jeske, O.; Lepleux, C.; Overmann, J.; Brönstrup, M. A Metabolomics-Based Toolbox to Assess and Compare the Metabolic Potential of Unexplored, Difficult-to-Grow Bacteria. Mar. Drugs 2022, 20, 713. https://doi.org/10.3390/md20110713
Fiorini F, Bajerski F, Jeske O, Lepleux C, Overmann J, Brönstrup M. A Metabolomics-Based Toolbox to Assess and Compare the Metabolic Potential of Unexplored, Difficult-to-Grow Bacteria. Marine Drugs. 2022; 20(11):713. https://doi.org/10.3390/md20110713
Chicago/Turabian StyleFiorini, Federica, Felizitas Bajerski, Olga Jeske, Cendrella Lepleux, Jörg Overmann, and Mark Brönstrup. 2022. "A Metabolomics-Based Toolbox to Assess and Compare the Metabolic Potential of Unexplored, Difficult-to-Grow Bacteria" Marine Drugs 20, no. 11: 713. https://doi.org/10.3390/md20110713