Recent Advances in Molecular Docking for the Research and Discovery of Potential Marine Drugs


Abstract
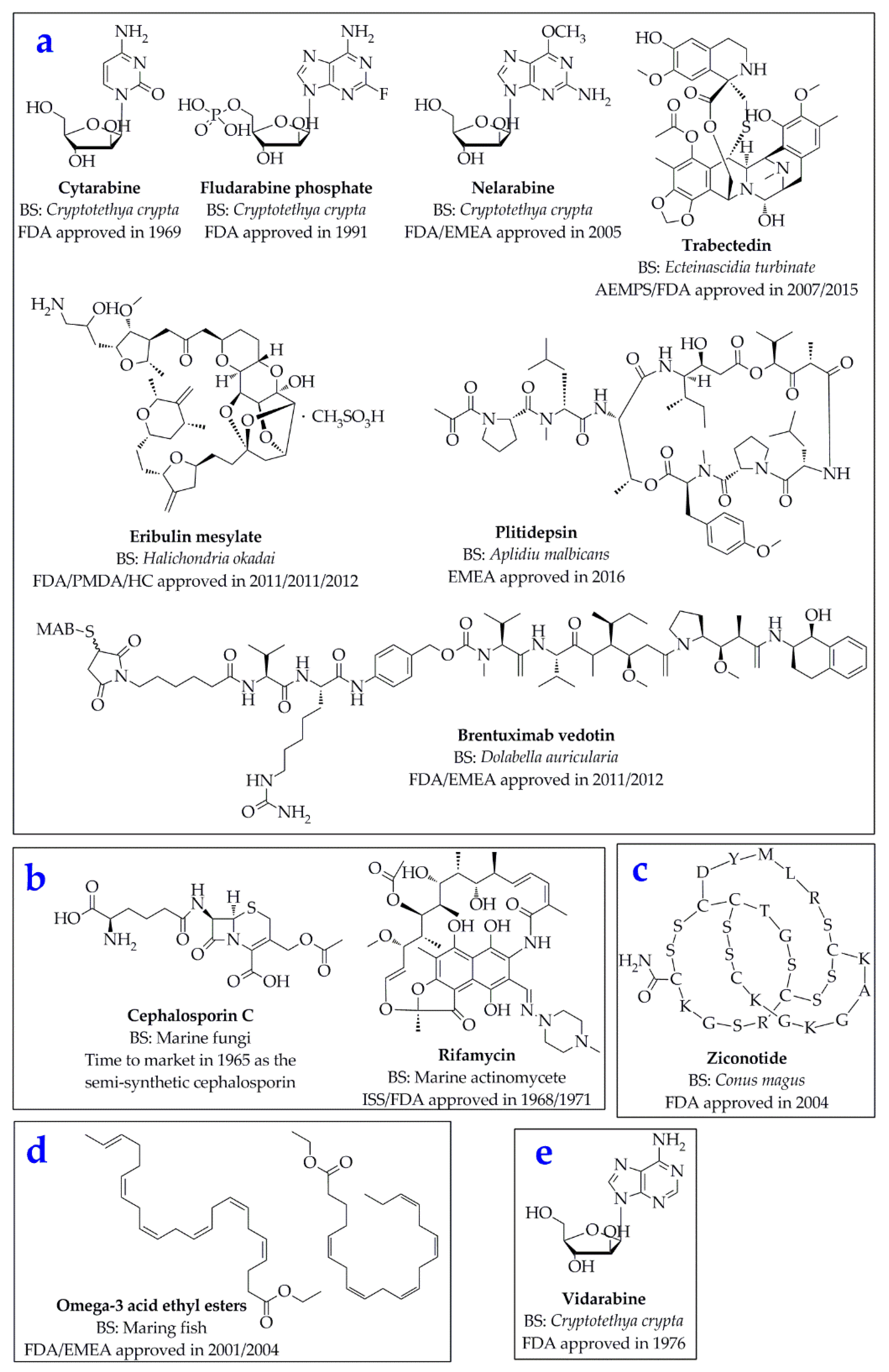
:1. Introduction
2. Principles of Molecular Docking
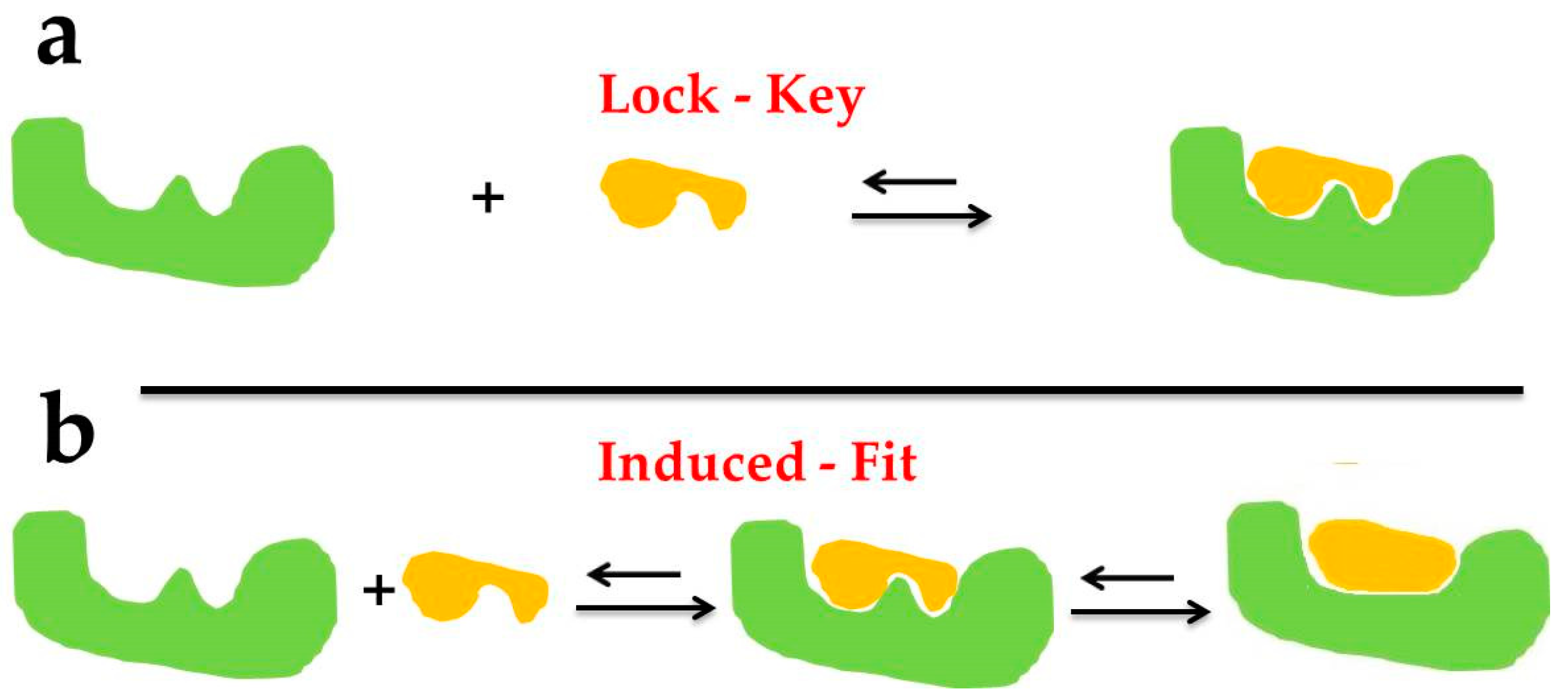
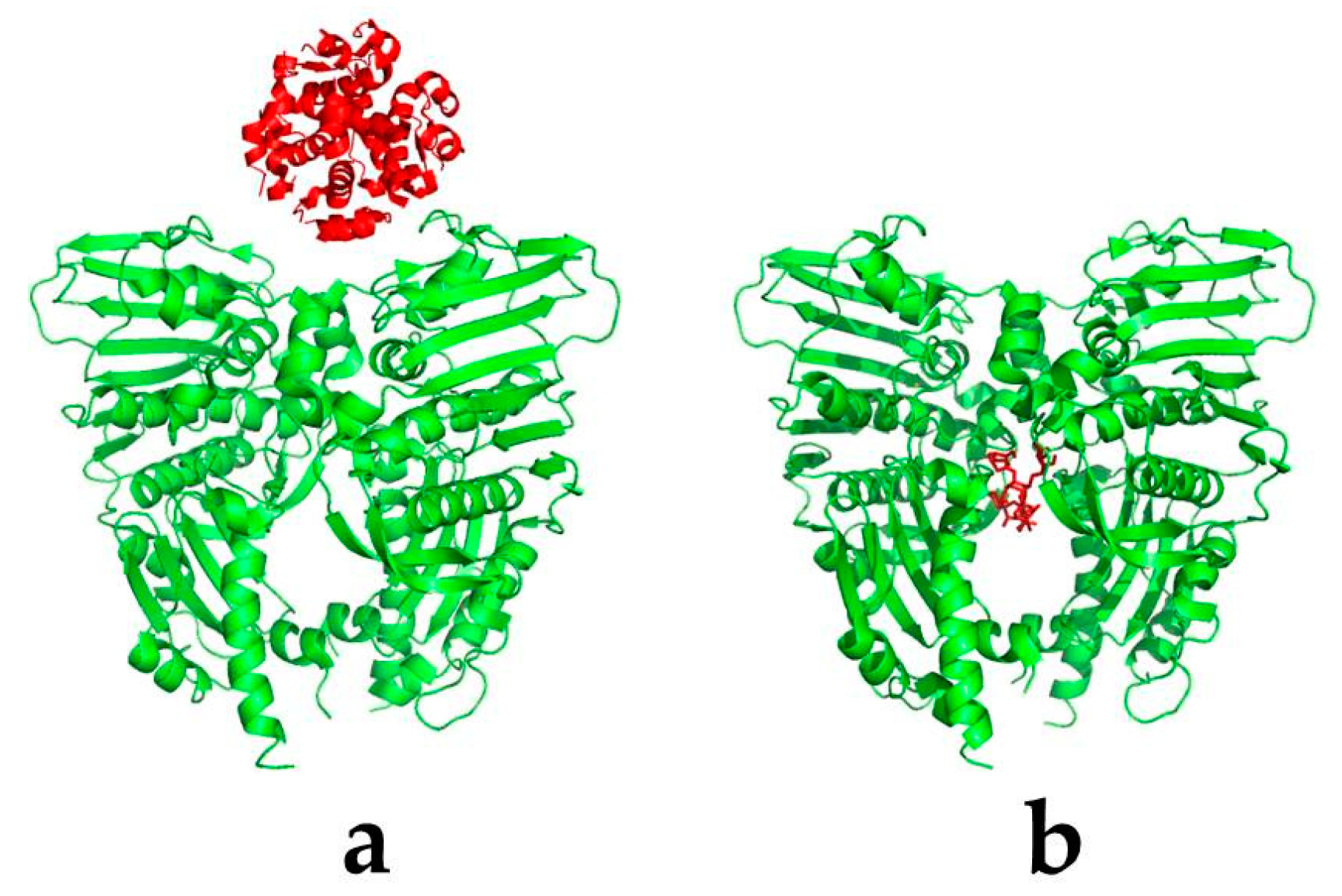
2.1. Basic Theories
2.2. Molecular Docking Methodologies
2.2.1. Rigid Docking
2.2.2. Flexible Docking
2.2.3. Semi-Flexible Docking
2.3. Molecular Docking Searching Algorithms
2.3.1. Exhaustive Searching Algorithms
2.3.2. Heuristic Searching Algorithms
2.4. Scoring Functions
2.4.1. Classifications of Scoring Functions
2.4.2. Classic Scoring Function Software
2.5. Molecular Docking Softwares
3. Applications of the Molecular Docking in the Research and Discovery of Potential Marine Drugs
3.1. Target Proteins of Melanin Formation
3.2. Target Proteins of Diabetes Mellitus
3.3. Target Proteins of Hypertension
3.4. Target Proteins of Inflammation
3.5. Target Proteins of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)
3.6. Target Proteins of Cancer
4. Conclusions and Outlooks
Author Contributions
Funding
Conflicts of Interest
References
- Ruggieri, G.D. Drugs from the sea. Science 1976, 194, 491–497. [Google Scholar] [CrossRef]
- Williams, D.E.; Andersen, R.J. Biologically active marine natural products and their molecular targets discovered using a chemical genetics approach. Nat. Prod. Rep. 2020, 37, 617–633. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F.; Aires-de-Sousa, J. Computational methodologies in the exploration of marine natural product leads. Mar. Drugs 2018, 16, 236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2017, 34, 235–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton-Miller, J.M.T. Sir Edward Abraham’s contribution to the development of the cephalosporins: A reassessment. Int. J. Antimicrob. Agents 2000, 15, 179–184. [Google Scholar] [CrossRef]
- Margalith, P.; Beretta, G. Rifomycin. XI. Taxonomic study on Streptomyces mediterranei nov. sp. Mycopathol. Mycol. Appl. 1960, 13, 321–330. [Google Scholar] [CrossRef]
- Sensi, P. History of the development of rifampin. Rev. Infect. Dis. 1983, 5, 402–406. [Google Scholar] [CrossRef]
- Zhang, S.W.; Huang, H.B.; Gui, C.; Ju, J.H. Progress on the research and development of marine drugs. Chin. J. Mar. Drugs 2018, 37, 77–92. [Google Scholar]
- Mulabagal, V.; Calderón, A.I. Development of an ultrafiltration-liquid chromatography/mass spectrometry (UF-LC/MS) based ligand-binding assay and an LC/MS based functional assay for mycobacterium tuberculosis shikimate kinase. Anal. Chem. 2010, 82, 3616–3621. [Google Scholar] [CrossRef]
- Suresh, P.S.; Kumar, A.; Kumar, R.; Singh, V.P. An in silico approach to bioremediation: Laccase as a case study. J. Mol. Graph. Model. 2008, 26, 845–849. [Google Scholar] [CrossRef]
- Chen, G.L.; Huang, B.X.; Guo, M.Q. Current advances in screening for bioactive components from medicinal plants by affinity ultrafiltration mass spectrometry. Phytochem. Anal. 2018, 29, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Kharkar, P.S.; Warrier, S.; Gaud, R.S. Reverse docking: A powerful tool for drug repositioning and drug rescue. Future Med. Chem. 2014, 6, 333–342. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Bruckner, R.J.; Paulo, J.A.; Cannon, J.R.; Ting, L.; Baltier, K.; Colby, G.; Gebreab, F.; Gygi, M.P.; Parzen, H.; et al. Architecture of the human interactome defines protein communities and disease networks. Nature 2017, 545, 505–509. [Google Scholar] [CrossRef]
- Li, X.H.; Chavali, P.L.; Babu, M.M. Capturing dynamic protein interactions. Science 2018, 359, 1105–1106. [Google Scholar] [CrossRef] [PubMed]
- Vakser, I.A. Protein-protein docking: From interaction to interactome. Biophys. J. 2014, 107, 1785–1793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villoutreix, B.O.; Bastard, K.; Sperandio, O.; Fahraeus, R.; Poyet, J.L.; Calvo, F.; Déprez, B.; Miteva, M.A. In silico-in vitro screening of protein-protein interactions: Towards the next generation of therapeutics. Curr. Pharm. Biotechnol. 2008, 9, 103–122. [Google Scholar] [CrossRef] [Green Version]
- Xue, L.C.; Dobbs, D.; Bonvin, A.M.; Honavar, V. Computational prediction of protein interfaces: A review of data driven methods. FEBS Lett. 2015, 589, 3516–3526. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L. Rusting of the lock and key model for protein-ligand binding. Science 1991, 254, 954–955. [Google Scholar] [CrossRef]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef]
- Fischer, E. Einfluss der configuration auf die wirkung der enzyme. Ber. Dtsch. Chem. Ges. 1894, 27, 3189–3232. [Google Scholar] [CrossRef] [Green Version]
- Koshland, D.E. Application of a theory of enzyme specificity to protein synthesis. Proc. Natl. Acad. Sci. USA 1958, 44, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful application for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Ewing, T.J.; Makino, S.; Skillman, A.G.; Kuntz, I.D. DOCK 4.0: Search strategies for automated molecular docking of flexible molecule databases. J. Comput. Aided Mol. Des. 2001, 15, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Stoddard, B.L.; Koshland, D.E. Prediction of the structure of a receptor–protein complex using a binary docking method. Nature 1992, 258, 774–776. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.X.; Jing, C.J.; Tang, K.L.; Tao, L.; Cao, Z.W.; Wu, W.Y.; Guan, S.H.; Jiang, B.H.; Yang, M.; Liu, X.; et al. Clarifying the signal network of salvianolic acid B using proteomic assay and bioinformatic analysis. Proteomics 2011, 11, 1473–1485. [Google Scholar] [CrossRef]
- Mangoni, M.; Roccatano, D.; Nola, A.D. Docking of flexible ligands to flexible receptors in solution by molecular dynamics simulation. Prot. Struct. Funct. Gent. 1999, 35, 153–162. [Google Scholar] [CrossRef]
- Bartuzi, D.; Kaczor, A.A.; Targowska-Duda, K.M.; Matosiuk, D. Recent advances and applications of molecular docking to G protein-coupled receptors. Molecules 2017, 22, 340. [Google Scholar] [CrossRef] [Green Version]
- Yuriev, E.; Holien, J.; Ramsland, P.A. Improvements, trends, and new ideas in molecular docking: 2012–2013 in review. J. Mol. Recognit. 2015, 28, 581–604. [Google Scholar] [CrossRef]
- Olsson, T.S.; Williams, M.A.; Pitt, W.R.; Ladbury, J.E. The thermodynamics of protein-ligand interaction and solvation: Insights for ligand design. J. Mol. Biol. 2008, 384, 1002–1017. [Google Scholar] [CrossRef]
- Huang, S.Y.; Zou, X. Advances and challenges in protein-ligand docking. Int. J. Mol. Sci. 2010, 11, 3016–3034. [Google Scholar] [CrossRef] [Green Version]
- Hogues, H.; Gaudreault, F.; Corbeil, C.R.; Deprez, C.; Sulea, T.; Purisima, E.O. ProPOSE: Direct exhaustive protein–protein docking with side chain flexibility. J. Chem. Theory Comput. 2018, 14, 4938–4947. [Google Scholar] [CrossRef] [PubMed]
- Katchalski-Katzir, E.S.; Shariv, I.; Eisenstein, M.; Friesem, A.A.; Aflalo, C.; Vakser, I.A. Molecular surface recognition: Determination of geometric fit between proteins and their ligands by correlation techniques. Proc. Natl. Acad. Sci. USA 1992, 89, 2195–2199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aloy, P.; Querol, E.; Aviles, F.X.; Sternberg, J.E. Automated structure-based prediction of functional sites in proteins: Applications to assessing the validity of inheriting protein function from homology in genome annotation and to protein docking. J. Mol. Biol. 2001, 311, 395–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alekseenko, A.; Kotelnikov, S.; Ignatov, M.; Egbert, M.; Kholodov, Y.; Vajda, S.; Kozakov, D. ClusPro LigTBM: Automated template-based small molecule docking. J. Mol. Biol. 2020, 432, 3404–3410. [Google Scholar] [CrossRef]
- Pierce, B.G.; Wiehe, K.; Hwang, H.; Kim, B.H.; Vreven, T.; Weng, Z. ZDOCK server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 2014, 30, 1771–1773. [Google Scholar] [CrossRef]
- Mandell, J.G.; Roberts, V.A.; Pique, M.; Kotlovyi, V.; Mitchell, J.C.; Nelson, E.; Tsigelny, I.; Ten Eyck, L.F. Protein docking using continuum electrostatics and geometric fit. Protein Eng. 2001, 14, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, D.W.; Kemp, G.J. Protein docking using spherical polar fourier correlations. Prot. Struct. Funct. Gent. 2000, 39, 178–194. [Google Scholar] [CrossRef]
- Garzon, J.I.; Lopez-Blanco, J.R.; Pons, C.; Kovacs, J.; Abagyan, R.; Fernandez-Recio, J.; Chacon, P. FRODOCK: A new approach for fast rotational protein-protein docking. Bioinformatics 2009, 25, 2544–2551. [Google Scholar] [CrossRef] [Green Version]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.H.; Teller, E. Equation of state calculations by fast computing machines. J. Chem. Phys. 1953, 21, 1087–1092. [Google Scholar]
- Holland, J.H. Adaptation in Natural and Artificial Systems; University of Michigan Press: Ann Arbor, MI, USA, 1975; pp. 1–183. [Google Scholar]
- Hinchey, M.G.S.; Sterritt, R.; Rouff, C. Swarms and swarm intelligence. Computer 2007, 40, 111–113. [Google Scholar] [CrossRef]
- Gray, J.J.; Moughon, S.; Wang, C.; Schueler-Furman, O.; Kuhlman, B.; Rohl, C.A.; Baker, D. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J. Mol. Biol. 2003, 331, 281–299. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef] [Green Version]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Prot. Struct. Funct. Gent. 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Torchala, M.; Moal, I.H.; Chaleil, R.A.G.; Fernandez-Recio, J.; Bates, P.A. SwarmDock: A server for flexible protein–protein docking. Bioinformatics 2013, 29, 807–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayik, S.A.; Dunbrack, R.; Merz, K.M. Mixed quantum mechanics/molecular mechanics scoring function to predict protein−ligand binding affinity. J. Chem. Theory Comput. 2010, 6, 3079–3091. [Google Scholar] [CrossRef]
- Hsieh, M.J.; Luo, R. Physical scoring function based on AMBER force field and Poisson-Boltzmann implicit solvent for protein structure prediction. Prot. Struct. Funct. Bioinform. 2004, 56, 475–486. [Google Scholar] [CrossRef]
- Wang, J.M.; Hou, T.J.; Xu, X.J. Recent advances in free energy calculations with a combination of molecular mechanics and continuum models. Curr. Comput. Aided Drug 2006, 2, 95–103. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Fang, Y.; Feinstein, W.P.; Ramanujam, J.; Koppelman, D.M.; Moreno, J.; Brylinski, M.; Jarrell, M. GeauxDock: A novel approach for mixed-resolution ligand docking using a descriptor-based force field. J. Comput. Chem. 2015, 36, 2013–2026. [Google Scholar] [CrossRef]
- Baek, M.; Shin, W.H.; Chung, H.W.; Seok, C. GalaxyDock BP2 score: A hybrid scoring function for accurate protein-ligand docking. J. Comput. Aided Mol. Des. 2017, 31, 653–666. [Google Scholar] [CrossRef]
- Bauer, A.; Kovári, Z.; Keserű, G.M. Optimization of virtual screening protocols: FlexX based virtual screening for COX-2 inhibitors reveals the importance of tailoring screen parameters. J. Mol. Struc. Theochem. 2004, 676, 1–5. [Google Scholar] [CrossRef]
- Fischer, M.J.; Kuipers, C.; Hofkes, R.P.; Hofmeyer, L.J.; Moret, E.E.; de Mol, N.J. Exploring computational lead optimisation with affinity constants obtained by surface plasmon resonance the interaction of PorA epitope peptides with antibody against Neisseria meningitidis. Biochim. Biophys. Acta 2001, 1568, 205–215. [Google Scholar] [CrossRef]
- Chen, R.; Li, L.; Weng, Z.P. ZDOCK: An initial-stage protein-docking algorithm. Prot. Struct. Funct. Bioinform. 2003, 52, 80–87. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, S.; Zhou, Y.Q. Accurate and efficient loop selections by the DFIRE-based all-atom statistical potential. Protein Sci. 2004, 13, 391–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.P.; Hsu, K.C.; Huang, J.W.; Chang, L.S.; Yang, J.M. Atrippi: An atom-residue preference scoring function for protein–protein interactions. Int. J. Artif. Intell. Tools 2011, 19, 251–266. [Google Scholar] [CrossRef]
- Pierce, B.; Weng, Z. ZRANK: Reranking protein docking predictions with an optimized energy function. Prot. Struct. Funct. Bioinform. 2007, 67, 1078–1086. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, R.; Weng, Z.P. RDOCK: Refinement of rigid-body protein docking predictions. Prot. Struct. Funct. Bioinform. 2003, 53, 693–707. [Google Scholar] [CrossRef] [Green Version]
- Palma, P.N.; Krippahl, L.; Wampler, J.E.; Moura, J.J.G. BIGGER: A new (soft) docking algorithm for predicting protein interactions. Prot. Struct. Funct. Bioinform. 2000, 39, 372–384. [Google Scholar] [CrossRef]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [Green Version]
- Dominguez, C.; Boelens, R.; Bonvin, A.M. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [Green Version]
- Van Dijk, M.V.K.M.; Kastritis, P.L.; Bonvin, A.M.J.J. Solvated protein—DNA docking using HADDOCK. J. Biomol. NMR 2013, 56, 51–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Fuentes, N.; de Vries, S.J.; Bonvin, A.M.J.J. CPORT: A consensus interface predictor and its performance in prediction-driven docking with HADDOCK. PLoS ONE 2011, 6, e17695. [Google Scholar]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: A review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Allen, W.J.; Balius, T.E.; Mukherjee, S.; Brozell, S.R.; Moustakas, D.T.; Lang, P.T.; Case, D.A.; Kuntz, I.D.; Rizzo, R.C. DOCK 6: Impact of new features and current docking performance. J. Comput. Chem. 2015, 36, 1132–1156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.Y.; Zou, X. Ensemble docking of multiple protein structures: Considering protein structural variations in molecular docking. Prot. Struct. Funct. Bioinform. 2007, 66, 399–421. [Google Scholar] [CrossRef]
- Spitzer, R.; Jain, A.N. Surflex-Dock: Docking benchmarks and real-world application. J. Comput. Aided Mol. Des. 2012, 26, 687–699. [Google Scholar] [CrossRef]
- Zsoldos, Z.; Reid, D.; Simon, A.; Sadjad, S.B.; Johnson, A.P. eHiTS: A new fast, exhaustive flexible ligand docking system. J. Mol. Graph. Model. 2007, 26, 198–212. [Google Scholar] [CrossRef]
- Grosdidier, A.; Zoete, V.; Michielin, O. EADock: Docking of small molecules into protein active sites with a multiobjective evolutionary optimization. Prot. Struct. Funct. Bioinform. 2007, 67, 1010–1025. [Google Scholar] [CrossRef]
- Kozakov, D.; Brenke, R.; Comeau, S.R.; Vajda, S. PIPER: An FFT-based protein docking program with pairwise potentials. Prot. Struct. Funct. Bioinform. 2006, 65, 392–406. [Google Scholar] [CrossRef] [Green Version]
- Welch, W.; Ruppert, J.; Jain, A.N. Hammerhead: Fast, fully automate docking of flexible ligands to protein binding sites. Chem. Biol. 1996, 3, 449–462. [Google Scholar] [CrossRef] [Green Version]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef]
- Zhao, Y.; Sanner, M.F. FLIPDock: Docking flexible ligands into flexible receptors. Prot. Struct. Funct. Bioinform. 2007, 68, 726–737. [Google Scholar] [CrossRef]
- Abagyan, R.; Totrov, M.; Kuznetsov, M. ICM—A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. Comput. Chem. 1994, 15, 488–506. [Google Scholar] [CrossRef]
- Miller, M.D.; Kearsley, S.K.; Underwood, D.J.; Sheridan, R.P. FLOG: A system to select ‘quasi-flexible’ ligands complementary to a receptor of known three-dimensional structure. J. Comput. Aided Mol. Des. 1994, 8, 153–174. [Google Scholar] [CrossRef]
- Sauton, N.; Lagorce, D.; Villoutreix, B.O.; Miteva, M.A. MS-DOCK: Accurate multiple conformation generator and rigid docking protocol for multi-step virtual ligand screening. BMC Bioinform. 2008, 9, 184. [Google Scholar] [CrossRef] [Green Version]
- Costin, G.E.; Hearing, V.J. Human skin pigmentation: Melanocytes modulate skin color in response to stress. FASEB J. 2007, 21, 976–994. [Google Scholar] [CrossRef]
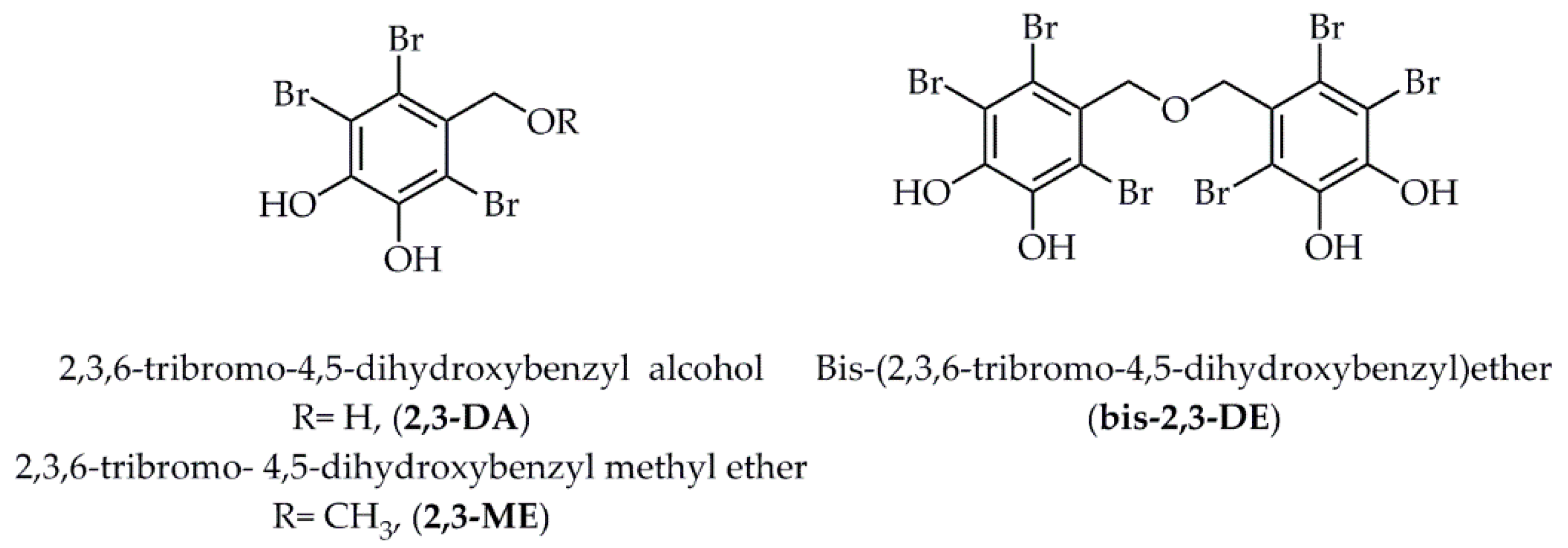
- Paudel, P.; Wagle, A.; Seong, S.H.; Park, H.J.; Jung, H.A.; Choi, J.S. A new tyrosinase inhibitor from the red alga Symphyocladia latiuscula (Harvey) Yamada (Rhodomelaceae). Mar. Drugs 2019, 17, 295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
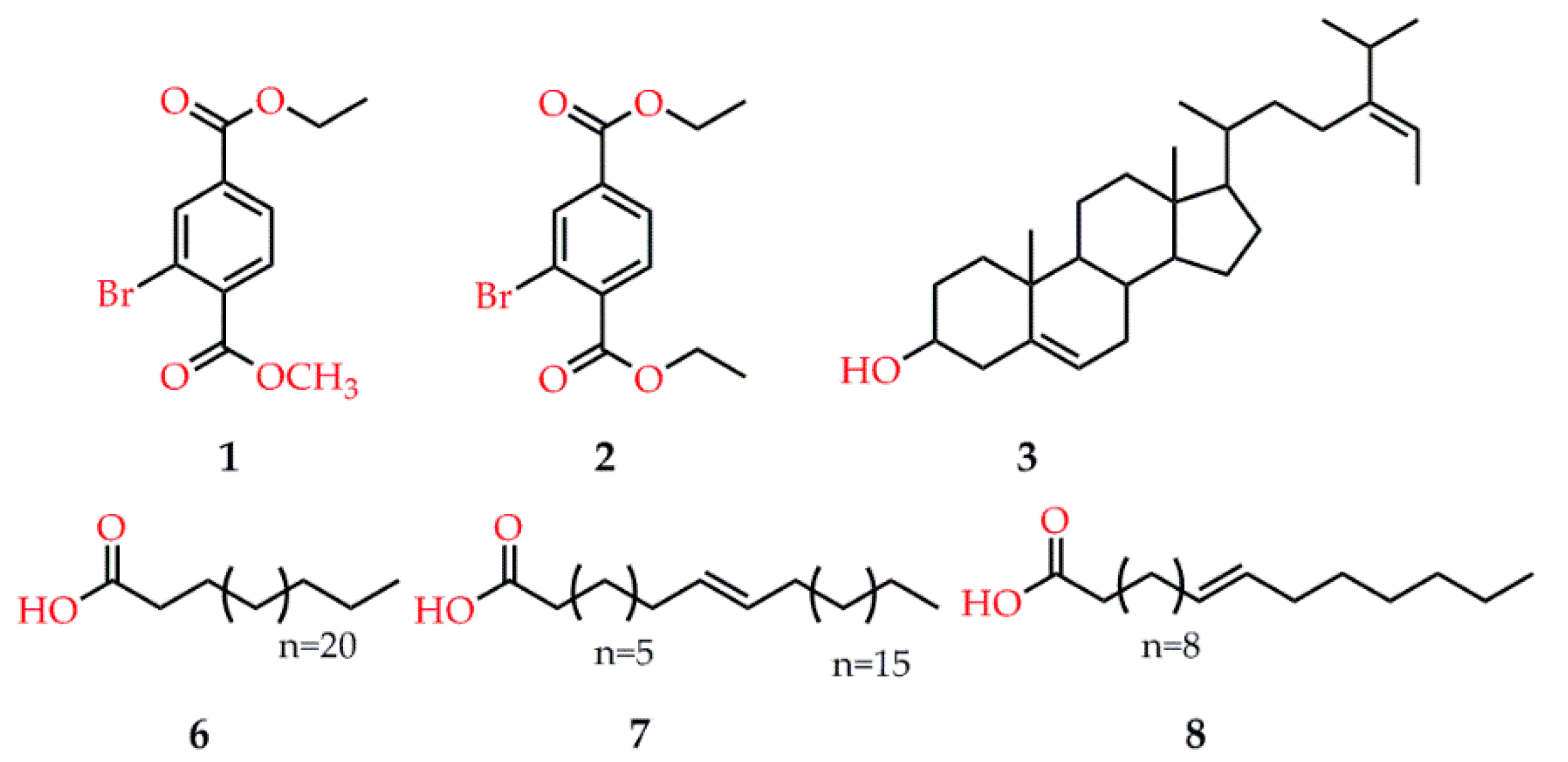
- Paudel, P.; Seong, S.H.; Park, H.J.; Jung, H.A.; Choi, J.S. Anti-diabetic activity of 2,3,6-tribromo-4,5-dihydroxybenzyl derivatives from Symphyocladia latiuscula through PTP1B downregulation and α-glucosidase inhibition. Mar. Drugs 2019, 17, 166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ur Rehman, N.; Rafiq, K.; Khan, A.; Ahsan Halim, S.; Ali, L.; Al-Saady, N.; Hilal Al-Balushi, A.; Al-Busaidi, H.K.; Al-Harrasi, A. α-Glucosidase inhibition and molecular docking studies of natural brominated metabolites from marine macro brown alga Dictyopteris hoytii. Mar. Drugs 2019, 17, 666. [Google Scholar] [CrossRef] [Green Version]
- Huang, B.B.; Lin, H.C.; Chang, Y.W. Analysis of proteins and potential bioactive peptides from tilapia (Oreochromis spp.) processing co-products using proteomic techniques coupled with BIOPEP database. J. Funct. Food 2015, 19, 629–640. [Google Scholar] [CrossRef]
- Sun, L.P.; Zhang, Y.F.; Zhuang, Y.L. Antiphotoaging effect and purification of an antioxidant peptide from tilapia (Oreochromis niloticus) gelatin peptides. J. Funct. Food 2013, 5, 154–162. [Google Scholar] [CrossRef]
- Ngo, D.H.; Ryu, B.; Kim, S.K. Active peptides from skate (Okamejei kenojei) skin gelatin diminish angiotensin-I converting enzyme activity and intracellular free radical-mediated oxidation. Food Chem. 2014, 143, 246–255. [Google Scholar] [CrossRef]
- Ngo, D.H.; Ryu, B.; Vo, T.S.; Himaya, S.W.A.; Wijesekara, I.; Kim, S.K. Free radical scavenging and angiotensin-I converting enzyme inhibitory peptides from pacific cod (Gadus macrocephalus) skin gelatin. Int. J. Biol. Macromol. 2011, 49, 1110–1116. [Google Scholar] [CrossRef] [PubMed]
- Kang, N.; Ko, S.C.; Kim, H.S.; Yang, H.W.; Ahn, G.; Lee, S.C.; Lee, T.G.; Lee, J.S.; Jeon, Y.J. Structural evidence for antihypertensive effect of an antioxidant peptide purified from the edible marine animal styela clava. J. Med. Food 2020, 23, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Mencarelli, A.; Migliorati, M.; Barbanti, M.; Cipriani, S.; Palladino, G.; Distrutti, E.; Renga, B.; Fiorucci, S. Pregnane-x-receptor mediates the anti-inflammatory activities of rifaximin on detoxification pathways in intestinal epithelial cells. Biochem. Pharmacol. 2010, 80, 1700–1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
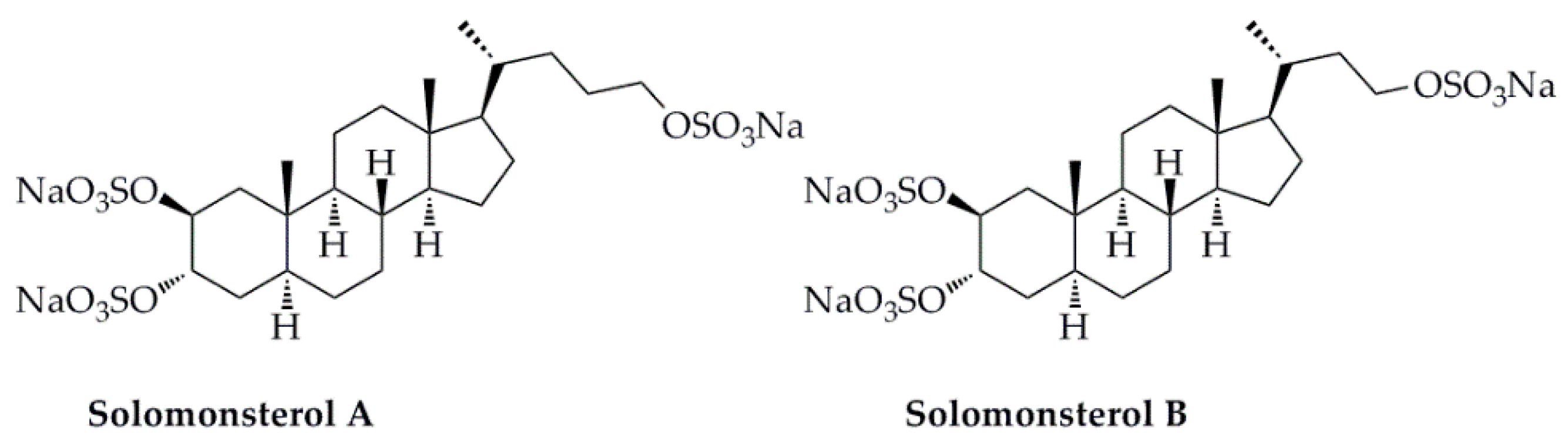
- Mencarelli, A.; D’Amore, C.; Renga, B.; Cipriani, S.; Carino, A.; Sepe, V.; Perissutti, E.; D’Auria, M.; Zampella, A.; Distrutti, E.; et al. Solomonsterol A, a marine pregnane-x-receptor agonist, attenuates inflammation and immune dysfunction in a mouse model of arthritis. Mar. Drugs 2013, 12, 36. [Google Scholar] [CrossRef] [PubMed]
- Sepe, V.; Ummarino, R.; D’Auria, M.V.; Lauro, G.; Bifulco, G.; D’Amore, C.; Renga, B.; Fiorucci, S.; Zampella, A. Modification in the side chain of solomonsterol a: Discovery of cholestan disulfate as a potent pregnane-x-receptor agonist. Org. Biomol. Chem. 2012, 10, 6350. [Google Scholar] [CrossRef]
- Martel-Pelletier, J.; Lajeunesse, D.L.; Reboul, P.; Pelletier, J.P. Therapeutic role of dual inhibitors of 5-LOX and COX, selective and non-selective non-steroidal anti-inflammatory drugs. Ann. Rheum. Dis. 2003, 62, 501–509. [Google Scholar] [CrossRef]
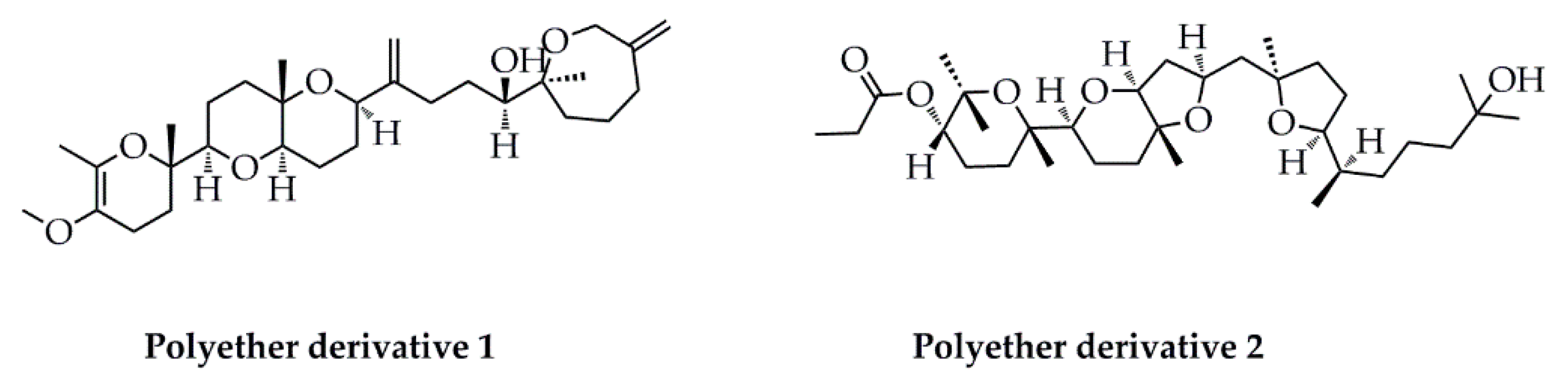
- Antony, T.; Chakraborty, K. Anti-inflammatory polyether triterpenoids from the marine macroalga Gracilaria salicornia: Newly described natural leads attenuate pro-inflammatory 5-lipoxygenase and cyclooxygenase-2. Algal Res. 2020, 47, 101791. [Google Scholar] [CrossRef]
- Anand, K. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS srugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Gentile, D.; Patamia, V.; Scala, A.; Sciortino, M.T.; Piperno, A.; Rescifina, A. Putative inhibitors of SARS-CoV-2 main protease from a library of marine natural products: A virtual screening and molecular modeling study. Mar. Drugs 2020, 18, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
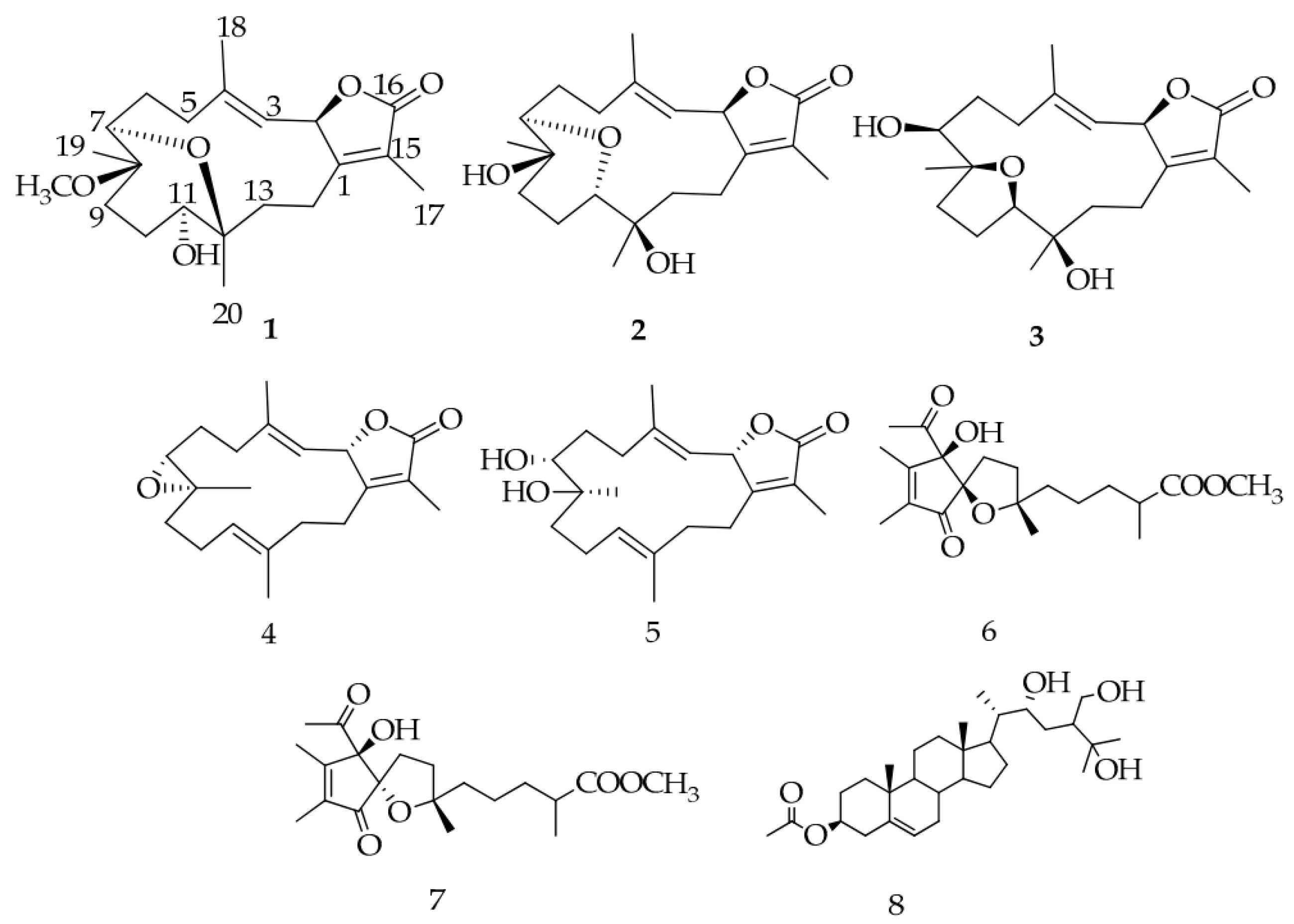
- Hegazy, M.F.; Elshamy, A.I.; Mohamed, T.A.; Hamed, A.R.; Ibrahim, M.A.A.; Ohta, S.; Pare, P.W. Cembrene diterpenoids with ether linkages from Sarcophyton ehrenbergi: An anti-proliferation and molecular-docking assessment. Mar. Drugs 2017, 15, 192. [Google Scholar] [CrossRef] [PubMed]
- Dutta, P.R.; Maity, A. Cellular responses to EGFR inhibitors and their relevance to cancer therapy. Cancer Lett. 2007, 254, 165–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Wang, X.Y.; Lv, P.C.; Zhu, H.L. Discovery of a series of novel phenylpiperazine derivatives as EGFR TK inhibitors. Sci. Rep. 2015, 5, 13934–13945. [Google Scholar] [CrossRef] [Green Version]
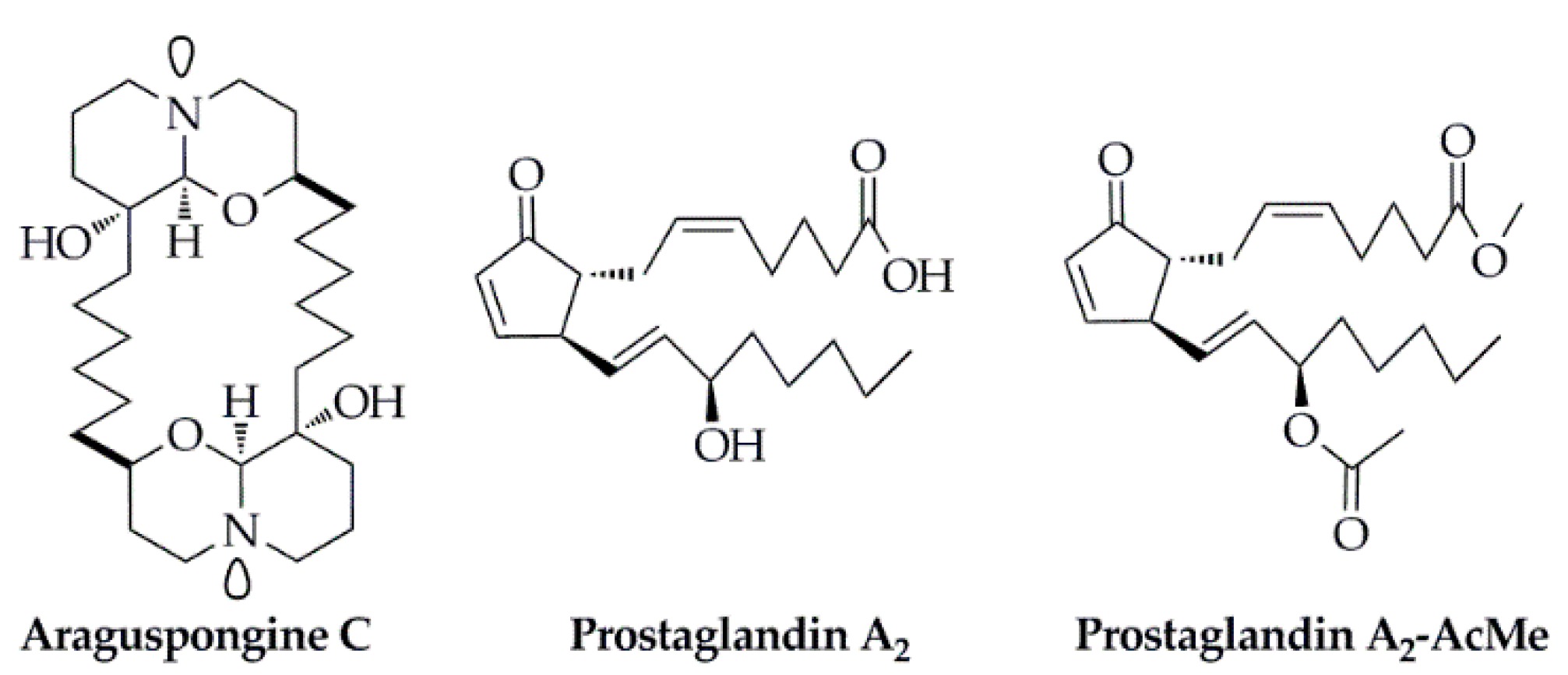
- Akl, M.R.; Ayoub, N.M.; Ebrahim, H.Y.; Mohyeldin, M.M.; Orabi, K.Y.; Foudah, A.I.; El Sayed, K.A. Araguspongine C induces autophagic death in breast cancer cells through suppression of c-Met and HER2 receptor tyrosine kinase signaling. Mar. Drugs 2015, 13, 288. [Google Scholar] [CrossRef] [Green Version]
- Hurtado, D.X.; Castellanos, F.A.; Coy-Barrera, E.; Tello, E. Prostaglandins isolated from the octocoral Plexaura homomalla: In silico and in vitro studies against different enzymes of cancer. Mar. Drugs 2020, 18, 141. [Google Scholar] [CrossRef] [Green Version]
- Levin, N.; Spencer, A.; Harrison, S.J.; Chauhan, D.; Burrows, F.J.; Anderson, K.C.; Reich, S.D.; Richardson, P.G.; Trikha, M. Marizomib irreversibly inhibits proteasome to overcome compensatory hyperactivation in multiple myeloma and solid tumour patients. Br. J. Haematol. 2016, 174, 711–720. [Google Scholar] [CrossRef]
- Nieto, F.R.; Cobos, E.J.; Tejada, M.A.; Sanchez-Fernandez, C.; Gonzalez-Cano, R.; Cendan, C.M. Tetrodotoxin (TTX) as a therapeutic agent for pain. Mar. Drugs 2012, 10, 281. [Google Scholar] [CrossRef] [PubMed]
- Le Tourneau, C.; Faivre, S.; Ciruelos, E.; Dominguez, M.J.; Lopez-Martin, J.A.; Izquierdo, M.A.; Jimeno, J.; Raymond, E. Reports of clinical benefit of plitidepsin (Aplidine), a new marine-derived anticancer agent, in patients with advanced medullary thyroid carcinoma. Am. J. Clin. Oncol. 2010, 33, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Dimou, A.; Syrigos, K.N.; Saif, M.W. Novel agents in the treatment of pancreatic adenocarcinoma. J. Pancreas 2013, 14, 138–140. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Program Name | Algorithm Characteristics | Typical Applications | Ref. |
|---|---|---|---|
| DOCK | Step-by-step geometric matching strategy; AMBER force field experience-based scoring function. As a kind of commonly used molecular docking software, it can be used for docking between flexible small-molecule ligands and flexible proteins. | Protein–small molecule | [65] |
| AutoDock | Lamarck genetic algorithm and experience-based scoring function; the flexibilities of small molecules and some residue side chains can be fully taken into consideration. | Protein–small molecule | [66] |
| AutoDock Vina | The upgraded version of AutoDock; the success rate and calculation speed are greatly improved compared to AutoDock; simple parameter setting, easy to use, and parallel operation on multi-core machines for docking flexible ligands and flexible protein side chains. | Protein–small molecule | [67] |
| MDock | Using the knowledge-based atomic–atomic contact potential scoring function, the flexibilities of proteins and small molecules are considered by using the conformations of the multiple proteins and small molecules during the docking process. | Protein–small molecule | [68] |
| FlexX | The best conformation is selected according to the size of the docking free energy, which has the advantages of fast speed, high efficiency, and easy operation. It is the representative software of the flexible docking and can also be used for the virtual screening of small molecule database. | Protein–small molecule | [25,52] |
| GOLD | Based on the GA docking program, the ligand is completely flexible, the receptor binding position is partially flexible; the automatic docking program can be used for virtual screening of the database. Its accuracy and reliability are highly evaluated in the molecular docking simulation. | Protein–small molecule | [45] |
| Surflex-Dock | The Hammerhead scoring function is used; it combines a large number of conformations from the intact molecules through a crossover process to achieve flexible docking. | Protein–small molecule | [69] |
| eHiTS | An accurate and fast molecular docking program, which can be used to study ligand and receptor interactions and perform high-throughput virtual screening. | Protein–small molecule | [70] |
| EADock | Multi-objective evolutionary optimization algorithm for docking small molecules with the active sites of proteins. | Protein–small molecule | [71] |
| Glide | Docking program based on search algorithms, including the modes of extra precision (XP), standard precision (SP), and a high-throughput virtual filter. It is mainly used for the flexible docking of small-molecule ligands and proteins. | Protein–small molecule | [43] |
| PIPER | FFT search algorithm; the knowledge-based atomic statistical potential scoring function, and applied to the ClusProServer | Protein–protein | [72] |
| ZDOCK | FFT search algorithm; filtering and sorting with RDOCK. | Protein–protein | [54] |
| Hammerhead | Fragment-based docking program for automated and rapid molecular docking of flexible ligands; the program uses an experience-based adjustment scoring function and a method to automatically identify and describe protein binding sites for molecular docking. | Protein–protein/small molecule | [73] |
| MOE | A comprehensive software system for the pharmaceutical and life science, which could fully support drug design and research through molecular simulation, protein structure analysis, small molecule database processing and protein and small-molecule docking research in a unified operating environment. | Protein–protein/small molecule | [74] |
| FLIPDock | A genetic algorithm-based docking program that uses the FlexTree data structure to represent the protein–ligand complex and enables docking of flexible ligands and flexible proteins. | Protein–protein/small molecule | [75] |
| ICM-Dock | User-friendly interactive image display, and the software also supports fast and accurate docking optimization. | Protein–protein/polypeptide/small molecule | [76] |
| HADDOCK | Docking program based on experimental data (such as NMR chemical shifts and point mutations), which was invented from protein–protein docking and can also be used for protein–ligand docking. | Protein–protein/DNA/RNA/small molecule | [61] |
| RosettaDock | MC search algorithm; the experience-based energy scoring function. | Protein–protein/DNA/RNA/small molecule | [42] |
| DOT | FFT search algorithm; the scoring function only has Van der Waals and electrostatic terms. | Protein–protein/DNA/RNA | [36] |
| FLOG | Rigid docking program using a pre-generated conformation database | Protein–protein/DNA/RNA | [77] |
| MS-Dock | The method consists of two main steps: first, generate a variety of 3D conformations; second, carry out the rigid docking of the conformations and multi-step virtual screening. | Protein–protein/DNA/RNA | [78] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, G.; Seukep, A.J.; Guo, M. Recent Advances in Molecular Docking for the Research and Discovery of Potential Marine Drugs. Mar. Drugs 2020, 18, 545. https://doi.org/10.3390/md18110545
Chen G, Seukep AJ, Guo M. Recent Advances in Molecular Docking for the Research and Discovery of Potential Marine Drugs. Marine Drugs. 2020; 18(11):545. https://doi.org/10.3390/md18110545
Chicago/Turabian StyleChen, Guilin, Armel Jackson Seukep, and Mingquan Guo. 2020. "Recent Advances in Molecular Docking for the Research and Discovery of Potential Marine Drugs" Marine Drugs 18, no. 11: 545. https://doi.org/10.3390/md18110545