

Addressing the Challenges in the Diagnosis and Management of Pediatric Wilson’s Disease—Case Report and Literature Review
 ,
,
Abstract
:1. Introduction
2. Case Presentation
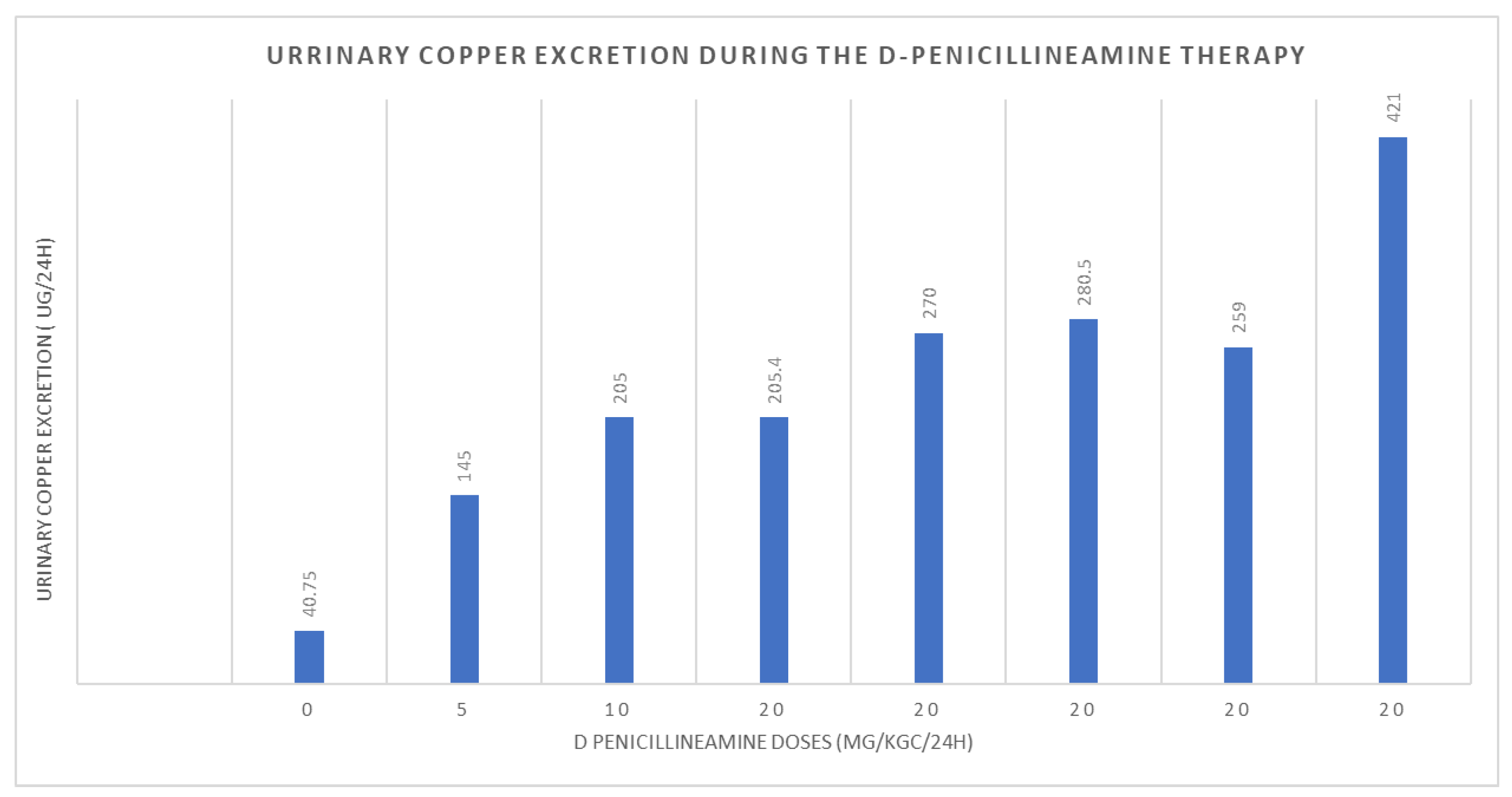
3. Discussion and Literature Review
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huster, D. Wilson Disease. Best Prac. Res. Clin. Gastroenterol. 2010, 24, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Chanpong, A.; Dhawan, A. Wilson Disease in Children and Young Adults—State of the Art. Saudi. J. Gastroenterol. 2022, 28, 21–31. [Google Scholar] [CrossRef]
- Gul, B.; Firasat, S.; Tehreem, R.; Shan, T.; Afshan, K. Analysis of Wilson Disease Mutations in Copper Binding Domain of ATP7B Gene. PLoS ONE 2022, 17, e0269833. [Google Scholar] [CrossRef] [PubMed]
- Abdel Ghaffar, T.Y.; Elsayed, S.M.; Elnaghy, S.; Shadeed, A.; Elsobky, E.S.; Schmidt, H. Phenotypic and Genetic Characterization of a Cohort of Pediatric Wilson Disease Patients. BMC Pediatr. 2011, 11, 56. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, K.; Thankappan, B. Wilson’s Disease Update: An Indian Perspective. Ann. Indian Acad. Neurol. 2022, 25, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Merle, U.; Schaefer, M.; Ferenci, P.; Stremmel, W. Clinical Presentation, Diagnosis and Long-term Outcome of Wilson’s Disease: A Cohort Study. Gut 2007, 56, 115–120. [Google Scholar] [CrossRef]
- Catana, A.M.; Medici, V. Liver Transplantation for Wilson Disease. World J. Hepatol. 2012, 4, 5–10. [Google Scholar] [CrossRef]
- Isac, T.; Isac, S.; Rababoc, R.; Cotorogea, M.; Iliescu, L. Epigenetics in Inflammatory Liver Diseases: A Clinical Perspective (Review). Exp. Ther. Med. 2022, 23, 366. [Google Scholar] [CrossRef]
- Poujois, A.; Woimant, F. Challenges in the Diagnosis of Wilson Disease. Ann. Transl. Med. 2019, 7, S67. [Google Scholar] [CrossRef]
- Cohen, I.P. A Case Report of a Hymenolepis Diminuta Infection in a Child in St James Parish, Jamaica. J. La. State Med. Soc. 1989, 141, 23–24. [Google Scholar]
- Miller, B.S.; Sarafoglou, K.; Addo, O.Y. Development of Tanner Stage-Age Adjusted CDC Height Curves for Research and Clinical Applications. J. Endocr. Soc. 2020, 4, bvaa098. [Google Scholar] [CrossRef] [PubMed]
- Socha, P.; Janczyk, W.; Dhawan, A.; Baumann, U.; D’Antiga, L.; Tanner, S.; Iorio, R.; Vajro, P.; Houwen, R.; Fischler, B.; et al. Wilson’s Disease in Children: A Position Paper by the Hepatology Committee of the European Society for Paediatric Gastroenterology, Hepatology and Nutrition. J. Pediatr. Gastroenterol. Nutr. 2018, 66, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Kathawala, M.; Hirschfield, G.M. Insights into the Management of Wilson’s Disease. Ther. Adv. Gastroenterol. 2017, 10, 889–905. [Google Scholar] [CrossRef] [PubMed]
- Kenney, S.M.; Cox, D.W. Sequence Variation Database for the Wilson Disease Copper Transporter, ATP7B. Hum. Mutat. 2007, 28, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Ma, D.; Cheng, J.; Zhang, J.; Luo, C.; Sun, Y.; Hu, P.; Wang, Y.; Jiang, T.; Xu, Z. Identification and Characterization of a Novel 43-Bp Deletion Mutation of the ATP7B Gene in a Chinese Patient with Wilson’s Disease: A Case Report. BMC Med. Genet. 2018, 19, 61. [Google Scholar] [CrossRef]
- Coffey, A.J.; Durkie, M.; Hague, S.; McLay, K.; Emmerson, J.; Lo, C.; Klaffke, S.; Joyce, C.J.; Dhawan, A.; Hadzic, N.; et al. A Genetic Study of Wilson’s Disease in the United Kingdom. Brain 2013, 136, 1476–1487. [Google Scholar] [CrossRef]
- Dedoussis, G.V.Z.; Genschel, J.; Sialvera, T.-E.; Bochow, B.; Manolaki, N.; Manios, Y.; Tsafantakis, E.; Schmidt, H. Wilson Disease: High Prevalence in a Mountainous Area of Crete. Ann. Hum. Genet. 2005, 69, 268–274. [Google Scholar] [CrossRef]
- Collet, C.; Laplanche, J.-L.; Page, J.; Morel, H.; Woimant, F.; Poujois, A. High Genetic Carrier Frequency of Wilson’s Disease in France: Discrepancies with Clinical Prevalence. BMC Med. Genet. 2018, 19, 143. [Google Scholar] [CrossRef]
- Lorente-Arencibia, P.; García-Villarreal, L.; González-Montelongo, R.; Rubio-Rodríguez, L.A.; Flores, C.; Garay-Sánchez, P.; delaCruz, T.; Santana-Verano, M.; Rodríguez-Esparragón, F.; Benitez-Reyes, J.N.; et al. Wilson Disease Prevalence: Discrepancy Between Clinical Records, Registries and Mutation Carrier Frequency. J. Pediatr. Gastroenterol. Nutr. 2022, 74, 192–199. [Google Scholar] [CrossRef]
- Lau, J.Y.; Lai, C.L.; Wu, P.C.; Pan, H.Y.; Lin, H.J.; Todd, D. Wilson’s Disease: 35 Years’ Experience. Q. J. Med. 1990, 75, 597–605. [Google Scholar]
- Emre, S.; Atillasoy, E.O.; Ozdemir, S.; Schilsky, M.; Rathna Varma, C.V.; Thung, S.N.; Sternlieb, I.; Guy, S.R.; Sheiner, P.A.; Schwartz, M.E.; et al. Orthotopic Liver Transplantation for Wilson’s Disease: A Single-Center Experience. Transplantation 2001, 72, 1232–1236. [Google Scholar] [CrossRef] [PubMed]
- Beinhardt, S.; Leiss, W.; Stättermayer, A.F.; Graziadei, I.; Zoller, H.; Stauber, R.; Maieron, A.; Datz, C.; Steindl-Munda, P.; Hofer, H.; et al. Long-Term Outcomes of Patients with Wilson Disease in a Large Austrian Cohort. Clin. Gastroenterol. Hepatol. 2014, 12, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.-J.; Wang, D.-X.; Ding, N.-N.; Lin, Y.; Jin, Y.; Zheng, C.-Q. Comprehensive Analysis on Clinical Features of Wilson’s Disease: An Experience over 28 Years with 133 Cases. Neurol. Res. 2014, 36, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Esezobor, C.I.; Banjoko, N.; Rotimi-Samuel, A.; Lesi, F.E.A. Wilson Disease in a Nigerian Child: A Case Report. J. Med. Case Rep. 2012, 6, 200. [Google Scholar] [CrossRef] [PubMed]
- Wadera, S.; Magid, M.S.; McOmber, M.; Carpentieri, D.; Miloh, T. Atypical Presentation of Wilson Disease. Semin. Liver Dis. 2011, 31, 319–326. [Google Scholar] [CrossRef]
- Wilson, D.C.; Phillips, M.J.; Cox, D.W.; Roberts, E.A. Severe Hepatic Wilson’s Disease in Preschool-Aged Children. J. Pediatr. 2000, 137, 719–722. [Google Scholar] [CrossRef]
- Beyersdorff, A.; Findeisen, A. Morbus Wilson: Case Report of a Two-Year-Old Child as First Manifestation. Scand. J. Gastroenterol. 2006, 41, 496–497. [Google Scholar] [CrossRef]
- Gitlin, J.D. Wilson Disease. Gastroenterology 2003, 125, 1868–1877. [Google Scholar] [CrossRef]
- Lorincz, M.T. Neurologic Wilson’s Disease. Ann. N. Y. Acad. Sci. 2010, 1184, 173–187. [Google Scholar] [CrossRef]
- Saito, T. Presenting Symptoms and Natural History of Wilson Disease. Eur. J. Pediatr. 1987, 146, 261–265. [Google Scholar] [CrossRef]
- Oder, W.; Grimm, G.; Kollegger, H.; Ferenci, P.; Schneider, B.; Deecke, L. Neurological and Neuropsychiatric Spectrum of Wilson’s Disease: A Prospective Study of 45 Cases. J. Neurol. 1991, 238, 281–287. [Google Scholar] [CrossRef]
- Ferenci, P.; Członkowska, A.; Merle, U.; Ferenc, S.; Gromadzka, G.; Yurdaydin, C.; Vogel, W.; Bruha, R.; Schmidt, H.T.; Stremmel, W. Late-Onset Wilson’s Disease. Gastroenterology 2007, 132, 1294–1298. [Google Scholar] [CrossRef] [PubMed]
- Manolaki, N.; Nikolopoulou, G.; Daikos, G.L.; Panagiotakaki, E.; Tzetis, M.; Roma, E.; Kanavakis, E.; Syriopoulou, V.P. Wilson Disease in Children: Analysis of 57 Cases. J. Pediatr. Gastroenterol. Nutr. 2009, 48, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.-J.; Xiao, P.; Lin, D.; Zhou, H.-M.; He, X.-X. Cirrhosis in Wilson Disease Is Characterized by Impaired Hepatic Synthesis, Leukopenia and Thrombocytopenia. Int. J. Med. Sci. 2020, 17, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, S.; Gulshan, J.; Baidya, M.; Rashid, R.; Tasneem, F.; Hasan, A.R.; Farhana, T.; Ahmed, S.S. Outcome of Wilson’s Disease in Bangladeshi Children: A Tertiary Center Experience. Egypt. Liver J. 2022, 12, 64. [Google Scholar] [CrossRef]
- Czlonkowska, A.; Litwin, T.; Dusek, P.; Ferenci, P.; Lutsenko, S.; Medici, V.; Rybakowski, J.K.; Weiss, K.H.; Schilsky, M.L. Nature Reviews Disease Primers Article: Wilson Disease. Nat. Rev. Dis. Prim. 2018, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Dafallah, M.A.; Habour, E.; Ragab, E.A.; Shouk, Z.M.; Hamad, F.; Ahmed, M.; Ahmed, M.H. Rare Presentation of Wilson Disease in an 11-Year-Old Sudanese Girl. Sudan J. Med. Sci. (SJMS) 2021, 16, 196–206. [Google Scholar] [CrossRef]
- Gow, P.J.; Smallwood, R.A.; Angus, P.W.; Smith, A.L.; Wall, A.J.; Sewell, R.B. Diagnosis of Wilson’s Disease: An Experience over Three Decades. Gut 2000, 46, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Kozić, D.; Svetel, M.; Petrović, B.; Dragasević, N.; Semnic, R.; Kostić, V.S. MR Imaging of the Brain in Patients with Hepatic Form of Wilson’s Disease. Eur. J. Neurol. 2003, 10, 587–592. [Google Scholar] [CrossRef]
- Litwin, T.; Dusek, P.; Szafrański, T.; Dzieżyc, K.; Członkowska, A.; Rybakowski, J.K. Psychiatric Manifestations in Wilson’s Disease: Possibilities and Difficulties for Treatment. Adv. Psychopharmacol. 2018, 8, 199–211. [Google Scholar] [CrossRef]
- Henao, J.A.; Valverde, K.; Ávila, M.L. Anemia hemolítica como presentación inicial de enfermedadde Wilson: Un caso pediátrico. Arch. Argent. Pediatr. 2016, 114, e436–e439. [Google Scholar] [CrossRef]
- Walshe, J.M. The Acute Haemolytic Syndrome in Wilson’s Disease—A Review of 22 Patients. QJM 2013, 106, 1003–1008. [Google Scholar] [CrossRef]
- Hogland, H.C.; Goldstein, N.P. Hematologic (Cytopenic) Manifestations of Wilson’s Disease (Hepatolenticular Degeneration). Mayo Clin. Proc. 1978, 53, 498–500. [Google Scholar]
- Kleine, R.T.; Mendes, R.; Pugliese, R.; Miura, I.; Danesi, V.; Porta, G. Wilson’s Disease: An Analysis of 28 Brazilian Children. Clinics 2012, 67, 231–235. [Google Scholar] [CrossRef] [PubMed]
- El Raziky, M.S.; Ali, A.; El Shahawy, A.; Hamdy, M.M. Acute Hemolytic Anemia as an Initial Presentation of Wilson Disease in Children. J. Pediatr. Hematol. Oncol. 2014, 36, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Toppo, A.; Rath, B.; Harbhajanka, A.; Lalita Jyotsna, P. Hemolytic Anemia as a Presenting Feature of Wilson’s Disease: A Case Report. Indian J. Hematol. Blood Transfus. 2010, 26, 101–102. [Google Scholar] [CrossRef]
- Ye, X.-N.; Mao, L.-P.; Lou, Y.-J.; Tong, H.-Y. Hemolytic Anemia as First Presentation of Wilson’s Disease with Uncommon ATP7B Mutation. Int. J. Clin. Exp. Med. 2015, 8, 4708–4711. [Google Scholar]
- Rukunuzzaman, M. Wilson’s Disease in Bangladeshi Children: Analysis of 100 Cases. Pediatr. Gastroenterol. Hepatol. Nutr. 2015, 18, 121–127. [Google Scholar] [CrossRef]
- Dhawan, A.; Taylor, R.M.; Cheeseman, P.; De Silva, P.; Katsiyiannakis, L.; Mieli-Vergani, G. Wilson’s Disease in Children: 37-Year Experience and Revised King’s Score for Liver Transplantation. Liver Transpl. 2005, 11, 441–448. [Google Scholar] [CrossRef]
- Sintusek, P.; Chongsrisawat, V.; Poovorawan, Y. Wilson’s Disease in Thai Children between 2000 and 2012 at King Chulalongkorn Memorial Hospital. J. Med. Assoc. Thai. 2016, 99, 182–187. [Google Scholar]
- Ostapowicz, G.; Fontana, R.J.; Schiødt, F.V.; Larson, A.; Davern, T.J.; Han, S.H.B.; McCashland, T.M.; Shakil, A.O.; Hay, J.E.; Hynan, L.; et al. Results of a Prospective Study of Acute Liver Failure at 17 Tertiary Care Centers in the United States. Ann. Intern. Med. 2002, 137, 947–954. [Google Scholar] [CrossRef]
- Porlas, R.V., Jr.; de Castillo, L.L.C.; Dioquino, C.P.C. Neurologic Wilson Disease: Case Series on a Diagnostic and Therapeutic Emergency. Dialogues Clin. Neurosci. 2018, 20, 341–345. [Google Scholar] [CrossRef]
- Gulhar, R.; Ashraf, M.A.; Jialal, I. Physiology, Acute Phase Reactants. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Menkes, J.H. CHAPTER 108—WILSON DISEASE. In Neurology and Clinical Neuroscience; Schapira, A.H.V., Byrne, E., DiMauro, S., Frackowiak, R.S.J., Johnson, R.T., Mizuno, Y., Samuels, M.A., Silberstein, S.D., Wszolek, Z.K., Eds.; Mosby: Philadelphia, PA, USA, 2007; pp. 1447–1453. ISBN 978-0-323-03354-1. [Google Scholar]
- Chen, C.; Shen, B.; Xiao, J.-J.; Wu, R.; Canning, S.J.D.; Wang, X.-P. Currently Clinical Views on Genetics of Wilson’s Disease. Chin. Med. J. 2015, 128, 1826–1830. [Google Scholar] [CrossRef] [PubMed]
- Kasztelan-Szczerbinska, B.; Cichoz-Lach, H. Wilson’s Disease: An Update on the Diagnostic Workup and Management. J. Clin. Med. 2021, 10, 5097. [Google Scholar] [CrossRef] [PubMed]
- Muro, S.-I.; Yasunaka, T.; Wada, N.; Morimoto, Y.; Ikeda, F.; Shiraha, H.; Takaki, A.; Noso, K.; Iwasaki, H.; Yamamoto, K. A Case of Wilson’s Disease with Characteristic Laparoscopic Findings. Clin. J. Gastroenterol. 2014, 7, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Oe, S.; Honma, Y.; Yabuki, K.; Morino, K.; Kumamoto, K.; Hayashi, T.; Kusanaga, M.; Ogino, N.; Minami, S.; Shibata, M.; et al. Importance of a Liver Biopsy in the Management of Wilson Disease. Intern. Med. 2020, 59, 77–81. [Google Scholar] [CrossRef]
- Güngör, Ş.; Selimoğlu, M.A.; Varol, F.İ.; Güngör, S. Pediatric Wilson’s Disease: Findings in Different Presentations. A Cross-Sectional Study. Sao Paulo Med. J. 2018, 136, 304–309. [Google Scholar] [CrossRef]
- Almeida, P.; Schreiber, R.A.; Liang, J.; Mujawar, Q.; Guttman, O.R. Clinical Characteristics and Complications of Pediatric Liver Biopsy: A Single Centre Experience. Ann. Hepatol. 2017, 16, 797–801. [Google Scholar] [CrossRef]
- Ludwig, J.; Moyer, T.P.; Rakela, J. The Liver Biopsy Diagnosis of Wilson’s Disease: Methods in Pathology. Am. J. Clin. Pathol. 1994, 102, 443–446. [Google Scholar] [CrossRef]
- Madakshira, M.G.; Das, A.; Umairv, M.; Dutta, U. Liver Histology and Histochemistry in Wilson Disease. Autops. Case Rep. 2018, 8, e2018026. [Google Scholar] [CrossRef]
- Schroeder, S.M.; Matsukuma, K.E.; Medici, V. Wilson Disease and the Differential Diagnosis of Its Hepatic Manifestations: A Narrative Review of Clinical, Laboratory, and Liver Histological Features. Ann. Transl. Med. 2021, 9, 1394. [Google Scholar] [CrossRef] [PubMed]
- Marcellini, M.; Di Ciommo, V.; Callea, F.; Devito, R.; Comparcola, D.; Sartorelli, M.R.; Carelli, G.; Nobili, V. Treatment of Wilson’s Disease with Zinc from the Time of Diagnosis in Pediatric Patients: A Single-Hospital, 10-Year Follow-up Study. J. Lab. Clin. Med. 2005, 145, 139–143. [Google Scholar] [CrossRef]
- Teufel-Schäfer, U.; Forster, C.; Schaefer, N. Low Copper Diet—A Therapeutic Option for Wilson Disease? Children 2022, 9, 1132. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.J.; Yuzbasiyan-Gurkan, V.; Johnson, V.; Dick, R.D.; Wang, Y. Treatment of Wilson’s Disease with Zinc: XI. Interaction with Other Anticopper Agents. J. Am. Coll. Nutr. 1993, 12, 26–30. [Google Scholar] [CrossRef] [PubMed]
- López-Méndez, E.; Uribe, M. Beta Blockers in Portal Hypertension. Are They Really a Good Option? Ann. Hepatol. 2006, 5, 86–91. [Google Scholar] [CrossRef]
- Kalita, J.; Kumar, V.; Misra, U.K.; Parashar, V.; Ranjan, A. Adjunctive Antioxidant Therapy in Neurologic Wilson’s Disease Improves the Outcomes. J. Mol. Neurosci. 2020, 70, 378–385. [Google Scholar] [CrossRef] [PubMed]





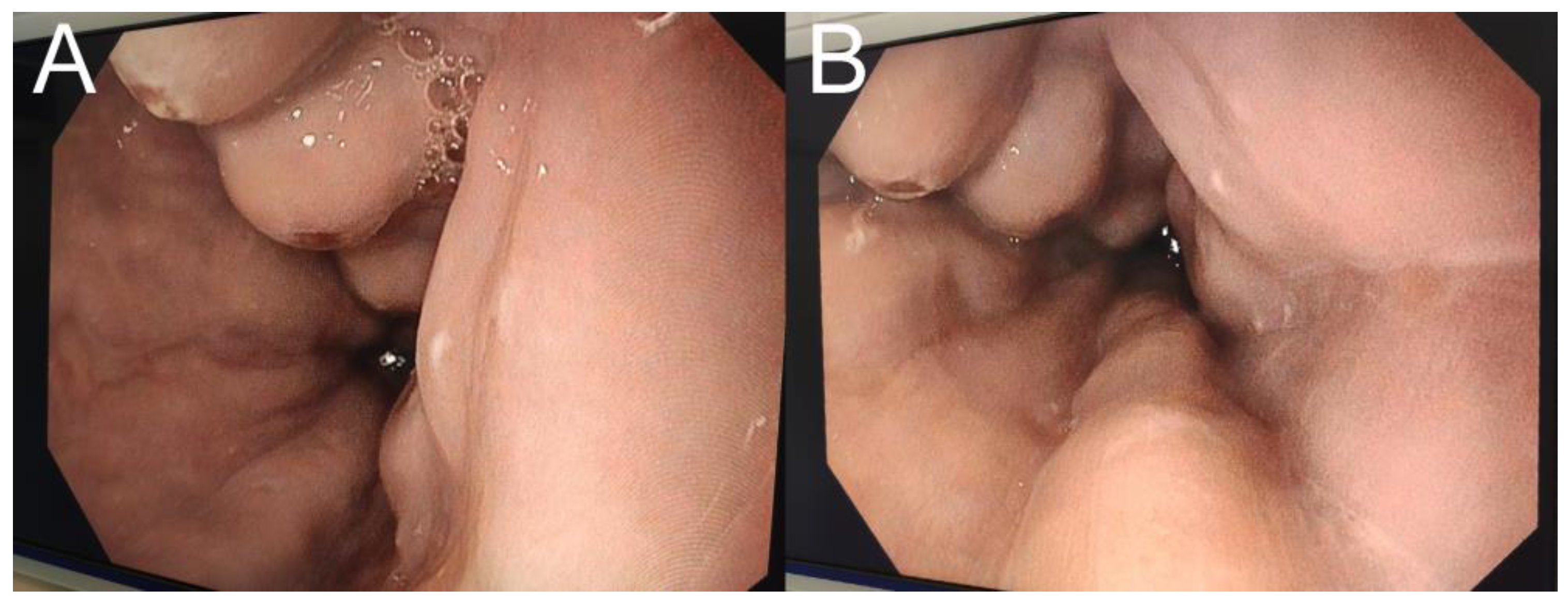{kind=link}
{kind=link}
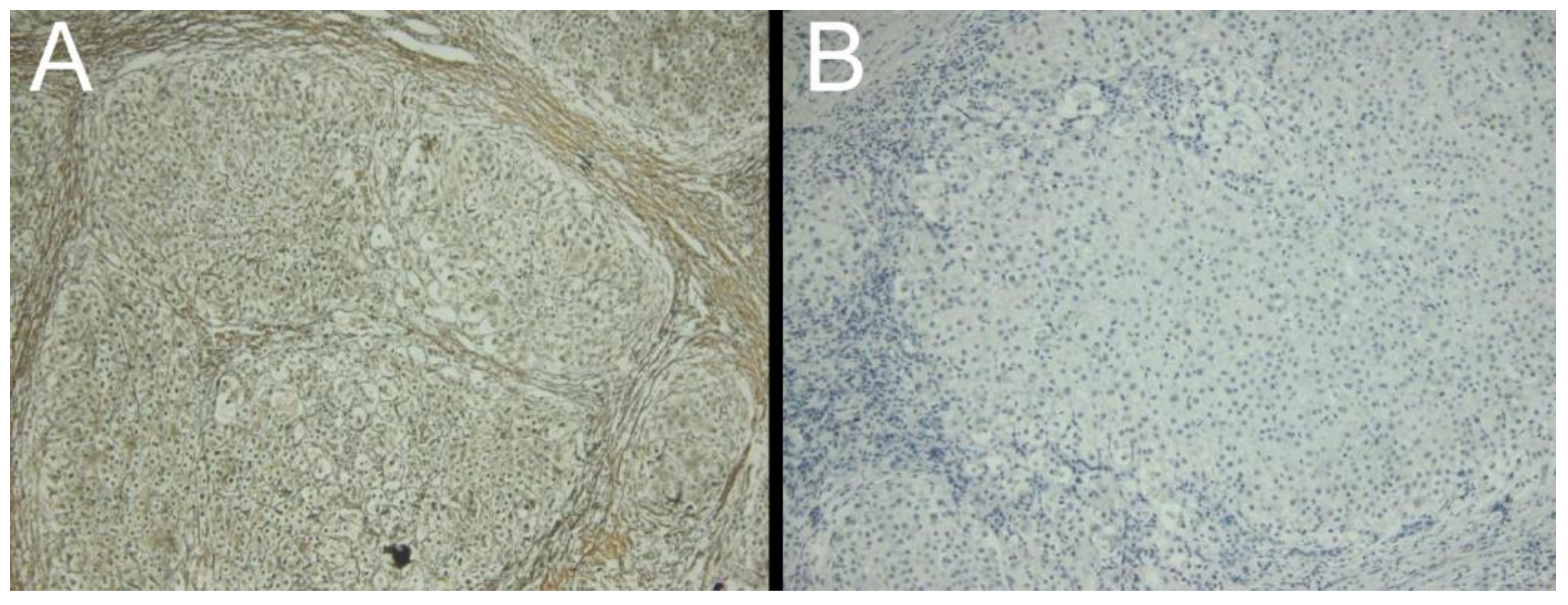{kind=link}
{kind=link}
| Test | Results | References |
|---|---|---|
| Ceruloplasmin | 0.469 | 0.20–0.60 g/L |
| Urinary copper/24 h | 58.86 | <60 ug/24h |
| Serum copper level | 193 | 90–190 ug/dL |
| Score | −1 | 0 | 1 | 2 | 4 |
|---|---|---|---|---|---|
| Keyser-Fleischer rings | Absent | Present | |||
| Neuropsychiatric symptoms suggestive of WD (or typical brain MRI) | Absent | Present | |||
| Coombs negative hemolytic anemia+ high serum copper | Absent | Present | |||
| Urinary copper (in the absence of acute hepatitis) | Normal | 1–2× ULN | >2× ULN or normal but >5× ULN 1 day after challenge with 2 × 0.5 g D-penicillamine | ||
| Liver copper quantitative | Normal | <5× ULN (<250 ug/g) | >5× ULN (>250 ug/g) | ||
| Rhodanine positive hepatocytes | Absent | Present | |||
| Serum ceruloplasmin (nephelometric assay) | >0.2 g/L | 0.1–0.2 g/L | <0.1 g/L | ||
| Disease-causing mutations detected | None | 1 | 2 | ||
| 0–1: Unlikely | 2–3: Probable | 4 or more: | High likely |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ungureanu, I.M.; Iesanu, M.I.; Boboc, C.; Cosoreanu, V.; Vatra, L.; Kadar, A.; Ignat, E.N.; Galos, F. Addressing the Challenges in the Diagnosis and Management of Pediatric Wilson’s Disease—Case Report and Literature Review. Medicina 2023, 59, 786. https://doi.org/10.3390/medicina59040786
Ungureanu IM, Iesanu MI, Boboc C, Cosoreanu V, Vatra L, Kadar A, Ignat EN, Galos F. Addressing the Challenges in the Diagnosis and Management of Pediatric Wilson’s Disease—Case Report and Literature Review. Medicina. 2023; 59(4):786. https://doi.org/10.3390/medicina59040786
Chicago/Turabian StyleUngureanu, Irene Maria, Mara Ioana Iesanu, Catalin Boboc, Vlad Cosoreanu, Lorena Vatra, Anna Kadar, Evelina Nicoleta Ignat, and Felicia Galos. 2023. "Addressing the Challenges in the Diagnosis and Management of Pediatric Wilson’s Disease—Case Report and Literature Review" Medicina 59, no. 4: 786. https://doi.org/10.3390/medicina59040786