
Oral Papillomatosis: Its Relation with Human Papilloma Virus Infection and Local Immunity—An Update


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Research Methods
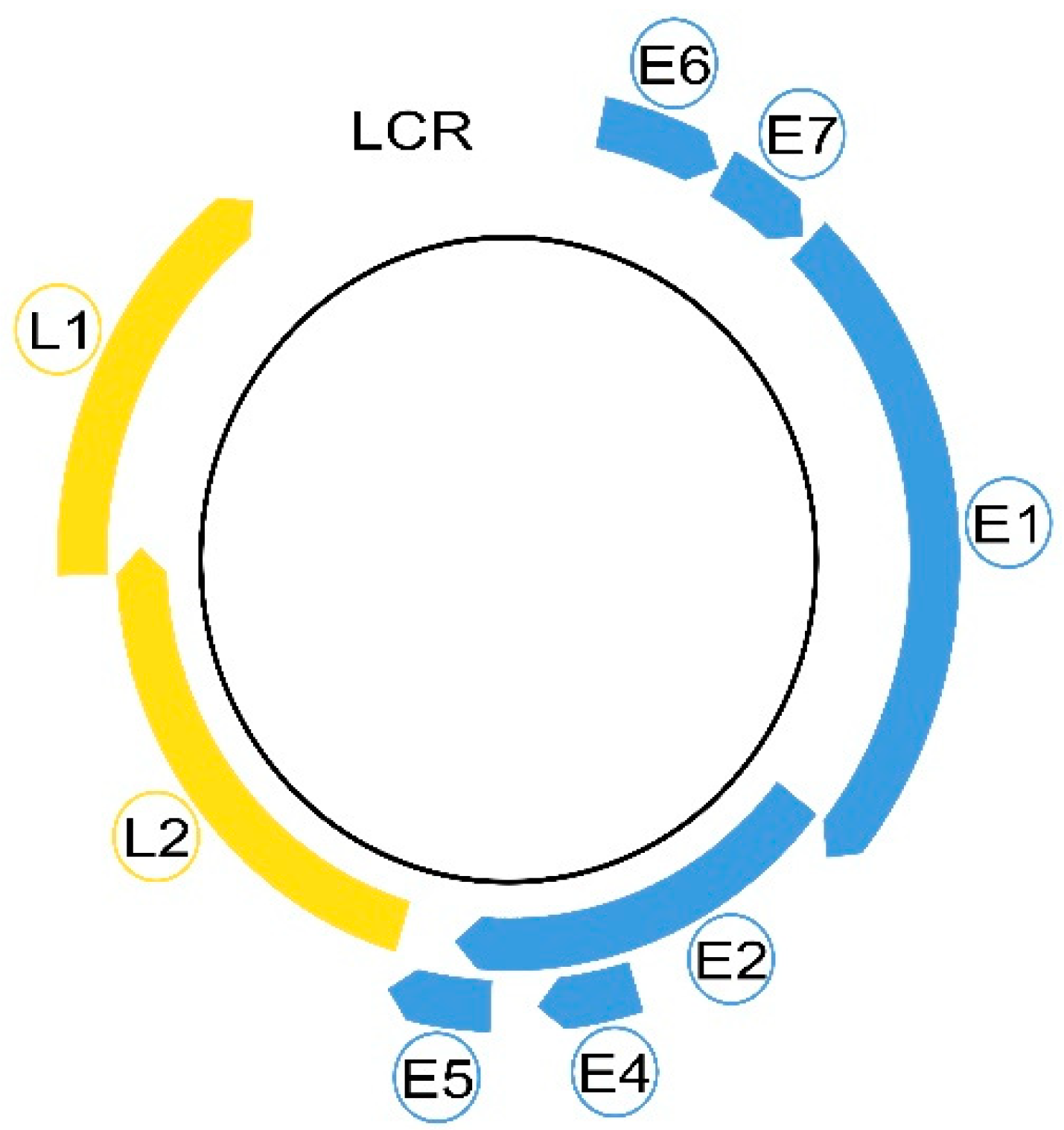
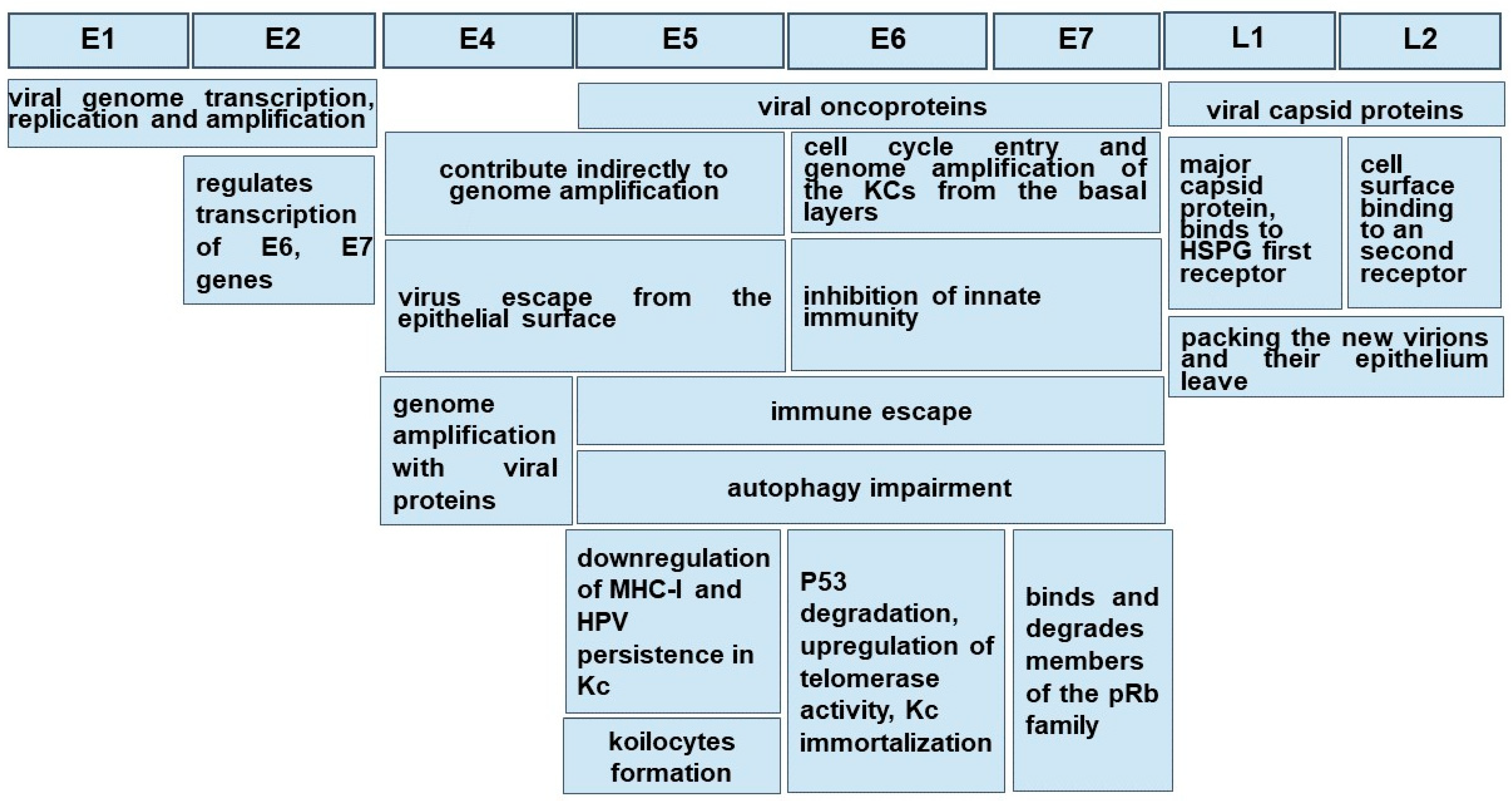
3. HPV Morphology
4. HPV Transmission
5. HPV Natural Infection
6. HPV Clearance
7. HPV Persistence
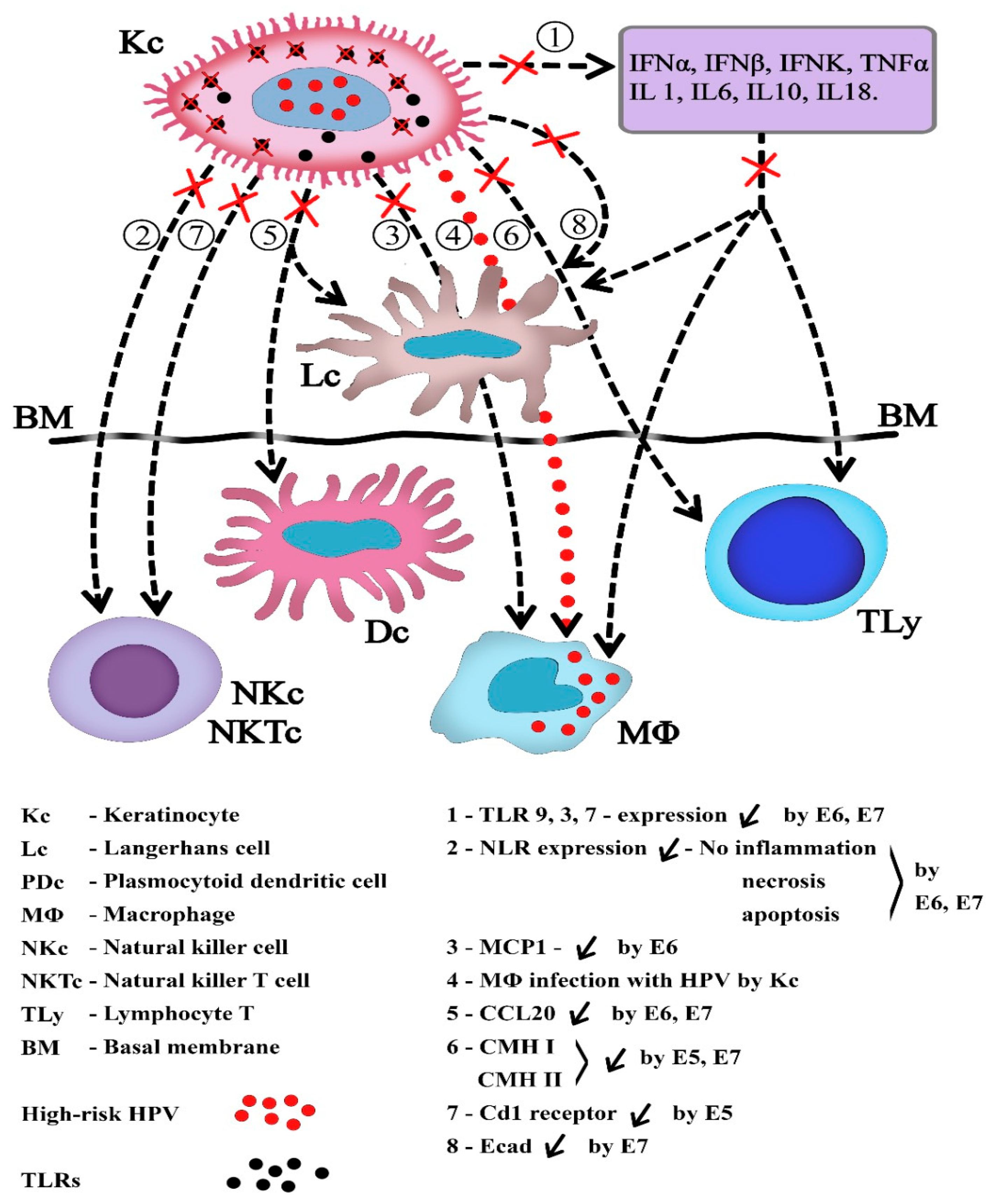
8. HPV and Local Immunity
9. HPV and Autophagy
10. HPV Oncogenicity
11. Clinical Aspects of Oral Papillomatous Lesions
12. Diagnostic of Oral Papillomatosis
13. HPV Vaccines
14. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Syrjänen, S. Oral manifestations of human papillomavirus infections. Eur. J. Oral Sci. 2018, 126 (Suppl. S1), 49–66. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 2015, 25 (Suppl. S1), 2–23. [Google Scholar] [CrossRef] [PubMed]
- Lisan, Q.; Laccourreye, O.; Bonfils, P. Sinonasal inverted papilloma: From diagnosis to treatment. Eur. Ann. Otorhinolaryngol. Head Neck. Dis. 2016, 133, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Sabry, A.O.; Patel, B.C. Papilloma. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK560737/ (accessed on 17 June 2022).
- Gupta, I.; Jabeen, A.; Al-Sarraf, R.; Farghaly, H.; Vranic, S.; Sultan, A.A.; Al Moustafa, A.-E.; Al-Thawadi, H. The co-presence of high-risk human papillomaviruses and Epstein-Barr virus is linked with tumor grade and stage in Qatari women with breast cancer. Hum. Vaccin. Immunother. 2021, 17, 982–989. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.R.; Graham, S.; Haughey, B.P.; Shedd, D.; O’Shea, R.; Brasure, J.; Wilkinson, G.S.; West, D. Smoking, alcohol, dentition and diet in the epidemiology of oral cancer. Eur. J. Cancer B Oral Oncol. 1992, 28, 9–15. [Google Scholar] [CrossRef]
- Behnoud, F.; Torabian, S.; Zargaran, M. Relationship between oral poor hygiene and broken teeth with oral tongue squamous cell carcinoma. Acta Med. Iran. 2011, 49, 159–162. [Google Scholar]
- Gupta, S.; Gupta, S. Role of human papillomavirus in oral squamous cell carcinoma and oral potentially malignant disorders: A review of the literature. Indian J. Dent. 2015, 6, 91–98. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, J.; Zhang, Y.; Lv, T.; Liu, J. The magnitude of the association between human papillomavirus and oral lichen planus: A meta-analysis. PLoS ONE 2016, 11, e0161339. [Google Scholar] [CrossRef]
- Razavi, S.M.; Ghalayani, P.; Salehi, M.R.; Attarzadeh, H.; Shahmoradi, M. Human papilloma virus as a possible factor in the pathogenesis of oral lichen planus. Dent. Res. J. 2009, 6, 82–86. [Google Scholar]
- Villa, T.G.; Sánchez-Pérez, Á.; Sieiro, C. Oral lichen planus: A microbiologist point of view. Int. Microbiol. 2021, 24, 275–289. [Google Scholar] [CrossRef]
- Johnson, D.E.; Burtness, B.; Leemans, C.R.; Lui, V.W.Y.; Bauman, J.E.; Grandis, J.R. Head and neck squamous cell carcinoma. Nat. Rev. Dis. Primers 2020, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Sun, X.; Chen, Z.; Du, J.; Wu, Y. Head and Neck Squamous Cell Carcinoma: Risk Factors, Molecular Alterations, Immunology and Peptide Vaccines. Int. J. Pept. Res. Ther. 2022, 28, 19. [Google Scholar] [CrossRef] [PubMed]
- Faraji, F.; Zaidi, M.; Fakhry, C.; Gaykalova, D.A. Molecular mechanisms of human papillomavirus-related carcinogenesis in head and neck cancer. Microbes Infect. 2017, 19, 464–475. [Google Scholar] [CrossRef] [PubMed]
- Chow, L.Q.M. Head and Neck Cancer. N. Engl. J. Med. 2020, 382, 60–72. [Google Scholar] [CrossRef]
- Zaravinos, A. An updated overview of HPV-associated head and neck carcinomas. Oncotarget 2014, 5, 3956–3969. [Google Scholar] [CrossRef]
- Camuzi, D.; Simão, T.A.; Dias, F.; Ribeiro Pinto, L.F.; Soares-Lima, S.C. Head and Neck Cancers Are Not Alike when Tarred with the Same Brush: An Epigenetic Perspective from the Cancerization Field to Prognosis. Cancers 2021, 13, 5630. [Google Scholar] [CrossRef]
- Marur, S.; Forastiere, A.A. Head and neck cancer: Changing epidemiology, diagnosis, and treatment. Mayo Clin. Proc. 2008, 83, 489–501. [Google Scholar] [CrossRef]
- Lechien, J.R.; Descamps, G.; Seminerio, I.; Furgiuele, S.; Dequanter, D.; Mouawad, F.; Badoual, C.; Journe, F.; Saussez, S. HPV Involvement in the Tumor Microenvironment and Immune Treatment in Head and Neck Squamous Cell Carcinomas. Cancers 2020, 12, 1060. [Google Scholar] [CrossRef]
- Shrestha, A.D.; Neupane, D.; Vedsted, P.; Kallestrup, P. Cervical Cancer Prevalence, Incidence and Mortality in Low and Middle Income Countries: A Systematic Review. Asian Pac. J. Cancer Prev. 2018, 19, 319–324. [Google Scholar]
- Taberna, M.; Mena, M.; Pavón, M.A.; Alemany, L.; Gillison, M.L.; Mesía, R. Human papillomavirus-related oropharyngeal cancer. Ann. Oncol. 2017, 28, 2386–2398. [Google Scholar] [CrossRef]
- Bednarczyk, R.A. Addressing HPV vaccine myths: Practical information for healthcare providers. Hum. Vaccin. Immunother. 2019, 15, 1628–1638. [Google Scholar] [CrossRef] [PubMed]
- Hirth, J. Disparities in HPV vaccination rates and HPV prevalence in the United States: A review of the literature. Hum. Vaccin. Immunother. 2019, 15, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef]
- Kumaraswamy, K.L.; Vidhya, M. Human papilloma virus and oral infections: An update. J. Cancer Res. Ther. 2011, 7, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Fiorillo, L.; Cervino, G.; Surace, G.; De Stefano, R.; Laino, L.; D’Amico, C.; Fiorillo, M.T.; Meto, A.; Herford, A.S.; Arzukanyan, A.V.; et al. Human Papilloma Virus: Current Knowledge and Focus on Oral Health. BioMed Res. Int. 2021, 2021, 6631757. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.F.; Wang, S.S.; Tang, Y.J.; Chen, Y.; Zheng, M.; Tang, Y.L.; Liang, X.H. The Double-Edged Sword-How Human Papillomaviruses Interact With Immunity in Head and Neck Cancer. Front. Immunol. 2019, 10, 653. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Ährlund-Richter, A.; Näsman, A.; Dalianis, T. Human papilloma virus (HPV) prevalence upon HPV vaccination in Swedish youth: A review based on our findings 2008–2018, and perspectives on cancer prevention. Arch. Gynecol. Obstet. 2021, 303, 329–335. [Google Scholar] [CrossRef]
- Doorbar, J.; Quint, W.; Banks, L.; Bravo, I.G.; Stoler, M.; Broker, T.R.; Stanley, M.A. The biology and life-cycle of human papillomaviruses. Vaccine 2012, 30 (Suppl. S5), F55–F70. [Google Scholar] [CrossRef]
- de Villiers, E.M.; Fauquet, C.; Broker, T.R.; Bernard, H.U.; zur Hausen, H. Classification of papillomaviruses. Virology 2004, 324, 17–27. [Google Scholar] [CrossRef]
- Bouvard, V.; Baan, R.; Straif, K.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Benbrahim-Tallaa, L.; Guha, N.; Freeman, C.; Galichet, L.; et al. A review of human carcinogens—Part B: Biological agents. Lancet Oncol. 2009, 10, 321–322. [Google Scholar] [CrossRef]
- Kreimer, A.R.; Clifford, G.M.; Boyle, P.; Franceschi, S. Human papillomavirus types in head and neck squamous cell carcinomas worldwide: A systematic review. Cancer Epidemiol. Biomark. Prev. 2005, 14, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Leemans, C.R.; Braakhuis, B.J.; Brakenhoff, R.H. The molecular biology of head and neck cancer. Nat. Rev. Cancer 2011, 11, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Melo, B.A.C.; Vilar, L.G.; Oliveira, N.R.; Lima, P.O.; Pinheiro, M.B.; Domingueti, C.P.; Pereira, M.C. Human papillomavirus infection and oral squamous cell carcinoma—A systematic review. Rev. Bras. Otorrinolaringol. 2021, 87, 346–352. [Google Scholar] [CrossRef]
- McCord, C.; Xu, J.; Xu, W.; Qiu, X.; Muhanna, N.; Irish, J.; Leong, I.; McComb, R.J.; Perez-Ordonez, B.; Bradley, G. Association of human papilloma virus with atypical and malignant oral papillary lesions. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2014, 117, 722–732. [Google Scholar] [CrossRef] [PubMed]
- Ivancic, R.; Iqbal, H.; de Silva, B.; Pan, Q.; Matrka, L. Immunological tolerance of low-risk HPV in recurrent respiratory papillomatosis. Clin. Exp. Immunol. 2020, 199, 131–142. [Google Scholar] [CrossRef]
- Sampaio, J.; Ferreira, J.; Santos, A.C.; Bicho, M.; Bicho, M.C. The Importance of the Extracellular Matrix in HPV-Associated Diseases. In Cervical Cancer—A Global Public Health Treatise; Rajkumar, R., Ed.; IntechOpen: London, UK, 2021; Available online: https://www.intechopen.com/chapters/78569 (accessed on 17 June 2022).
- Münger, K.; Howley, P.M. Human papillomavirus immortalization and transformation functions. Virus Res. 2002, 89, 213–228. [Google Scholar] [CrossRef]
- Candotto, V.; Lauritano, D.; Nardone, M.; Baggi, L.; Arcuri, C.; Gatto, R.; Gaudio, R.M.; Spadari, F.; Carinci, F. HPV infection in the oral cavity: Epidemiology, clinical manifestations and relationship with oral cancer. Oral Implantol. 2017, 10, 209–220. [Google Scholar] [CrossRef]
- Bharti, A.H.; Chotaliya, K.; Marfatia, Y.S. An update on oral human papillomavirus infection. Indian J. Sex. Transm. Dis. AIDS 2013, 34, 77–82. [Google Scholar] [CrossRef]
- Castro, T.P.; Bussoloti Filho, I. Prevalence of human papillomavirus (HPV) in oral cavity and oropharynx. Rev. Bras. Otorrinolaringol. 2006, 72, 272–282. [Google Scholar] [CrossRef]
- Winning, T.A.; Townsend, G.C. Oral mucosal embryology and histology. Clin. Dermatol. 2000, 18, 499–511. [Google Scholar] [CrossRef]
- Brizuela, M.; Winters, R. Histology, Oral Mucosa. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK572115/ (accessed on 17 June 2022).
- Nikoloudaki, G.; Creber, K.; Hamilton, D.W. Wound healing and fibrosis: A contrasting role for periostin in skin and the oral mucosa. Am. J. Physiol. Cell Physiol. 2020, 318, C1065–C1077. [Google Scholar] [CrossRef] [PubMed]
- Groeger, S.; Meyle, J. Oral Mucosal Epithelial Cells. Front. Immunol. 2019, 10, 208. [Google Scholar] [CrossRef]
- Otsuka-Tanaka, Y.; Oommen, S.; Kawasaki, M.; Kawasaki, K.; Imam, N.; Jalani-Ghazani, F.; Hindges, R.; Sharpe, P.T.; Ohazama, A. Oral lining mucosa development depends on mesenchymal microRNAs. J. Dent. Res. 2013, 92, 229–234. [Google Scholar] [CrossRef]
- Zhou, C.; Tuong, Z.K.; Frazer, I.H. Papillomavirus immune evasion strategies target the infected cell and the local immune system. Front. Oncol. 2019, 9, 682. [Google Scholar] [CrossRef] [PubMed]
- Kines, R.C.; Thompson, C.D.; Lowy, D.R.; Schiller, J.T.; Day, P.M. The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. Proc. Natl. Acad. Sci. USA 2009, 106, 20458–20463. [Google Scholar] [CrossRef] [PubMed]
- Mattoscio, D.; Medda, A.; Chiocca, S. Human Papilloma Virus and Autophagy. Int. J. Mol. Sci. 2018, 19, 1775. [Google Scholar] [CrossRef] [PubMed]
- Stanley, M.A. Epithelial cell responses to infection with human papillomavirus. Clin. Microbiol. Rev. 2012, 25, 215–222. [Google Scholar] [CrossRef]
- Rodríguez, A.C.; Schiffman, M.; Herrero, R.; Wacholder, S.; Hildesheim, A.; Castle, P.E.; Solomon, D.; Burk, R.; Proyecto Epidemiológico Guanacaste Group. Rapid clearance of human papillomavirus and implications for clinical focus on persistent infections. J. Natl. Cancer Inst. 2008, 100, 513–517. [Google Scholar] [CrossRef]
- Mishra, G.A.; Pimple, S.A.; Shastri, S.S. An overview of prevention and early detection of cervical cancers. Indian J. Med. Paediatr. Oncol. 2011, 32, 125–132. [Google Scholar] [CrossRef]
- Markowitz, L.E.; Schiller, J.T. Human Papillomavirus Vaccines. J. Infect. Dis. 2021, 224 (Suppl. S2), S367–S378. [Google Scholar] [CrossRef]
- Giuliano, A.R.; Lee, J.H.; Fulp, W.; Villa, L.L.; Lazcano, E.; Papenfuss, M.R.; Abrahamsen, M.; Salmeron, J.; Anic, G.M.; Rollison, D.E.; et al. Incidence and clearance of genital human papillomavirus infection in men (HIM): A cohort study. Lancet 2011, 377, 932–940. [Google Scholar] [CrossRef]
- Ryser, M.D.; Myers, E.R.; Durrett, R. HPV clearance and the neglected role of stochasticity. PLoS Comput. Biol. 2015, 11, e1004113. [Google Scholar] [CrossRef] [PubMed]
- Shanmugasundaram, S.; You, J. Targeting Persistent Human Papillomavirus Infection. Viruses 2017, 9, 229. [Google Scholar] [CrossRef] [PubMed]
- Huebbers, C.U.; Preuss, S.F.; Kolligs, J.; Vent, J.; Stenner, M.; Wieland, U.; Silling, S.; Drebber, U.; Speel, E.J.; Klussmann, J.P. Integration of HPV6 and downregulation of AKR1C3 expression mark malignant transformation in a patient with juvenile-onset laryngeal papillomatosis. PLoS ONE 2013, 8, e57207. [Google Scholar] [CrossRef] [PubMed]
- Pim, D.; Banks, L. Interaction of viral oncoproteins with cellular target molecules: Infection with high-risk vs. low-risk human papillomaviruses. Apmis 2010, 118, 471–493. [Google Scholar] [CrossRef]
- Liaw, K.L.; Hildesheim, A.; Burk, R.D.; Gravitt, P.; Wacholder, S.; Manos, M.M.; Scott, D.R.; Sherman, M.E.; Kurman, R.J.; Glass, A.G.; et al. A prospective study of human papillomavirus (HPV) type 16 DNA detection by polymerase chain reaction and its association with acquisition and persistence of other HPV types. J. Infect. Dis. 2001, 183, 8–15. [Google Scholar] [CrossRef]
- Maglennon, G.A.; McIntosh, P.; Doorbar, J. Persistence of viral DNA in the epithelial basal layer suggests a model for papillomavirus latency following immune regression. Virology 2011, 414, 153–163. [Google Scholar] [CrossRef]
- Myers, J.E.; Guidry, J.T.; Scott, M.L.; Zwolinska, K.; Raikhy, G.; Prasai, K.; Bienkowska-Haba, M.; Bodily, J.M.; Sapp, M.J.; Scott, R.S. Detecting episomal or integrated human papillomavirus 16 DNA using an exonuclease V-qPCR-based assay. Virology 2019, 537, 149–156. [Google Scholar] [CrossRef]
- Vescovo, T.; Pagni, B.; Piacentini, M.; Fimia, G.M.; Antonioli, M. Regulation of Autophagy in Cells Infected with Oncogenic Human Viruses and Its Impact on Cancer Development. Front. Cell Dev. Biol. 2020, 8, 47. [Google Scholar] [CrossRef]
- Chang, Y.; Moore, P.S.; Weiss, R.A. Human oncogenic viruses: Nature and discovery. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 20160264. [Google Scholar] [CrossRef]
- Mikuličić, S.; Florin, L. The endocytic trafficking pathway of oncogenic papillomaviruses. Papillomavirus Res. 2019, 7, 135–137. [Google Scholar] [CrossRef] [PubMed]
- Wittekindt, C.; Wagner, S.; Sharma, S.J.; Würdemann, N.; Knuth, J.; Reder, H.; Klußmann, J.P. HPV—A different view on Head and Neck Cancer. HPV—Das andere Kopf-Hals-Karzinom. Laryngo-Rhino-Otologie 2018, 97 (Suppl. S1), S48. [Google Scholar] [PubMed]
- Ashrafi, G.H.; Brown, D.R.; Fife, K.H.; Campo, M.S. Down-regulation of MHC class I is a property common to papillomavirus E5 proteins. Virus Res. 2006, 120, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Cortese, M.S.; Ashrafi, G.H.; Campo, M.S. All 4 di-leucine motifs in the first hydrophobic domain of the E5 oncoprotein of human papillomavirus type 16 are essential for surface MHC class I downregulation activity and E5 endomembrane localization. Int. J. Cancer 2010, 126, 1675–1682. [Google Scholar] [CrossRef]
- Franceschi, S.; Muñoz, N.; Bosch, X.F.; Snijders, P.J.; Walboomers, J.M. Human papillomavirus and cancers of the upper aerodigestive tract: A review of epidemiological and experimental evidence. Cancer Epidemiol. Biomarkers Prev. 1996, 5, 567–575. [Google Scholar]
- Wilson, S.S.; Wiens, M.E.; Smith, J.G. Antiviral mechanisms of human defensins. J. Mol. Biol. 2013, 425, 4965–4980. [Google Scholar] [CrossRef]
- Paz, I.B.; Cook, N.; Odom-Maryon, T.; Xie, Y.; Wilczynski, S.P. Human papillomavirus (HPV) in head and neck cancer. An association of HPV 16 with squamous cell carcinoma of Waldeyer’s tonsillar ring. Cancer 1997, 79, 595–604. [Google Scholar] [CrossRef]
- Fehrenbach, M.J.; Popowics, T. Illustrated Dental Embryology, Histology, and Anatomy, 4th ed.; Elssevier Saunders: Maryland Heights, MO, USA, 2016; pp. 9–17. [Google Scholar]
- Hormia, M.; Willberg, J.; Ruokonen, H.; Syrjänen, S. Marginal periodontium as a potential reservoir of human papillomavirus in oral mucosa. J. Periodontol. 2005, 76, 358–363. [Google Scholar] [CrossRef]
- Tezal, M.; Sullivan Nasca, M.; Stoler, D.L.; Melendy, T.; Hyland, A.; Smaldino, P.J.; Rigual, N.R.; Loree, T.R. Chronic periodontitis-human papillomavirus synergy in base of tongue cancers. Arch. Otolaryngol. Head Neck Surg. 2009, 135, 391–396. [Google Scholar] [CrossRef]
- Sasagawa, T.; Takagi, H.; Makinoda, S. Immune responses against human papillomavirus (HPV) infection and evasion of host defense in cervical cancer. J. Infect. Chemother. 2012, 18, 807–815. [Google Scholar] [CrossRef]
- Nestle, F.O.; Di Meglio, P.; Qin, J.Z.; Nickoloff, B.J. Skin immune sentinels in health and disease. Nat. Rev. Immunol. 2009, 9, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMPs and DAMPs: Signal 0s that spur autophagy and immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef] [PubMed]
- Andrei, A.M.; Andrei, E.C.; Stănciulescu, E.C.; Mehedinți, M.C.; Țuculină, M.J.; Baniță, I.M.; Buteică, S.A.; Pisoschi, C.G. Innate Immune Response as a New Challenge in Periodontal Inflammation. In Periodontology—Fundamentals and Clinical Features; Surlin, P., Ed.; IntechOpen: London, UK, 2021; Available online: https://www.intechopen.com/chapters/75894 (accessed on 17 June 2022).
- McClure, R.; Massari, P. TLR-Dependent Human Mucosal Epithelial Cell Responses to Microbial Pathogens. Front. Immunol. 2014, 5, 386. [Google Scholar] [CrossRef] [PubMed]
- Kaisho, T.; Akira, S. Toll-like receptor function and signaling. J. Allergy Clin. Immunol. 2006, 117, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Hibma, M.H. The immune response to papillomavirus during infection persistence and regression. Open Virol. J. 2012, 6, 241–248. [Google Scholar] [CrossRef]
- Montero Vega, M.T.; de Andrés Martín, A. The significance of toll-like receptors in human diseases. Allergol. Immunopathol. 2009, 37, 252–263. [Google Scholar] [CrossRef]
- Daud, I.I.; Scott, M.E.; Ma, Y.; Shiboski, S.; Farhat, S.; Moscicki, A.B. Association between toll-like receptor expression and human papillomavirus type 16 persistence. Int. J. Cancer 2011, 128, 879–886. [Google Scholar] [CrossRef]
- Britto, A.M.A.; Goes, L.R.; Sivro, A.; Policarpo, C.; Meirelles, A.R.; Furtado, Y.; Almeida, G.; Arthos, J.; Cicala, C.; Soares, M.A.; et al. HPV Induces Changes in Innate Immune and Adhesion Molecule Markers in Cervical Mucosa with Potential Impact on HIV Infection. Front. Immunol. 2020, 11, 2078. [Google Scholar] [CrossRef]
- Gonzalez, S.M.; Aguilar-Jimenez, W.; Su, R.C.; Rugeles, M.T. Mucosa: Key Interactions Determining Sexual Transmission of the HIV Infection. Front. Immunol. 2019, 10, 144. [Google Scholar] [CrossRef]
- Miller, L.S.; Modlin, R.L. Human keratinocyte Toll-like receptors promote distinct immune responses. J. Investig. Dermatol. 2007, 127, 262–263. [Google Scholar] [CrossRef]
- Le Bon, A.; Tough, D.F. Links between innate and adaptive immunity via type I interferon. Curr. Opin. Immunol. 2002, 14, 432–436. [Google Scholar] [CrossRef]
- Arend, W.P.; Palmer, G.; Gabay, C. IL-1, IL-18, and IL-33 families of cytokines. Immunol. Rev. 2008, 223, 20–38. [Google Scholar] [CrossRef] [PubMed]
- LaFleur, D.W.; Nardelli, B.; Tsareva, T.; Mather, D.; Feng, P.; Semenuk, M.; Taylor, K.; Buergin, M.; Chinchilla, D.; Roshke, V.; et al. Interferon-kappa, a novel type I interferon expressed in human keratinocytes. J. Biol. Chem. 2001, 276, 39765–39771. [Google Scholar] [CrossRef]
- Scott, M.L.; Woodby, B.L.; Ulicny, J.; Raikhy, G.; Orr, A.W.; Songock, W.K.; Bodily, J.M. Human Papillomavirus 16 E5 Inhibits Interferon Signaling and Supports Episomal Viral Maintenance. J. Virol. 2020, 94, e01582-19. [Google Scholar] [CrossRef] [PubMed]
- Reiser, J.; Hurst, J.; Voges, M.; Krauss, P.; Münch, P.; Iftner, T.; Stubenrauch, F. High-risk human papillomaviruses repress constitutive kappa interferon transcription via E6 to prevent pathogen recognition receptor and antiviral-gene expression. J. Virol. 2011, 85, 11372–11380. [Google Scholar] [CrossRef]
- DeCarlo, C.A.; Severini, A.; Edler, L.; Escott, N.G.; Lambert, P.F.; Ulanova, M.; Zehbe, I. IFN-κ, a novel type I IFN, is undetectable in HPV-positive human cervical Keratinocytes. Lab. Investig. 2010, 90, 1482–1491. [Google Scholar] [CrossRef]
- Kanodia, S.; Fahey, L.M.; Kast, W.M. Mechanisms used by human papillomaviruses to escape the host immune response. Curr. Cancer Drug Targets 2007, 7, 79–89. [Google Scholar] [CrossRef]
- Routes, J.M.; Morris, K.; Ellison, M.C.; Ryan, S. Macrophages kill human papillomavirus type 16 E6-expressing tumor cells by tumor necrosis factor alpha- and nitric oxide-dependent mechanisms. J. Virol. 2005, 79, 116–123. [Google Scholar] [CrossRef]
- Bedoui, S.; Whitney, P.G.; Waithman, J.; Eidsmo, L.; Wakim, L.; Caminschi, I.; Allan, R.S.; Wojtasiak, M.; Shortman, K.; Carbone, F.R.; et al. Cross-presentation of viral and self antigens by skin-derived CD103+ dendritic cells. Nat. Immunol. 2009, 10, 488–495. [Google Scholar] [CrossRef]
- Tamoutounour, S.; Guilliams, M.; Montanana Sanchis, F.; Liu, H.; Terhorst, D.; Malosse, C.; Pollet, E.; Ardouin, L.; Luche, H.; Sanchez, C.; et al. Origins and functional specialization of macrophages and of conventional and monocyte-derived dendritic cells in mouse skin. Immunity 2013, 39, 925–938. [Google Scholar] [CrossRef]
- Cao, Y.; Liu, C.; Gu, Z.; Zhang, Y.; Duan, Y.; Zhang, Y.; Zhang, H.; Tang, K.; Huang, B. Microparticles mediate human papillomavirus type 6 or 11 infection of human macrophages. Cell. Mol. Immunol. 2017, 14, 395–397. [Google Scholar] [CrossRef] [PubMed]
- Hovav, A.H. Dendritic cells of the oral mucosa. Mucosal Immunol. 2014, 7, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Séguier, S.; Bodineau, A.; Godeau, G.; Pellat, B.; Brousse, N. Langerin+ versus CD1a+ Langerhans cells in human gingival tissue: A comparative and quantitative immunohistochemical study. Arch. Oral Biol. 2003, 48, 255–262. [Google Scholar] [CrossRef]
- Jotwani, R.; Cutler, C.W. Multiple dendritic cell (DC) subpopulations in human gingiva and association of mature DCs with CD4+ T-cells in situ. J. Dent. Res. 2003, 82, 736–741. [Google Scholar] [CrossRef]
- Wollenberg, A.; Wagner, M.; Günther, S.; Towarowski, A.; Tuma, E.; Moderer, M.; Rothenfusser, S.; Wetzel, S.; Endres, S.; Hartmann, G. Plasmacytoid dendritic cells: A new cutaneous dendritic cell subset with distinct role in inflammatory skin diseases. J. Investig. Dermatol. 2002, 119, 1096–1102. [Google Scholar] [CrossRef]
- Sallusto, F.; Lanzavecchia, A. Heterogeneity of CD4+ memory T cells: Functional modules for tailored immunity. Eur. J. Immunol. 2009, 39, 2076–2082. [Google Scholar] [CrossRef]
- Joffre, O.P.; Segura, E.; Savina, A.; Amigorena, S. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 2012, 12, 557–569. [Google Scholar] [CrossRef]
- Schutyser, E.; Struyf, S.; Van Damme, J. The CC chemokine CCL20 and its receptor CCR6. Cytokine Growth Factor Rev. 2003, 14, 409–426. [Google Scholar] [CrossRef]
- Le Borgne, M.; Etchart, N.; Goubier, A.; Lira, S.A.; Sirard, J.C.; van Rooijen, N.; Caux, C.; Aït-Yahia, S.; Vicari, A.; Kaiserlian, D.; et al. Dendritic cells rapidly recruited into epithelial tissues via CCR6/CCL20 are responsible for CD8+ T cell crosspriming in vivo. Immunity 2006, 24, 191–201. [Google Scholar] [CrossRef]
- Guess, J.C.; McCance, D.J. Decreased migration of Langerhans precursor-like cells in response to human Keratinocytes expressing human papillomavirus type 16 E6/E7 is related to reduced macrophage inflammatory protein-3alpha production. J. Virol. 2005, 79, 14852–14862. [Google Scholar] [CrossRef]
- Hubert, P.; Caberg, J.H.; Gilles, C.; Bousarghin, L.; Franzen-Detrooz, E.; Boniver, J.; Delvenne, P. E-cadherin-dependent adhesion of dendritic and Langerhans cells to Keratinocytes is defective in cervical human papillomavirus-associated (pre)neoplastic lesions. J. Pathol. 2005, 206, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Laurson, J.; Khan, S.; Chung, R.; Cross, K.; Raj, K. Epigenetic repression of E-cadherin by human papillomavirus 16 E7 protein. Carcinogenesis 2010, 31, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Shannon, B.; Yi, T.J.; Perusini, S.; Gajer, P.; Ma, B.; Humphrys, M.S.; Thomas-Pavanel, J.; Chieza, L.; Janakiram, P.; Saunders, M.; et al. Association of HPV infection and clearance with cervicovaginal immunology and the vaginal microbiota. Mucosal Immunol. 2017, 10, 1310–1319. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F.; Wild, C.M.; Mittelberger, J.; Dobler, F.; Schneider, M.; Ansorge, N.; Köpke, M.; Strieder, A.; Ditsch, N.; Jeschke, U.; et al. The Role of Chemokines in Cervical Cancers. Medicina 2021, 57, 1141. [Google Scholar] [CrossRef]
- Pahne-Zeppenfeld, J.; Schröer, N.; Walch-Rückheim, B.; Oldak, M.; Gorter, A.; Hegde, S.; Smola, S. Cervical cancer cell-derived interleukin-6 impairs CCR7-dependent migration of MMP-9-expressing dendritic cells. Int. J. Cancer 2014, 134, 2061–2073. [Google Scholar] [CrossRef]
- Vicari, A.P.; Vanbervliet, B.; Massacrier, C.; Chiodoni, C.; Vaure, C.; Aït-Yahia, S.; Dercamp, C.; Matsos, F.; Reynard, O.; Taverne, C.; et al. In vivo manipulation of dendritic cell migration and activation to elicit antitumour immunity. Novartis Found. Symp. 2004, 256, 241–254. [Google Scholar]
- Bashaw, A.A.; Leggatt, G.R.; Chandra, J.; Tuong, Z.K.; Frazer, I.H. Modulation of antigen presenting cell functions during chronic HPV infection. Papillomavirus Res. 2017, 4, 58–65. [Google Scholar] [CrossRef]
- Rees, R.C. MHC restricted and non-restricted killer lymphocytes. Blood Rev. 1990, 4, 204–210. [Google Scholar] [CrossRef]
- Cioni, B.; Jordanova, E.S.; Hooijberg, E.; van der Linden, R.; de Menezes, R.X.; Tan, K.; Willems, S.; Elbers, J.B.W.; Broeks, A.; Bergman, A.M.; et al. HLA class II expression on tumor cells and low numbers of tumor-associated macrophages predict clinical outcome in oropharyngeal cancer. Head Neck 2019, 41, 463–478. [Google Scholar] [CrossRef]
- Zhao, H.; Zhang, J.X. Natural killer cells: The first defense against human papilloma virus early infection. J. Public Health Emerg. 2017, 1, 5. [Google Scholar] [CrossRef]
- Campo, M.S.; Graham, S.V.; Cortese, M.S.; Ashrafi, G.H.; Araibi, E.H.; Dornan, E.S.; Miners, K.; Nunes, C.; Man, S. HPV-16 E5 down-regulates expression of surface HLA class I and reduces recognition by CD8 T cells. Virology 2010, 407, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Sutlu, T.; Alici, E. Natural killer cell-based immunotherapy in cancer: Current insights and future prospects. J. Intern. Med. 2009, 266, 154–181. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.T.; Irving, A.T.; Lambley, E.H.; Payne, E.; McMillan, N.A. A novel method for screening viral interferon-resistance genes. J. Interferon Cytokine Res. 2004, 24, 470–477. [Google Scholar] [CrossRef] [PubMed]
- DeVoti, J.A.; Steinberg, B.M.; Rosenthal, D.W.; Hatam, L.; Vambutas, A.; Abramson, A.L.; Shikowitz, M.J.; Bonagura, V.R. Failure of gamma interferon but not interleukin-10 expression in response to human papillomavirus type 11 E6 protein in respiratory papillomatosis. Clin. Diagn. Lab. Immunol. 2004, 11, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Brownlie, R.J.; Zamoyska, R. T cell receptor signalling networks: Branched, diversified and bounded. Nat. Rev. Immunol. 2013, 13, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Shintani, T.; Klionsky, D.J. Autophagy in health and disease: A double-edged sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Levine, B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe 2009, 5, 527–549. [Google Scholar] [CrossRef]
- Yin, Z.; Pascual, C.; Klionsky, D.J. Autophagy: Machinery and regulation. Microb. Cell 2016, 3, 588–596. [Google Scholar] [CrossRef]
- Nakamura, S.; Yoshimori, T. New insights into autophagosome-lysosome fusion. J. Cell Sci. 2017, 130, 1209–1216. [Google Scholar] [CrossRef]
- Zhao, Y.G.; Zhang, H. Autophagosome maturation: An epic journey from the ER to lysosomes. J. Cell Biol. 2019, 218, 757–770. [Google Scholar] [CrossRef]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Belleudi, F.; Nanni, M.; Raffa, S.; Torrisi, M.R. HPV16 E5 deregulates the autophagic process in human keratinocytes. Oncotarget 2015, 6, 9370–9386. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [PubMed]
- Warnakulasuriya, S. Global epidemiology of oral and oropharyngeal cancer. Oral Oncol. 2009, 45, 309–316. [Google Scholar] [CrossRef]
- Syrjänen, S.; Lodi, G.; von Bültzingslöwen, I.; Aliko, A.; Arduino, P.; Campisi, G.; Challacombe, S.; Ficarra, G.; Flaitz, C.; Zhou, H.M.; et al. Human papillomaviruses in oral carcinoma and oral potentially malignant disorders: A systematic review. Oral Dis. 2011, 17 (Suppl. S1), 58–72. [Google Scholar]
- Betz, S.J. HPV-Related Papillary Lesions of the Oral Mucosa: A Review. Head Neck Pathol. 2019, 13, 80–90. [Google Scholar] [CrossRef]
- Babaji, P.; Singh, V.; Chaurasia, V.R.; Masamatti, V.S.; Sharma, A.M. Squamous papilloma of the hard palate. Indian J. Dent. 2014, 5, 211–213. [Google Scholar] [CrossRef]
- Hassan Saad, R.; Halawa, S.M.; Zidan, A.M.; Emara, N.M.; Abdelghany, O.A. Malignant transformation of oral squamous cell papilloma: A case report. Int. J. Surg. Case Rep. 2020, 75, 348–351. [Google Scholar] [CrossRef]
- Chaturvedi, A.K.; Anderson, W.F.; Lortet-Tieulent, J.; Curado, M.P.; Ferlay, J.; Franceschi, S.; Rosenberg, P.S.; Bray, F.; Gillison, M.L. Worldwide trends in incidence rates for oral cavity and oropharyngeal cancers. J. Clin. Oncol. 2013, 31, 4550–4559. [Google Scholar] [CrossRef]
- Kanellou, P.; Zaravinos, A.; Zioga, M.; Stratigos, A.; Baritaki, S.; Soufla, G.; Zoras, O.; Spandidos, D.A. Genomic instability, mutations and expression analysis of the tumour suppressor genes p14(ARF), p15(INK4b), p16(INK4a) and p53 in actinic keratosis. Cancer Lett. 2008, 264, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Stephen, J.K.; Divine, G.; Chen, K.M.; Chitale, D.; Havard, S.; Worsham, M.J. Significance of p16 in Site-specific HPV Positive and HPV Negative Head and Neck Squamous Cell Carcinoma. Cancer Clin. Oncol. 2013, 2, 51–61. [Google Scholar] [PubMed]
- Ghittoni, R.; Accardi, R.; Hasan, U.; Gheit, T.; Sylla, B.; Tommasino, M. The biological properties of E6 and E7 oncoproteins from human papillomaviruses. Virus Genes 2010, 40, 1–13. [Google Scholar] [CrossRef]
- Boscolo-Rizzo, P.; Da Mosto, M.C.; Rampazzo, E.; Giunco, S.; Del Mistro, A.; Menegaldo, A.; Baboci, L.; Mantovani, M.; Tirelli, G.; De Rossi, A. Telomeres and telomerase in head and neck squamous cell carcinoma: From pathogenesis to clinical implications. Cancer Metastasis Rev. 2016, 35, 457–474. [Google Scholar] [CrossRef]
- Münger, K.; Baldwin, A.; Edwards, K.M.; Hayakawa, H.; Nguyen, C.L.; Owens, M.; Grace, M.; Huh, K. Mechanisms of human papillomavirus-induced oncogenesis. J. Virol. 2004, 78, 11451–11460. [Google Scholar] [CrossRef] [PubMed]
- Bunz, F.; Dutriaux, A.; Lengauer, C.; Waldman, T.; Zhou, S.; Brown, J.P.; Sedivy, J.M.; Kinzler, K.W.; Vogelstein, B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998, 282, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. p53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Perri, F.; Pacelli, R.; Della Vittoria Scarpati, G.; Cella, L.; Giuliano, M.; Caponigro, F.; Pepe, S. Radioresistance in head and neck squamous cell carcinoma: Biological bases and therapeutic implications. Head Neck 2015, 37, 763–770. [Google Scholar] [PubMed]
- Boscolo-Rizzo, P.; Rampazzo, E.; Perissinotto, E.; Piano, M.A.; Giunco, S.; Baboci, L.; Spinato, G.; Spinato, R.; Tirelli, G.; Da Mosto, M.C.; et al. Telomere shortening in mucosa surrounding the tumor: Biosensor of field cancerization and prognostic marker of mucosal failure in head and neck squamous cell carcinoma. Oral Oncol. 2015, 51, 500–507. [Google Scholar] [CrossRef]
- Hock, A.K.; Vousden, K.H. The role of ubiquitin modification in the regulation of p53. Biochim. Biophys. Acta 2014, 1843, 137–149. [Google Scholar] [CrossRef]
- Klingelhutz, A.J.; Roman, A. Cellular transformation by human papillomaviruses: Lessons learned by comparing high- and low-risk viruses. Virology 2012, 424, 77–98. [Google Scholar] [CrossRef] [PubMed]
- Namazie, A.; Alavi, S.; Olopade, O.I.; Pauletti, G.; Aghamohammadi, N.; Aghamohammadi, M.; Gornbein, J.A.; Calcaterra, T.C.; Slamon, D.J.; Wang, M.B.; et al. Cyclin D1 amplification and p16(MTS1/CDK4I) deletion correlate with poor prognosis in head and neck tumors. Laryngoscope 2002, 112, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Psyrri, A.; DiMaio, D. Human papillomavirus in cervical and head-and-neck cancer. Nat. Clin. Pract. Oncol. 2008, 5, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.H.; Zhang, Q.; Kong, C.S.; Harris, J.; Fertig, E.J.; Harari, P.M.; Wang, D.; Redmond, K.P.; Shenouda, G.; Trotti, A.; et al. p16 protein expression and human papillomavirus status as prognostic biomarkers of nonoropharyngeal head and neck squamous cell carcinoma. J. Clin. Oncol. 2014, 32, 3930–3938. [Google Scholar] [CrossRef]
- Aida, J.; Kobayashi, T.; Saku, T.; Yamaguchi, M.; Shimomura, N.; Nakamura, K.; Ishikawa, N.; Maruyama, S.; Cheng, J.; Poon, S.S.; et al. Short telomeres in an oral precancerous lesion: Q-FISH analysis of leukoplakia. J. Oral Pathol. Med. 2012, 41, 372–378. [Google Scholar] [CrossRef]
- Deng, Y.; Chan, S.S.; Chang, S. Telomere dysfunction and tumour suppression: The senescence connection. Nat. Rev. Cancer 2008, 8, 450–458. [Google Scholar] [CrossRef]
- Fan, H.C.; Chang, F.W.; Tsai, J.D.; Lin, K.M.; Chen, C.M.; Lin, S.Z.; Liu, C.A.; Harn, H.J. Telomeres and Cancer. Life 2021, 11, 1405. [Google Scholar] [CrossRef]
- Chen, C.H.; Chen, R.J. Prevalence of telomerase activity in human cancer. J. Formos. Med. Assoc. 2011, 110, 275–289. [Google Scholar] [CrossRef]
- Baillie, R.; Tan, S.T.; Itinteang, T. Cancer Stem Cells in Oral Cavity Squamous Cell Carcinoma: A Review. Front. Oncol. 2017, 7, 112. [Google Scholar] [CrossRef]
- Prince, M.E.; Sivanandan, R.; Kaczorowski, A.; Wolf, G.T.; Kaplan, M.J.; Dalerba, P.; Weissman, I.L.; Clarke, M.F.; Ailles, L.E. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2007, 104, 973–978. [Google Scholar] [CrossRef]
- Cirillo, N.; Wu, C.; Prime, S.S. Heterogeneity of Cancer Stem Cells in Tumorigenesis, Metastasis, and Resistance to Antineoplastic Treatment of Head and Neck Tumours. Cells 2021, 10, 3068. [Google Scholar] [CrossRef] [PubMed]
- Mărgăritescu, C.; Pirici, D.; Simionescu, C.; Stepan, A. The utility of CD44, CD117 and CD133 in identification of cancer stem cells (CSC) in oral squamous cell carcinomas (OSCC). Rom. J. Morphol. Embryol. 2011, 52 (Suppl. S3), 985–993. [Google Scholar] [PubMed]
- Mannelli, G.; Magnelli, L.; Deganello, A.; Busoni, M.; Meccariello, G.; Parrinello, G.; Gallo, O. Detection of putative stem cell markers, CD44/CD133, in primary and lymph node metastases in head and neck squamous cell carcinomas. A preliminary immunohistochemical and in vitro study. Clin. Otolaryngol. 2015, 40, 312–320. [Google Scholar] [CrossRef]
- Nakanishi, Y.; Squamous Papilloma. PathologyOutlines.com Website. Available online: https://www.pathologyoutlines.com/topic/esophagussquamouspapilloma.html (accessed on 16 July 2022).
- Kerge, S.; Vuorinen, J.; Hurme, S.; Soukka, T.; Gheit, T.; Tommasino, M.; Syrjänen, S.; Rautava, J. Benign proliferative epithelial lesions of oral mucosa are infrequently associated with α-, β-, or γ human papillomaviruses. Laryngoscope Investig. Otolaryngol. 2018, 4, 43–48. [Google Scholar] [CrossRef]
- Orenuga, O.O.; Oluwo, A.; Oluwakuyide, R.T.; Olawuyi, A.B. Recurrent oral squamous papilloma in a pediatric patient: Case report and review of the literature. Niger. J. Clin. Pract. 2018, 21, 1674–1677. [Google Scholar]
- Orrù, G.; Mameli, A.; Demontis, C.; Rossi, P.; Ratto, D.; Occhinegro, A.; Piras, V.; Kuqi, L.; Berretta, M.; Taibi, R.; et al. Oral human papilloma virus infection: An overview of clinical-laboratory diagnosis and treatment. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 8148–8157. [Google Scholar] [PubMed]
- Pringle, G.A. The role of human papillomavirus in oral disease. Dent. Clin. N. Am. 2014, 58, 385–399. [Google Scholar] [CrossRef]
- Abbey, L.M.; Page, D.G.; Sawyer, D.R. The clinical and histopathologic features of a series of 464 oral squamous cell papillomas. Oral Surg. Oral Med. Oral Pathol. 1980, 49, 419–428. [Google Scholar] [CrossRef]
- Syrjänen, S. Human papillomavirus infections and oral tumors. Med. Microbiol. Immunol. 2003, 192, 123–128. [Google Scholar] [CrossRef]
- Stojanov, I.J.; Squamous Papilloma. PathologyOutlines.com Website. Available online: https://www.pathologyoutlines.com/topic/oralcavitysquamouspapilloma.html (accessed on 4 April 2022).
- Eversole, L.R.; Laipis, P.J.; Merrell, P.; Choi, E. Demonstration of human papillomavirus DNA in oral condyloma acuminatum. J. Oral Pathol. 1987, 16, 266–272. [Google Scholar] [CrossRef]
- Bennett, L.K.; Hinshaw, M. Heck’s disease: Diagnosis and susceptibility. Pediatr. Dermatol. 2009, 26, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Said, A.K.; Leao, J.C.; Fedele, S.; Porter, S.R. Focal epithelial hyperplasia—An update. J. Oral Pathol. Med. 2013, 42, 435–442. [Google Scholar] [CrossRef] [PubMed]
- García-Corona, C.; Vega-Memije, E.; Mosqueda-Taylor, A.; Yamamoto-Furusho, J.K.; Rodríguez-Carreón, A.A.; Ruiz-Morales, J.A.; Salgado, N.; Granados, J. Association of HLA-DR4 (DRB1*0404) with human papillomavirus infection in patients with focal epithelial hyperplasia. Arch. Dermatol. 2004, 140, 1227–1231. [Google Scholar] [CrossRef] [PubMed]
- Syrjänen, S.; Syrjänen, K. HPV infections of the oral mucosa. In Papillomavirus Infections in Human Pathology; Syrjänen, K., Syrjänen, S., Eds.; J. Wiley & Sons: New York, NY, USA, 2000; pp. 379–412. [Google Scholar]
- Gillenwater, A.M.; Vigneswaran, N.; Fatani, H.; Saintigny, P.; El-Naggar, A.K. Proliferative verrucous leukoplakia (PVL): A review of an elusive pathologic entity! Adv. Anat. Pathol. 2013, 20, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Mete, O.; Keskin, Y.; Hafiz, G.; Kayhan, K.B.; Unur, M. Oral proliferative verrucous leukoplakia: Underdiagnosed oral precursor lesion that requires retrospective clinicopathological correlation. Dermatol. Online J. 2010, 16, 6. [Google Scholar] [CrossRef]
- van der Waal, I. Potentially malignant disorders of the oral and oropharyngeal mucosa; present concepts of management. Oral Oncol. 2010, 46, 423–425. [Google Scholar] [CrossRef]
- Boch, K.; Langan, E.A.; Kridin, K.; Zillikens, D.; Ludwig, R.J.; Bieber, K. Lichen Planus. Front. Med. 2021, 8, 737813. [Google Scholar] [CrossRef]
- Gorouhi, F.; Davari, P.; Fazel, N. Cutaneous and mucosal lichen planus: A comprehensive review of clinical subtypes, risk factors, diagnosis, and prognosis. Sci. World J. 2014, 2014, 742826. [Google Scholar] [CrossRef]
- Epstein, J.B.; Wan, L.S.; Gorsky, M.; Zhang, L. Oral lichen planus: Progress in understanding its malignant potential and the implications for clinical management. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2003, 96, 32–37. [Google Scholar] [CrossRef]
- Balighi, K.; Daneshpazhooh, M.; Lajevardi, V.; Mahmoudi, H.; Azizzadeh-Roodpishi, S.; Tavakolpour, S.; Shyaeganmehr, M. Association of human herpes virus 6 infection with lichen planopilaris. Mymensingh Med. J. 2020, 29, 977–982. [Google Scholar]
- Bombeccari, G.P.; Giannì, A.B.; Spadari, F. Oral Candida colonization and oral lichen planus. Oral Dis. 2017, 23, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Shang, Q.; Peng, J.; Zhou, Y.; Chen, Q.; Xu, H. Association of Human Papillomavirus with Oral Lichen Planus and Oral Leukoplakia: A Meta-analysis. J. Evid. Based Dent. Pract. 2020, 20, 101485. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A. Fundamentals of Cytological Diagnosis and Its Biological Basis. In Pathobiology of Human Disease; McManus, L.M., Mitchell, R.N., Eds.; Academic Press: Cambridge, MA, USA, 2014; pp. 3311–3344. [Google Scholar]
- Krause, K.A.; Neelon, D.; Butler, S.L. Koilocytosis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK532958/ (accessed on 17 June 2022).
- Bean, S.M.; Chhieng, D.C.; Cytology, A.R. The Other Pap Test. Lab. Med. 2010, 41, 168–171. [Google Scholar] [CrossRef]
- Toki, T.; Yajima, A. “HPV score”, a scoring system for histological diagnosis of human papillomavirus infection in dysplasia of the uterine cervix. Acta Pathol. Jpn. 1987, 37, 449–455. [Google Scholar] [CrossRef]
- Fletcher, S. Histopathology of papilloma virus infection of the cervix uteri: The history, taxonomy, nomenclature and reporting of koilocytic dysplasias. J. Clin. Pathol. 1983, 36, 616–624. [Google Scholar] [CrossRef]
- Meisels, A.; Fortin, R. Condylomatous lesions of the cervix and vagina. I. Cytologic patterns. Acta Cytol. 1976, 20, 505–509. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.M.; Perez-Montiel, D.; Miles, L.; Allen, C.M.; Nuovo, G.J. The histologic differentiation of oral condyloma acuminatum from its mimics. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2003, 96, 420–428. [Google Scholar] [CrossRef]
- Gültekin, S.E.; Sengüven, B.; Klussmann, J.P.; Dienes, H.P. P16(INK 4a) and Ki-67 expression in human papilloma virus-related head and neck mucosal lesions. Investig. Clin. 2015, 56, 47–59. [Google Scholar]
- Zaravinos, A.; Mammas, I.N.; Sourvinos, G.; Spandidos, D.A. Molecular detection methods of human papillomavirus (HPV). Int. J. Biol. Markers 2009, 24, 215–222. [Google Scholar] [CrossRef]
- Chandarana, S.P.; Lee, J.S.; Chanowski, E.J.; Sacco, A.G.; Bradford, C.R.; Wolf, G.T.; Prince, M.E.; Moyer, J.S.; Eisbruch, A.; Worden, F.P.; et al. Prevalence and predictive role of p16 and epidermal growth factor receptor in surgically treated oropharyngeal and oral cavity cancer. Head Neck 2013, 35, 1083–1090. [Google Scholar] [CrossRef]
- Prigge, E.S.; Arbyn, M.; von Knebel Doeberitz, M.; Reuschenbach, M. Diagnostic accuracy of p16INK4a immunohistochemistry in oropharyngeal squamous cell carcinomas: A systematic review and meta-analysis. Int. J. Cancer 2017, 140, 1186–1198. [Google Scholar] [CrossRef] [PubMed]
- Geraets, D.T.; Struijk, L.; Kleter, B.; Molijn, A.; van Doorn, L.J.; Quint, W.G.; Colau, B. The original SPF10 LiPA25 algorithm is more sensitive and suitable for epidemiologic HPV research than the SPF10 INNO-LiPA Extra. J. Virol. Methods 2015, 215, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Smeets, S.J.; Hesselink, A.T.; Speel, E.J.; Haesevoets, A.; Snijders, P.J.; Pawlita, M.; Meijer, C.J.; Braakhuis, B.J.; Leemans, C.R.; Brakenhoff, R.H. A novel algorithm for reliable detection of human papillomavirus in paraffin embedded head and neck cancer specimen. Int. J. Cancer 2007, 121, 2465–2472. [Google Scholar] [CrossRef]
- von Knebel Doeberitz, M. The causal role of human papillomavirus infections in non-anogenital cancers. It’s time to ask for the functional evidence. Int. J. Cancer 2016, 139, 9–11. [Google Scholar] [CrossRef]
- de Araujo, M.R.; De Marco, L.; Santos, C.F.; Rubira-Bullen, I.R.; Ronco, G.; Pennini, I.; Vizzini, L.; Merletti, F.; Gillio-Tos, A. GP5+/6+ SYBR Green methodology for simultaneous screening and quantification of human papillomavirus. J. Clin. Virol. 2009, 45, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Kreimer, A.R.; Johansson, M.; Waterboer, T.; Kaaks, R.; Chang-Claude, J.; Drogen, D.; Tjønneland, A.; Overvad, K.; Quirós, J.R.; González, C.A.; et al. Evaluation of human papillomavirus antibodies and risk of subsequent head and neck cancer. J. Clin. Oncol. 2013, 31, 2708–2715. [Google Scholar] [CrossRef] [PubMed]
- Veress, G.; Kónya, J.; Csiky-Mészáros, T.; Czeglédy, J.; Gergely, L. Human papillomavirus DNA and anti-HPV secretory IgA antibodies in cytologically normal cervical specimens. J. Med. Virol. 1994, 43, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Elfgren, K.; Bistoletti, P.; Dillner, L.; Walboomers, J.M.; Meijer, C.J.; Dillner, J. Conization for cervical intraepithelial neoplasia is followed by disappearance of human papillomavirus deoxyribonucleic acid and a decline in serum and cervical mucus antibodies against human papillomavirus antigens. Am. J. Obstet. Gynecol. 1996, 174, 937–942. [Google Scholar] [CrossRef]
- Tjiong, M.Y.; Zumbach, K.; Schegget, J.T.; van der Vange, N.; Out, T.A.; Pawlita, M.; Struyk, L. Antibodies against human papillomavirus type 16 and 18 E6 and E7 proteins in cervicovaginal washings and serum of patients with cervical neoplasia. Viral Immunol. 2001, 14, 415–424. [Google Scholar] [CrossRef]
- Nguyen, H.H.; Broker, T.R.; Chow, L.T.; Alvarez, R.D.; Vu, H.L.; Andrasi, J.; Brewer, L.R.; Jin, G.; Mestecky, J. Immune responses to human papillomavirus in genital tract of women with cervical cancer. Gynecol. Oncol. 2005, 96, 452–461. [Google Scholar] [CrossRef]
- Pattyn, J.; Van Keer, S.; Tjalma, W.; Matheeussen, V.; Van Damme, P.; Vorsters, A. Infection and vaccine-induced HPV-specific antibodies in cervicovaginal secretions. A review of the literature. Papillomavirus Res. 2019, 8, 100185. [Google Scholar] [CrossRef] [PubMed]
- Meites, E.; Szilagyi, P.G.; Chesson, H.W.; Unger, E.R.; Romero, J.R.; Markowitz, L.E. Human Papillomavirus Vaccination for Adults: Updated Recommendations of the Advisory Committee on Immunization Practices. MMWR Morb. Mortal. Wkly. Rep. 2019, 68, 698–702. [Google Scholar] [CrossRef] [PubMed]
- Stanley, M. HPV—Immune response to infection and vaccination. Infect. Agent. Cancer 2010, 5, 19. [Google Scholar] [CrossRef] [PubMed]
- Harper, D.M.; DeMars, L.R. HPV vaccines—A review of the first decade. Gynecol. Oncol. 2017, 146, 196–204. [Google Scholar] [CrossRef]
- Gallagher, K.E.; Erio, T.; Baisley, K.; Lees, S.; Watson-Jones, D. The impact of a human papillomavirus (HPV) vaccination campaign on routine primary health service provision and health workers in Tanzania: A controlled before and after study. BMC Health Serv. Res. 2018, 18, 173. [Google Scholar] [CrossRef]
- Kunda, N.K.; Peabody, J.; Zhai, L.; Price, D.N.; Chackerian, B.; Tumban, E.; Muttil, P. Evaluation of the thermal stability and the protective efficacy of spray-dried HPV vaccine, Gardasil® 9. Hum. Vaccin. Immunother. 2019, 15, 1995–2002. [Google Scholar] [CrossRef]
- Cheng, L.; Wang, Y.; Du, J. Human Papillomavirus Vaccines: An Updated Review. Vaccines 2020, 8, 391. [Google Scholar] [CrossRef]
- Schauner, S.; Lyon, C. Bivalent HPV Recombinant Vaccine (Cervarix) for the Prevention of Cervical Cancer. Am. Fam. Physician 2010, 82, 1541–1542. [Google Scholar]
- Joura, E.A.; Leodolter, S.; Hernandez-Avila, M.; Wheeler, C.M.; Perez, G.; Koutsky, L.A.; Garland, S.M.; Harper, D.M.; Tang, G.W.; Ferris, D.G.; et al. Efficacy of a quadrivalent prophylactic human papillomavirus (types 6, 11, 16, and 18) L1 virus-like-particle vaccine against high-grade vulval and vaginal lesions: A combined analysis of three randomised clinical trials. Lancet 2007, 369, 1693–1702. [Google Scholar] [CrossRef]
- Paavonen, J.; Jenkins, D.; Bosch, F.X.; Naud, P.; Salmerón, J.; Wheeler, C.M.; Chow, S.N.; Apter, D.L.; Kitchener, H.C.; Castellsague, X.; et al. HPV PATRICIA study group. Efficacy of a prophylactic adjuvanted bivalent L1 virus-like-particle vaccine against infection with human papillomavirus types 16 and 18 in young women: An interim analysis of a phase III double-blind, randomised controlled trial. Lancet 2007, 369, 2161–2170. [Google Scholar] [CrossRef]
- Nicol, A.F.; Andrade, C.V.; Russomano, F.B.; Rodrigues, L.L.; Oliveira, N.S.; Provance, D.W., Jr. HPV vaccines: A controversial issue? Braz. J. Med. Biol. Res. 2016, 49, e5060. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrei, E.C.; Baniță, I.M.; Munteanu, M.C.; Busuioc, C.J.; Mateescu, G.O.; Mălin, R.D.; Pisoschi, C.G. Oral Papillomatosis: Its Relation with Human Papilloma Virus Infection and Local Immunity—An Update. Medicina 2022, 58, 1103. https://doi.org/10.3390/medicina58081103
Andrei EC, Baniță IM, Munteanu MC, Busuioc CJ, Mateescu GO, Mălin RD, Pisoschi CG. Oral Papillomatosis: Its Relation with Human Papilloma Virus Infection and Local Immunity—An Update. Medicina. 2022; 58(8):1103. https://doi.org/10.3390/medicina58081103
Chicago/Turabian StyleAndrei, Elena Cristina, Ileana Monica Baniță, Maria Cristina Munteanu, Cristina Jana Busuioc, Garofița Olivia Mateescu, Ramona Denise Mălin, and Cătălina Gabriela Pisoschi. 2022. "Oral Papillomatosis: Its Relation with Human Papilloma Virus Infection and Local Immunity—An Update" Medicina 58, no. 8: 1103. https://doi.org/10.3390/medicina58081103