
SIRT1 Activator E1231 Alleviates Nonalcoholic Fatty Liver Disease by Regulating Lipid Metabolism
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
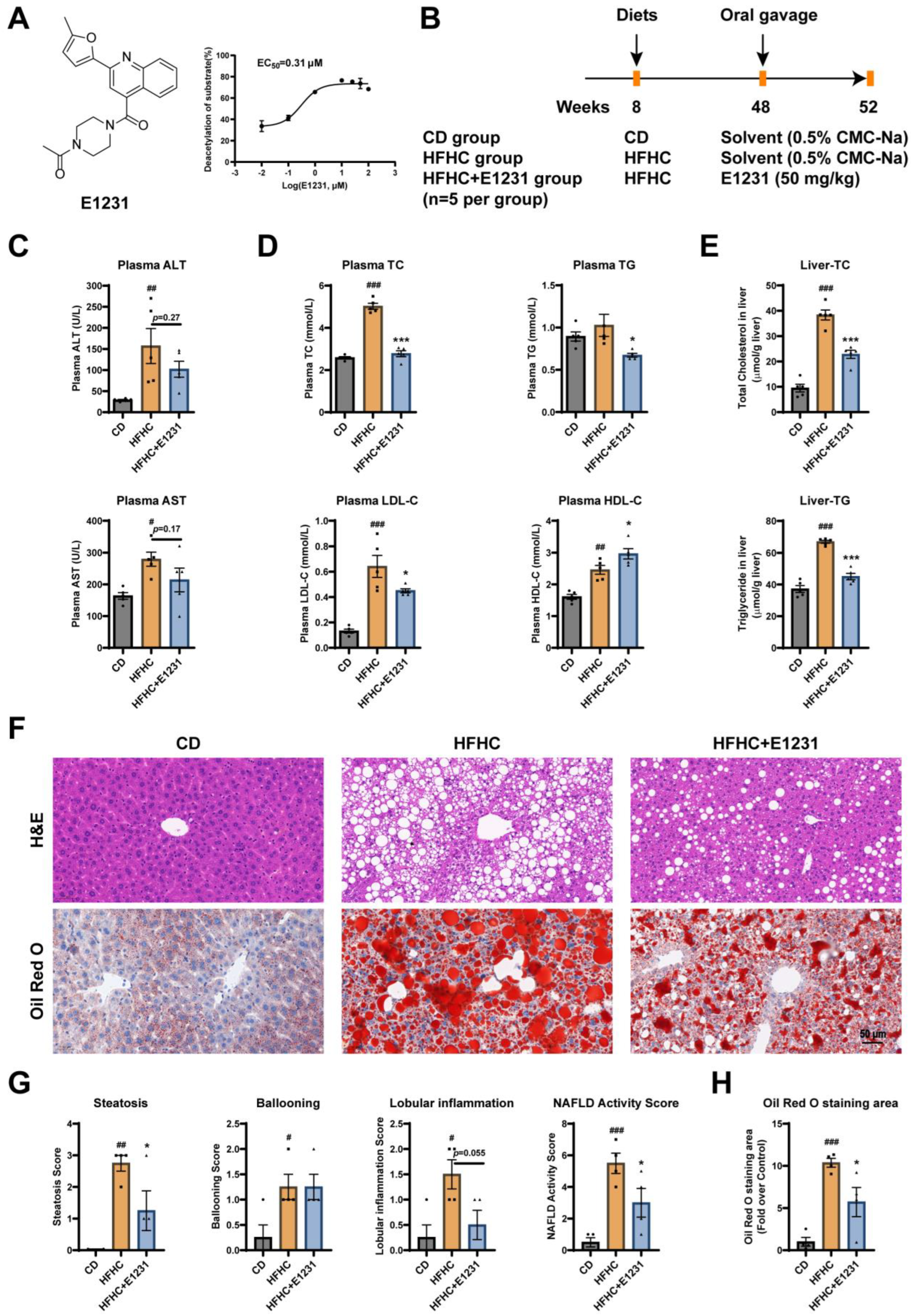
2.1. E1231 Treatment Ameliorated Plasma Biochemistry Parameter Abnormalities and Hepatic Steatosis in HFHC-Induced NAFLD Mice
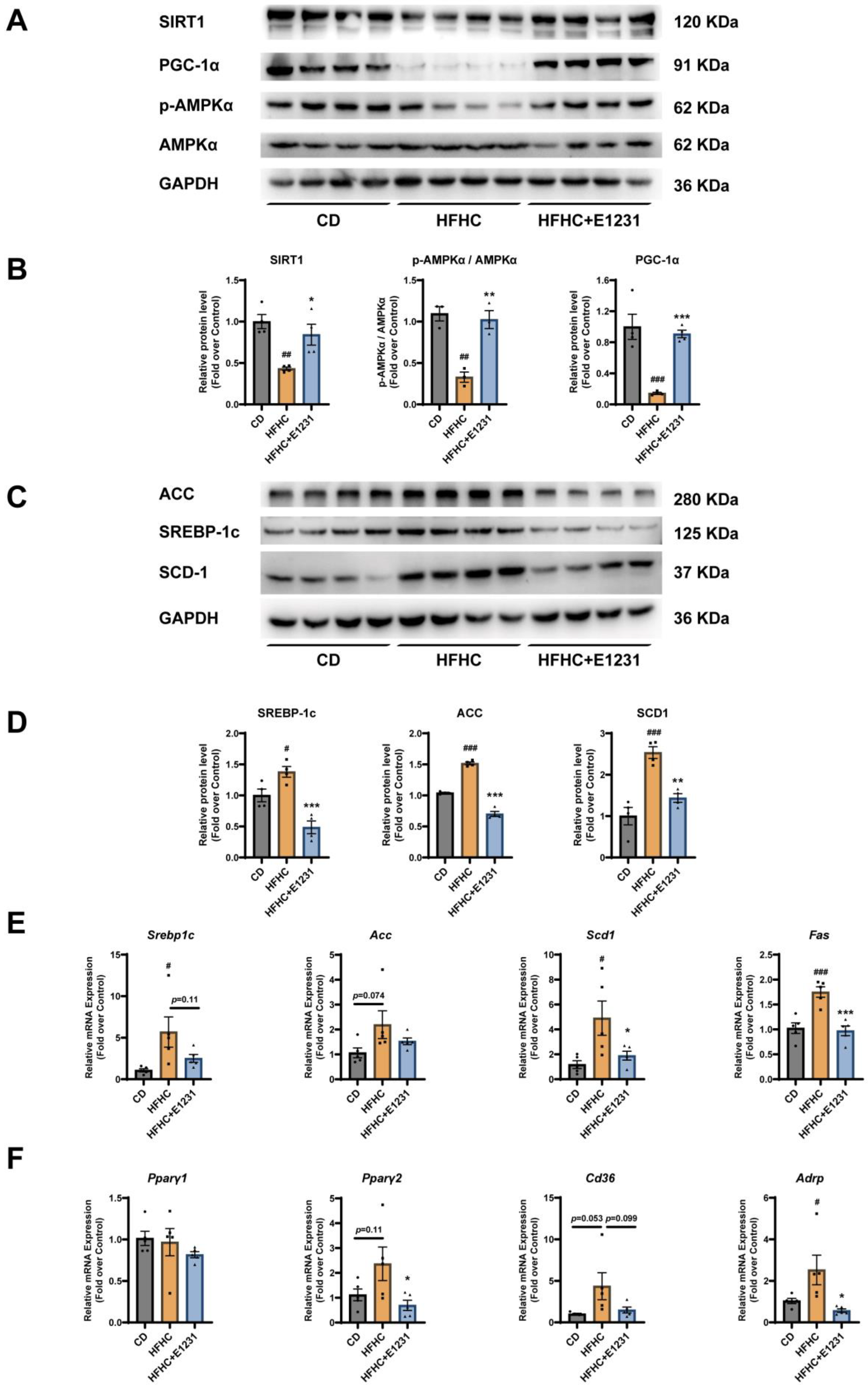
2.2. E1231 Regulates SIRT1 and Other Lipid Metabolism Regulators in the Liver
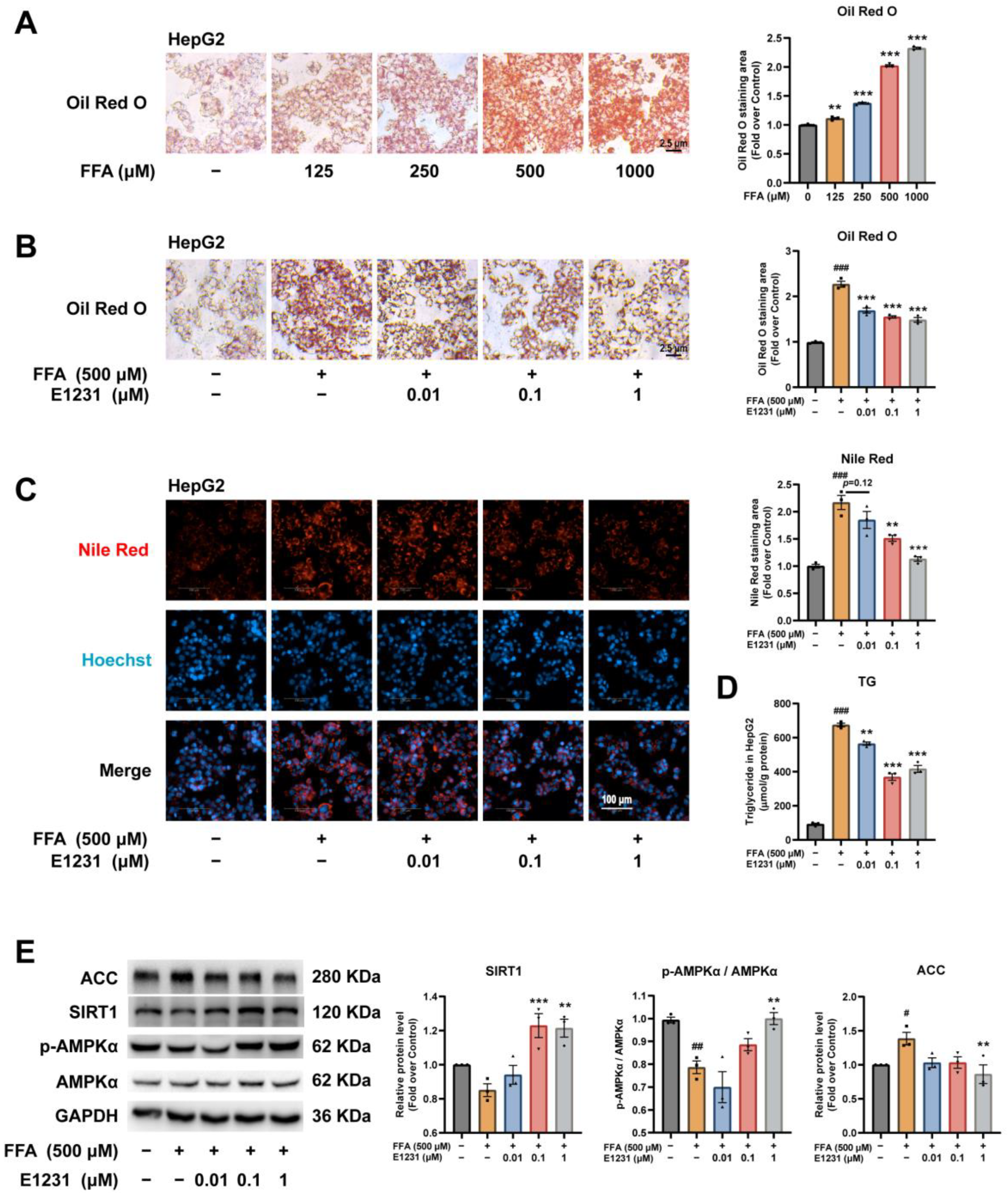
2.3. E1231 Suppresses FFA-Induced Lipid Accumulation in the Human Hepatoma Cell Line HepG2
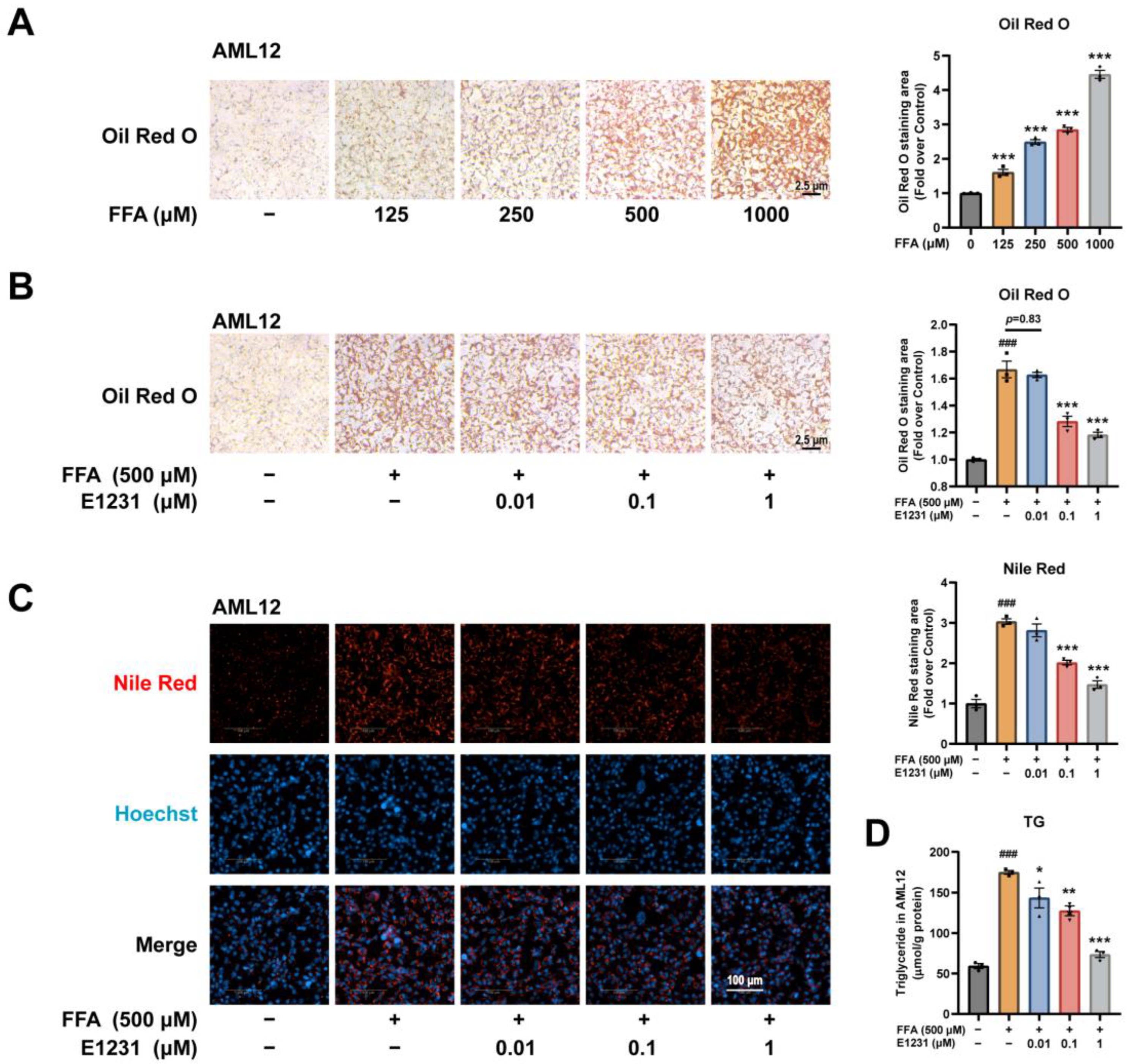
2.4. E1231 Suppresses FFA-Challenged Lipid Accumulation in Alpha Mouse Liver 12 (AML12) Cells
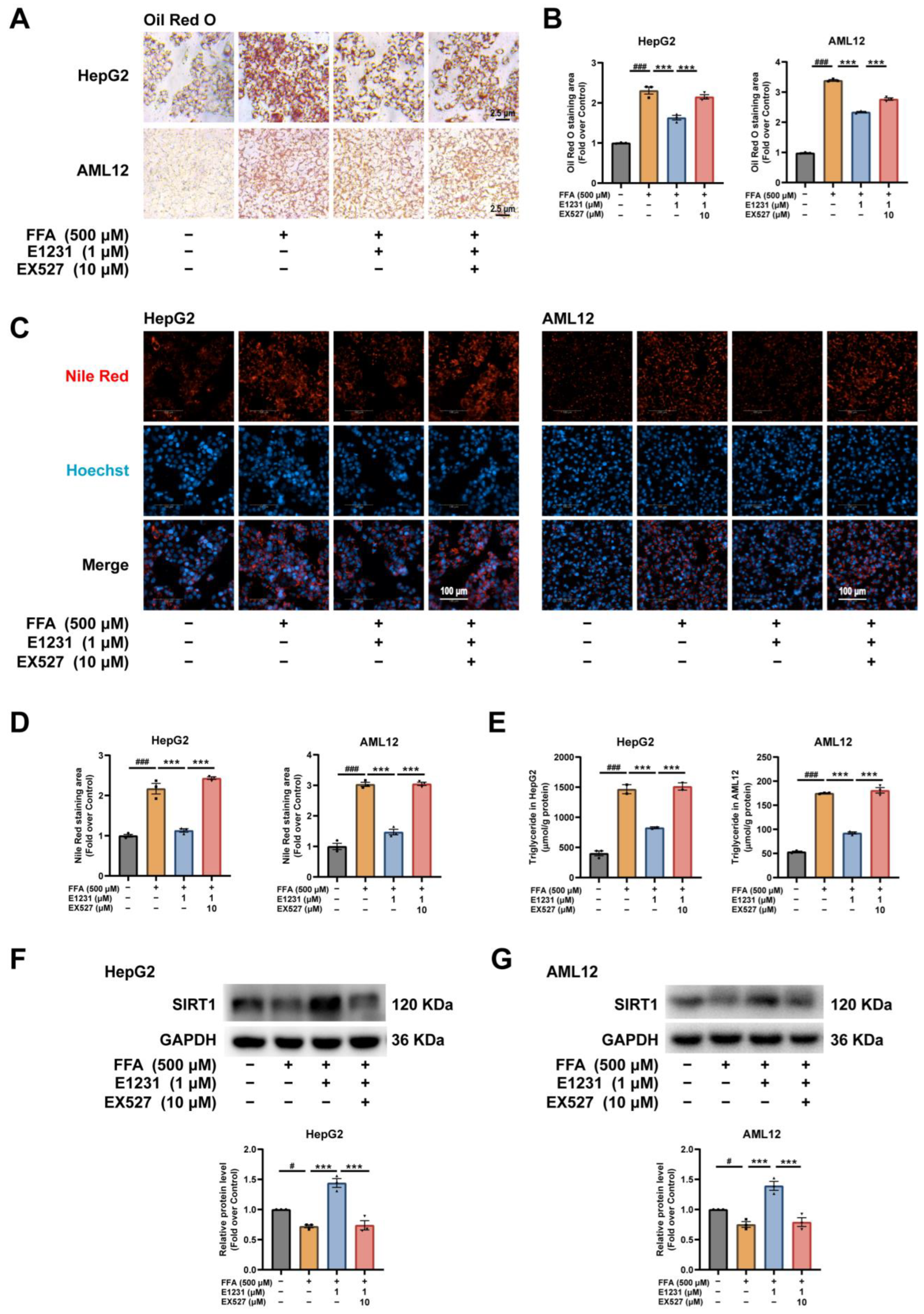
2.5. E1231 Represses Lipid Accumulation in FFA-Challenged Hepatocytes by Activating SIRT1
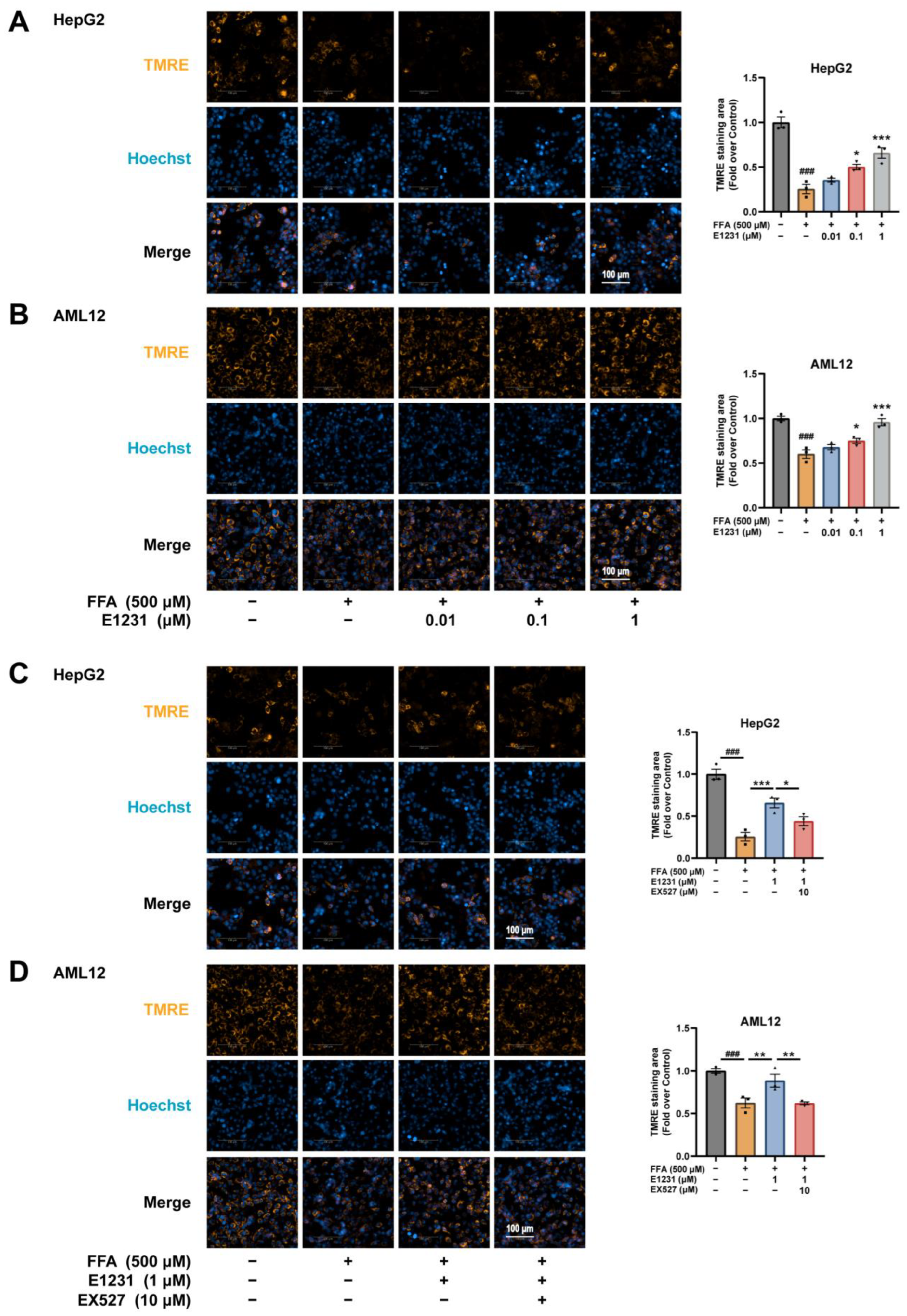
2.6. E1231 Improves Mitochondrial Function in FFA-Challenged Hepatocytes by Activating SIRT1
3. Discussion
4. Materials and Methods
4.1. Establishment of an HFHC-Induced Mouse Model of NAFLD
4.2. Plasma Assay and Hepatic Lipid Assay
4.3. Histological Analysis
4.4. Cell Culture
4.5. FFA-Challenged Lipid Accumulation Model in Hepatocytes
4.6. E1231 Treatment in FFA-Challenged Hepatocytes
4.7. Nile Red Fluorescent Staining
4.8. Tetramethylrhodamine Ethyl Ester (TMRE) Staining
4.9. Western Blot Analysis
4.10. RNA Isolation and Real–Time Quantitative PCR
4.11. Statistical Analysis
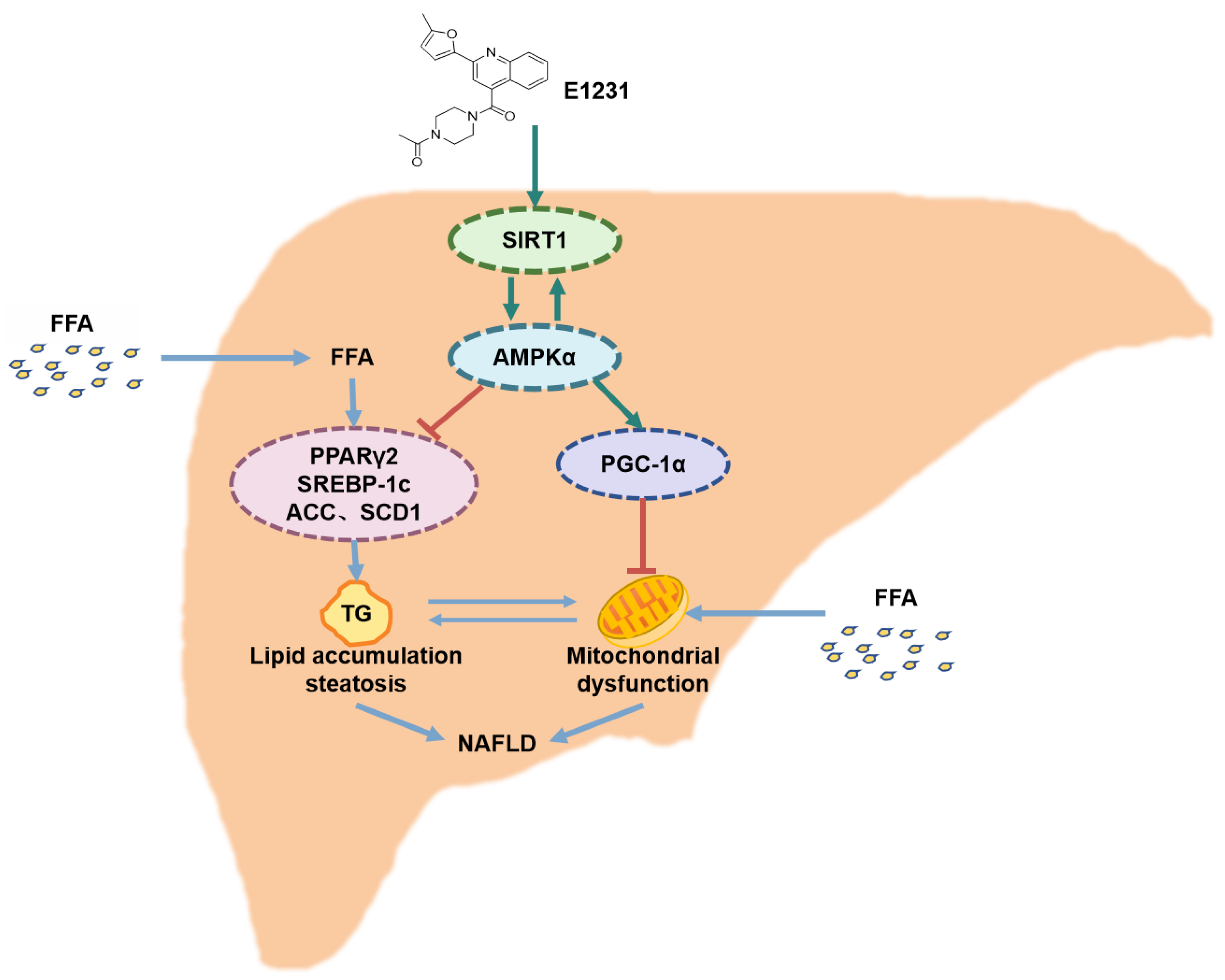
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eslam, M.; Sanyal, A.J.; George, J.; International Consensus Panel. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Powell, E.E.; Wong, V.W.; Rinella, M. Non-alcoholic fatty liver disease. Lancet (Lond. Engl.) 2021, 397, 2212–2224. [Google Scholar] [CrossRef]
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 2021, 184, 2537–2564. [Google Scholar] [CrossRef]
- Sheka, A.C.; Adeyi, O.; Thompson, J.; Hameed, B.; Crawford, P.A.; Ikramuddin, S. Nonalcoholic Steatohepatitis: A Review. JAMA 2020, 323, 1175–1183. [Google Scholar] [CrossRef]
- Khairnar, R.; Islam, M.A.; Fleishman, J.; Kumar, S. Shedding light on non-alcoholic fatty liver disease: Pathogenesis, molecular mechanisms, models, and emerging therapeutics. Life Sci. 2023, 312, 121185. [Google Scholar] [CrossRef]
- Geier, A.; Tiniakos, D.; Denk, H.; Trauner, M. From the origin of NASH to the future of metabolic fatty liver disease. Gut 2021, 70, 1570–1579. [Google Scholar] [CrossRef]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology (Baltim. Md.) 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Jaishy, B.; Abel, E.D. Lipids, lysosomes, and autophagy. J. Lipid Res. 2016, 57, 1619–1635. [Google Scholar] [CrossRef] [Green Version]
- Geng, Y.; Faber, K.N.; de Meijer, V.E.; Blokzijl, H.; Moshage, H. How does hepatic lipid accumulation lead to lipotoxicity in non-alcoholic fatty liver disease? Hepatol. Int. 2021, 15, 21–35. [Google Scholar] [CrossRef]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. CMLS 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.J.; Zhang, T.N.; Chen, H.H.; Yu, X.F.; Lv, J.L.; Liu, Y.Y.; Liu, Y.S.; Zheng, G.; Zhao, J.Q.; Wei, Y.F.; et al. The sirtuin family in health and disease. Signal Transduct. Target. Ther. 2022, 7, 402. [Google Scholar] [CrossRef]
- Ding, R.B.; Bao, J.; Deng, C.X. Emerging roles of SIRT1 in fatty liver diseases. Int. J. Biol. Sci. 2017, 13, 852–867. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F.; Ibdah, J.A. Sirtuins and nonalcoholic fatty liver disease. World J. Gastroenterol. 2016, 22, 10084–10092. [Google Scholar] [CrossRef] [PubMed]
- Mariani, S.; Fiore, D.; Basciani, S.; Persichetti, A.; Contini, S.; Lubrano, C.; Salvatori, L.; Lenzi, A.; Gnessi, L. Plasma levels of SIRT1 associate with non-alcoholic fatty liver disease in obese patients. Endocrine 2015, 49, 711–716. [Google Scholar] [CrossRef]
- Wu, T.; Liu, Y.H.; Fu, Y.C.; Liu, X.M.; Zhou, X.H. Direct evidence of sirtuin downregulation in the liver of non-alcoholic fatty liver disease patients. Ann. Clin. Lab. Sci. 2014, 44, 410–418. [Google Scholar]
- Purushotham, A.; Schug, T.T.; Xu, Q.; Surapureddi, S.; Guo, X.; Li, X. Hepatocyte-specific deletion of SIRT1 alters fatty acid metabolism and results in hepatic steatosis and inflammation. Cell Metab. 2009, 9, 327–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, X.; Xu, S.; Maitland-Toolan, K.A.; Sato, K.; Jiang, B.; Ido, Y.; Lan, F.; Walsh, K.; Wierzbicki, M.; Verbeuren, T.J.; et al. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. J. Biol. Chem. 2008, 283, 20015–20026. [Google Scholar] [CrossRef] [Green Version]
- Feige, J.N.; Lagouge, M.; Canto, C.; Strehle, A.; Houten, S.M.; Milne, J.C.; Lambert, P.D.; Mataki, C.; Elliott, P.J.; Auwerx, J. Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab. 2008, 8, 347–358. [Google Scholar] [CrossRef]
- Feng, T.; Liu, P.; Wang, X.; Luo, J.; Zuo, X.; Jiang, X.; Liu, C.; Li, Y.; Li, N.; Chen, M.; et al. SIRT1 activator E1231 protects from experimental atherosclerosis and lowers plasma cholesterol and triglycerides by enhancing ABCA1 expression. Atherosclerosis 2018, 274, 172–181. [Google Scholar] [CrossRef]
- Pouwels, S.; Sakran, N.; Graham, Y.; Leal, A.; Pintar, T.; Yang, W.; Kassir, R.; Singhal, R.; Mahawar, K.; Ramnarain, D. Non-alcoholic fatty liver disease (NAFLD): A review of pathophysiology, clinical management and effects of weight loss. BMC Endocr. Disord. 2022, 22, 63. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology (Baltim. Md.) 2005, 41, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Strable, M.S.; Ntambi, J.M. Genetic control of de novo lipogenesis: Role in diet-induced obesity. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 199–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruderman, N.B.; Xu, X.J.; Nelson, L.; Cacicedo, J.M.; Saha, A.K.; Lan, F.; Ido, Y. AMPK and SIRT1: A long-standing partnership? Am. J. Physiol. Endocrinol. Metab. 2010, 298, E751–E760. [Google Scholar] [CrossRef]
- Ferré, P.; Foufelle, F. Hepatic steatosis: A role for de novo lipogenesis and the transcription factor SREBP-1c. Diabetes Obes. Metab. 2010, 12 (Suppl. 2), 83–92. [Google Scholar] [CrossRef]
- Ponugoti, B.; Kim, D.H.; Xiao, Z.; Smith, Z.; Miao, J.; Zang, M.; Wu, S.Y.; Chiang, C.M.; Veenstra, T.D.; Kemper, J.K. SIRT1 deacetylates and inhibits SREBP-1C activity in regulation of hepatic lipid metabolism. J. Biol. Chem. 2010, 285, 33959–33970. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.F.; Ku, H.C.; Lin, H. PGC-1alpha as a Pivotal Factor in Lipid and Metabolic Regulation. Int. J. Mol. Sci. 2018, 19, 3447. [Google Scholar] [CrossRef] [Green Version]
- Dusabimana, T.; Park, E.J.; Je, J.; Jeong, K.; Yun, S.P.; Kim, H.J.; Kim, H.; Park, S.W. P2Y2R Deficiency Ameliorates Hepatic Steatosis by Reducing Lipogenesis and Enhancing Fatty Acid beta-Oxidation through AMPK and PGC-1alpha Induction in High-Fat Diet-Fed Mice. Int. J. Mol. Sci. 2021, 22, 5528. [Google Scholar] [CrossRef]
- Jiang, Y.; Chen, D.; Gong, Q.; Xu, Q.; Pan, D.; Lu, F.; Tang, Q. Elucidation of SIRT-1/PGC-1α-associated mitochondrial dysfunction and autophagy in nonalcoholic fatty liver disease. Lipids Health Dis. 2021, 20, 40. [Google Scholar] [CrossRef]
- Zeng, C.; Chen, M. Progress in Nonalcoholic Fatty Liver Disease: SIRT Family Regulates Mitochondrial Biogenesis. Biomolecules 2022, 12, 1079. [Google Scholar] [CrossRef]
- Yamazaki, T.; Shiraishi, S.; Kishimoto, K.; Miura, S.; Ezaki, O. An increase in liver PPARγ2 is an initial event to induce fatty liver in response to a diet high in butter: PPARγ2 knockdown improves fatty liver induced by high-saturated fat. J. Nutr. Biochem. 2011, 22, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Malhi, H.; Gores, G.J. Molecular mechanisms of lipotoxicity in nonalcoholic fatty liver disease. Semin. Liver Dis. 2008, 28, 360–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noureddin, M.; Sanyal, A.J. Pathogenesis of NASH: The Impact of Multiple Pathways. Curr. Hepatol. Rep. 2018, 17, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Li, X.X.; Lu, X.Y.; Zhang, S.J.; Chiu, A.P.; Lo, L.H.; Largaespada, D.A.; Chen, Q.B.; Keng, V.W. Sodium tanshinone IIA sulfonate ameliorates hepatic steatosis by inhibiting lipogenesis and inflammation. Biomed. Pharmacother. Biomed. Pharmacother. 2019, 111, 68–75. [Google Scholar] [CrossRef]
- Colak, Y.; Yesil, A.; Mutlu, H.H.; Caklili, O.T.; Ulasoglu, C.; Senates, E.; Takir, M.; Kostek, O.; Yilmaz, Y.; Yilmaz Enc, F.; et al. A potential treatment of non-alcoholic fatty liver disease with SIRT1 activators. J. Gastrointest. Liver Dis. JGLD 2014, 23, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Napper, A.D.; Hixon, J.; McDonagh, T.; Keavey, K.; Pons, J.F.; Barker, J.; Yau, W.T.; Amouzegh, P.; Flegg, A.; Hamelin, E.; et al. Discovery of indoles as potent and selective inhibitors of the deacetylase SIRT1. J. Med. Chem. 2005, 48, 8045–8054. [Google Scholar] [CrossRef]
- Broussy, S.; Laaroussi, H.; Vidal, M. Biochemical mechanism and biological effects of the inhibition of silent information regulator 1 (SIRT1) by EX-527 (SEN0014196 or selisistat). J. Enzym. Inhib. Med. Chem. 2020, 35, 1124–1136. [Google Scholar] [CrossRef]
- Li, Q.; Tan, J.X.; He, Y.; Bai, F.; Li, S.W.; Hou, Y.W.; Ji, L.S.; Gao, Y.T.; Zhang, X.; Zhou, Z.H.; et al. Atractylenolide III ameliorates Non-Alcoholic Fatty Liver Disease by activating Hepatic Adiponectin Receptor 1-Mediated AMPK Pathway. Int. J. Biol. Sci. 2022, 18, 1594–1611. [Google Scholar] [CrossRef]
- Arroyave-Ospina, J.C.; Wu, Z.; Geng, Y.; Moshage, H. Role of Oxidative Stress in the Pathogenesis of Non-Alcoholic Fatty Liver Disease: Implications for Prevention and Therapy. Antioxidants (Basel Switz.) 2021, 10, 174. [Google Scholar] [CrossRef]
- Jornayvaz, F.R.; Shulman, G.I. Regulation of mitochondrial biogenesis. Essays Biochem. 2010, 47, 69–84. [Google Scholar] [CrossRef] [Green Version]
- Montandon, S.A.; Somm, E.; Loizides-Mangold, U.; de Vito, C.; Dibner, C.; Jornayvaz, F.R. Multi-technique comparison of atherogenic and MCD NASH models highlights changes in sphingolipid metabolism. Sci. Rep. 2019, 9, 16810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimano, H.; Sato, R. SREBP-regulated lipid metabolism: Convergent physiology—Divergent pathophysiology. Nat. Rev. Endocrinol. 2017, 13, 710–730. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F.; Ibdah, J.A. Role of mitochondria in nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2014, 15, 8713–8742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Liu, M.; Yu, H.; Wang, W.; Han, L.; Chen, Q.; Ruan, J.; Wen, S.; Zhang, Y.; Wang, T. Mangiferin Improves Hepatic Lipid Metabolism Mainly Through Its Metabolite-Norathyriol by Modulating SIRT-1/AMPK/SREBP-1c Signaling. Front. Pharm. 2018, 9, 201. [Google Scholar] [CrossRef]
- Poornima, M.S.; Sindhu, G.; Billu, A.; Sruthi, C.R.; Nisha, P.; Gogoi, P.; Baishya, G.; Raghu, K.G. Pretreatment of hydroethanolic extract of Dillenia indica L. attenuates oleic acid induced NAFLD in HepG2 cells via modulating SIRT-1/p-LKB-1/AMPK, HMGCR & PPAR-α signaling pathways. J. Ethnopharmacol. 2022, 292, 115237. [Google Scholar] [CrossRef] [PubMed]
- Legaki, A.I.; Moustakas, I.I.; Sikorska, M.; Papadopoulos, G.; Velliou, R.I.; Chatzigeorgiou, A. Hepatocyte Mitochondrial Dynamics and Bioenergetics in Obesity-Related Non-Alcoholic Fatty Liver Disease. Curr. Obes. Rep. 2022, 11, 126–143. [Google Scholar] [CrossRef]
- Lee, E.; Korf, H.; Vidal-Puig, A. An adipocentric perspective on the development and progression of non-alcoholic fatty liver disease. J. Hepatol. 2023, 78, 1048–1062. [Google Scholar] [CrossRef]
- Majeed, Y.; Halabi, N.; Madani, A.Y.; Engelke, R.; Bhagwat, A.M.; Abdesselem, H.; Agha, M.V.; Vakayil, M.; Courjaret, R.; Goswami, N.; et al. SIRT1 promotes lipid metabolism and mitochondrial biogenesis in adipocytes and coordinates adipogenesis by targeting key enzymatic pathways. Sci. Rep. 2021, 11, 8177. [Google Scholar] [CrossRef]
- Giblin, W.; Skinner, M.E.; Lombard, D.B. Sirtuins: Guardians of mammalian healthspan. Trends Genet. 2014, 30, 271–286. [Google Scholar] [CrossRef] [Green Version]
- Baur, J.A.; Ungvari, Z.; Minor, R.K.; Le Couteur, D.G.; de Cabo, R. Are sirtuins viable targets for improving healthspan and lifespan? Nat. Rev. Drug Discov. 2012, 11, 443–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinclair, D.A.; Guarente, L. Small-molecule allosteric activators of sirtuins. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 363–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, Y.; Usui, I.; Kanatani, Y.; Matsuya, Y.; Tsuneyama, K.; Fujisaka, S.; Bukhari, A.; Suzuki, H.; Senda, S.; Imanishi, S.; et al. Treatment with SRT1720, a SIRT1 activator, ameliorates fatty liver with reduced expression of lipogenic enzymes in MSG mice. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1179–E1186. [Google Scholar] [CrossRef] [Green Version]
- Sodum, N.; Kumar, G.; Bojja, S.L.; Kumar, N.; Rao, C.M. Epigenetics in NAFLD/NASH: Targets and therapy. Pharmacol. Res. 2021, 167, 105484. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, S.; Mihaylova, M.M.; Zheng, B.; Hou, X.; Jiang, B.; Park, O.; Luo, Z.; Lefai, E.; Shyy, J.Y.; et al. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011, 13, 376–388. [Google Scholar] [CrossRef] [Green Version]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, J.; Li, S.; Wang, W.; Jiang, X.; Liu, C.; Lei, L.; Li, Y.; Sheng, R.; Zhang, Y.; Wu, Y.; et al. SIRT1 Activator E1231 Alleviates Nonalcoholic Fatty Liver Disease by Regulating Lipid Metabolism. Curr. Issues Mol. Biol. 2023, 45, 5052-5070. https://doi.org/10.3390/cimb45060321
Han J, Li S, Wang W, Jiang X, Liu C, Lei L, Li Y, Sheng R, Zhang Y, Wu Y, et al. SIRT1 Activator E1231 Alleviates Nonalcoholic Fatty Liver Disease by Regulating Lipid Metabolism. Current Issues in Molecular Biology. 2023; 45(6):5052-5070. https://doi.org/10.3390/cimb45060321
Chicago/Turabian StyleHan, Jiangxue, Shunwang Li, Weizhi Wang, Xinhai Jiang, Chao Liu, Lijuan Lei, Yining Li, Ren Sheng, Yuyan Zhang, Yexiang Wu, and et al. 2023. "SIRT1 Activator E1231 Alleviates Nonalcoholic Fatty Liver Disease by Regulating Lipid Metabolism" Current Issues in Molecular Biology 45, no. 6: 5052-5070. https://doi.org/10.3390/cimb45060321