


Natural Mitochondria Targeting Substances and Their Effect on Cellular Antioxidant System as a Potential Benefit in Mitochondrial Medicine for Prevention and Remediation of Mitochondrial Dysfunctions

{kind=link}
{kind=link}
Abstract
:1. Introduction
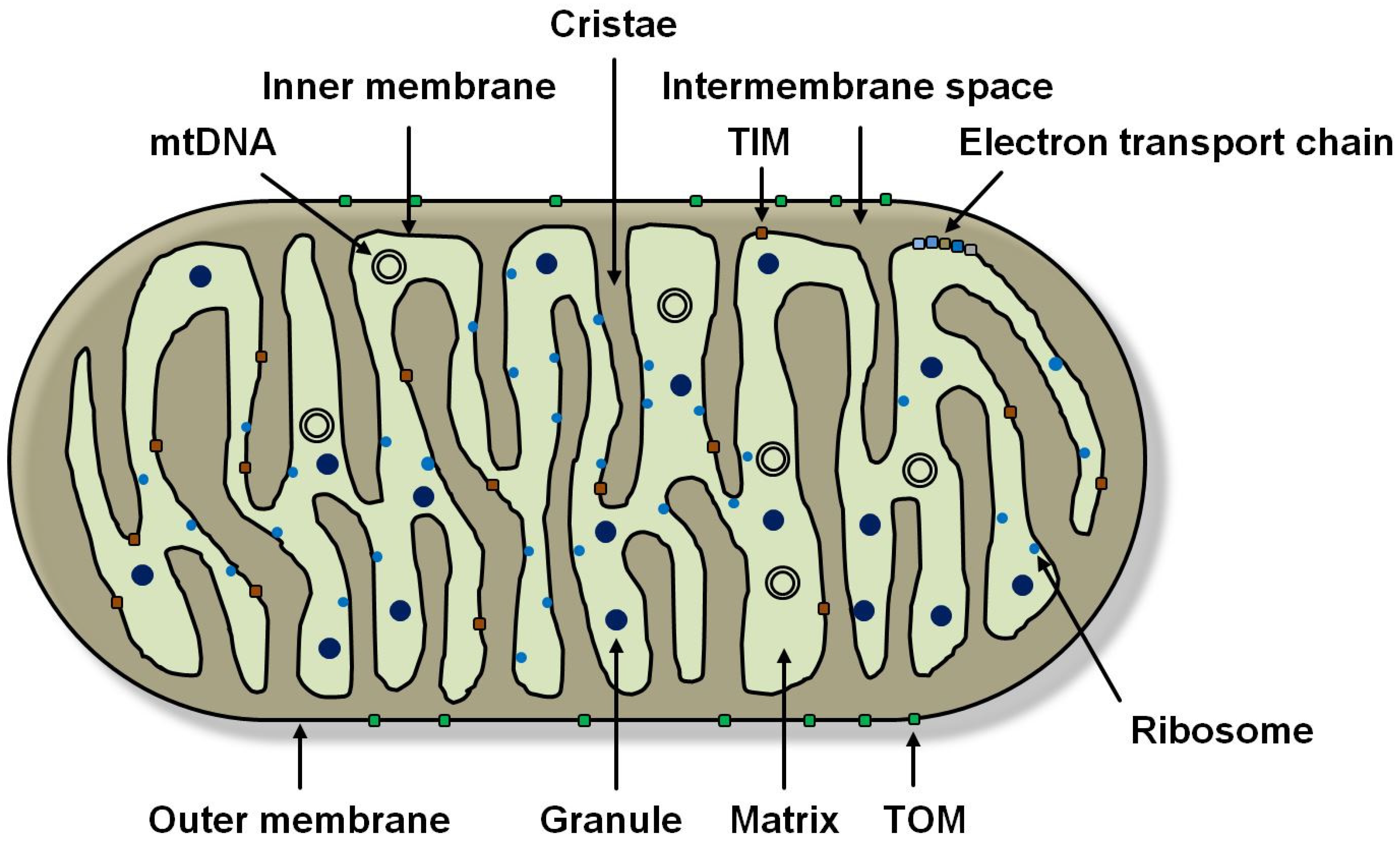
1.1. Mitochondrial Function and Genetics
1.2. The Respiratory Chain
1.3. Reactive Oxygen Species (ROS)
1.4. Mitochondria in the Process of Aging
2. Mitochondrial Dysfunctions—Clinical Relevance
3. Mitotropic Substances
3.1. Electron and Proton Carrier
3.1.1. Coenzyme Q10
- Molecular mechanism/Biological function—Pleiotropic effects of coenzyme Q10
- Studies on the efficacy of coenzyme Q10
3.1.2. NMN/NAD+/NADH
- Molecular mechanism/Biological function
- Studies on the efficacy of NMN/NAD+/NADH
3.2. Vitamins
- Molecular mechanism/Biological function
- Studies on the efficacy of Vitamins
3.3. Minerals
- Molecular mechanism/Biological function
- Studies on the efficacy of Minerals
3.4. Further Mitotropic Substances
3.4.1. Resveratrol
- Molecular mechanism/Biological function
- Studies on the efficacy of Resveratrol
3.4.2. Spermidine
- Molecular mechanism/Biological function
- Studies on the efficacy of Spermidine
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Klinke, R.; Pape, H.C.; Kurtz, A.; Silbernagl, S. Physiologie, 6th ed.; Georg Thieme Verlag: Stuttgart, Germany, 2009; ISBN 978-3137960065. [Google Scholar]
- Nordheim, A.; Knippers, R. Molekulare Genetik, 8th ed.; Georg Thieme Verlag: Stuttgart, Germany, 2018; ISBN 978-3132426375. [Google Scholar]
- Schatz, G. The Magic Garden. Annu. Rev. Biochem. 2007, 76, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Krasnikov, B.F.; Kuzminova, A.E.; Vysokikh, M.Y.; Zorova, L.D. Mitochondria Revisited. Alternative Functions of Mitochondria. Biosci. Rep. 1997, 17, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Alberts, B. Lehrbuch der Molekularen Zellbiologie, 5th ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2015; ISBN 978-3-527-32384-5. [Google Scholar]
- Madigan, M.T.; Martinko, J.M.; Stahl, D.A.; Clark, D.P. Brock Mikrobiologie, 13th ed.; Pearson: München, Germany, 2013; ISBN 978-3868941449. [Google Scholar]
- Adrain, C.; Martin, S.J. The mitochondrial apoptosome: A killer unleashed by the cytochrome seas. Trends Biochem. Sci. 2001, 26, 390–397. [Google Scholar] [CrossRef]
- Gvozdjáková, A. Mitochondrial Medicine: Mitochondrial Metabolism, Diseases, Diagnosis and Therapy, 2008th ed.; Springer: Berlin, Germany, 2008; ISBN 978-1402067136. [Google Scholar]
- Lill, R.; Diekert, K.; Kaut, A.; Lange, H.; Pelzer, W.; Prohl, C.; Kispal, G. The Essential Role of Mitochondria in the Biogenesis of Cellular Iron-Sulfur Proteins. Biol. Chem. 1999, 380, 1157–1166. [Google Scholar] [CrossRef]
- DiMauro, S.; Schon, E.A. Mitochondrial DNA mutations in human disease. Am. J. Med. Genet. 2001, 106, 18–26. [Google Scholar] [CrossRef]
- Al Amir Dache, Z.; Otandault, A.; Tanos, R.; Pastor, B.; Meddeb, R.; Sanchez, C.; Arena, G.; Lasorsa, L.; Bennett, A.; Grange, T.; et al. Blood contains circulating cell-free respiratory competent mitochondria. FASEB J. 2020, 34, 3616–3630. [Google Scholar] [CrossRef]
- Meinecke, M.; Wagner, R.; Kovermann, P.; Guiard, B.; Mick, D.U.; Hutu, D.P.; Voos, W.; Truscott, K.N.; Chacinska, A.; Pfanner, N.; et al. Tim50 Maintains the Permeability Barrier of the Mitochondrial Inner Membrane. Science 2006, 312, 1523–1526. [Google Scholar] [CrossRef]
- Nelson, D.; Cox, M. Lehninger Biochemie, 2011st ed.; Springer: Berlin, Germany, 2011; ISBN 978-3540686378. [Google Scholar]
- Nedergaard, J.; Ricquier, D.; Kozak, L.P. Uncoupling proteins: Current status and therapeutic prospects. EMBO Rep. 2005, 6, 917–921. [Google Scholar] [CrossRef]
- Turunen, M.; Olsson, J.; Dallner, G. Metabolism and function of coenzyme Q. Biochim. Biophys. Acta 2004, 1660, 171–199. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Lian, G. ROS and diseases: Role in metabolism and energy supply. Mol. Cell. Biochem. 2020, 467, 1–12, Erratum in Mol. Cell. Biochem. 2020, 467, 13. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjørås, M. Base Excision Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative Stress and Antioxidant Defense. World Allerg. Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.J.; Brand, M.D. Reactive Oxygen Species Production by Mitochondria. Methods Mol. Biol. 2009, 554, 165–181. [Google Scholar] [CrossRef] [PubMed]
- Bottje, W. Oxidative metabolism and efficiency: The delicate balancing act of mitochondria. Poult. Sci. 2019, 98, 4223–4230. [Google Scholar] [CrossRef]
- Hunte, C.; Zickermann, V.; Brandt, U. Functional Modules and Structural Basis of Conformational Coupling in Mitochondrial Complex I. Science 2010, 329, 448–451. [Google Scholar] [CrossRef] [PubMed]
- Wirth, C.J.; Zichner, L. Orthopädie und Orthopädische Chirurgie: Stoffwechel-und Systemerkrankungen, 1st ed.; Georg Thieme Verlag: Stuttgart, Germany, 2003; ISBN 9783131269317. [Google Scholar]
- Furda, A.M.; Marrangoni, A.M.; Lokshin, A.; Van Houten, B. Oxidants and not alkylating agents induce rapid mtDNA loss and mitochondrial dysfunction. DNA Repair 2012, 11, 684–692. [Google Scholar] [CrossRef]
- Yoshida, T.; Goto, S.; Kawakatsu, M.; Urata, Y.; Li, T.-S. Mitochondrial dysfunction, a probable cause of persistent oxidative stress after exposure to ionizing radiation. Free. Radic. Res. 2012, 46, 147–153. [Google Scholar] [CrossRef]
- Chinnery, P.F.; Elliott, H.R.; Hudson, G.; Samuels, D.C.; Relton, C.L. Epigenetics, epidemiology and mitochondrial DNA diseases. Int. J. Epidemiol. 2012, 41, 177–187. [Google Scholar] [CrossRef]
- DeBalsi, K.L.; Hoff, K.E.; Copeland, W.C. Role of the mitochondrial DNA replication machinery in mitochondrial DNA mutagenesis, aging and age-related diseases. Ageing Res. Rev. 2017, 33, 89–104. [Google Scholar] [CrossRef]
- Schniertshauer, D.; Gebhard, D.; Bergemann, J. Einfluss von Solarer Strahlung auf die Mitochondrien. OM & Ernährung. 2018, SH08, pp. 50–57. Available online: https://www.omundernaehrung.com/einfluss-von-solarer-strahlung-auf-die-mitochondrien.html (accessed on 19 October 2022).
- Lagouge, M.; Larsson, N. The role of mitochondrial DNA mutations and free radicals in disease and ageing. J. Intern. Med. 2013, 273, 529–543. [Google Scholar] [CrossRef]
- Grivennikova, V.G.; Vinogradov, A.D. Mitochondrial production of reactive oxygen species. Biochemisry 2013, 78, 1490–1511. [Google Scholar] [CrossRef]
- Richter, C.; Park, J.W.; Ames, B.N. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc. Natl. Acad. Sci. USA 1988, 85, 6465–6467. [Google Scholar] [CrossRef]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. The Biologic Clock: The Mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Prinzinger, R. Programmed ageing: The theory of maximal metabolic scope. How does the biological clock tick? EMBO Rep. 2005, 6 (Suppl. 1), S14–S19. [Google Scholar] [CrossRef]
- Krutmann, J.; Schroeder, P. Role of Mitochondria in Photoaging of Human Skin: The Defective Powerhouse Model. J. Investig. Dermatol. Symp. Proc. 2009, 14, 44–49. [Google Scholar] [CrossRef]
- Damian, M.S.; Ellenberg, D.; Gildemeister, R.; Lauermann, J.; Simonis, G.; Sauter, W.; Georgi, C. Coenzyme Q10 Combined With Mild Hypothermia After Cardiac Arrest: A preliminary study. Circulation 2004, 110, 3011–3016. [Google Scholar] [CrossRef] [PubMed]
- Hazane, F.; Sauvaigo, S.; Douki, T.; Favier, A.; Beani, J.-C. Age-dependent DNA repair and cell cycle distribution of human skin fibroblasts in response to UVA irradiation. J. Photochem. Photobiol. B 2006, 82, 214–223. [Google Scholar] [CrossRef]
- Prahl, S.; Kueper, T.; Biernoth, T.; Wöhrmann, Y.; Münster, A.; Fürstenau, M.; Schmidt, M.; Schulze, C.; Wittern, K.-P.; Wenck, H.; et al. Aging skin is functionally anaerobic: Importance of coenzyme Q10 for anti aging skin care. Biofactors 2008, 32, 245–255. [Google Scholar] [CrossRef]
- Sauvaigo, S.; Bonnet-Duquennoy, M.; Odin, F.; Hazane-Puch, F.; Lachmann, N.; Bonté, F.; Kurfürst, R.; Favier, A. DNA repair capacities of cutaneous fibroblasts: Effect of sun exposure, age and smoking on response to an acute oxidative stress. Br. J. Dermatol. 2007, 157, 26–32. [Google Scholar] [CrossRef]
- Wei, Y.-H.; Wu, S.-B.; Ma, Y.-S.; Lee, H.-C. Respiratory function decline and DNA mutation in mitochondria, oxidative stress and altered gene expression during aging. Chang Gung Med. J. 2009, 32, 113–132. [Google Scholar] [PubMed]
- Schniertshauer, D.; Gebhard, D.; Bergemann, J. Age-Dependent Loss of Mitochondrial Function in Epithelial Tissue Can Be Reversed by Coenzyme Q10. J. Aging Res. 2018, 2018, 6354680. [Google Scholar] [CrossRef]
- Bentinger, M.; Tekle, M.; Dallner, G. Coenzyme Q—Biosynthesis and functions. Biochem. Biophys. Res. Commun. 2010, 396, 74–79. [Google Scholar] [CrossRef]
- Holt, J.I.; Harding, A.E.; Morgan-Hughes, J.A. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 1988, 331, 717–719. [Google Scholar] [CrossRef]
- Holt, I.J.; Harding, E.A.; Petty, R.K.; Morgan-Hughes, A.J. A new mitochondrial disease associated with mitochondrial DNA heteroplasmy. Am. J. Hum. Genet. 1990, 46, 428–433. [Google Scholar]
- Macmillan, C.; Lach, B.; Shoubridge, E.A. Variable distribution of mutant mitochondria1 DNAs (tRNALeu[3243]) in tissues of symptomatic relatives with MELAS: The role of mitotic segregation. Neurology 1993, 43, 1586. [Google Scholar] [CrossRef]
- Koopman, W.J.; Willems, P.H.; Smeitink, J.A. Monogenic Mitochondrial Disorders. N. Engl. J. Med. 2012, 366, 1132–1141. [Google Scholar] [CrossRef] [PubMed]
- Bogan, K.L.; Brenner, C. Nicotinic Acid, Nicotinamide, and Nicotinamide Riboside: A Molecular Evaluation of NAD+ Precursor Vitamins in Human Nutrition. Annu. Rev. Nutr. 2008, 28, 115–130. [Google Scholar] [CrossRef]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Gopalan, V.; Holland, O.; Neuzil, J. Mitocans Revisited: Mitochondrial Targeting as Efficient Anti-cancer Therapy. Int. J. Mol. Sci. 2020, 21, 7941. [Google Scholar] [CrossRef] [PubMed]
- Crane, F.L. Biochemical Functions of Coenzyme Q10. J. Am. Coll. Nutr. 2001, 20, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P. Protonmotive redox mechanism of the cytochrome b-c 1 complex in the respiratory chain: Protonmotive ubiquinone cycle. FEBS Lett. 1975, 56, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lenaz, G.; Genova, M.L. Mobility and function of Coenzyme Q (ubiquinone) in the mitochondrial respiratory chain. Biochim. Biophys. Acta 2009, 1787, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Bentinger, M.; Brismar, K.; Dallner, G. The antioxidant role of coenzyme Q. Mitochondrion 2007, 7, S41–S50. [Google Scholar] [CrossRef]
- Tomasetti, M.; Alleva, R.; Collins, A.R. In vivo supplementation with coenzyme Q10 enhances the recovery of human lymphocytes from oxidative DNA damage. FASEB J. 2001, 15, 1425–1427. [Google Scholar] [CrossRef]
- Evans, M.; Baisley, J.; Barss, S.; Guthrie, N. A randomized, double-blind trial on the bioavailability of two CoQ10 formulations. J. Funct. Foods 2009, 1, 65–73. [Google Scholar] [CrossRef]
- Cleren, C.; Yang, L.; Lorenzo, B.; Calingasan, N.Y.; Schomer, A.; Sireci, A.; Wille, E.J.; Beal, M.F. Therapeutic effects of coenzyme Q10 (CoQ10) and reduced CoQ10 in the MPTP model of Parkinsonism. J. Neurochem. 2008, 104, 1613–1621. [Google Scholar] [CrossRef]
- Sawaddiruk, P.; Apaijai, N.; Paiboonworachat, S.; Kaewchur, T.; Kasitanon, N.; Jaiwongkam, T.; Kerdphoo, S.; Chattipakorn, N.; Chattipakorn, S.C. Coenzyme Q10 supplementation alleviates pain in pregabalin-treated fibromyalgia patients via reducing brain activity and mitochondrial dysfunction. Free. Radic. Res. 2019, 53, 901–909. [Google Scholar] [CrossRef]
- Sharp, J.; Farha, S.; Park, M.M.; Comhair, S.A.; Lundgrin, E.L.; Tang, W.W.; Bongard, R.D.; Merker, M.P.; Erzurum, S.C. Coenzyme Q supplementation in pulmonary arterial hypertension. Redox Biol. 2014, 2, 884–891. [Google Scholar] [CrossRef]
- Estornell, E.; Fato, R.; Castelluccio, C.; Cavazzoni, M.; Castelli, G.; Lenaz, G. Saturation kinetics of coenzyme Q in NADH and succinate oxidation in beef heart mitochondria. FEBS Lett. 1992, 311, 107–109. [Google Scholar] [CrossRef]
- Littarru, G.P.; Tiano, L. Bioenergetic and Antioxidant Properties of Coenzyme Q10: Recent Developments. Mol. Biotechnol. 2007, 37, 31–37. [Google Scholar] [CrossRef]
- Schniertshauer, D.; Müller, S.; Mayr, T.; Sonntag, T.; Gebhard, D.; Bergemann, J. Accelerated Regeneration of ATP Level after Irradiation in Human Skin Fibroblasts by Coenzyme Q10. Photochem. Photobiol. 2016, 92, 488–494. [Google Scholar] [CrossRef]
- Schniertshauer, D.; Gebhard, D.; van Beek, H.; Nöth, V.; Schon, J.; Bergemann, J. The activity of the DNA repair enzyme hOGG1 can be directly modulated by ubiquinol. DNA Repair 2020, 87, 102784. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.-H.; Lee, Y.-L.; Chen, S.-F.; Lee, Y.-P.; Hsieh, Y.-H.; Tsai, J.-H.; Hsu, J.-L.; Tian, W.-T.; Huang, W. Essential role of β-human 8-oxoguanine DNA glycosylase 1 in mitochondrial oxidative DNA repair. Environ. Mol. Mutagen. 2013, 54, 54–64. [Google Scholar] [CrossRef]
- Edeas, M. Strategies to Target Mitochondria and Oxidative Stress by Antioxidants: Key Points and Perspectives. Pharm. Res. 2011, 28, 2771–2779. [Google Scholar] [CrossRef] [PubMed]
- Perumal, S.S.; Shanthi, P.; Sachdanandam, P. Augmented efficacy of tamoxifen in rat breast tumorigenesis when gavaged along with riboflavin, niacin, and CoQ10: Effects on lipid peroxidation and antioxidants in mitochondria. Chem. Biol. Interact. 2005, 152, 49–58. [Google Scholar] [CrossRef]
- Echtay, K.S.; Winkler, E.; Klingenberg, M. Coenzyme Q is an obligatory cofactor for uncoupling protein function. Nature 2000, 408, 609–613. [Google Scholar] [CrossRef]
- Echtay, K.S.; Winkler, E.; Frischmuth, K.; Klingenberg, M. Uncoupling proteins 2 and 3 are highly active H+ transporters and highly nucleotide sensitive when activated by coenzyme Q (ubiquinone). Proc. Natl. Acad. Sci. USA 2001, 98, 1416–1421. [Google Scholar] [CrossRef] [PubMed]
- Pu, Q.; Guo, X.-X.; Hu, J.-J.; Li, A.-L.; Li, G.-G.; Li, X.-Y. Nicotinamide mononucleotide increases cell viability and restores tight junctions in high-glucose-treated human corneal epithelial cells via the SIRT1/Nrf2/HO-1 pathway. Biomed. Pharmacother. 2022, 147, 112659. [Google Scholar] [CrossRef]
- Mills, K.F.; Yoshida, S.; Stein, L.R.; Grozio, A.; Kubota, S.; Sasaki, Y.; Redpath, P.; Migaud, M.E.; Apte, R.S.; Uchida, K.; et al. Long-Term Administration of Nicotinamide Mononucleotide Mitigates Age-Associated Physiological Decline in Mice. Cell Metab. 2016, 24, 795–806. [Google Scholar] [CrossRef]
- Hou, Y.; Lautrup, S.; Cordonnier, S.; Wang, Y.; Croteau, D.L.; Zavala, E.; Zhang, Y.; Moritoh, K.; O’connell, J.F.; Baptiste, B.A.; et al. NAD+ supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl. Acad. Sci. USA 2018, 115, E1876–E1885. [Google Scholar] [CrossRef]
- Brazill, J.M.; Li, C.; Zhu, Y.; Zhai, R.G. NMNAT: It’s an NAD+ synthase… It’s a chaperone… It’s a neuroprotector. Curr. Opin. Genet. Dev. 2017, 44, 156–162. [Google Scholar] [CrossRef]
- Imai, S.-I.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef]
- Strømland, Ø.; Diab, J.; Ferrario, E.; Sverkeli, L.J.; Ziegler, M. The balance between NAD+ biosynthesis and consumption in ageing. Mech. Ageing Dev. 2021, 199, 111569. [Google Scholar] [CrossRef]
- Zhou, B.; Wang, D.D.-H.; Qiu, Y.; Airhart, S.; Liu, Y.; Stempien-Otero, A.; O’brien, K.D.; Tian, R. Boosting NAD level suppresses inflammatory activation of PBMCs in heart failure. J. Clin. Investig. 2020, 130, 6054–6063. [Google Scholar] [CrossRef] [PubMed]
- Enzmann, F. Das MitoMed-Konzept—Mse Pharmazeutika GmbH. Available online: https://www.mse-pharma.de/mitomed-konzept/ (accessed on 8 January 2023).
- Mora, J.R.; Iwata, M.; von Andrian, U.H. Vitamin effects on the immune system: Vitamins A and D take centre stage. Nat. Rev. Immunol. 2008, 8, 685–698. [Google Scholar] [CrossRef]
- Thieme Verlag. Available online: https://viamedici.thieme.de/lernmodul/548911/538843/vitamine+und+vitaminosen+grundlagen (accessed on 22 December 2022).
- Vitamin E: Physiologie, Funktionen, Vorkommen, Referenzwerte und Versorgung in Deutschland. Available online: https://www.ernaehrungs-umschau.de/fileadmin/Ernaehrungs-Umschau/pdfs/pdf_2010/11_10/EU11_2010_608_615.qxd.pdf (accessed on 14 December 2022).
- Pietrzik, K.; Golly, I.; Loew, D. Handbuch Vitamine: Für Prophylaxe, Therapie und Beratung, 1st ed.; Urban&Fischer, Elsevier: München, Germany, 2008; ISBN 9783437553615. [Google Scholar]
- EFSA Panel on Dietetic Products, Nutrition, and Allergies (NDA). Scientific Opinion on Dietary Reference Values for vitamin E as α-tocopherol. EFSA J. 2015, 13, 4149–4221. [Google Scholar] [CrossRef]
- Napolitano, G.; Fasciolo, G.; Di Meo, S.; Venditti, P. Vitamin E Supplementation and Mitochondria in Experimental and Functional Hyperthyroidism: A Mini-Review. Nutrients 2019, 11, 2900. [Google Scholar] [CrossRef] [PubMed]
- Majima, H.J.; Indo, H.P.; Suenaga, S.; Matsui, H.; Yen, H.-C.; Ozawa, T. Mitochondria as Possible Pharmaceutical Targets for the Effects of Vitamin E and its Homologues in Oxidative Stress-Related Diseases. Curr. Pharm. Des. 2011, 17, 2190–2195. [Google Scholar] [CrossRef]
- Fiorillo, M.; Tóth, F.; Sotgia, F.; Lisanti, M.P. Doxycycline, Azithromycin and Vitamin C (DAV): A potent combination therapy for targeting mitochondria and eradicating cancer stem cells (CSCs). Aging 2019, 11, 2202–2216. [Google Scholar] [CrossRef] [PubMed]
- Nan, Y.; Lin, J.; Cui, Y.; Yao, J.; Yang, Y.; Li, Q. Protective role of vitamin B6 against mitochondria damage in Drosophila models of SCA. Neurochem. Int. 2021, 144, 104979. [Google Scholar] [CrossRef]
- Zoroddu, M.A.; Aaseth, J.; Crisponi, G.; Medici, S.; Peana, M.; Nurchi, V.M. The essential metals for humans: A brief overview. J. Inorg. Biochem. 2019, 195, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Rehner, G.; Daniel, H. Biochemie Der Ernährung, 3rd ed.; Springer: Berlin, Germany, 2010; ISBN 9783827422170. [Google Scholar]
- Boyle, N.B.; Lawton, C.; Dye, L. The Effects of Magnesium Supplementation on Subjective Anxiety and Stress—A Systematic Review. Nutrients 2017, 9, 429. [Google Scholar] [CrossRef]
- Li, F.-Y.; Chaigne-Delalande, B.; Kanellopoulou, C.; Davis, J.C.; Matthews, H.F.; Douek, D.C.; Cohen, J.I.; Uzel, G.; Su, H.C.; Lenardo, M.J. Second messenger role for Mg2+ revealed by human T-cell immunodeficiency. Nature 2011, 475, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Pilchova, I.; Klacanova, K.; Tatarkova, Z.; Kaplan, P.; Racay, P. The Involvement of Mg2+ in Regulation of Cellular and Mitochondrial Functions. Oxid. Med. Cell. Longev. 2017, 2017, 6797460. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Dudley, S.C., Jr. Magnesium, Oxidative Stress, Inflammation, and Cardiovascular Disease. Antioxidants 2020, 9, 907. [Google Scholar] [CrossRef] [PubMed]
- Barbagallo, M.; Veronese, N.; Dominguez, L.J. Magnesium in Aging, Health and Diseases. Nutrients 2021, 13, 463. [Google Scholar] [CrossRef]
- Resveratrol—Schlüssel für ein Langes Leben. Available online: https://www.pharmazeutische-zeitung.de/ausgabe-292007/schluessel-fuer-ein-langes-leben/ (accessed on 4 January 2023).
- Zhou, D.-D.; Luo, M.; Huang, S.-Y.; Saimaiti, A.; Shang, A.; Gan, R.-Y.; Li, H.-B. Effects and Mechanisms of Resveratrol on Aging and Age-Related Diseases. Oxid. Med. Cell. Longev. 2021, 2021, 9932218. [Google Scholar] [CrossRef]
- Ingólfsson, H.I.; Thakur, P.; Herold, K.F.; Hobart, E.A.; Ramsey, N.B.; Periole, X.; de Jong, D.H.; Zwama, M.; Yilmaz, D.; Hall, K.; et al. Phytochemicals Perturb Membranes and Promiscuously Alter Protein Function. ACS Chem. Biol. 2014, 9, 1788–1798. [Google Scholar] [CrossRef]
- Leonard, S.S.; Xia, C.; Jiang, B.-H.; Stinefelt, B.; Klandorf, H.; Harris, G.K.; Shi, X. Resveratrol scavenges reactive oxygen species and effects radical-induced cellular responses. Biochem. Biophys. Res. Commun. 2003, 309, 1017–1026. [Google Scholar] [CrossRef]
- Martinez, J.; Moreno, J.J. Effect of resveratrol, a natural polyphenolic compound, on reactive oxygen species and prostaglandin production. Biochem. Pharmacol. 2000, 59, 865–870. [Google Scholar] [CrossRef] [PubMed]
- Matt, K.; Hochecker, B.; Schöller-Mann, A.; Bergemann, J. mRNA expression of ageing-associated genes in calorie reduction is subject to donor variability and can be induced by calorie restriction mimetics. Nutr. Health 2020, 26, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Lagouge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P.; et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1α. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef]
- Lu, Z.; Xu, X.; Hu, X.; Fassett, J.; Zhu, G.; Tao, Y.; Li, J.; Huang, Y.; Zhang, P.; Zhao, B.; et al. PGC-1α Regulates Expression of Myocardial Mitochondrial Antioxidants and Myocardial Oxidative Stress After Chronic Systolic Overload. Antioxid. Redox Signal. 2010, 13, 1011–1022. [Google Scholar] [CrossRef]
- Nijland, P.G.; Witte, E.M.; Hof, B.V.H.; Van Der Pol, S.; Bauer, J.S.; Lassmann, H.; Van Der Valk, P.; De Vries, E.H.; Van Horssen, J. Astroglial PGC-1alpha increases mitochondrial antioxidant capacity and suppresses inflammation: Implications for multiple sclerosis. Acta Neuropathol. Commun. 2014, 2, 170. [Google Scholar] [CrossRef]
- Supinski, G.S.; Schroder, E.A.; Callahan, L.A. Mitochondria and Critical Illness. Chest 2020, 157, 310–322. [Google Scholar] [CrossRef]
- Jardim, F.R.; De Rossi, F.T.; Nascimento, M.X.; Barros, R.G.d.S.; Borges, P.A.; Prescilio, I.C.; De Oliveira, M.R. Resveratrol and Brain Mitochondria: A Review. Mol. Neurobiol. 2018, 55, 2085–2101. [Google Scholar] [CrossRef]
- Sheu, S.-J.; Liu, N.-C.; Ou, C.-C.; Bee, Y.-S.; Chen, S.-C.; Lin, H.-C.; Chan, J.Y.H. Resveratrol Stimulates Mitochondrial Bioenergetics to Protect Retinal Pigment Epithelial Cells From Oxidative Damage. Investig. Opthalmol. Vis. Sci. 2013, 54, 6426–6438. [Google Scholar] [CrossRef]
- Miao, L.; St Clair, D.K. Regulation of superoxide dismutase genes: Implications in disease. Free Radic. Biol. Med. 2009, 47, 344–356. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Mao, P.; Calkins, M.J.; Cornea, A.; Reddy, A.P.; Murphy, M.P.; Szeto, H.H.; Park, B.; Reddy, P.H. Mitochondria-Targeted Antioxidants Protect Against Amyloid-β Toxicity in Alzheimer’s Disease Neurons. J. Alzheimer’s Dis. 2010, 20 (Suppl. 2), S609–S631. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Tao, Y.; Zhang, J.; Wang, J.; Ye, F.; Dan, G.; Zhao, Y.; Cai, Y.; Zhao, J.; Wu, Q.; et al. Resveratrol Regulates Mitochondrial Biogenesis and Fission/Fusion to Attenuate Rotenone-Induced Neurotoxicity. Oxid. Med. Cell. Longev. 2016, 2016, 6705621. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, M.R.; Nabavi, S.F.; Manayi, A.; Daglia, M.; Hajheidari, Z.; Nabavi, S.M. Resveratrol and the mitochondria: From triggering the intrinsic apoptotic pathway to inducing mitochondrial biogenesis, a mechanistic view. Biochim. Biophys. Acta 2016, 1860, 727–745. [Google Scholar] [CrossRef] [PubMed]
- Roggerio, A.; Strunz, C.M.C.; Pacanaro, A.P.; Leal, D.P.; Takada, J.Y.; Avakian, S.D.; Mansur, A.D.P. Gene Expression of Sirtuin-1 and Endogenous Secretory Receptor for Advanced Glycation End Products in Healthy and Slightly Overweight Subjects after Caloric Restriction and Resveratrol Administration. Nutrients 2018, 10, 937. [Google Scholar] [CrossRef]
- Bass, T.M.; Weinkove, D.; Houthoofd, K.; Gems, D.; Partridge, L. Effects of resveratrol on lifespan in Drosophila melanogaster and Caenorhabditis elegans. Mech. Ageing Dev. 2007, 128, 546–552. [Google Scholar] [CrossRef]
- Hofer, S.J.; Liang, Y.; Zimmermann, A.; Schroeder, S.; Dengjel, J.; Kroemer, G.; Eisenberg, T.; Sigrist, S.J.; Madeo, F. Spermidine-induced hypusination preserves mitochondrial and cognitive function during aging. Autophagy 2021, 17, 2037–2039. [Google Scholar] [CrossRef]
- Partridge, L.; Fuentealba, M.; Kennedy, B.K. The quest to slow ageing through drug discovery. Nat. Rev. Drug Discov. 2020, 19, 513–532. [Google Scholar] [CrossRef]
- Ren, J.; Zhang, Y. Targeting Autophagy in Aging and Aging-Related Cardiovascular Diseases. Trends Pharmacol. Sci. 2018, 39, 1064–1076. [Google Scholar] [CrossRef]
- Wang, J.; Li, S.; Wang, J.; Wu, F.; Chen, Y.; Zhang, H.; Guo, Y.; Lin, Y.; Li, L.; Yu, X.; et al. Spermidine alleviates cardiac aging by improving mitochondrial biogenesis and function. Aging 2020, 12, 650–671. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, X.; Ma, H.; Yang, Q.; Shang, Q.; Song, L.; Zheng, Z.; Zhang, S.; Pan, Y.; Huang, P.; et al. Spermidine endows macrophages anti-inflammatory properties by inducing mitochondrial superoxide-dependent AMPK activation, Hif-1α upregulation and autophagy. Free. Radic. Biol. Med. 2020, 161, 339–350. [Google Scholar] [CrossRef]
- Liu, X.; Li, D.; Liu, Z.; Song, Y.; Zhang, B.; Zang, Y.; Zhang, W.; Niu, Y.; Shen, C. Nicotinamide mononucleotide promotes pancreatic islet function through the SIRT1 pathway in mice after severe burns. Burns 2022, 48, 1922–1932. [Google Scholar] [CrossRef] [PubMed]
- Madeo, F.; Eisenberg, T.; Pietrocola, F.; Kroemer, G. Spermidine in health and disease. Science 2018, 359, eaan2788. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.-T.; Li, H.; Dai, Z.; Lau, G.K.; Li, B.-Y.; Zhu, W.-L.; Liu, X.-Q.; Liu, H.-F.; Cai, W.-W.; Huang, S.-Q.; et al. Spermidine and spermine delay brain aging by inducing autophagy in SAMP8 mice. Aging 2020, 12, 6401–6414. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, T.; Abdellatif, M.; Schroeder, S.; Primessnig, U.; Stekovic, S.; Pendl, T.; Harger, A.; Schipke, J.; Zimmermann, A.; Schmidt, A.; et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat. Med. 2016, 22, 1428–1438. [Google Scholar] [CrossRef]
- Gabandé-Rodríguez, E.; Gomez de Las Heras, M.M.; Mittelbrunn, M. Control of Inflammation by Calorie Restriction Mimetics: On the Crossroad of Autophagy and Mitochondria. Cells 2019, 9, 82. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schniertshauer, D.; Wespel, S.; Bergemann, J. Natural Mitochondria Targeting Substances and Their Effect on Cellular Antioxidant System as a Potential Benefit in Mitochondrial Medicine for Prevention and Remediation of Mitochondrial Dysfunctions. Curr. Issues Mol. Biol. 2023, 45, 3911-3932. https://doi.org/10.3390/cimb45050250
Schniertshauer D, Wespel S, Bergemann J. Natural Mitochondria Targeting Substances and Their Effect on Cellular Antioxidant System as a Potential Benefit in Mitochondrial Medicine for Prevention and Remediation of Mitochondrial Dysfunctions. Current Issues in Molecular Biology. 2023; 45(5):3911-3932. https://doi.org/10.3390/cimb45050250
Chicago/Turabian StyleSchniertshauer, Daniel, Susanne Wespel, and Jörg Bergemann. 2023. "Natural Mitochondria Targeting Substances and Their Effect on Cellular Antioxidant System as a Potential Benefit in Mitochondrial Medicine for Prevention and Remediation of Mitochondrial Dysfunctions" Current Issues in Molecular Biology 45, no. 5: 3911-3932. https://doi.org/10.3390/cimb45050250