
A Feedback Loop between TGF-β1 and ATG5 Mediated by miR-122-5p Regulates Fibrosis and EMT in Human Trabecular Meshwork Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. siRNA Transfection
2.2. Stimulation of HTM Cells with TGF-β1
2.3. RNA Isolation and Quantitative Polymerase Chain Reaction (qPCR)
2.4. Annexin V/propidium Iodide (PI) Staining Assay
2.5. Protein Whole Cell Lysate and Immunoblotting
2.6. Electron Micrograph and Immunofluorescence Staining
2.7. Statistical Analysis
3. Results
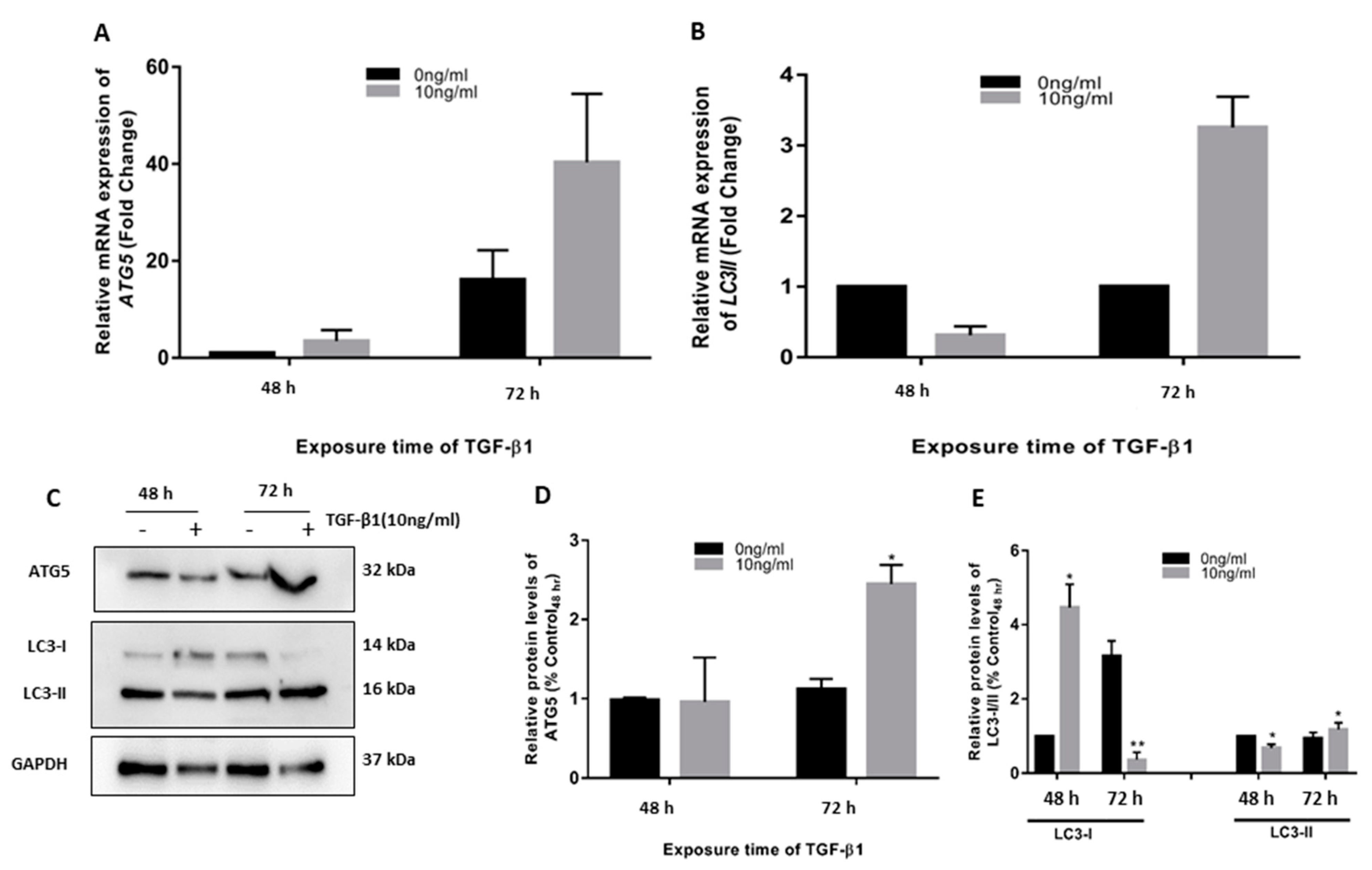
3.1. TGF-β1 Induced ATG5 Activation, Fibrosis, and Epithelial to Mesenchymal Transition in HTM
3.2. TGF-β1-Induced Fibrosis and Epithelial to Mesenchymal Transition Is Regulated by Autophagy Induction
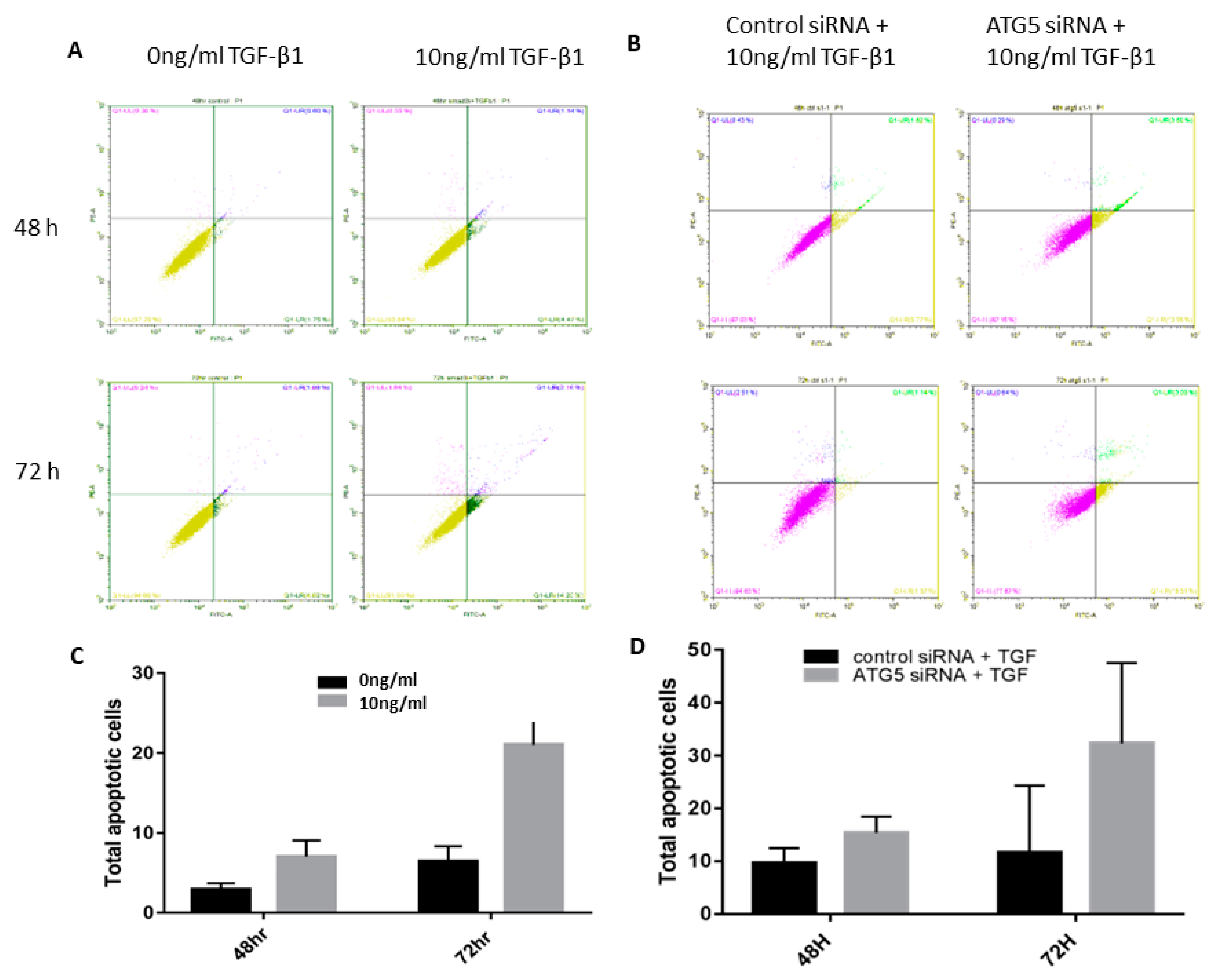
3.3. TGF-Autophagy Crosstalk Regulates Apoptosis in HTM Cells
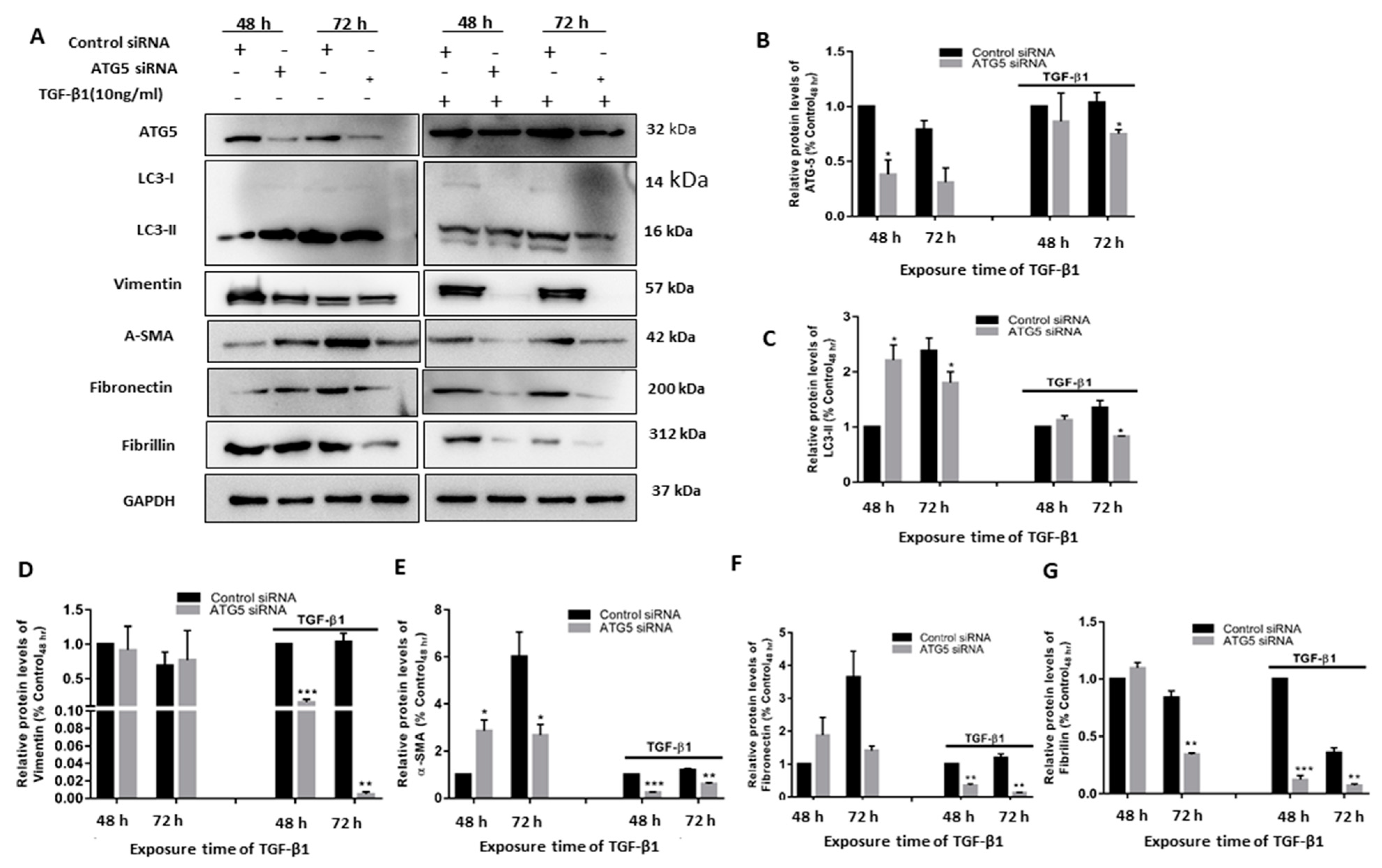
3.4. TGF Induced Autophagy Involves a Positive Feedback Loop of ATG5 on the TGF Pathway
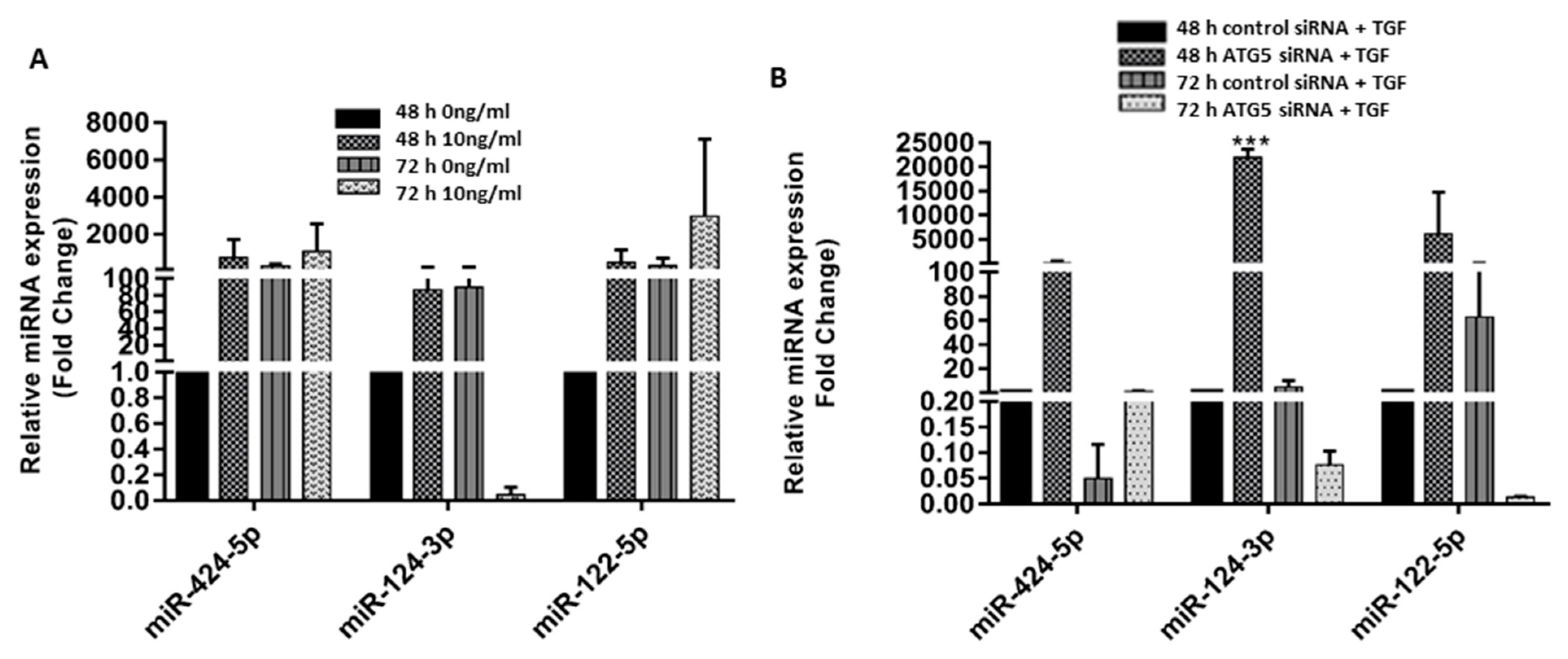
3.5. Crosstalk between ATG5 and TGF May Be Mediated by miR 122-5p
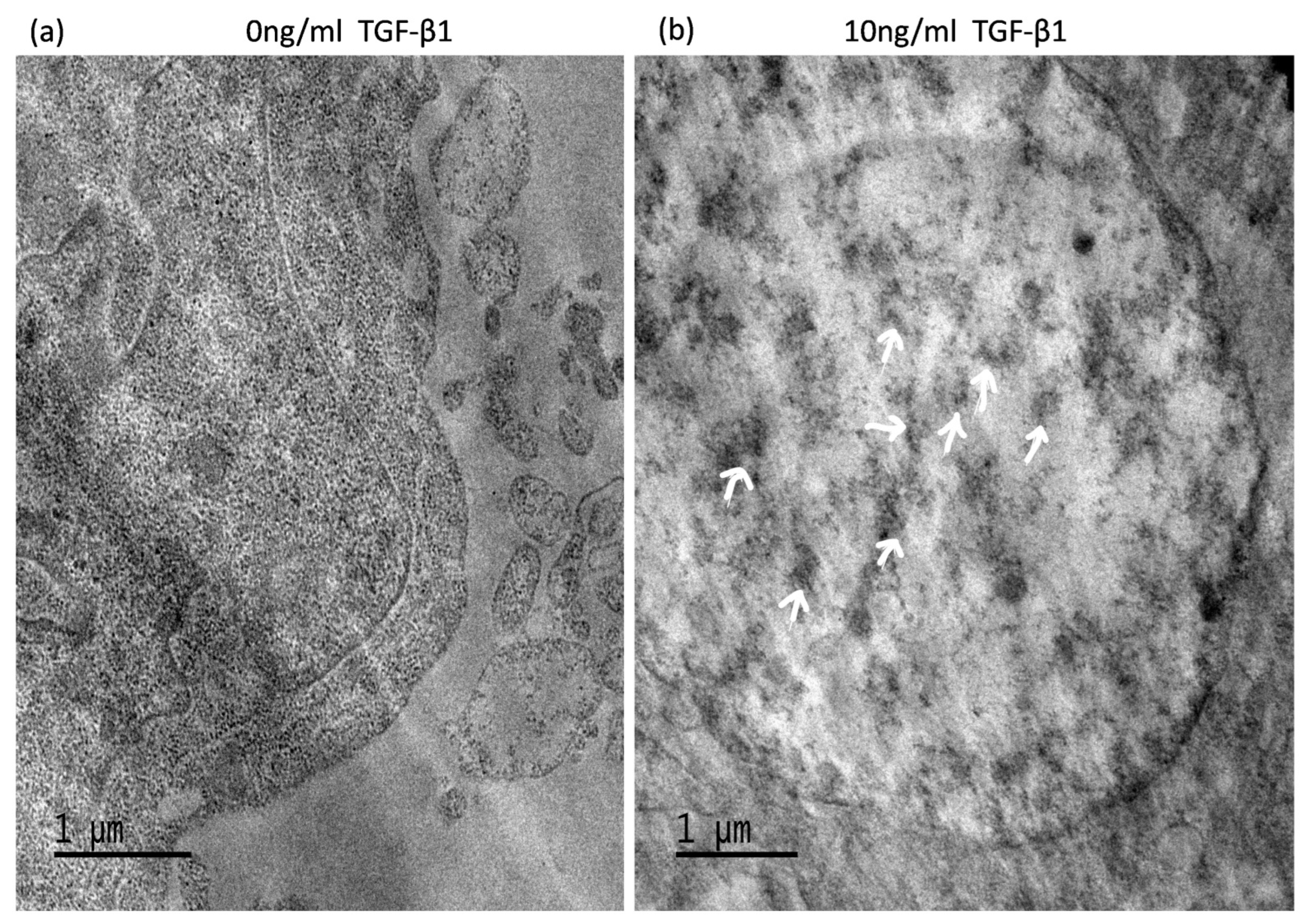
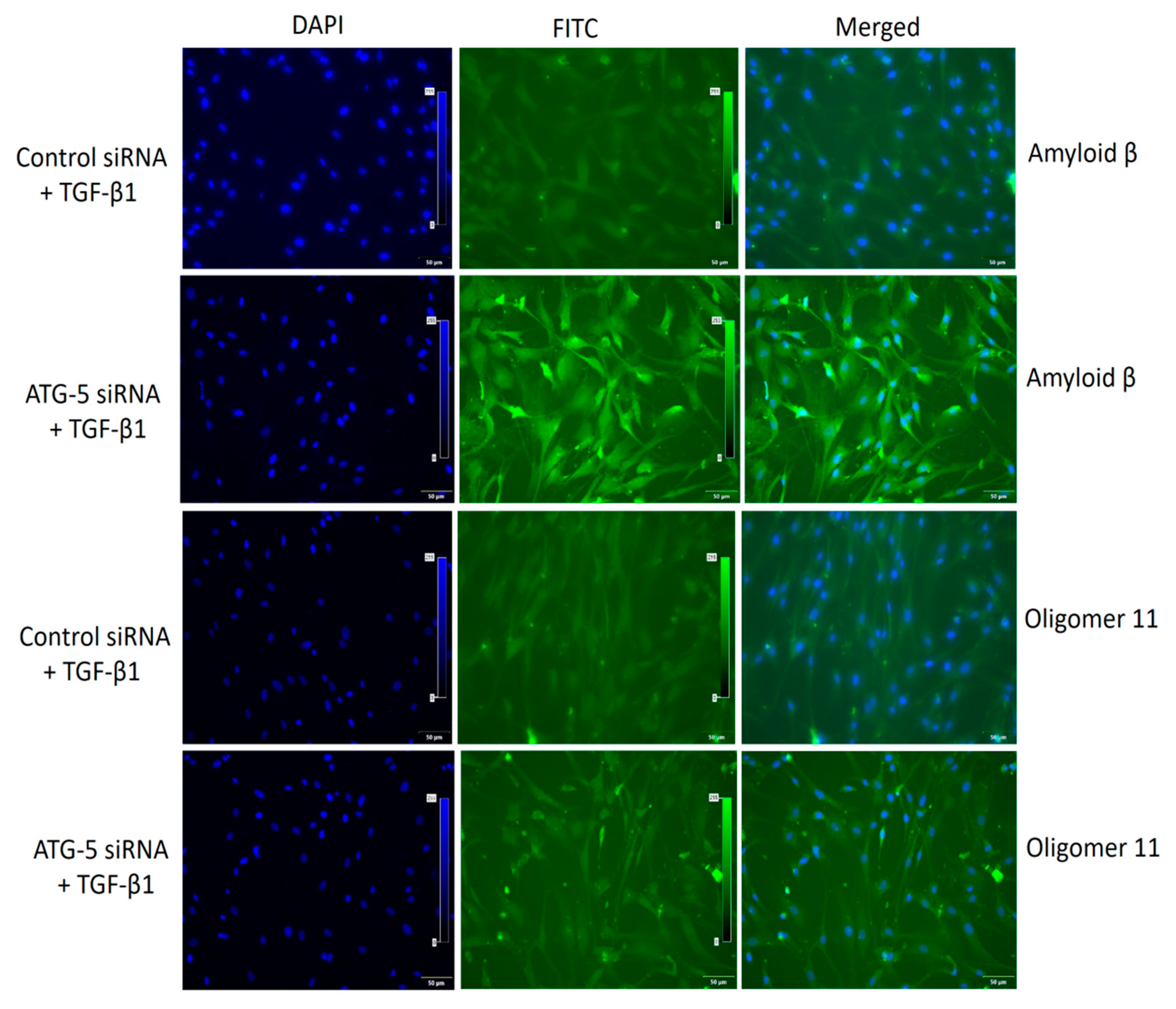
3.6. TGF-Induced ATG5 Activation Is Essential to Prevent TGF-Induced Aggregate Formation in HTM Cells
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rao, A.; Chakraborty, M.; Roy, A.; Sahay, P.; Pradhan, A.; Raj, N. Differential miRNA Expression: Signature for Glaucoma in Pseudoexfoliation. Clin. Ophthalmol. 2020, 14, 3025–3038. [Google Scholar] [CrossRef]
- Sahay, P.; Reddy, S.; Prusty, B.K.; Modak, R.; Rao, A. TGFβ1, MMPs and cytokines profiles in ocular surface: Possible tear biomarkers for pseudoexfoliation. PLoS ONE 2021, 16, e0249759. [Google Scholar] [CrossRef]
- Heydarpour, F.; Sajadimajd, S.; Mirzarazi, E.; Haratipour, P.; Joshi, T.; Farzaei, M.H.; Khan, H.; Echeverría, J. Involvement of TGF-β and Autophagy Pathways in Pathogenesis of Diabetes: A Comprehensive Review on Biological and Pharmacological Insights. Front. Pharmacol. 2020, 11, 498758. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Choi, M.E. Regulation of autophagy by TGF-β: Emerging role in kidney fibrosis. Semin. Nephrol. 2014, 34, 62–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quan, W.; Lim, Y.M.; Lee, M.S. Role of autophagy in diabetes and endoplasmic reticulum stress of pancreatic β-cells. Exp. Mol. Med. 2012, 44, 81–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, M.; Sahay, P.; Rao, A. Primary Human Trabecular Meshwork Model for Pseudoexfoliation. Cells 2021, 10, 3448. [Google Scholar] [CrossRef]
- Chakraborty, M.; Rao, A. Alternate Causes for Pathogenesis of Exfoliation Glaucoma, a Multifactorial Elastotic Disorder: A Literature Review. Curr. Issues Mol. Biol. 2022, 44, 1191–1202. [Google Scholar] [CrossRef]
- Sharma, S.; Chataway, T.; Klebe, S.; Griggs, K.; Martin, S.; Chegeni, N.; Dave, A.; Zhou, T.; Ronci, M.; Voelcker, N.H.; et al. Novel protein constituents of pathological ocular exfoliation syndrome deposits identified with mass spectrometry. Mol. Vis. 2018, 24, 801–917. [Google Scholar] [PubMed]
- Wolosin, J.M.; Ritch, R.; Bernstein, A.M. Is Autophagy Dysfunction a Key to Exfoliation Glaucoma? J. Glaucoma 2018, 27, 197–201. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Sahay, P.; Chakraborty, M.; Prusty, B.K.; Srinivasan, S.; Jhingan, G.D.; Mishra, P.; Modak, R.; Suar, M. Switch to Autophagy the Key Mechanism for Trabecular Meshwork Death in Severe Glaucoma. Clin. Ophthalmol. 2021, 15, 3027–3039. [Google Scholar] [CrossRef] [PubMed]
- De Juan-Marcos, L.; Escudero-Domínguez, F.A.; Hernández-Galilea, E.; Cruz-González, F.; Follana-Neira, I.; González-Sarmiento, R. Investigation of Association between Autophagy-Related Gene Polymorphisms and Exfoliation Syndrome and Exfoliation Glaucoma in a Spanish Population. Semin. Ophthalmol. 2018, 33, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Want, A.; Gillespie, S.R.; Wang, Z.; Gordon, R.; Iomini, C.; Ritch, R.; Wolosin, J.M.; Bernstein, A.M. Autophagy and Mitochondrial Dysfunction in Tenon Fibroblasts from Exfoliation Glaucoma Patients. PLoS ONE 2016, 11, e0157404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ecker, N.; Mor, A.; Journo, D.; Abeliovich, H. Induction of autophagic flux by amino acid deprivation is distinct from nitrogen starvation-induced macroautophagy. Autophagy 2010, 6, 879–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Shao, S.H.; Xu, Z.X.; Hennessy, B.; Ding, Z.; Larrea, M.; Kondo, S.; Dumont, D.J.; Gutterman, J.U.; Walker, C.L.; et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat. Cell Biol. 2007, 9, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Meijer, A.J.; Codogno, P. Autophagy: Regulation by energy sensing. Curr. Biol. 2011, 21, R227–R229. [Google Scholar] [CrossRef] [Green Version]
- Nijholt, D.A.; de Graaf, T.R.; van Haastert, E.S.; Oliveira, A.O.; Berkers, C.R.; Zwart, R.; Ovaa, H.; Baas, F.; Hoozemans, J.J.; Scheper, W. Endoplasmic reticulum stress activates autophagy but not the proteasome in neuronal cells: Implications for Alzheimer’s disease. Cell Death Differ. 2011, 18, 1071–1081. [Google Scholar] [CrossRef] [Green Version]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 40, 294–309. [Google Scholar] [CrossRef] [Green Version]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Das, G.; Shravage, B.V.; Baehrecke, E.H. Regulation and function of autophagy during cell survival and cell death. Cold Spring Harb. Perspect. Biol. 2012, 4, a008813. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Karantza, V. Autophagy as a therapeutic target in cancer. Cancer Biol. Ther. 2011, 11, 157–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uddin, M.S.; Stachowiak, A.; Mamun, A.A.; Tzvetkov, N.T.; Takeda, S.; Atanasov, A.G.; Bergantin, L.B.; Abdel-Daim, M.M.; Stankiewicz, A.M. Autophagy and Alzheimer’s Disease: From Molecular Mechanisms to Therapeutic Implications. Front. Aging Neurosci. 2018, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortun, J.; Dunn, W.A., Jr.; Joy, S.; Li, J.; Notterpek, L. Emerging role for autophagy in the removal of aggresomes in Schwann cells. J. Neurosci. 2003, 23, 10672–10680. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Yang, S.; Huang, J.; Ruan, S.; Zheng, Z.; Lin, J. Tgf-beta1 induces autophagy and promotes apoptosis in renal tubular epithelial cells. Int. J. Mol. Med. 2012, 29, 781–790. [Google Scholar] [PubMed]
- Bolt, A.M.; Klimecki, W.T. Autophagy in toxicology: Self-consumption in times of stress and plenty. J. Appl. Toxicol. 2012, 32, 465–479. [Google Scholar] [CrossRef] [Green Version]
- Poon, A.H.; Chouiali, F.; Tse, S.M.; Litonjua, A.A.; Hussain, S.N.; Baglole, C.J.; Eidelman, D.H.; Olivenstein, R.; Martin, J.G.; Weiss, S.T.; et al. Genetic and histologic evidence for autophagy in asthma pathogenesis. J. Allergy Clin. Immunol. 2012, 129, 569–571. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Gea, V.; Ghiassi-Nejad, Z.; Rozenfeld, R.; Gordon, R.; Fiel, M.I.; Yue, Z.; Czaja, M.J.; Friedman, S.L. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology 2012, 142, 938–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lijnen, P.J.; Petrov, V.V.; Fagard, R.H. Induction of cardiac fibrosis by transforming growth factor-beta(1). Mol. Genet. Metab. 2000, 71, 418–435. [Google Scholar] [CrossRef]
- Zhang, J.; Jiang, N.; Ping, J.; Xu, L. TGF-β1-induced autophagy activates hepatic stellate cells via the ERK and JNK signaling pathways. Int. J. Mol. Med. 2021, 47, 256–266. [Google Scholar] [CrossRef] [PubMed]
- Tabor-Godwin, J.M.; Tsueng, G.; Sayen, M.R.; Gottlieb, R.A.; Feuer, R. The role of autophagy during coxsackievirus infection of neural progenitor and stem cells. Autophagy 2012, 8, 938–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghavami, S.; Cunnington, R.H.; Gupta, S.; Yeganeh, B.; Filomeno, K.L.; Freed, D.H.; Chen, S.; Klonisch, T.; Halayko, A.J.; Ambrose, E.; et al. Autophagy is a regulator of TGF-[beta]1-induced fibrogenesis in primary human atrial myofibroblasts. Cell Death Dis. 2015, 6, e1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, K.M.; Jeyabalan, N.; Liton, P.B. MTOR-independent induction of autophagy in trabecular meshwork cells subjected to biaxial stretch. Biochim. Biophys. Acta 2014, 1843, 1054–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nettesheim, A.; Shim, M.S.; Hirt, J.; Liton, P.B. Transcriptome analysis reveals autophagy as regulator of TGFβ/Smad-induced fibrogenesis in trabecular meshwork cells. Sci. Rep. 2019, 9, 16092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matoba, R.; Morizane, Y.; Shiode, Y.; Hirano, M.; Doi, S.; Toshima, S.; Araki, R.; Hosogi, M.; Yonezawa, T.; Shiraga, F. Suppressive effect of AMP-activated protein kinase on the epithelial-mesenchymal transition in retinal pigment epithelial cells. PloS ONE 2017, 12, e0181481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yam, G.H.; Wang, K.; Jhanji, V.; Choy, K.W.; Baum, L.; Pang, C.P. In vitro amyloid aggregate forming ability of TGFBI mutants that cause corneal dystrophies. Investig. Ophthalmol. Vis. Sci. 2012, 53, 5890–5898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Z.; Lin, J.; Sun, Y.; Cong, S.; Liu, S.; Zhang, Y.; Chen, J. miR-122-5p negatively regulates the transforming growth factor-β/Smad signaling pathway in skeletal muscle myogenesis. Cell Biochem. Funct. 2020, 38, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wang, H.; Li, Y.; Liu, S.; Chen, J.; Ying, H. miR-24 and miR-122 Negatively Regulate the Transforming Growth Factor-β/Smad Signaling Pathway in Skeletal Muscle Fibrosis. Mol. Ther. Nucleic Acids 2018, 11, 528–537. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhao, H.; Guo, M.; Fei, D.; Zhang, L.; Xing, M. Targeting the miR-122/PKM2 autophagy axis relieves arsenic stress. J. Hazard. Mater. 2020, 383, 121217. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chakraborthy, M.; Rao, A. A Feedback Loop between TGF-β1 and ATG5 Mediated by miR-122-5p Regulates Fibrosis and EMT in Human Trabecular Meshwork Cells. Curr. Issues Mol. Biol. 2023, 45, 2381-2392. https://doi.org/10.3390/cimb45030154
Chakraborthy M, Rao A. A Feedback Loop between TGF-β1 and ATG5 Mediated by miR-122-5p Regulates Fibrosis and EMT in Human Trabecular Meshwork Cells. Current Issues in Molecular Biology. 2023; 45(3):2381-2392. https://doi.org/10.3390/cimb45030154
Chicago/Turabian StyleChakraborthy, Munmun, and Aparna Rao. 2023. "A Feedback Loop between TGF-β1 and ATG5 Mediated by miR-122-5p Regulates Fibrosis and EMT in Human Trabecular Meshwork Cells" Current Issues in Molecular Biology 45, no. 3: 2381-2392. https://doi.org/10.3390/cimb45030154