
Antimicrobial Peptides Targeting Gram-Positive Bacteria


Abstract
:1. Introduction
2. Structural Characteristics of AMPs
3. Architecture of Gram-Positive Cell Envelopes
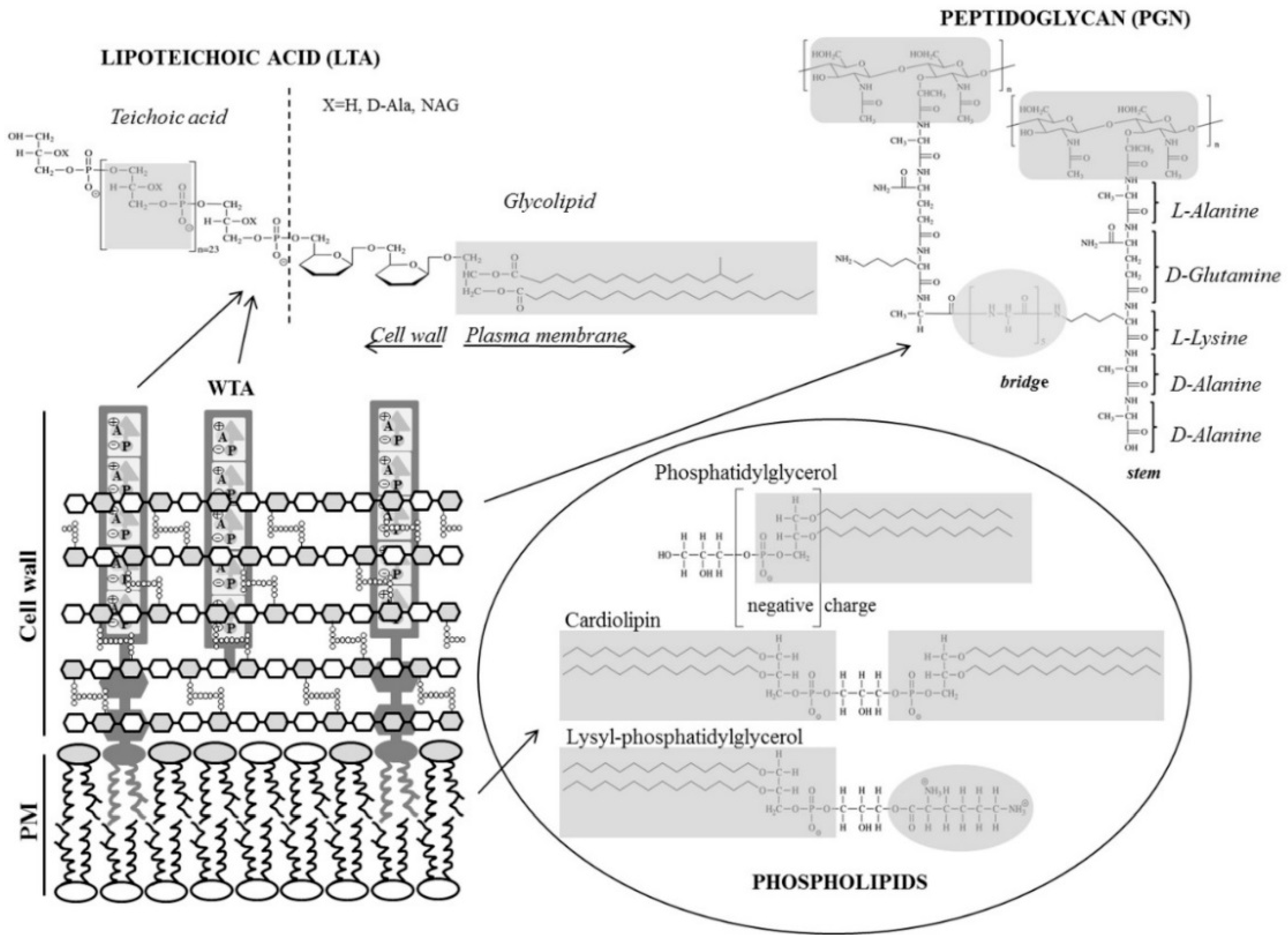
3.1. Peptidoglycan, a Cell Wall Mesh
3.2. Lipoteichoic Acid, an Anionic Polymer Matrix
3.3. Plasma Membrane Phospholipids
4. Mode of Action of AMPs
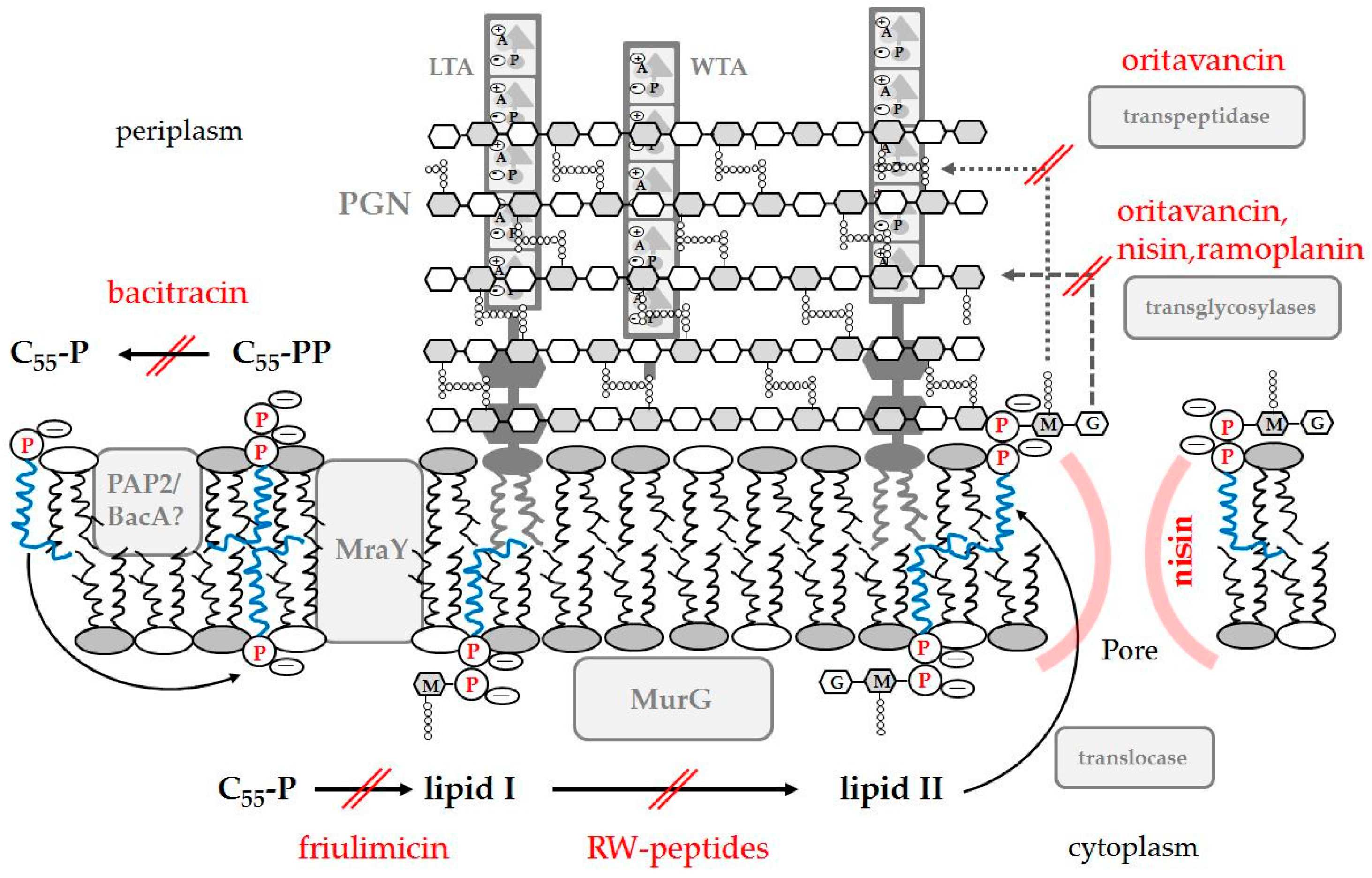
4.1. AMPs Targeting Peptidoglycan
4.1.1. Inhibition of PGN Biosynthesis
4.1.2. Binding to PGN: Recognition and Elimination of Pathogens
4.2. Teichoic Acid: Enhancing or Blocking AMP Activity
4.2.1. Direct Killing by Binding to LTA
4.3. Interaction of AMPs with the Cytoplasmic Membrane
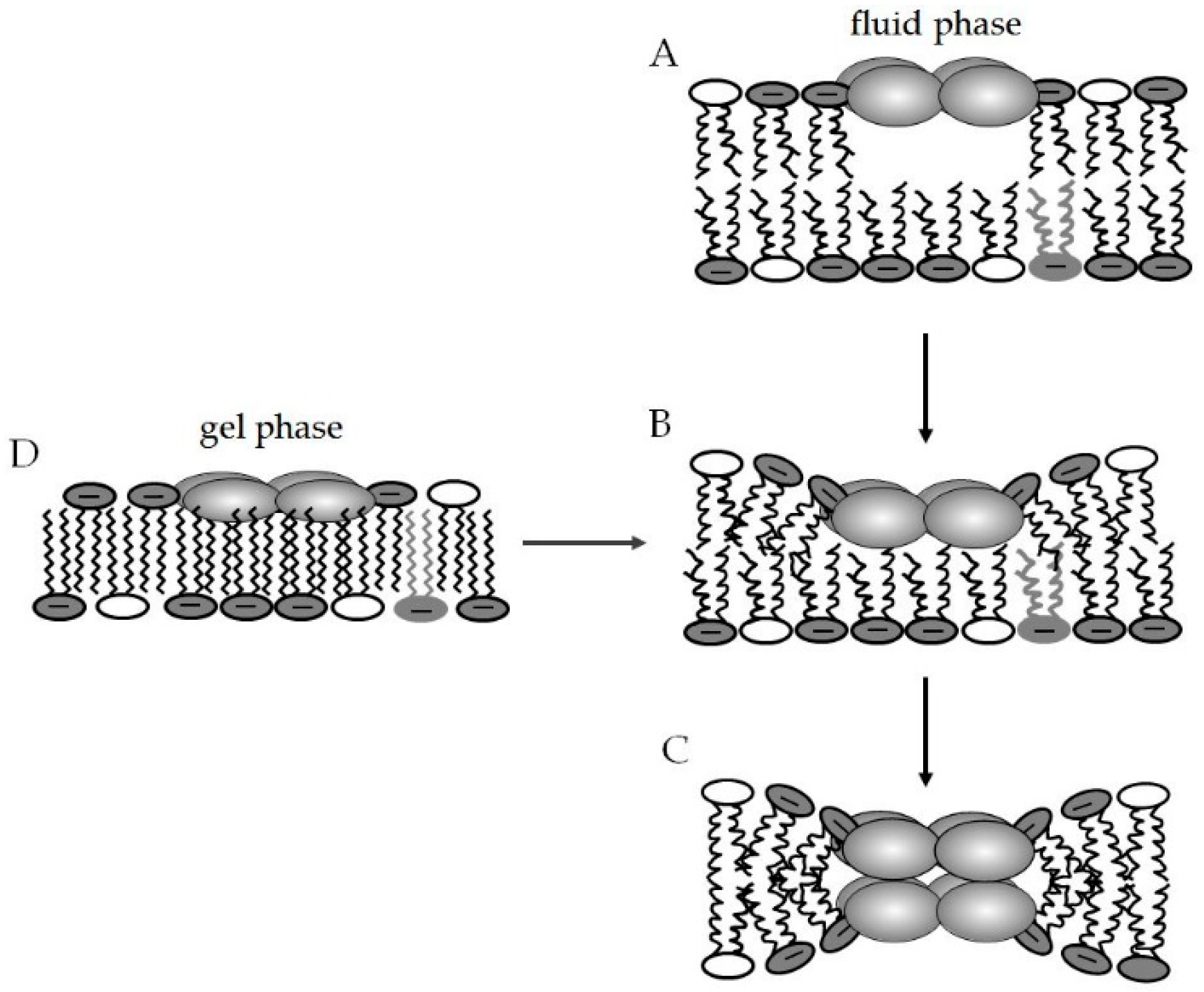
4.3.1. Lipid Segregation and Alteration of Membrane Domains as a Mode of Action of AMPs
4.3.2. A Role Model for AMPs: The Multifaceted Actions of Daptomycin
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AMPs | antimicrobial peptides |
| CL | cardiolipin |
| DPPE | 1,2-dipamitoyl-sn-glycero-3-phospho-rac-ethanolamine |
| DPPG | 1,2-dipamitoyl-sn-glycero-3-phospho-rac-glycerol |
| LPS | Lipopolysaccharide |
| LTA | lipoteichoic acid |
| LUV | large unilamellar vesicle |
| lysyl-PG | lysyl-phosphatidylglycerol |
| MLVs | multilamellar vesicles |
| NAM | N-acetyl glucosamine |
| NAG | N-acetyl muramic acid |
| PA | phosphatidic acid |
| PC | phosphatidylcholine |
| PE | phosphatidylethanolamine |
| PGN | peptidoglycan |
| POPC | 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine |
| POPE | 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-rac-ethanolamine |
| POPG | 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-rac-glycerol |
| PS | phosphatidylserine |
| PI | phosphatidylinositol |
| WTA | wall teichoic acid |
References
- World Health Organization. Antimicrobial Resistance Global Report on Surveillance: 2014 Summary; WHO: Geneva, Switzerland, 2014. [Google Scholar]
- Fish, D.N. Optimal antimicrobial therapy for sepsis. Am. J. Health Syst. Pharm. 2002, 59, S13–S19. [Google Scholar] [PubMed]
- Linder, K.E.; Nicolau, D.P.; Nailor, M.D. Predicting and preventing antimicrobial resistance utilizing pharmacodynamics: Part I Gram positive bacteria. Expert Opin. Drug Metab. Toxicol. 2016, 12, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Kali, A.; Charles, M.V.; Srirangaraj, S. Cadazolid: A new hope in the treatment of Clostridium difficile infection. Australas. Med. J. 2015, 8, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Edelsberg, J.; Weycker, D.; Barron, R.; Li, X.; Wu, H.; Oster, G.; Badre, S.; Langeberg, W.J.; Weber, D.J. Prevalence of antibiotic resistance in US hospitals. Diagn. Microbiol. Infect. Dis. 2014, 78, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Camargo, A.C.; Woodward, J.J.; Nero, L.A. The Continuous Challenge of Characterizing the Foodborne Pathogen Listeria monocytogenes. Foodborne Pathog. Dis. 2016, 13, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Cannon, J.P.; Lee, T.A.; Bolanos, J.T.; Danziger, L.H. Pathogenic relevance of Lactobacillus: A retrospective review of over 200 cases. Eur. J. Clin. Microbiol. Infect. Dis. 2005, 24, 31–40. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Global Tuberculosis Report 2014; WHO Press: Geneva, Switzerland, 2014. [Google Scholar]
- World Health Organization. Global Tuberculosis Report 2015; WHO Press: Geneva, Switzerland, 2015. [Google Scholar]
- Gutsmann, T. Interaction between antimicrobial peptides and mycobacteria. Biochim. Biophys. Acta Biomembr. 2016, 1858, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Lohner, K. Development of Novel Antimicrobial Agents: Emerging Strategies; Horizon Scientific Press: Wymondham, UK, 2001; p. 270. [Google Scholar]
- Boman, H.G. Antibacterial peptides: Basic facts and emerging concepts. J. Intern. Med. 2003, 254, 197–215. [Google Scholar] [CrossRef] [PubMed]
- Marr, A.K.; Gooderham, W.J.; Hancock, R.E. Antibacterial peptides for therapeutic use: Obstacles and realistic outlook. Curr. Opin. Pharmacol. 2006, 6, 468–472. [Google Scholar] [CrossRef] [PubMed]
- Steckbeck, J.D.; Deslouches, B.; Montelaro, R.C. Antimicrobial peptides: New drugs for bad bugs? Expert Opin. Biol. Ther. 2014, 14, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Lohner, K.; Staudegger, E. Are we on the threshold of the post-antibiotic era? In Development of Novel Antimicrobial Agents: Emerging Strategies; Lohner, K., Ed.; Horizon Scientific Press: Wymondham, UK, 2001; pp. 1–15. [Google Scholar]
- Wang, G.; Li, X.; Wang, Z. APD2: The updated antimicrobial peptide database and its application in peptide design. Nucleic Acids Res. 2009, 37, D933–D937. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Ruiz, A.; Seaton, R.A.; Hamed, K. Daptomycin: An evidence-based review of its role in the treatment of Gram-positive infections. Infect. Drug Resist. 2016, 9, 47–58. [Google Scholar] [PubMed]
- Brade, K.D.; Rybak, J.M.; Rybak, M.J. Oritavancin: A New Lipoglycopeptide Antibiotic in the Treatment of Gram-Positive Infections. Infect. Dis. Ther. 2016, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T.; Lehrer, R.I. Antimicrobial peptides in innate immunity. In Development of Novel Antimicrobial Agents: Emerging Strategies; Lohner, K., Ed.; Horizon Scientific Press: Wymondham, UK, 2001; pp. 139–147. [Google Scholar]
- Hancock, R.E. Cationic peptides: effectors in innate immunity and novel antimicrobials. Lancet Infect. Dis. 2001, 1, 156–164. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Haney, E.F.; Vogel, H.J. The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 2011, 29, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Steinstraesser, L.; Kraneburg, U.; Jacobsen, F.; Al-Benna, S. Host defense peptides and their antimicrobial-immunomodulatory duality. Immunobiology 2011, 216, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.; Sahl, H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Mansour, S.C.; Pena, O.M.; Hancock, R.E. Host defense peptides: Front-line immunomodulators. Trends Immunol. 2014, 35, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Fjell, C.D.; Hiss, J.A.; Hancock, R.E.; Schneider, G. Designing antimicrobial peptides: Form follows function. Nat. Rev. Drug Discov. 2012, 11, 37–51. [Google Scholar]
- Malanovic, N.; Lohner, K. Gram-positive bacterial cell envelopes: The impact on the activity of antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 2016, 1858, 936–946. [Google Scholar] [CrossRef] [PubMed]
- Roversi, D.; Luca, V.; Aureli, S.; Park, Y.; Mangoni, M.L.; Stella, L. How many antimicrobial peptide molecules kill a bacterium? The case of PMAP-23. ACS Chem. Biol. 2014, 9, 2003–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohner, K. New strategies for novel antibiotics: Peptides targeting bacterial cell membranes. Gen. Physiol. Biophys. 2009, 28, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Lohner, K.; Blondelle, S.E. Molecular mechanisms of membrane perturbation by antimicrobial peptides and the use of biophysical studies in the design of novel peptide antibiotics. Comb. Chem. High Throughput Screen 2005, 8, 241–256. [Google Scholar]
- Lohner, K.; Latal, A.; Lehrer, R.I.; Ganz, T. Differential scanning microcalorimetry indicates that human defensin, HNP-2, interacts specifically with biomembrane mimetic systems. Biochemistry 1997, 36, 1525–1531. [Google Scholar] [CrossRef] [PubMed]
- Latal, A.; Degovics, G.; Epand, R.F.; Epand, R.M.; Lohner, K. Structural aspects of the interaction of peptidyl-glycylleucine-carboxyamide, a highly potent antimicrobial peptide from frog skin, with lipids. Eur. J. Biochem. 1997, 248, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.K.; Bhunia, A.; Kotler, S.A.; Ramamoorthy, A. Detergent-type membrane fragmentation by MSI-78, MSI-367, MSI-594, and MSI-843 antimicrobial peptides and inhibition by cholesterol: A solid-state nuclear magnetic resonance study. Biochemistry 2015, 54, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Sugishita, K.; Fujii, N.; Miyajima, K. Molecular basis for membrane selectivity of an antimicrobial peptide, magainin 2. Biochemistry 1995, 34, 3423–3429. [Google Scholar] [CrossRef] [PubMed]
- Wang, G. Structures of human host defense cathelicidin LL-37 and its smallest antimicrobial peptide KR-12 in lipid micelles. J. Biol. Chem. 2008, 283, 32637–32643. [Google Scholar] [CrossRef] [PubMed]
- Porcelli, F.; Verardi, R.; Shi, L.; Henzler-Wildman, K.A.; Ramamoorthy, A.; Veglia, G. NMR structure of the cathelicidin-derived human antimicrobial peptide LL-37 in dodecylphosphocholine micelles. Biochemistry 2008, 47, 5565–5572. [Google Scholar] [CrossRef] [PubMed]
- Porcelli, F.; Buck-Koehntop, B.A.; Thennarasu, S.; Ramamoorthy, A.; Veglia, G. Structures of the dimeric and monomeric variants of magainin antimicrobial peptides (MSI-78 and MSI-594) in micelles and bilayers, determined by NMR spectroscopy. Biochemistry 2006, 45, 5793–5799. [Google Scholar] [CrossRef] [PubMed]
- Haney, E.F.; Hunter, H.N.; Matsuzaki, K.; Vogel, H.J. Solution NMR studies of amphibian antimicrobial peptides: Linking structure to function? Biochim. Biophys. Acta Biomembr. 2009, 1788, 1639–1655. [Google Scholar] [CrossRef] [PubMed]
- Henzler Wildman, K.A.; Lee, D.K.; Ramamoorthy, A. Mechanism of lipid bilayer disruption by the human antimicrobial peptide, LL-37. Biochemistry 2003, 42, 6545–6558. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B. The structure, dynamics and orientation of antimicrobial peptides in membranes by multidimensional solid-state NMR spectroscopy. Biochim. Biophys. Acta Biomembr. 1999, 1462, 157–183. [Google Scholar] [CrossRef]
- Bechinger, B.; Resende, J.M.; Aisenbrey, C. The structural and topological analysis of membrane-associated polypeptides by oriented solid-state NMR spectroscopy: Established concepts and novel developments. Biophys. Chem. 2011, 153, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Strandberg, E.; Kanithasen, N.; Tiltak, D.; Burck, J.; Wadhwani, P.; Zwernemann, O.; Ulrich, A.S. Solid-state NMR analysis comparing the designer-made antibiotic MSI-103 with its parent peptide PGLa in lipid bilayers. Biochemistry 2008, 47, 2601–2616. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Mishra, B.; Epand, R.F.; Epand, R.M. High-quality 3D structures shine light on antibacterial, anti-biofilm and antiviral activities of human cathelicidin LL-37 and its fragments. Biochim. Biophys. Acta Biomembr. 2014, 1838, 2160–2172. [Google Scholar] [CrossRef] [PubMed]
- Lohner, K. Membrane-active antimicrobial peptides as template structures for novel antibiotic agents. Curr. Top. Med. Chem. 2016, in press. [Google Scholar]
- Melo, M.N.; Ferre, R.; Castanho, M.A. Antimicrobial peptides: Linking partition, activity and high membrane-bound concentrations. Nat. Rev. Microbiol. 2009, 7, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Findlay, B.; Zhanel, G.G.; Schweizer, F. Cationic amphiphiles, a new generation of antimicrobials inspired by the natural antimicrobial peptide scaffold. Antimicrob. Agents Chemother. 2010, 54, 4049–4058. [Google Scholar] [CrossRef] [PubMed]
- Henderson, J.M.; Lee, K.Y.C. Promising antimicrobial agents designed from natural peptide templates. Curr. Opin. Solid State Mater. Sci. 2013, 17, 175–192. [Google Scholar] [CrossRef]
- Leitgeb, B.; Szekeres, A.; Manczinger, L.; Vagvolgyi, C.; Kredics, L. The history of alamethicin: A review of the most extensively studied peptaibol. Chem. Biodivers. 2007, 4, 1027–1051. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, W.M.; Wilkinson, D.A. Gram-positive bacteria. In Microbial Lipids, 1st ed.; Ratledge, C., Wilkinson, S.G., Eds.; Academic Press: London, UK, 1988; Volume 1, pp. 117–201. [Google Scholar]
- Wilkinson, S.G. Gram-negative bacteria. In Microbial Lipids, 1st ed.; Ratledge, C., Wilkinson, S.G., Eds.; Academic Press: London, UK, 1988; Volume 1, pp. 299–488. [Google Scholar]
- Vollmer, W.; Holtje, J.V. The architecture of the murein (peptidoglycan) in Gram-negative bacteria: Vertical scaffold or horizontal layer(s)? J. Bacteriol. 2004, 186, 5978–5987. [Google Scholar] [CrossRef] [PubMed]
- Rogers, H.J.; Perkins, H.R.; Ward, J.B. Membranes of bacteria lacking peptidoglycan. In Microbial Cell Walls and Membranes; Rogers, H.J., Perkins, H.R., Ward, J.B., Eds.; Springer: Dordrecht, The Netherlands, 1980; pp. 176–189. [Google Scholar]
- Hayami, M.; Okabe, A.; Kariyama, R.; Abe, M.; Kanemasa, Y. Lipid-composition of Staphylococcus aureus and its derived L-forms. Microbiol. Immunol. 1979, 23, 435–442. [Google Scholar] [CrossRef] [PubMed]
- White, D.A.; Frerman, F.E. Fatty Acid Composition of the complex lipids of Staphylococcus aureus During the Formation of the Membran-bound Electron Transport System. J. Bacteriol. 1968, 95, 2198–2209. [Google Scholar]
- Mishra, N.N.; Bayer, A.S.; Tran, T.T.; Shamoo, Y.; Mileykovskaya, E.; Dowhan, W.; Guan, Z.; Arias, C.A. Daptomycin resistance in enterococci is associated with distinct alterations of cell membrane phospholipid content. PLoS ONE 2012, 7, e43958. [Google Scholar] [CrossRef] [PubMed]
- Chiu, T.H.; Hung, S.A. Effect of age on the membrane lipid composition of Streptococcus sanguis. Biochim. Biophys. Acta Biomembr. 1979, 558, 267–272. [Google Scholar] [CrossRef]
- Fozo, E.M.; Kajfasz, J.K.; Quivey, R.G. Low pH-induced membrane fatty acid alterations in oral bacteria. FEMS Microbiol. Lett. 2004, 238, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Trombe, M.C.; Laneelle, M.A.; Laneelle, G. Lipid composition of aminopterin-resistant and sensitive strains of Streptococcus pneumoniae. Effect of aminopterin inhibition. Biochim. Biophys. Acta Biomembr. 1979, 574, 290–300. [Google Scholar]
- Nampoothiri, K.M.; Hoischen, C.; Bathe, B.; Mockel, B.; Pfefferle, W.; Krumbach, K.; Sahm, H.; Eggeling, L. Expression of genes of lipid synthesis and altered lipid composition modulates l-glutamate efflux of Corynebacterium glutamicum. Appl. Microbiol. Biotechnol. 2002, 58, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Hoischen, C.; Kramer, R. Membrane-alteration is necessary but not sufficient for effective glutamate secretion in Corynebacterium glutamicum. J. Bacteriol. 1990, 172, 3409–3416. [Google Scholar] [PubMed]
- Hoischen, C.; Gura, K.; Luge, C.; Gumpert, J. Lipid and fatty acid composition of cytoplasmic membranes from Streptomyces hygroscopicus and its stable protoplast-type L form. J. Bacteriol. 1997, 179, 3430–3436. [Google Scholar] [PubMed]
- Nandedkar, A.K.N. Comparative study of the lipid-composition of particular pathogenic and non-pathogenic species of Mycobacterium. J. Natl. Med. Assoc. 1983, 75, 69–74. [Google Scholar] [PubMed]
- Sareen, M.; Khuller, G.K. Cell wall and membrane changes associated with ethambutol resistance in Mycobacterium tuberculosis H37Ra. Antimicrob. Agents Chemother. 1990, 34, 1773–1776. [Google Scholar] [CrossRef] [PubMed]
- Rottem, S.; Razin, S. Membrane lipids of Mycoplasma hominis. J. Bacteriol. 1973, 113, 565–571. [Google Scholar] [PubMed]
- Ward, J.B.; Perkins, H.R. The chemical composition of the membranes of protoplasts and L-forms of Staphylococcus. Biochem. J. 1968, 106, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.B.; Wardle, H.M.; Boote, V. Phospholipid profiles of Clostridium difficile. J. Bacteriol. 1996, 178, 5844–5846. [Google Scholar] [PubMed]
- Clejan, S.; Krulwich, T.A; Mondrus, K.R.; Seto-Young, D. Membrane lipid composition of obligately and facultatively alkalophilic strains of Bacillus spp. J. Bacteriol. 1986, 168, 334–340. [Google Scholar]
- den Kamp, J.A.; Redai, I.; van Deenen, L.L. Phospholipid composition of Bacillus subtilis. J. Bacteriol. 1969, 99, 298–303. [Google Scholar] [PubMed]
- Bishop, D.G.; Rutberg, L.; Samuelsson, B. The chemical composition of the cytoplasmic membrane of Bacillus subtilis. Eur. J. Biochem. 1967, 2, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Crandall, A.D.; Montville, T.J. Nisin resistance in Listeria monocytogenes ATCC 700302 is a complex phenotype. Appl. Environ. Microbiol. 1998, 64, 231–237. [Google Scholar] [PubMed]
- Errington, J. L-form bacteria, cell walls and the origins of life. Open Biol. 2013, 3, 120143. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Chen, S.; Jensen, G.J. Molecular organization of Gram-negative peptidoglycan. Proc. Natl. Acad. Sci. USA 2008, 105, 18953–18957. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Chang, J.; Singh, M. Peptidoglycan architecture of Gram-positive bacteria by solid-state NMR. Biochim. Biophys. Acta Biomembr. 2015, 1848, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Breukink, E.; de Kruijff, B. Lipid II as a target for antibiotics. Nat. Rev. Drug Discov. 2006, 5, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Beeby, M.; Gumbart, J.C.; Roux, B.; Jensen, G.J. Architecture and assembly of the Gram-positive cell wall. Mol. Microbiol. 2013, 88, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Hayhurst, E.J.; Kailas, L.; Hobbs, J.K.; Foster, S.J. Cell wall peptidoglycan architecture in Bacillus subtilis. Proc. Natl. Acad. Sci. USA 2008, 105, 14603–14608. [Google Scholar] [CrossRef] [PubMed]
- Percy, M.G.; Grundling, A. Lipoteichoic acid synthesis and function in Gram-positive bacteria. Annu. Rev. Microbiol. 2014, 68, 81–100. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.; Santa, M.J., Jr.; Walker, S. Wall teichoic acids of Gram-positive bacteria. Annu. Rev. Microbiol. 2013, 67, 313–336. [Google Scholar] [CrossRef] [PubMed]
- Schneewind, O.; Missiakas, D. Lipoteichoic acids, phosphate-containing polymers in the envelope of Gram-positive bacteria. J. Bacteriol. 2014, 196, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Reichmann, N.T.; Grundling, A. Location, synthesis and function of glycolipids and polyglycerolphosphate lipoteichoic acid in Gram-positive bacteria of the phylum Firmicutes. FEMS Microbiol. Lett. 2011, 319, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Naumova, I.B.; Shashkov, A.S.; Tul’skaya, E.M.; Streshinskaya, G.M.; Kozlova, Y.I.; Potekhina, N.V.; Evtushenko, L.I.; Stackebrandt, E. Cell wall teichoic acids: structural diversity, species specificity in the genus Nocardiopsis, and chemotaxonomic perspective. FEMS Microbiol. Rev. 2001, 25, 269–283. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W. Physiology of lipoteichoic acids in bacteria. Adv. Microb. Physiol. 1988, 29, 233–302. [Google Scholar] [PubMed]
- Gutberlet, T.; Frank, J.; Bradaczek, H.; Fischer, W. Effect of lipoteichoic acid on thermotropic membrane properties. J. Bacteriol. 1997, 179, 2879–2883. [Google Scholar] [PubMed]
- Oku, Y.; Kurokawa, K.; Matsuo, M.; Yamada, S.; Lee, B.L.; Sekimizu, K. Pleiotropic roles of polyglycerolphosphate synthase of lipoteichoic acid in growth of Staphylococcus aureus cells. J. Bacteriol. 2009, 191, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Huff, E. Lipoteichoic acid, a major amphiphile of Gram-positive bacteria that is not readily extractable. J. Bacteriol. 1982, 149, 399–402. [Google Scholar] [PubMed]
- Koch, H.U.; Haas, R.; Fischer, W. The role of lipoteichoic acid biosynthesis in membrane lipid metabolism of growing Staphylococcus aureus. Eur. J. Biochem. 1984, 138, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Gutberlet, T.; Markwitz, S.; Labischinski, H.; Bradaczek, H. Monolayer investigations on the bacterial amphiphile lipoteichoic acid and on lipoteichoic acid/dipalmitoyl-phosphatidylglycerol mixtures. Makromol. Chem. Macromol. Symp. 1991, 46, 283–287. [Google Scholar] [CrossRef]
- Labischinski, H.; Naumann, D.; Fischer, W. Small and medium-angle X-ray analysis of bacterial lipoteichoic acid phase structure. Eur. J. Biochem. 1991, 202, 1269–1274. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W.; Markwitz, S.; Labischinski, H. Small-angle X-ray scattering analysis of pneumococcal lipoteichoic acid phase structure. Eur. J. Biochem. 1997, 244, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Courtney, H.S.; Simpson, W.A.; Beachey, E.H. Relationship of critical micelle concentrations of bacterial lipoteichoic acids to biological activities. Infect. Immun. 1986, 51, 414–418. [Google Scholar] [PubMed]
- Mitchell, N.J.; Seaton, P.; Pokorny, A. Branched phospholipids render lipid vesicles more susceptible to membrane-active peptides. Biochim. Biophys. Acta Biomembr. 2016, 1858, 988–994. [Google Scholar] [CrossRef] [PubMed]
- Rappolt, M.; Hickel, A.; Bringezu, F.; Lohner, K. Mechanism of the lamellar/inverse hexagonal phase transition examined by high resolution X-ray diffraction. Biophys. J. 2003, 84, 3111–3122. [Google Scholar] [CrossRef]
- Seddon, J.M. Structure of the inverted hexagonal (HII) phase, and non-lamellar phase transitions of lipids. Biochim. Biophys. Acta Biomembr. 1990, 1031, 1–69. [Google Scholar] [CrossRef]
- Epand, R.M.; Hui, S.W. Effect of electrostatic repulsion on the morphology and thermotropic transitions of anionic phospholipids. FEBS Lett. 1986, 209, 257–260. [Google Scholar] [CrossRef]
- Prossnigg, F.; Hickel, A.; Pabst, G.; Lohner, K. Packing behaviour of two predominant anionic phospholipids of bacterial cytoplasmic membranes. Biophys. Chem. 2010, 150, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Israelachvili, J.N.; Marcelja, S.; Horn, R.G. Physical principles of membrane organization. Q. Rev. Biophys. 1980, 13, 121–200. [Google Scholar] [CrossRef] [PubMed]
- Pink, D.A.; McNeil, S.; Quinn, B.; Zuckermann, M.J. A model of hydrogen bond formation in phosphatidylethanolamine bilayers. Biochim. Biophys. Acta Biomembr. 1998, 1368, 289–305. [Google Scholar] [CrossRef]
- Boggs, J.M. Lipid intermolecular hydrogen bonding: Influence on structural organization and membrane-function. Biochim. Biophys. Acta Biomembr. 1987, 906, 353–404. [Google Scholar] [CrossRef]
- Lohner, K.; Latal, A.; Degovics, G.; Garidel, P. Packing characteristics of a model system mimicking cytoplasmic bacterial membranes. Chem. Phys. Lipids 2001, 111, 177–192. [Google Scholar] [CrossRef]
- Pozo, N.B.; Lohner, K.; Deutsch, G.; Sevcsik, E.; Riske, K.A.; Dimova, R.; Garidel, P.; Pabst, G. Composition dependence of vesicle morphology and mixing properties in a bacterial model membrane system. Biochim. Biophys. Acta Biomembr. 2005, 1716, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Haiens, T.H.; Dencher, N.A. Cardiolipin: A proton trap for oxidative phosphorylation. FEBS Lett. 2002, 528, 35–39. [Google Scholar] [CrossRef]
- Lewis, R.N.; McElhaney, R.N. The physicochemical properties of cardiolipin bilayers and cardiolipin-containing lipid membranes. Biochim. Biophys. Acta Biomembr. 2009, 1788, 2069–2079. [Google Scholar] [CrossRef] [PubMed]
- Rand, R.P.; SenGupta, S. Cardiolipin forms hexagonal structures with divalent cations. Biochim. Biophys. Acta Biomembr. 1972, 255, 484–492. [Google Scholar] [CrossRef]
- Mileykovskaya, E.; Dowhan, W. Visualization of phospholipid domains in Escherichia coli by using the cardiolipin-specific fluorescent dye 10-N-nonyl acridine orange. J. Bacteriol. 2000, 182, 1172–1175. [Google Scholar] [CrossRef] [PubMed]
- Kawai, F.; Shoda, M.; Harashima, R.; Sadaie, Y.; Hara, H.; Matsumoto, K. Cardiolipin domains in Bacillus subtilis marburg membranes. J. Bacteriol. 2004, 186, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Mileykovskaya, E.; Dowhan, W. Cardiolipin membrane domains in prokaryotes and eukaryotes. Biochim. Biophys. Acta Biomembr. 2009, 1788, 2084–2091. [Google Scholar] [CrossRef] [PubMed]
- Renner, L.D.; Weibel, D.B. Cardiolipin microdomains localize to negatively curved regions of Escherichia coli membranes. Proc. Natl. Acad. Sci. USA 2011, 108, 6264–6269. [Google Scholar] [CrossRef] [PubMed]
- Frias, M.; Benesch, M.G.; Lewis, R.N.; McElhaney, R.N. On the miscibility of cardiolipin with 1,2-diacyl phosphoglycerides: Binary mixtures of dimyristoylphosphatidylethanolamine and tetramyristoylcardiolipin. Biochim. Biophys. Acta Biomembr. 2011, 1808, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Benesch, M.G.; Lewis, R.N.; McElhaney, R.N. On the miscibility of cardiolipin with 1,2-diacyl phosphoglycerides: Binary mixtures of dimyristoylphosphatidylglycerol and tetramyristoylcardiolipin. Biochim. Biophys. Acta Biomembr. 2015, 1848, 2878–2888. [Google Scholar] [CrossRef] [PubMed]
- Barak, I.; Muchova, K.; Wilkinson, A.J.; O’Toole, P.J.; Pavlendova, N. Lipid spirals in Bacillus subtilis and their role in cell division. Mol. Microbiol. 2008, 68, 1315–1327. [Google Scholar] [CrossRef] [PubMed]
- Pomorski, T.G.; Nylander, T.; Cardenas, M. Model cell membranes: Discerning lipid and protein contributions in shaping the cell. Adv. Colloid Interface Sci. 2014, 205, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Barak, I.; Muchova, K. The role of lipid domains in bacterial cell processes. Int. J. Mol. Sci. 2013, 14, 4050–4065. [Google Scholar] [CrossRef] [PubMed]
- White, D.C.; Frerman, F.E. Fatty acid composition of the complex lipids of Staphylococcus aureus during the formation of the membrane-bound electron transport system. J. Bacteriol. 1968, 95, 2198–2209. [Google Scholar] [PubMed]
- Kuhn, S.; Slavetinsky, C.J.; Peschel, A. Synthesis and function of phospholipids in Staphylococcus aureus. Int. J. Med. Microbiol. 2015, 305, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.N.; Bayer, A.S. Correlation of cell membrane lipid profiles with daptomycin resistance in methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2013, 57, 1082–1085. [Google Scholar] [CrossRef] [PubMed]
- Hebeler, B.H.; Chatterjee, A.N.; Young, F.E. Regulation of the bacterial cell wall: Effect of antibiotics on lipid biosynthesis. Antimicrob. Agents Chemother. 1973, 4, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Peschel, A.; Jack, R.W.; Otto, M.; Collins, L.V.; Staubitz, P.; Nicholson, G.; Kalbacher, H.; Nieuwenhuizen, W.F.; Jung, G.; Tarkowski, A.; et al. Staphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor mprF is based on modification of membrane lipids with l-lysine. J. Exp. Med. 2001, 193, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Kristian, S.A.; Durr, M.; Van Strijp, J.A.; Neumeister, B.; Peschel, A. MprF-mediated lysinylation of phospholipids in Staphylococcus aureus leads to protection against oxygen-independent neutrophil killing. Infect. Immun. 2003, 71, 546–549. [Google Scholar] [CrossRef] [PubMed]
- Danner, S.; Pabst, G.; Lohner, K.; Hickel, A. Structure and thermotropic behavior of the Staphylococcus aureus lipid lysyl-dipalmitoylphosphatidylglycerol. Biophys. J. 2008, 94, 2150–2159. [Google Scholar] [CrossRef] [PubMed]
- Gago, G.; Diacovich, L.; Arabolaza, A.; Tsai, S.C.; Gramajo, H. Fatty acid biosynthesis in actinomycetes. FEMS Microbiol. Rev. 2011, 35, 475–497. [Google Scholar] [CrossRef] [PubMed]
- Hwang, F.; Wen, D.C.; Wu, Y.W.; Li, Y.Z.; Dong, Q.H.; Wang, S.M. Studies on the phospholipid-composition of pathogenic cell-membranes of Mycoplasma hyopneumoniae. FEBS Lett. 1986, 195, 323–326. [Google Scholar] [CrossRef]
- Rance, M.; Jeffrey, K.R.; Tulloch, A.P.; Butler, K.W.; Smith, I.C.P. Effects of cholesterol on the orientational order of unsaturated lipids in the membranes of Acholeplasma laidlawii: A 2H-NMR study. Biochim. Biophys. Acta Biomembr. 1982, 688, 191–200. [Google Scholar] [CrossRef]
- Stockton, G.W.; Johnson, K.G.; Butler, K.W.; Tulloch, A.P.; Boulanger, Y.; Smith, I.C.P.; Davis, J.H.; Bloom, M. Deuterium NMR Study of lipid organization in Acholeplasma laidlawii membranes. Nature 1977, 269, 267–268. [Google Scholar]
- Mercier, R.; Dominguez-Cuevas, P.; Errington, J. Crucial role for membrane fluidity in proliferation of primitive cells. Cell Rep. 2012, 1, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Bozic, B.; Svetina, S. Vesicle self-reproduction: The involvement of membrane hydraulic and solute permeabilities. Eur. Phys. J. E 2007, 24, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Peterlin, P.; Arrigler, V.; Kogej, K.; Svetina, S.; Walde, P. Growth and shape transformations of giant phospholipid vesicles upon interaction with an aqueous oleic acid suspension. Chem. Phys. Lipids 2009, 159, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Yeung, A.T.; Gellatly, S.L.; Hancock, R.E. Multifunctional cationic host defence peptides and their clinical applications. Cell Mol. Life Sci. 2011, 68, 2161–2176. [Google Scholar] [CrossRef] [PubMed]
- Otvos, L. Antibacterial peptides and proteins with multiple cellular targets. J. Pept. Sci. 2005, 11, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Park, C.B.; Kim, H.S.; Kim, S.C. Mechanism of action of the antimicrobial peptide buforin II: Buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem. Biophys. Res. Commun. 1998, 244, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Krizsan, A.; Prahl, C.; Goldbach, T.; Knappe, D.; Hoffmann, R. Short proline-rich antimicrobial peptides inhibit either the bacterial 70S ribosome or the assembly of its large 50S subunit. Chembiochem 2015, 16, 2304–2308. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, C.L.; Moyles, D.; Beveridge, T.J.; Hancock, R.E.W. Antibacterial action of structurally diverse cationic peptides on Gram-positive bacteria. Antimicrob. Agents Chemother. 2000, 44, 2086–2092. [Google Scholar] [CrossRef] [PubMed]
- Demchick, P.; Koch, A.L. The permeability of the wall fabric of Escherichia coli and Bacillus subtilis. J. Bacteriol. 1996, 178, 768–773. [Google Scholar] [PubMed]
- Torrent, M.; Navarro, S.; Moussaoui, M.; Nogues, M.V.; Boix, E. Eosinophil cationic protein high-affinity binding to bacteria-wall lipopolysaccharides and peptidoglycans. Biochemistry 2008, 47, 3544–3555. [Google Scholar] [CrossRef] [PubMed]
- Melo, M.N.; Dugourd, D.; Castanho, M.A. Omiganan pentahydrochloride in the front line of clinical applications of antimicrobial peptides. Recent Pat. Antiinfect. Drug Discov. 2006, 1, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Boix, E.; Nogues, M.V. Mammalian antimicrobial proteins and peptides: Overview on the RNase A superfamily members involved in innate host defence. Mol. Biosyst. 2007, 3, 317–335. [Google Scholar] [CrossRef] [PubMed]
- Torrent, M.; Cuyas, E.; Carreras, E.; Navarro, S.; Lopez, O.; De La Maza, A.; Nogués, M.V.; Reshetnyak, Y.K.; Boix, E. Topography studies on the membrane interaction mechanism of the eosinophil cationic protein. Biochemistry 2007, 46, 720–733. [Google Scholar] [CrossRef] [PubMed]
- Carreras, E.; Boix, E.; Rosenberg, H.F.; Cuchillo, C.M.; Nogues, M.V. Both aromatic and cationic residues contribute to the membrane-lytic and bactericidal activity of eosinophil cationic protein. Biochemistry 2003, 42, 6636–6644. [Google Scholar] [CrossRef] [PubMed]
- Marchand, C.; Krajewski, K.; Lee, H.F.; Antony, S.; Johnson, A.A.; Amin, R.; Roller, P.; Kvaratskhelia, M.; Pommier, Y. Covalent binding of the natural antimicrobial peptide indolicidin to DNA abasic sites. Nucleic Acids Res. 2006, 34, 5157–5165. [Google Scholar] [CrossRef] [PubMed]
- Van Bambeke, F.; Mingeot-Leclercq, M.P.; Struelens, M.J.; Tulkens, P.M. The bacterial envelope as a target for novel anti-MRSA antibiotics. Trends Pharmacol. Sci. 2008, 29, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.E. From vancomycin to oritavancin: The discovery and development of a novel lipoglycopeptide antibiotic. Antiinfect Agents Med. Chem. 2010, 9, 23–47. [Google Scholar] [CrossRef]
- Bionda, N.; Pitteloud, J.P.; Cudic, P. Cyclic lipodepsipeptides: A new class of antibacterial agents in the battle against resistant bacteria. Future Med. Chem. 2013, 5, 1311–1330. [Google Scholar] [CrossRef] [PubMed]
- Walker, S.; Chen, L.; Hu, Y.; Rew, Y.; Shin, D.; Boger, D.L. Chemistry and biology of ramoplanin: A lipoglycodepsipeptide with potent antibiotic activity. Chem. Rev. 2005, 105, 449–476. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.; Shin, D.; Rew, Y.; Boger, D.L. Alanine scan of [l-Dap2]ramoplanin A2 aglycon: Assessment of the importance of each residue. J. Am. Chem. Soc. 2007, 129, 8747–8755. [Google Scholar] [CrossRef] [PubMed]
- Somner, E.A.; Reynolds, P.E. Inhibition of peptidoglycan biosynthesis by ramoplanin. Antimicrob. Agents Chemother. 1990, 34, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Cudic, P.; Kranz, J.K.; Behenna, D.C.; Kruger, R.G.; Tadesse, H.; Wand, A.J.; Veklich, Y.I.; Weisel, J.W.; McCafferty, D.G. Complexation of peptidoglycan intermediates by the lipoglycodepsipeptide antibiotic ramoplanin: minimal structural requirements for intermolecular complexation and fibril formation. Proc. Natl. Acad. Sci. USA 2002, 99, 7384–7389. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Tiyanont, K.; Zhang, Y.; Wanner, J.; Boger, D.; Walker, S. The mechanism of action of ramoplanin and enduracidin. Mol. Biosyst. 2006, 2, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Huang, J.X.; Ramu, S.; Butler, M.S.; Cooper, M.A. Ramoplanin at bactericidal concentrations induces bacterial membrane depolarization in Staphylococcus aureus. Antimicrob. Agents Chemother. 2014, 58, 6819–6827. [Google Scholar] [CrossRef] [PubMed]
- Oppedijk, S.F.; Martin, N.I.; Breukink, E. Hit’em where it hurts: The growing and structurally diverse family of peptides that target lipid-II. Biochim. Biophys. Acta Biomembr. 2016, 1858, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, I.; Breukink, E.; van Kraaij, C.; Kuipers, O.P.; Bierbaum, G.; de Kruijff, B.; Sahl, H.G. Specific binding of nisin to the peptidoglycan precursor lipid II combines pore formation and inhibition of cell wall biosynthesis for potent antibiotic activity. J. Biol. Chem. 2001, 276, 1772–1779. [Google Scholar] [CrossRef] [PubMed]
- Hasper, H.E.; Kramer, N.E.; Smith, J.L.; Hillman, J.D.; Zachariah, C.; Kuipers, O.P.; De Kruijff, B.; Breukink, E. An alternative bactericidal mechanism of action for lantibiotic peptides that target lipid II. Science 2006, 313, 1636–1637. [Google Scholar]
- Hsu, S.T.; Breukink, E.; Tischenko, E.; Lutters, M.A.; de Kruijff, B.; Kaptein, R.; Bonvin, A.M.; van Nuland, N.A. The nisin-lipid II complex reveals a pyrophosphate cage that provides a blueprint for novel antibiotics. Nat. Struct. Mol. Biol. 2004, 11, 963–967. [Google Scholar] [CrossRef] [PubMed]
- Breukink, E.; van Heusden, H.E.; Vollmerhaus, P.J.; Swiezewska, E.; Brunner, L.; Walker, S.; Heck, A.J.; de Kruijff, B. Lipid II is an intrinsic component of the pore induced by nisin in bacterial membranes. J. Biol. Chem. 2003, 278, 19898–19903. [Google Scholar] [CrossRef] [PubMed]
- Brotz, H.; Josten, M.; Wiedemann, I.; Schneider, U.; Gotz, F.; Bierbaum, G.; Sahl, H.G. Role of lipid-bound peptidoglycan precursors in the formation of pores by nisin, epidermin and other lantibiotics. Mol. Microbiol. 1998, 30, 317–327. [Google Scholar] [CrossRef] [PubMed]
- de Kruijff, B.; van Dam, V.; Breukink, E. Lipid II: A central component in bacterial cell wall synthesis and a target for antibiotics. Prostaglandins Leukot. Essent. Fatty Acids 2008, 79, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Martin, N.I.; Breukink, E. Expanding role of lipid II as a target for lantibiotics. Future Microbiol. 2007, 2, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Brotz, H.; Bierbaum, G.; Leopold, K.; Reynolds, P.E.; Sahl, H.G. The lantibiotic mersacidin inhibits peptidoglycan synthesis by targeting lipid II. Antimicrob. Agents Chemother. 1998, 42, 154–160. [Google Scholar] [PubMed]
- Schmitt, P.; Wilmes, M.; Pugniere, M.; Aumelas, A.; Bachere, E.; Sahl, H.G.; Schneider, T.; Destoumieux-Garzón, D. Insight into invertebrate defensin mechanism of action: Oyster defensins inhibit peptidoglycan biosynthesis by binding to lipid II. J. Biol. Chem. 2010, 285, 29208–29216. [Google Scholar] [CrossRef] [PubMed]
- Mygind, P.H.; Fischer, R.L.; Schnorr, K.M.; Hansen, M.T.; Sonksen, C.P.; Ludvigsen, S.; Raventós, D.; Buskov, S.; Christensen, B.; De Maria, L.; et al. Plectasin is a peptide antibiotic with therapeutic potential from a saprophytic fungus. Nature 2005, 437, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, P.; Gueguen, Y.; Desmarais, E.; Bachere, E.; de Lorgeril, J. Molecular diversity of antimicrobial effectors in the oyster Crassostrea gigas. BMC Evol. Biol. 2010, 10, 23. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, P.; Rosa, R.D.; Destoumieux-Garzon, D. An intimate link between antimicrobial peptide sequence diversity and binding to essential components of bacterial membranes. Biochim. Biophys. Acta Biomembr. 2016, 1858, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Zhanel, G.G.; Schweizer, F.; Karlowsky, J.A. Oritavancin: Mechanism of action. Clin. Infect. Dis. 2012, 54, S214–S219. [Google Scholar] [CrossRef] [PubMed]
- Belley, A.; McKay, G.A.; Arhin, F.F.; Sarmiento, I.; Beaulieu, S.; Fadhil, I.; Parr, T.R.; Moeck, G. Oritavancin disrupts membrane integrity of Staphylococcus aureus and vancomycin-resistant enterococci to effect rapid bacterial killing. Antimicrob. Agents Chemother. 2010, 54, 5369–5371. [Google Scholar] [CrossRef] [PubMed]
- Belley, A.; Neesham-Grenon, E.; McKay, G.; Arhin, F.F.; Harris, R.; Beveridge, T.; Parr, T.R.; Moeck, G. Oritavancin kills stationary-phase and biofilm Staphylococcus aureus cells in vitro. Antimicrob. Agents Chemother. 2009, 53, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Domenech, O.; Dufrene, Y.F.; Van Bambeke, F.; Tukens, P.M.; Mingeot-Leclercq, M.P. Interactions of oritavancin, a new semi-synthetic lipoglycopeptide, with lipids extracted from Staphylococcus aureus. Biochim. Biophys. Acta Biomembr. 2010, 1798, 1876–1885. [Google Scholar] [CrossRef] [PubMed]
- Domenech, O.; Francius, G.; Tulkens, P.M.; Van Bambeke, F.; Dufrene, Y.; Mingeot-Leclercq, M.P. Interactions of oritavancin, a new lipoglycopeptide derived from vancomycin, with phospholipid bilayers: Effect on membrane permeability and nanoscale lipid membrane organization. Biochim. Biophys. Acta Biomembr. 2009, 1788, 1832–1840. [Google Scholar] [CrossRef] [PubMed]
- Arthur, M.; Depardieu, F.; Reynolds, P.; Courvalin, P. Moderate-level resistance to glycopeptide LY333328 mediated by genes of the vanA and vanB clusters in enterococci. Antimicrob. Agents Chemother. 1999, 43, 1875–1880. [Google Scholar] [PubMed]
- Stone, K.J.; Strominger, J.L. Mechanism of action of bacitracin: Complexation with metal ion and C55-isoprenyl pyrophosphate. Proc. Natl. Acad. Sci. USA 1971, 68, 3223–3227. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.; Gries, K.; Josten, M.; Wiedemann, I.; Pelzer, S.; Labischinski, H.; Sahl, H.G. The lipopeptide antibiotic friulimicin B inhibits cell wall biosynthesis through complex formation with bactoprenol phosphate. Antimicrob. Agents Chemother. 2009, 53, 1610–1618. [Google Scholar] [CrossRef] [PubMed]
- Straus, S.K.; Hancock, R.E.W. Mode of action of the new antibiotic for Gram-positive pathogens daptomycin: Comparison with cationic antimicrobial peptides and lipopeptides. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Guan, R.; Mariuzza, R.A. Peptidoglycan recognition proteins of the innate immune system. Trends Microbiol. 2007, 15, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Lehotzky, R.E.; Partch, C.L.; Mukherjee, S.; Cash, H.L.; Goldman, W.E.; Gardner, K.H.; Hooper, L.V. Molecular basis for peptidoglycan recognition by a bactericidal lectin. Proc. Natl. Acad. Sci. USA 2010, 107, 7722–7727. [Google Scholar] [CrossRef] [PubMed]
- Svajger, U.; Anderluh, M.; Jeras, M.; Obermajer, N. C-type lectin DC-SIGN: An adhesion, signalling and antigen-uptake molecule that guides dendritic cells in immunity. Cell. Signal. 2010, 22, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Zheng, H.; Derebe, M.G.; Callenberg, K.M.; Partch, C.L.; Rollins, D.; Propheter, D.C.; Rizo, J.; Grabe, M.; Jiang, Q.X. Antibacterial membrane attack by a pore-forming intestinal C-type lectin. Nature 2014, 505, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement system part I—molecular mechanisms of activation and regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef] [PubMed]
- Merle, N.S.; Noe, R.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement system part II: Role in immunity. Front. Immunol. 2015, 6, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevcsik, E.; Pabst, G.; Richter, W.; Danner, S.; Amenitsch, H.; Lohner, K. Interaction of LL-37 with model membrane systems of different complexity: Influence of the lipid matrix. Biophys. J. 2008, 94, 4688–4699. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.G.; Gold, M.R.; Hancock, R.E. Interaction of cationic peptides with lipoteichoic acid and Gram-positive bacteria. Infect. Immun. 1999, 67, 6445–6453. [Google Scholar] [PubMed]
- Bucki, R.; Janmey, P.A. Interaction of the gelsolin-derived antibacterial PBP 10 peptide with lipid bilayers and cell membranes. Antimicrob. Agents Chemother. 2006, 50, 2932–2940. [Google Scholar] [CrossRef] [PubMed]
- Vadyvaloo, V.; Arous, S.; Gravesen, A.; Hechard, Y.; Chauhan-Haubrock, R.; Hastings, J.W.; Rautenbach, M. Cell-surface alterations in class IIa bacteriocin-resistant Listeria monocytogenes strains. Microbiology 2004, 150, 3025–3033. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, S.; Zhang, J.; Liu, M.; Liu, Z. Vitellogenin is a cidal factor capable of killing bacteria via interaction with lipopolysaccharide and lipoteichoic acid. Mol. Immunol. 2009, 46, 3232–3239. [Google Scholar] [CrossRef] [PubMed]
- Malanovic, N.; Leber, R.; Schmuck, M.; Kriechbaum, M.; Cordfunke, R.A.; Drijfhout, J.W.; de Breij, A.; Nibbering, P.H.; Kolb, D.; Lohner, K. Phospholipid-driven differences determine the action of the synthetic antimicrobial peptide OP-145 on Gram-positive bacterial and mammalian membrane model systems. Biochim. Biophys. Acta Biomembr. 2015, 1848, 2437–2447. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.H.; Kao, P.H.; Lin, S.R.; Chang, L.S. Membrane-damaging activity with A chain and B chain of β-bungarotoxin. Toxicon 2009, 53, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.L.; Wu, B.J.; Kao, P.H.; Fu, Y.S.; Chang, L.S. Antibacterial and membrane-damaging activities of β-bungarotoxin B chain. J. Pept. Sci. 2013, 19, 1–8. [Google Scholar] [PubMed]
- Koprivnjak, T.; Weidenmaier, C.; Peschel, A.; Weiss, J.P. Wall teichoic acid deficiency in Staphylococcus aureus confers selective resistance to mammalian group IIA phospholipase A2 and human β-defensin 3. Infect. Immun. 2008, 76, 2169–2176. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W.; Rosel, P. The alanine ester substitution of lipoteichoic acid (LTA) in Staphylococcus aureus. FEBS Lett. 1980, 119, 224–226. [Google Scholar] [CrossRef]
- Collins, L.V.; Kristian, S.A.; Weidenmaier, C.; Faigle, M.; van Kessel, K.P.; Van Strijp, J.A.; Gotz, F.; Neumeister, B.; Peschel, A. Staphylococcus aureus strains lacking d-alanine modifications of teichoic acids are highly susceptible to human neutrophil killing and are virulence attenuated in mice. J. Infect. Dis. 2002, 186, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Peschel, A.; Vuong, C.; Otto, M.; Gotz, F. The d-alanine residues of Staphylococcus aureus teichoic acids alter the susceptibility to vancomycin and the activity of autolytic enzymes. Antimicrob. Agents Chemother. 2000, 44, 2845–2847. [Google Scholar] [CrossRef] [PubMed]
- Peschel, A.; Otto, M.; Jack, R.W.; Kalbacher, H.; Jung, G.; Gotz, F. Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J. Biol. Chem. 1999, 274, 8405–8410. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Gill, C.J.; Lee, S.H.; Mann, P.; Zuck, P.; Meredith, T.C.; Murgolo, N.; She, X.; Kales, S.; Liang, L.; et al. Discovery of wall teichoic acid inhibitors as potential anti-MRSA β-lactam combination agents. Chem. Biol. 2013, 20, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Saar-Dover, R.; Bitler, A.; Nezer, R.; Shmuel-Galia, L.; Firon, A.; Shimoni, E.; Trieu-Cuot, P.; Shai, Y. d-alanylation of lipoteichoic acids confers resistance to cationic peptides in group B Streptococcus by increasing the cell wall density. PLoS Pathog. 2012, 8, e1002891. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, S.; Li, H.; Li, L. Vitellogenin, a multivalent sensor and an antimicrobial effector. Int. J. Biochem. Cell Biol. 2011, 43, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Liu, Z.; Zhou, L.; Ji, G.; Yang, A. Molecular cloning, expression, purification and characterization of vitellogenin in scallop Patinopecten yessoensis with special emphasis on its antibacterial activity. Dev. Comp. Immunol. 2015, 49, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Hoernke, M.; Schwieger, C.; Kerth, A.; Blume, A. Binding of cationic pentapeptides with modified side chain lengths to negatively charged lipid membranes: Complex interplay of electrostatic and hydrophobic interactions. Biochim. Biophys. Acta Biomembr. 2012, 1818, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Lohner, K. DSC Studies on the modulation of membrane lipid polymorphism and domain organization by antimicrobial peptides. In Biocalorimetry: Foundations and Contemporary Approaches; Bastos, M., Ed.; CRC Press: Boca Raton, FL, USA, 2016; pp. 169–190. [Google Scholar]
- Shai, Y. Mode of action of membrane active antimicrobial peptides. Biopolymers 2002, 66, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Murase, O.; Fujii, N.; Miyajima, K. An antimicrobal peptide, magainin 2, induced rapid flip-flop of phospholipids coupled with pore formation and peptide translocation. Biochemistry 1996, 35, 11361–11368. [Google Scholar] [CrossRef] [PubMed]
- Bartucci, R.; Guzzi, R.; Sportelli, L.; Marsh, D. Intramembrane water associated with TOAC spin-labeled alamethicin: Electron spin-echo envelope modulation by D2O. Biophys. J. 2009, 96, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Qian, S.; Wang, W.C.; Yang, L.; Huang, H.W. Structure of the alamethicin pore reconstructed by X-ray diffraction analysis. Biophys. J. 2008, 94, 3512–3522. [Google Scholar] [CrossRef] [PubMed]
- Pieta, P.; Mirza, J.; Lipkowski, J. Direct visualization of the alamethicin pore formed in a planar phospholipid matrix. Proc. Natl. Acad. Sci. USA 2012, 109, 21223–21227. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Ludtke, S.J.; Heller, W.T.; Huang, H.W. Mechanism of alamethicin insertion into lipid bilayers. Biophys. J. 1996, 71, 2669–2679. [Google Scholar] [CrossRef]
- Yang, L.; Harroun, T.A.; Weiss, T.M.; Ding, L.; Huang, H.W. Barrel-stave model or toroidal model? A case study on melittin pores. Biophys. J. 2001, 81, 1475–1485. [Google Scholar] [PubMed]
- Ludtke, S.J.; He, K.; Heller, W.T.; Harroun, T.A.; Yang, L.; Huang, H.W. Membrane pores induced by magainin. Biochemistry 1996, 35, 13723–13728. [Google Scholar] [CrossRef] [PubMed]
- Bechinger, B.; Lohner, K. Detergent-like actions of linear amphipathic cationic antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- Wimley, W.C. Describing the mechanism of antimicrobial peptide action with the interfacial activity model. ACS Chem. Biol. 2010, 5, 905–917. [Google Scholar] [CrossRef] [PubMed]
- Kandaswamy, K.; Liew, T.H.; Wang, C.Y.; Huston-Warren, E.; Meyer-Hoffert, U.; Hultenby, K.; Schroder, J.M.; Caparon, M.G.; Normark, S.; Henriques-Normark, B.; et al. Focal targeting by human β-defensin 2 disrupts localized virulence factor assembly sites in Enterococcus faecalis. Proc. Natl. Acad. Sci. USA 2013, 110, 20230–20235. [Google Scholar] [CrossRef] [PubMed]
- Vega, L.A.; Caparon, M.G. Cationic antimicrobial peptides disrupt the Streptococcus pyogenes ExPortal. Mol. Microbiol. 2012, 85, 1119–1132. [Google Scholar] [CrossRef] [PubMed]
- Rangarajan, N.; Bakshi, S.; Weisshaar, J.C. Localized permeabilization of E. coli membranes by the antimicrobial peptide Cecropin A. Biochemistry 2013, 52, 6584–6594. [Google Scholar] [PubMed]
- Zweytick, D.; Japelj, B.; Mileykovskaya, E.; Zorko, M.; Dowhan, W.; Blondelle, S.E.; Riedl, S.; Jerala, R.; Lohner, K. N-acylated peptides derived from human lactoferricin perturb organization of cardiolipin and phosphatidylethanolamine in cell membranes and induce Defects in Escherichia coli cell division. PLoS ONE 2014, 9, e90228. [Google Scholar] [CrossRef] [PubMed]
- Finger, S.; Kerth, A.; Dathe, M.; Blume, A. The efficacy of trivalent cyclic hexapeptides to induce lipid clustering in PG/PE membranes correlates with their antimicrobial activity. Biochim. Biophys. Acta Biomembr. 2015, 1848, 2998–3006. [Google Scholar] [CrossRef] [PubMed]
- Scheinpflug, K.; Krylova, O.; Nikolenko, H.; Thurm, C.; Dathe, M. Evidence for a novel mechanism of antimicrobial action of a cyclic R-, W-rich hexapeptide. PLoS ONE 2015, 10, e0125056. [Google Scholar] [CrossRef] [PubMed]
- Dowhan, W. Molecular basis for membrane phospholipid diversity: Why are there so many lipids? Annu. Rev. Biochem. 1997, 66, 199–232. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Kusaka, J.; Nishibori, A.; Hara, H. Lipid domains in bacterial membranes. Mol. Microbiol. 2006, 61, 1110–1117. [Google Scholar] [CrossRef] [PubMed]
- Renner, L.D.; Weibel, D.B. MinD and MinE interact with anionic phospholipids and regulate division plane formation in Escherichia coli. J. Biol. Chem. 2012, 287, 38835–38844. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, M.; Chiriac, A.I.; Otto, A.; Zweytick, D.; May, C.; Schumacher, C.; Gust, R.; Albada, H.B.; Penkova, M.; Kramer, U.; et al. Small cationic antimicrobial peptides delocalize peripheral membrane proteins. Proc. Natl. Acad. Sci. USA 2014, 111, E1409–E1418. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, M.; Schriek, P.; Prochnow, P.; Albada, H.B.; Metzler-Nolte, N.; Bandow, J.E. Influence of lipidation on the mode of action of a small RW-rich antimicrobial peptide. Biochim. Biophys. Acta Biomembr. 2016, 1858, 1004–1011. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.M.; Epand, R.F. Lipid domains in bacterial membranes and the action of antimicrobial agents. Biochim. Biophys. Acta Biomembr. 2009, 1788, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.F.; Wang, G.; Berno, B.; Epand, R.M. Lipid segregation explains selective toxicity of a series of fragments derived from the human cathelicidin LL-37. Antimicrob. Agents Chemother. 2009, 53, 3705–3714. [Google Scholar] [CrossRef] [PubMed]
- Lohner, K.; Prenner, E.J. Differential scanning calorimetry and X-ray diffraction studies of the specificity of the interaction of antimicrobial peptides with membrane-mimetic systems. Biochim. Biophys. Acta Biomembr. 1999, 1462, 141–156. [Google Scholar] [CrossRef]
- Epand, R.M.; Epand, R.F.; Arnusch, C.J.; Papahadjopoulos-Sternberg, B.; Wang, G.; Shai, Y. Lipid clustering by three homologous arginine-rich antimicrobial peptides is insensitive to amino acid arrangement and induced secondary structure. Biochim. Biophys. Acta Biomembr. 2010, 1798, 1272–1280. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.M.; Epand, R.F. Bacterial membrane lipids in the action of antimicrobial agents. J. Pept. Sci. 2011, 17, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.F.; Schmitt, M.A.; Gellman, S.H.; Epand, R.M. Role of membrane lipids in the mechanism of bacterial species selective toxicity by two α/β-antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.F.; Mowery, B.P.; Lee, S.E.; Stahl, S.S.; Lehrer, R.I.; Gellman, S.H.; Epand, R.M. Dual mechanism of bacterial lethality for a cationic sequence-random copolymer that mimics host-defense antimicrobial peptides. J. Mol. Biol. 2008, 379, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Radzishevsky, I.S.; Kovachi, T.; Porat, Y.; Ziserman, L.; Zaknoon, F.; Danino, D.; Mor, A. Structure-activity relationships of antibacterial acyl-lysine oligomers. Chem. Biol. 2008, 15, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Zaknoon, F.; Sarig, H.; Rotem, S.; Livne, L.; Ivankin, A.; Gidalevitz, D.; Mor, A. Antibacterial properties and mode of action of a short acyl-lysyl oligomer. Antimicrob. Agents Chemother. 2009, 53, 3422–3429. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.F.; Maloy, W.L.; Ramamoorthy, A.; Epand, R.M. Probing the “charge cluster mechanism” in amphipathic helical cationic antimicrobial peptides. Biochemistry 2010, 49, 4076–4084. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, J.K.; Miller, K.; O’Neill, A.J.; Chopra, I. Consequences of daptomycin-mediated membrane damage in Staphylococcus aureus. J. Antimicrob. Chemother. 2008, 62, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.A.; Perlmutter, N.G.; Shapiro, H.M. Correlation of daptomycin bactericidal activity and membrane depolarization in Staphylococcus aureus. Antimicrob. Agents Chemother. 2003, 47, 2538–2544. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Muraih, J.K.; MacCormick, B.; Silverman, J.; Palmer, M. Daptomycin forms cation- and size-selective pores in model membranes. Biochim. Biophys. Acta Biomembr. 2014, 1838, 2425–2430. [Google Scholar] [CrossRef] [PubMed]
- Haney, E.F.; Nathoo, S.; Vogel, H.J.; Prenner, E.J. Induction of non-lamellar lipid phases by antimicrobial peptides: A potential link to mode of action. Chem. Phys. Lipids 2010, 163, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Muraih, J.K.; Tishbi, N.; Herskowitz, J.; Victor, R.L.; Silverman, J.; Uwumarenogie, S.; Taylor, S.D.; Palmer, M.; Mintzer, E. Cardiolipin prevents membrane translocation and permeabilization by daptomycin. J. Biol. Chem. 2014, 289, 11584–11591. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.W. Molecular mechanism of antimicrobial peptides: The origin of cooperativity. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1292–1302. [Google Scholar] [CrossRef] [PubMed]
- Koller, D.; Lohner, K. The role of spontaneous lipid curvature in the interaction of interfacially active peptides with membranes. Biochim. Biophys. Acta Biomembr. 2014, 1838, 2250–2259. [Google Scholar] [CrossRef] [PubMed]
- Sevcsik, E.; Pabst, G.; Jilek, A.; Lohner, K. How lipids influence the mode of action of membrane-active peptides. Biochim. Biophys. Acta Biomembr. 2007, 1768, 2586–2595. [Google Scholar] [CrossRef] [PubMed]
- Staudegger, E.; Prenner, E.J.; Kriechbaum, M.; Degovics, G.; Lewis, R.N.; McElhaney, R.N.; Lohner, K. X-ray studies on the interaction of the antimicrobial peptide gramicidin S with microbial lipid extracts: Evidence for cubic phase formation. Biochim. Biophys. Acta Biomembr. 2000, 1468, 213–230. [Google Scholar] [CrossRef]
- Ludtke, S.; He, K.; Huang, H. Membrane thinning caused by magainin 2. Biochemistry 1995, 34, 16764–16769. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.F.; Sun, T.L.; Sun, Y.; Huang, H.W. Interaction of daptomycin with lipid bilayers: A lipid extracting effect. Biochemistry 2014, 53, 5384–5392. [Google Scholar] [CrossRef] [PubMed]
- Pogliano, J.; Pogliano, N.; Silverman, J.A. Daptomycin-mediated reorganization of membrane architecture causes mislocalization of essential cell division proteins. J. Bacteriol. 2012, 194, 4494–4504. [Google Scholar] [CrossRef] [PubMed]
- Anderson, T.M.; Clay, M.C.; Cioffi, A.G.; Diaz, K.A.; Hisao, G.S.; Tuttle, M.D.; Nieuwkoop, A.J.; Comellas, G.; Maryum, N.; Wang, S.; et al. Amphotericin forms an extramembranous and fungicidal sterol sponge. Nat. Chem. Biol. 2014, 10, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Lohner, K. Antimicrobial mechanisms: A sponge against fungal infections. Nat. Chem. Biol. 2014, 10, 411–412. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.J.; Xiong, Y.Q.; Dunman, P.M.; Schrenzel, J.; Francois, P.; Peschel, A.; Bayer, A.S. Regulation of mprF in daptomycin-nonsusceptible Staphylococcus aureus strains. Antimicrob. Agents Chemother. 2009, 53, 2636–2637. [Google Scholar] [CrossRef] [PubMed]
- Arias, C.A.; Panesso, D.; McGrath, D.M.; Qin, X.; Mojica, M.F.; Miller, C.; Diaz, L.; Tran, T.T.; Rincon, S.; Barbu, E.M.; et al. Genetic basis for in vivo daptomycin resistance in enterococci. N. Engl. J. Med. 2011, 365, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Hachmann, A.; Sevim, E.; Gaballa, A.; Popham, D.L.; Antelmann, H.; Helmann, J.D. Reduction in membrane phosphatidylglycerol content leads to daptomycin resistance in Bacillus subtilis. Antimicrob. Agents Chemother. 2011, 55, 4326–4337. [Google Scholar] [CrossRef] [PubMed]
- Lenarcic, R.; Halbedel, S.; Visser, L.; Shaw, M.; Wu, L.J.; Errington, J.; Marenduzzo, D.; Hamoen, L.W. Localisation of DivIVA by targeting to negatively curved membranes. EMBO J. 2009, 28, 2272–2282. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.T.; Panesso, D.; Mishra, N.N.; Mileykovskaya, E.; Guan, Z.; Munita, J.M.; Reyes, J.; Diaz, L.; Weinstock, G.M.; Murray, B.E.; et al. Daptomycin-resistant Enterococcus faecalis diverts the antibiotic molecule from the division septum and remodels cell membrane phospholipids. MBio 2013, 4, e00281–e002813. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antimicrobial Activity | |||
|---|---|---|---|
| Anti Gram-Positive | Anti Gram-Negative | Broad Spectrum | |
| peptide length | |||
| <10 AA | 16 | 5 | 63 |
| 10–50 AA | 361 | 153 | 1890 |
| 50–100 AA | 49 | 37 | 211 |
| 100–150 AA | 6 | 10 | 35 |
| net charge | |||
| >20 | 0 | 0 | 11 |
| 11–20 | 3 | 4 | 57 |
| 6–10 | 51 | 28 | 415 |
| 1–5 | 308 | 145 | 1212 |
| 0 | 44 | 14 | 112 |
| <0 | 26 | 17 | 100 |
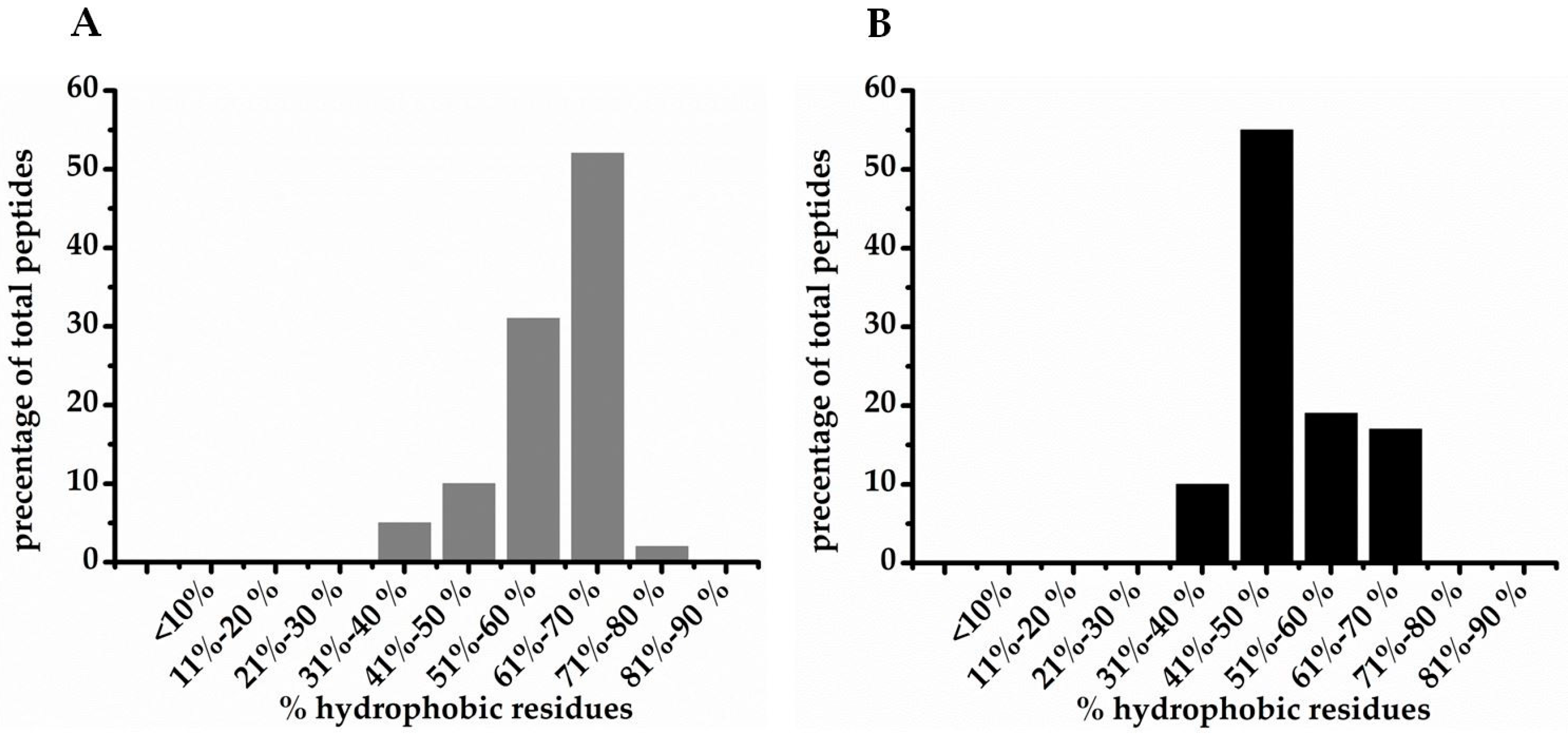
| % hydrophobic residues | |||
| <10 | 1 | 6 | 23 |
| 11–20 | 3 | 4 | 57 |
| 21–30 | 27 | 26 | 155 |
| 31–40 | 136 | 41 | 544 |
| 41–50 | 103 | 73 | 704 |
| 51–60 | 76 | 26 | 440 |
| 61–70 | 76 | 10 | 264 |
| 71–80 | 3 | 1 | 20 |
| 81–90 | 1 | 1 | 3 |
| >90 | 1 | 0 | 3 |
| secondary structure | |||
| unknown | 305 | 127 | 1385 |
| helix | 38 | 19 | 348 |
| beta-strand | 8 | 7 | 77 |
| helix & beta strand (unpacked) | 4 | 0 | 3 |
| helix & beta strand (packed) | 16 | 3 | 59 |
| disulfide bonds | 52 | 23 | 236 |
| rich in unusual AA | 0 | 23 | 70 |
| Total AMP entries 1 | 432 | 205 | 2199 |
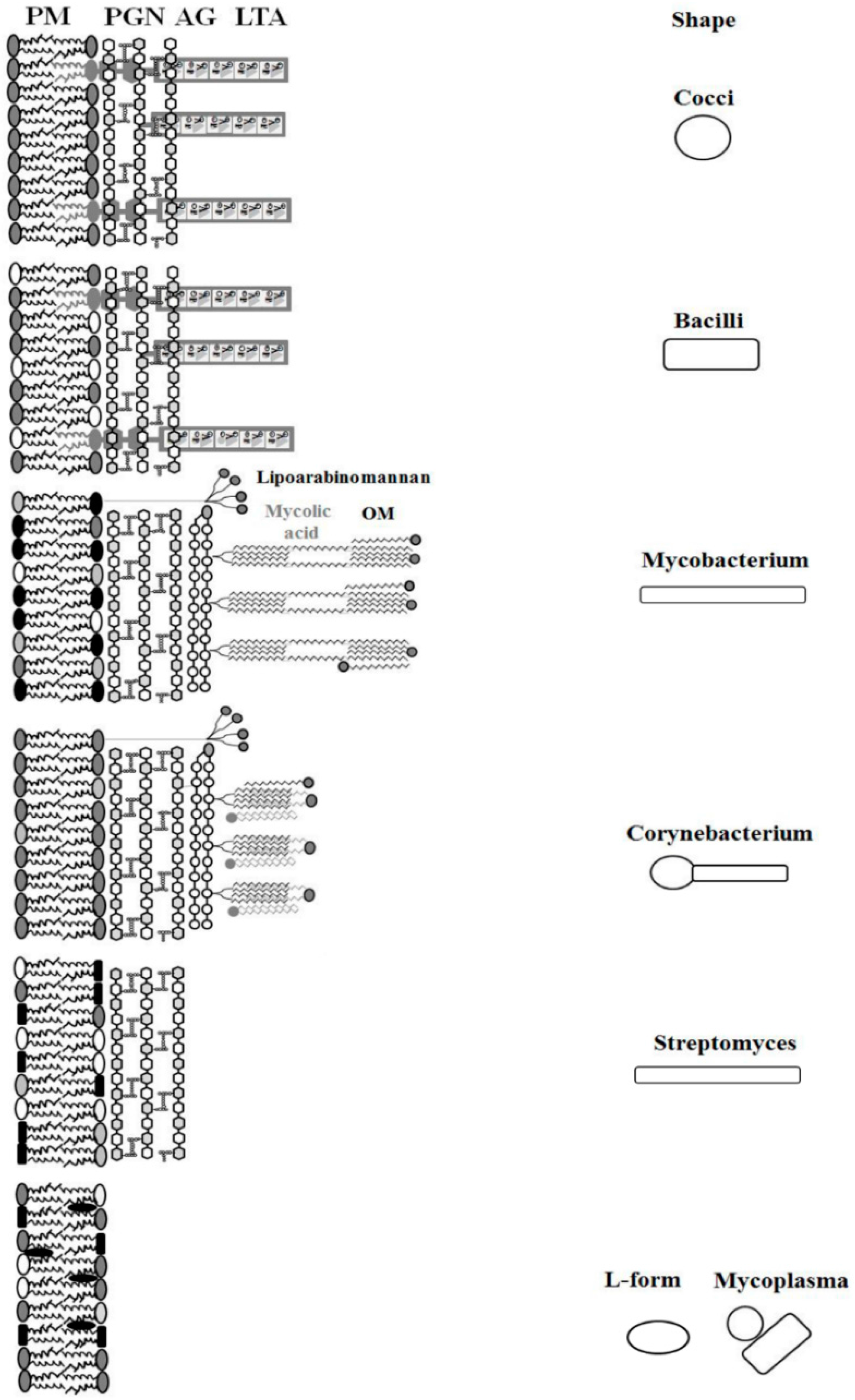
| Organism | Morphology Cell/Colony | Weight Percentage of Total Lipid of Cell Membrane | Major Fatty Acids | References | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PG | lysyl-PG | PI a | PE | CL | NL | Others b | ||||
| Cocci | round/chain | |||||||||
| Staphylococcus aureus | round/cluster | 43 | 30 | - | - | 22 | - | 5 | ai-C15:0; C18:0 ai-C17:0; C20:0 | [53,54] |
| Enterococcus faecium | round/diplococci | 33.8 | 14.4 | - | - | 39 | - | 12.8 | C16:0; C16:1w7c C18:1w7c | [55] |
| Streptococcus sanguis | round/diplococci | 82 | - | - | - | 18 | - | - | C18:1; C16:1; C16:0 | [56,57] |
| Streptococcus pneumoniae | round/cocci lancet shape | 50 | - | - | - | 50 | - | - | C16:0; C16:1; C18:0 | [58] |
| Actinomycetes | spherical pleomorph/filamentous | |||||||||
| Corynebacterium glutamicum | rods/V-shaped | 72.4 | - | 13.1 | - | 14.1 | - | 0,4 | C16:0; C18:1 | [59,60] |
| Streptomyces hygroscopicus | rods, mycelium | - | - | 7 | 36 | 16 c | 30 | 11 | i-C16:0; i-C14:0; i-C15:0 | [61] |
| Mycobacterium tuberculosis | long, slender rods/filamentous | - | - | 13 | 5 | 6 | 54 | 22 | C16:1, C18:Me | [62,63] |
| Cell wall deficient | polymorph/fried egg | |||||||||
| Mycoplasma hominis d | round to oblong/fried egg | 33 | - | - | - | - | 60 | 7 | C16:0; C18:0; C18:1 | [64] |
| L-form S. aureus | polymorph/fried-egg | 26 | 17 | - | - | 54 | 3 | ai-C15:0; C18:0 ai-C17:0; C20:0 | [53,65] | |
| L-form S. hygroscopicus | polymorph/fried-egg | - | - | 13 | 37 | 22 | 16 | 12 | ai-C15:0; ai-C17:0, i-C16:0; C18:2 e | [61] |
| Bacilli | round-ended cylinders/single or in chains | |||||||||
| Clostridium difficile | large, blunt-ended rods/pairs or chains | 100 | - | - | - | - | - | - | C16:0; C16:1 C18:0; C18:1 | [66] |
| Bacillus subtilis | Rods/chain | 70 | - | - | 12 | 4 | - | 14 | ai-C15:0; i-C17:0 | [67,68,69] |
| Listeria monocytogenes | slender, short rods/single or in chains | 29 | 23 | - | 9 | 22 | - | 17 | ai-C15:0; i-C15:0; i-C17:0 | [70] |
| PG | lysyl-PG | CL | PE | |
|---|---|---|---|---|
| Net charge | −1 | +1 | −2 (−1) * | 0 |
| H-bonding ability | no | no | yes | yes |
| Molecular shape | cylindrical | truncated cone | inverted truncated cone | inverted truncated cone |
| Intrinsic curvature | zero | positive | negative | negative |
| Organization | bilayer | bilayer | inverse micelles | inverse micelles |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malanovic, N.; Lohner, K. Antimicrobial Peptides Targeting Gram-Positive Bacteria. Pharmaceuticals 2016, 9, 59. https://doi.org/10.3390/ph9030059
Malanovic N, Lohner K. Antimicrobial Peptides Targeting Gram-Positive Bacteria. Pharmaceuticals. 2016; 9(3):59. https://doi.org/10.3390/ph9030059
Chicago/Turabian StyleMalanovic, Nermina, and Karl Lohner. 2016. "Antimicrobial Peptides Targeting Gram-Positive Bacteria" Pharmaceuticals 9, no. 3: 59. https://doi.org/10.3390/ph9030059