
A Three-Step Process to Isolate Large Quantities of Bioactive Sesquiterpene Lactones from Cichorium intybus L. Roots and Semisynthesis of Chicory STLs Standards

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
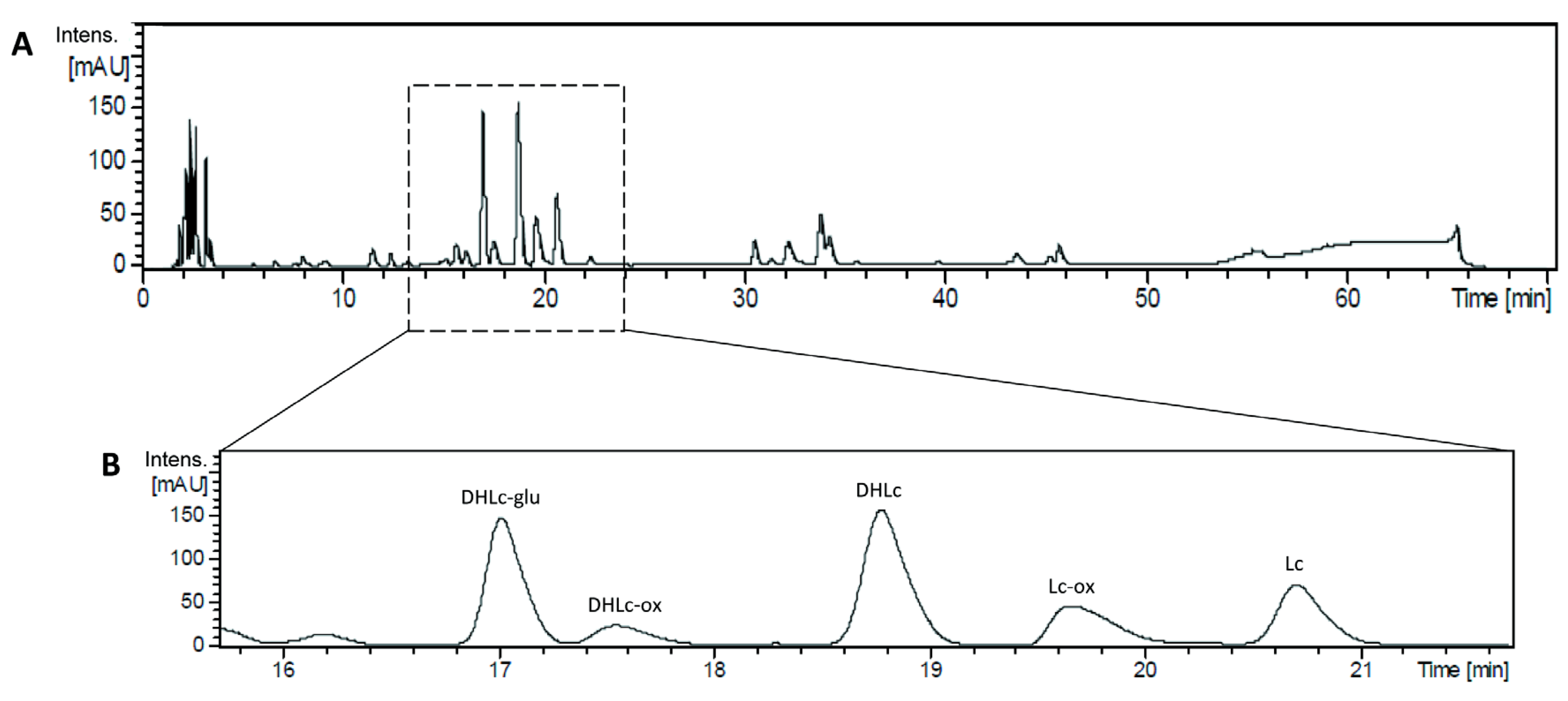
2.1. Identification of the Main STLs
2.2. Method Development
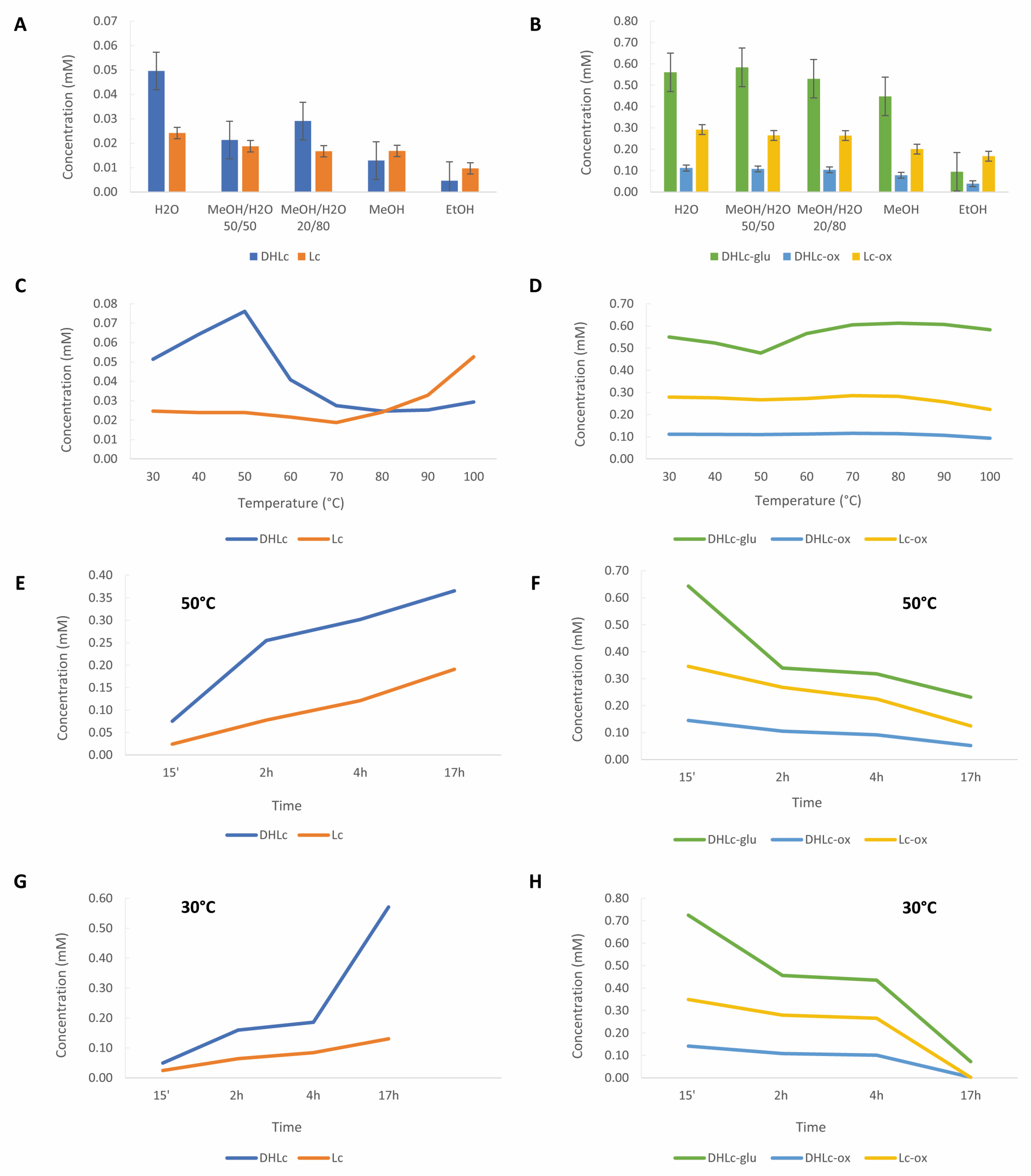
Extraction Conditions Determination
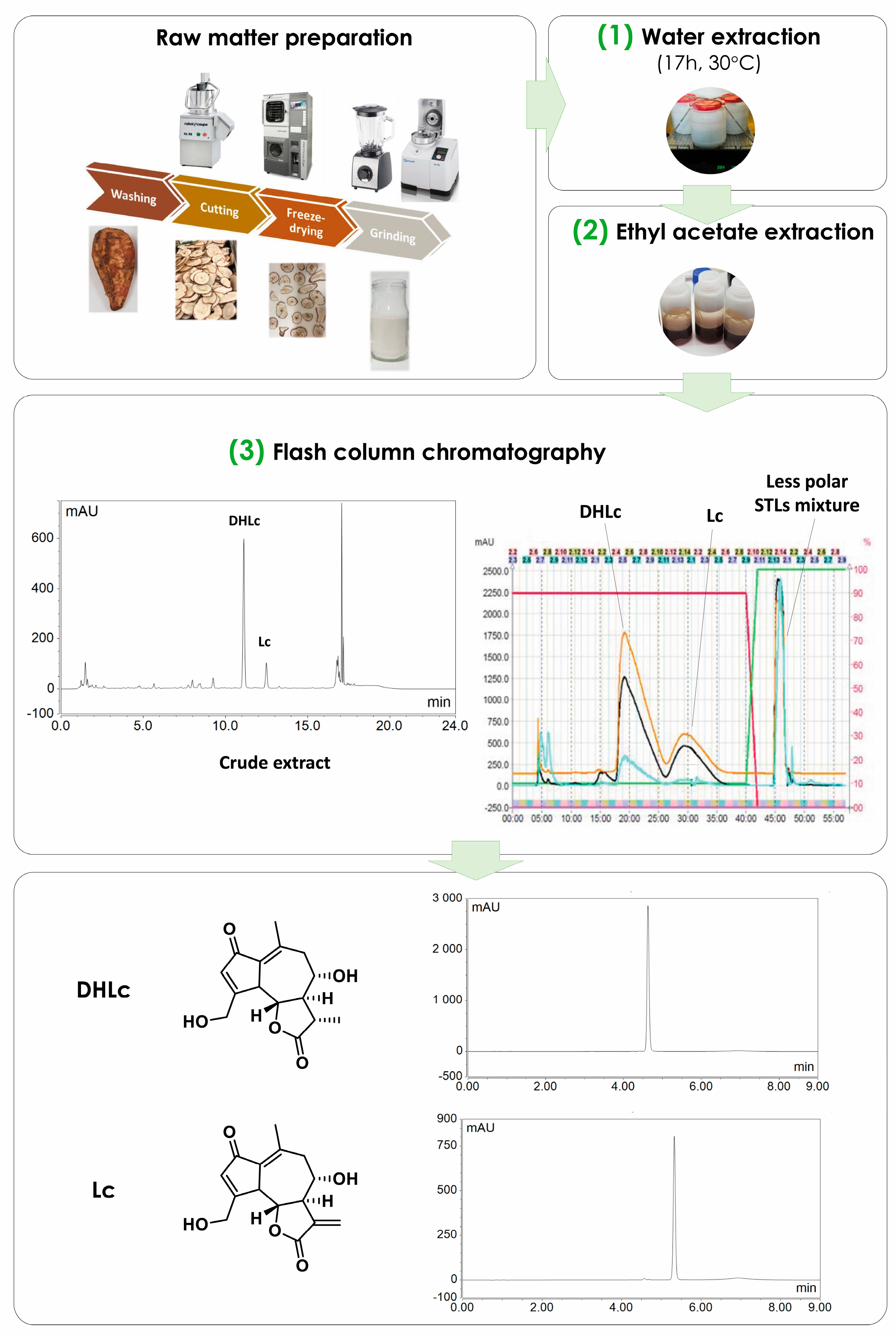
2.3. Scaling-Up
2.4. Analytical Validation of STLs
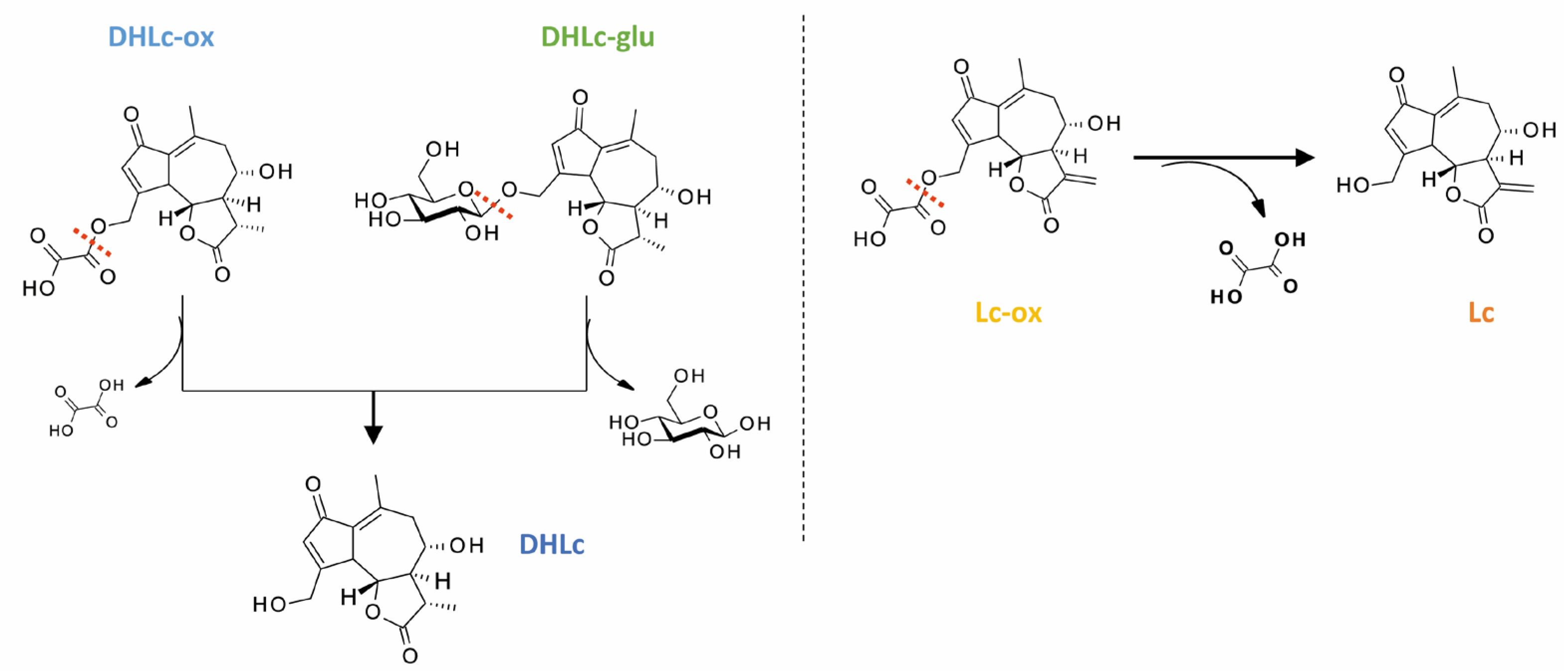
2.5. Isolation of 11β,13-Dihydrolactucin-glucoside
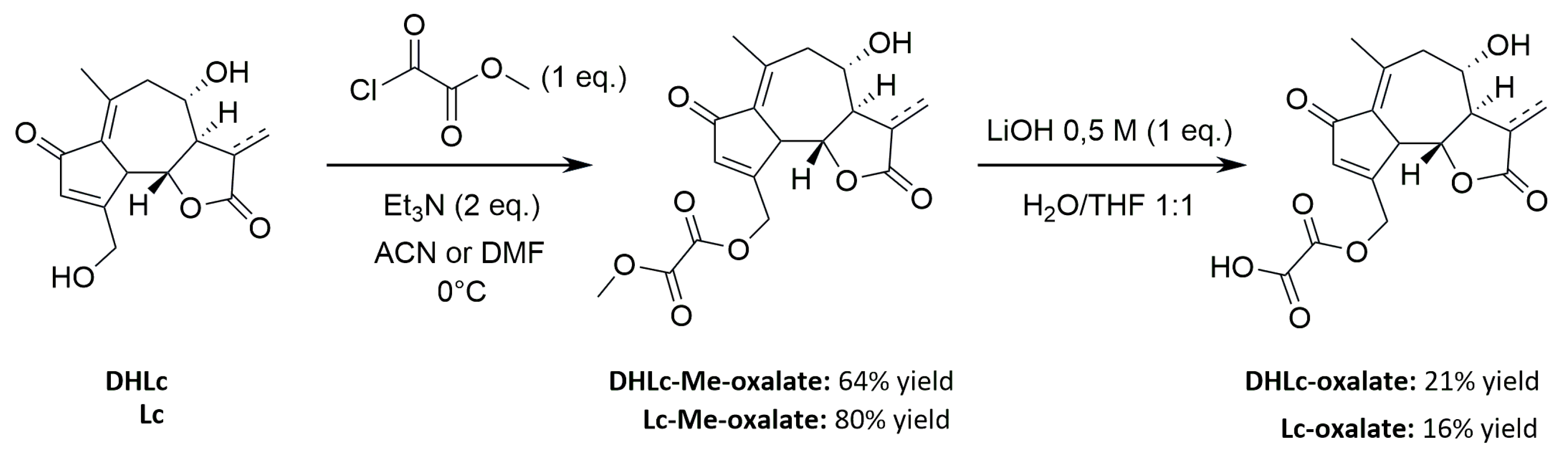
2.6. Synthesis of STLs Standards
3. Discussion
4. Materials and Methods
4.1. Plant Material
4.2. Reagents and Chemicals
4.3. Method Development
4.3.1. Extraction of STLs from Freeze-Dried Chicory Roots
4.3.2. Scaling-Up
Selective Fractionation of Free STLs and Removal of Sugars by Liquid–Liquid Extraction
4.3.3. The Standardized Big-Scale Process
4.4. 11β,13-Dihydrolactucin-glucoside Isolation
4.5. STLs Analysis
4.5.1. Quantification of STLs by UPLC-DAD
4.5.2. LC-QTOF-HRMS Analysis of STLs
4.5.3. NMR Analysis
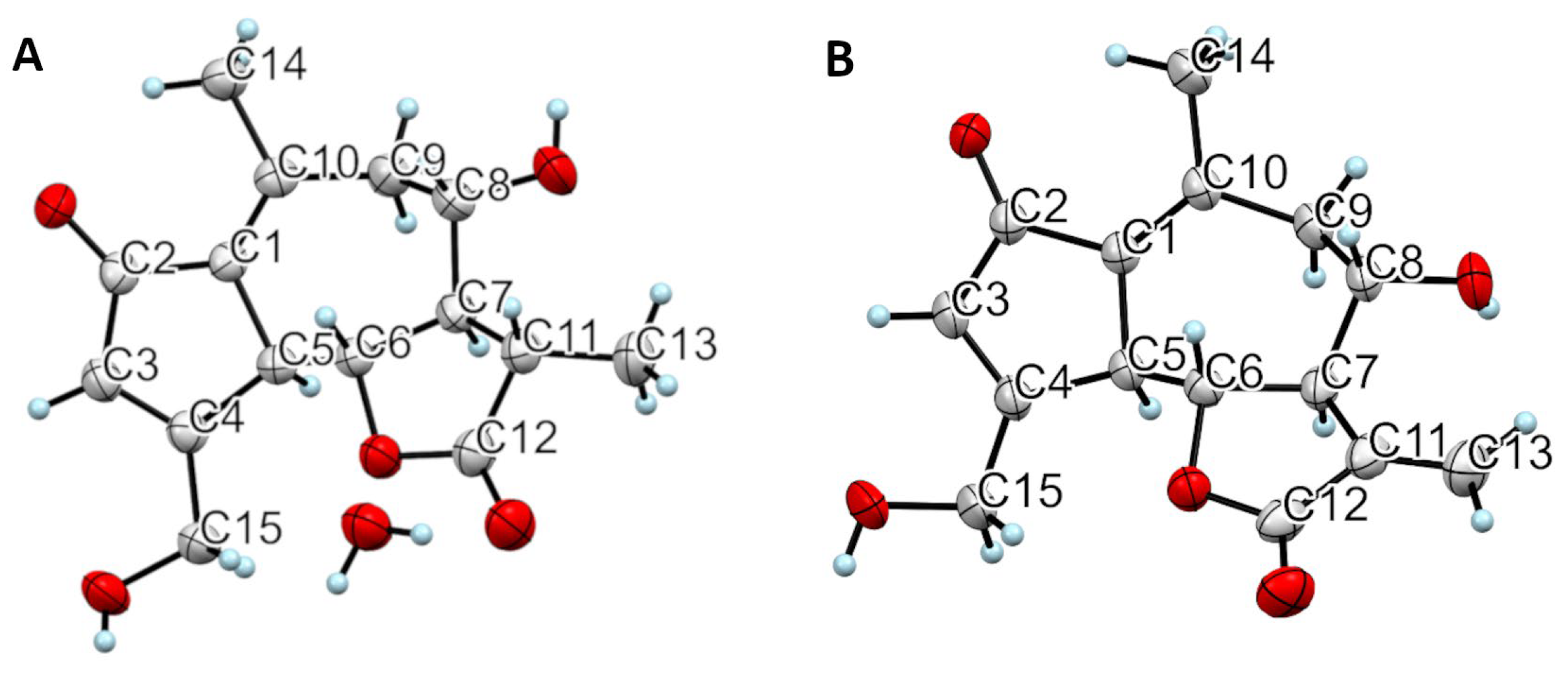
4.5.4. X-ray Structural Determination
4.6. Chemistry
4.6.1. Chemical Synthesis
General Procedure for the Synthesis of DHLc-Me-oxalate and Lc-Me-oxalate
General Procedure for the Synthesis of DHLc-oxalate and Lc-oxalate
4.7. Statistical Assays
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Padilla-Gonzalez, G.F.; dos Santos, F.A.; Da Costa, F.B. Sesquiterpene Lactones: More Than Protective Plant Compounds with High Toxicity. Crit. Rev. Plant Sci. 2016, 35, 18–37. [Google Scholar] [CrossRef]
- Van Beek, T.A.; Maas, P.; King, B.M.; Leclercq, E.; Voragen, A.G.J.; De Groot, A. Bitter Sesquiterpene Lactones from Chicory Roots. J. Agric. Food Chem. 1990, 38, 1035–1038. [Google Scholar] [CrossRef]
- Graziani, G.; Ferracane, R.; Sambo, P.; Santagata, S.; Nicoletto, C.; Fogliano, V. Profiling Chicory Sesquiterpene Lactones by High Resolution Mass Spectrometry. Food Res. Int. 2015, 67, 193–198. [Google Scholar] [CrossRef]
- Ivanescu, B.; Miron, A.; Corciova, A. Sesquiterpene Lactones from Artemisia Genus: Biological Activities and Methods of Analysis. J. Anal. Methods Chem. 2015, 2015, 247685. [Google Scholar] [CrossRef]
- Schmidt, T.J. Structure-Activity Relationships of Sesquiterpene Lactones. In Studies in Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 2006; Volume 33, pp. 309–392. ISBN 978-0-444-52717-2. [Google Scholar]
- Häkkinen, S.T.; Nohynek, L.; Ivanov, M.; Tsitko, I.; Matos, M.; Baixinho, J.P.; Ivasiv, V.; Fernández, N. Chicory Extracts and Sesquiterpene Lactones Show Potent Activity against Bacterial and Fungal Pathogens. Pharmaceuticals 2021, 14, 941. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, M.; Cai, G.H.; Chen, Y.; Shi, X.C.; Zhang, C.C.; Xia, B.; Xie, B.C.; Liu, H.; Zhang, R.X.; et al. A Potential Nutraceutical Candidate Lactucin Inhibits Adipogenesis through Downregulation of JAK2/STAT3 Signaling Pathway-Mediated Mitotic Clonal Expansion. Cells 2020, 9, 331. [Google Scholar] [CrossRef] [PubMed]
- Imam, K.M.S.U.; Tian, Y.; Xin, F.; Xie, Y.; Wen, B. Lactucin, a Bitter Sesquiterpene from Cichorium Intybus, Inhibits Cancer Cell Proliferation by Downregulating the MAPK and Central Carbon Metabolism Pathway. Molecules 2022, 27, 7358. [Google Scholar] [CrossRef]
- Bischoff, T.A.; Kelley, C.J.; Karchesy, Y.; Laurantos, M.; Nguyen-Dinh, P.; Arefi, A.G. Antimalarial Activity of Lactucin and Lactucopicrin: Sesquiterpene Lactones Isolated from Cichorium Intybus L. J. Ethnopharmacol. 2004, 95, 455–457. [Google Scholar] [CrossRef]
- Wesołowska, A.; Nikiforuk, A.; Michalska, K.; Kisiel, W.; Chojnacka-Wójcik, E. Analgesic and Sedative Activities of Lactucin and Some Lactucin-like Guaianolides in Mice. J. Ethnopharmacol. 2006, 107, 254–258. [Google Scholar] [CrossRef]
- Rojas-Silva, P. Leishmanicidal, Anti-Inflammatory and Anti-Obesity Properties of Natural Products from Common Medicinal and Edible Plants. Ph.D. Thesis, Rutgers University, New Brunswick, NJ, USA, 2014. [Google Scholar] [CrossRef]
- Jaśkiewicz, A.; Budryn, G.; Carmena-Bargueño, M.; Pérez-Sánchez, H. Evaluation of Activity of Sesquiterpene Lactones and Chicory Extracts as Acetylcholinesterase Inhibitors Assayed in Calorimetric and Docking Simulation Studies. Nutrients 2022, 14, 3633. [Google Scholar] [CrossRef]
- Sessa, R.A.; Bennett, M.H.; Lewis, M.J.; Mansfield, J.W.; Beale, M.H. Metabolite Profiling of Sesquiterpene Lactones from Lactuca Species. J. Biol. Chem. 2000, 275, 26877–26884. [Google Scholar] [CrossRef] [PubMed]
- Willeman, H.; Hance, P.; Fertin, A.; Voedts, N.; Duhal, N.; Goossens, J.-F.; Hilbert, J.-L. A Method for the Simultaneous Determination of Chlorogenic Acid and Sesquiterpene lactone Content in Industrial Chicory Root Foodstuffs. Sci. World J. 2014, 2014, 583180. [Google Scholar] [CrossRef] [PubMed]
- Chadni, M.; Isidore, E.; Diemer, E.; Ouguir, O.; Brunois, F.; Catteau, R.; Cassan, L.; Ioannou, I. Optimization of Extraction Conditions to Improve Chlorogenic Acid Content and Antioxidant Activity of Extracts from Forced Witloof Chicory Roots. Foods 2022, 11, 1217. [Google Scholar] [CrossRef] [PubMed]
- Milala, J.; Grzelak, K.; Król, B.; Juśkiewicz, J.; Zduńczyk, Z. Composition and Properties of Chicory Extracts Rich in Fructans and Polyphenols. Pol. J. Food Nutr. Sci. 2009, 59, 35–43. [Google Scholar]
- Sinkovič, L.; Hribar, J.; Žnidarčič, D.; Treutter, D.; Vidrih, R. Comparison of the Phenolics Profiles of Forced and Unforced Chicory (Cichorium Intybus L.) Cultivars. Agr. Conspec. Sci. 2014, 79, 133–137. [Google Scholar]
- Deng, Y.; Scott, L.; Swanson, D.; Snyder, J.K.; Sari, N.; Dogan, H. Guaianolide Sesquiterpene Lactones from Cichorium Intybus (Asteraceae) [1]. Z. Für Nat. B 2001, 56, 787–796. [Google Scholar] [CrossRef]
- Michalska, K.; Szneler, E.; Kisiel, W. Complete NMR Spectral Assignments of Two Lactucin-Type Sesquiterpene Lactone Glycosides from Picris Conyzoides: Sesquiterpene lactone Glycosides from Picris conyzoides. Magn. Reson. Chem. 2011, 49, 753–756. [Google Scholar] [CrossRef]
- Dang, T.; Zheng, G.; Zhang, Q.; Jin, P.; Zhang, H.; Su, L.; Qin, D.; Yao, G. Sesquiterpenoids with Diverse Carbon Skeletons from the Roots of Cichorium glandulosum and Their Anti-Inflammatory Activities. Fitoterapia 2019, 136, 104170. [Google Scholar] [CrossRef]
- Ruban, G.; Zabel, V.; Gensch, K.H.; Smalla, H. The Crystal Structure and Absolute Configuration of Lactucin. Acta Crystallogr. B Struct. Sci. 1978, 34, 1163–1167. [Google Scholar] [CrossRef]
- Escobar-Ledesma, F.R.; Sánchez-Moreno, V.E.; Vera, E.; Ciobotă, V.; Jentzsch, P.V.; Jaramillo, L.I. Extraction of Inulin from Andean Plants: An Approach to Non-Traditional Crops of Ecuador. Molecules 2020, 25, 5067. [Google Scholar] [CrossRef]
- 11beta,13-Dihydrolactucin 3810, CAS 83117-63-9-SESQUITERPENE. Available online: https://www.extrasynthese.com/sesquiterpene/290-11beta13-dihydrolactucin.html (accessed on 5 March 2023).
- Leclercq, E.; Netjes, J.J. Release of Sesquiterpene Lactones by Enzymatic Liquefaction of Chicory Roots. Z Leb. Unters. 1985, 181, 475–477. [Google Scholar] [CrossRef]
- Baixinho, J.P.; Anastácio, J.D.; Ivasiv, V.; Cankar, K.; Bosch, D.; Menezes, R.; de Roode, M.; dos Santos, C.N.; Matias, A.A.; Fernández, N. Supercritical CO2 Extraction as a Tool to Isolate Anti-Inflammatory Sesquiterpene Lactones from Cichorium intybus L. Roots. Molecules 2021, 26, 2583. [Google Scholar] [CrossRef] [PubMed]
- Ajiaikebaier, A.; Wei, Z.; Sun, G. Preparation Method of Lactucin. Chinese Patent CN111689935A, 22 September 2020. [Google Scholar]
- Katiyar, K.C.; Duggar, R.; Yadav, S.S.; Sharma, N.; Ray, A.; Shirumalla, R.; Bajpai, M.; Shukla, G.; Subhadra, B.; Kontham, L.; et al. Preparation Method and Application of Lactucin Extracted from Cichorium Intybus. Chinese Patent CN112939912A, 11 June 2021. [Google Scholar]
- Xin, X.; Ajiaikebaier, A.; Li, Y. Lactucin and Its Preparation Method and Application. Chinese Patent CN101099566A, 9 January 2008. [Google Scholar]
- Qin, D.; Zou, N.; Cai, G.; Wang, S.; Su, L.; Dang, T. Lactucin and Application Thereof as Anti-Inflammatory Component. Chinese Patent CN112079804A, 15 December 2020. [Google Scholar]
- De Oliveira, D.M.P.; Forde, B.M.; Kidd, T.J.; Harris, P.N.A.; Schembri, M.A.; Beatson, S.A.; Paterson, D.L.; Walker, M.J. Antimicrobial Resistance in ESKAPE Pathogens. Clin. Microbiol. Rev. 2020, 33, e00181-19. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| R | R1 | R2 | |
| Lactucin | OH | CH2 | OH |
| Lactucin-15-oxalate | OH | CH2 | OCOCOOH |
| Lactucin-15-glucoside | OH | CH2 | OGlucose |
| 11β,13-dihydrolactucin | OH | CH3 | OH |
| 11β,13-dihydrolactucin-15-oxalate | OH | CH3 | OCOCOOH |
| 11β,13-dihydrolactucin-15-glucoside | OH | CH3 | OGlucose |
| 8-deoxylactucin | H | CH2 | OH |
| 8-deoxylactucin-15-oxalate | H | CH2 | OCOCOOH |
| 8-deoxylactucin-15-glucoside | H | CH2 | OGlucose |
| 11β,13-dihydro-8-deoxylactucin | H | CH3 | OH |
| 11β,13-dihydro-8-deoxylactucin-oxalate | H | CH3 | OCOCOOH |
| 11β,13-dihydro-8-deoxylactucin-glucoside | H | CH3 | OGlucose |
| Lactucopicrin |  | CH2 | OH |
| Lactucopicrin-15-oxalate | CH2 | OCOCOOH | |
| Lactucopicrin-15-glucoside | CH2 | OGlucose | |
| 11β,13-dihydrolactucopicrin |  | CH3 | OH |
| 11β,13-dihydrolactucopicrin-15-oxalate | CH3 | OCOCOOH | |
| 11β,13-dihydrolactucopicrin-15-glucoside | CH3 | OGlucose | |
| Compound | Molecular Weight | Retention Time (min) | ESI Major Fragment Ions (m/z) |
|---|---|---|---|
| DHLc-glu | 440 | 16.9 | 463 (100) [M+Na]+; 441 (13) [M+H]+; 279 (18) [M-glucosyl+H]+; 261 (9) [M-glucosyl-H2O+H]+; 243 (10) [M-glucosyl-2H2O+H]+; 215 (8) [M-glucosyl-H2O-HCO2H+H]+ |
| DHLc-ox | 350 | 17.7 | 373 (57) [M+Na]+; 351 (100) [M+H]+; 261 (94) [M-oxaloyl-H2O+H]+; 243 (56) [M-oxaloyl-2H2O+H]+; 215 (39) [M-oxaloyl-H2O-HCO2H+H]+ |
| DHLc | 278 | 18.8 | 301 (100) [M+Na]+; 279 (59) [M+H]+; 261 (7) [M-H2O+H]+; 243 (8) [M-2H2O+H]+; 215 (17) [M-HCO2H-H2O+H]+ |
| Lc-ox | 348 | 19.7 | 371 (73) [M+Na]+; 349 (86) [M+H]+; 259 (100) [M-oxaloyl-H2O+H]+; 241 (96) [M-oxaloyl-2H2O+H]+; 213 (60) [M-oxaloyl-H2O-HCO2H+H]+ |
| Lc | 276 | 20.7 | 299 (100) [M+Na]+; 277 (44) [M+H]+; 259 (8) [M-H2O+H]+; 241 (17) [M-2H2O+H]+; 213 (18) [M-HCO2H-H2O+H]+ |
| STLs Concentration (mM) ± SD | ||||||||
|---|---|---|---|---|---|---|---|---|
| Parameter | DHLc-glu | DHLc-ox | DHLc | Lc-ox | Lc | TOTAL Conjugated STLs | TOTAL Free STLs | |
| Solvent (15’, 30 °C) | H2O | 0.561 ± 0.012 | 0.112 ± 0.004 | 0.050 ± 0.003 | 0.292 ± 0.008 | 0.024 ± 0.002 | 0.965 ± 0.023 c | 0.0737 ± 0.005 d |
| MeOH/H2O 20/80 | 0.530 ± 0.099 | 0.104 ± 0.019 | 0.029 ± 0.004 | 0.264 ± 0.048 | 0.017 ± 0.004 | 0.898 ± 0.167 bc | 0.0457 ± 0.008 c | |
| MeOH/H2O 50/50 | 0.584 ± 0.010 | 0.108 ± 0.003 | 0.021 ± 0.001 | 0.265 ± 0.005 | 0.019 ± 0.002 | 0.957 ± 0.018 c | 0.0400 ± 0.003 bc | |
| MeOH | 0.448 ± 0.029 | 0.078 ± 0.005 | 0.013 ± 0.000 | 0.201 ± 0.016 | 0.017 ± 0.003 | 0.727 ± 0.049 b | 0.0297 ± 0.003 b | |
| EtOH | 0.095 ± 0.002 | 0.039 ± 0.006 | 0.005 ± 0.000 | 0.167 ± 0.005 | 0.009 ± 0.000 | 0.301 ± 0.008 a | 0.0143 ± 0.000 a | |
| Temperature (15’, H2O) | 30 °C | 0.550 ± 0.006 | 0.112 ± 0.002 | 0.051 ± 0.001 | 0.279 ± 0.006 | 0.025 ± 0.001 | 0.940 ± 0.011 ab | 0.076 ± 0.001 a |
| 40 °C | 0.523 ± 0.011 | 0.111 ± 0.004 | 0.064 ± 0.001 | 0.276 ± 0.008 | 0.024 ± 0.000 | 0.909 ± 0.023 bc | 0.088 ± 0.001 b | |
| 50 °C | 0.478 ± 0.010 | 0.110 ± 0.001 | 0.076 ± 0.001 | 0.267 ± 0.003 | 0.024 ± 0.000 | 0.855 ± 0.015 d | 0.100 ± 0.001 c | |
| 60 °C | 0.565 ± 0.013 | 0.112 ± 0.002 | 0.041 ± 0.000 | 0.272 ± 0.005 | 0.022 ± 0.001 | 0.950 ± 0.014 ac | 0.062 ± 0.002 d | |
| 70 °C | 0.605 ± 0.011 | 0.116 ± 0.004 | 0.028 ± 0.000 | 0.286 ± 0.004 | 0.019 ± 0.000 | 1.007 ± 0.022 e | 0.046 ± 0.001 e | |
| 80 °C | 0.612 ± 0.005 | 0.114 ± 0.002 | 0.025 ± 0.000 | 0.282 ± 0.004 | 0.024 ± 0.000 | 1.009 ± 0.006 e | 0.049 ± 0.001 e | |
| 90 °C | 0.607 ± 0.013 | 0.107 ± 0.003 | 0.025 ± 0.000 | 0.258 ± 0.002 | 0.033 ± 0.001 | 0.971 ± 0.017 ae | 0.058 ± 0.001 f | |
| 100 °C | 0.583 ± 0.008 | 0.093 ± 0.002 | 0.029 ± 0.001 | 0.223 ± 0.002 | 0.053 ± 0.053 | 0.900 ± 0.008 bd | 0.082 ± 0.001 g | |
| Time (H2O, 30 °C) | 15’ | 0.552 ± 0.005 | 0.113 ± 0.026 | 0.049 ± 0.006 | 0.291 ± 0.066 | 0.024 ± 0.003 | 0.955 ± 0.015 * | 0.074 ± 0.026 ns |
| 2 h | 0.456 ± 0.039 | 0.108 ± 0.008 | 0.160 ± 0.016 | 0.280 ± 0.022 | 0.064 ± 0.007 | 0.844 ± 0.069 ** | 0.224 ± 0.023 *** | |
| 4 h | 0.435 ± 0.048 | 0.101 ± 0.010 | 0.185 ± 0.011 | 0.265 ± 0.022 | 0.084 ± 0.008 | 0.801 ± 0.080 *** | 0.270 ± 0.019 **** | |
| 17 h | 0.072 ± 0.013 | 0.003 ± 0.000 | 0.571 ± 0.066 | 0.002 ± 0.000 | 0.131 ± 0.006 | 0.077 ± 0.014 **** | 0.701 ± 0.070 **** | |
| Time (H2O, 50 °C) | 15’ | 0.485 ± 0.014 | 0.110 ± 0.002 | 0.075 ± 0.001 | 0.269 ± 0.006 | 0.024 ± 0.001 | 0.864 ± 0.019 | 0.100 ± 0.001 |
| 2 h | 0.340 ± 0.017 | 0.105 ± 0.004 | 0.255 ± 0.008 | 0.268 ± 0.014 | 0.078 ± 0.004 | 0.712 ± 0.035 | 0.333 ± 0.012 | |
| 4 h | 0.318 ± 0.016 | 0.091 ± 0.001 | 0.302 ± 0.003 | 0.225 ± 0.003 | 0.121 ± 0.003 | 0.634 ± 0.020 | 0.423 ± 0.003 | |
| 17 h | 0.231 ± 0.011 | 0.052 ± 0.003 | 0.366 ± 0.015 | 0.125 ± 0.008 | 0.191 ± 0.011 | 0.408 ± 0.021 | 0.556 ± 0.025 | |
| STLs Concentration (mM) ± SD | ||
|---|---|---|
| DHLc | Lc | |
| Initial H2O | 0.571 ± 0.066 | 0.134 ± 0.005 |
| EtOAc 1 | 0.345 ± 0.010 | 0.081 ± 0.002 |
| EtOAc 2 | 0.153 ± 0.004 | 0.033 ± 0.003 |
| EtOAc 3 | 0.066 ± 0.000 | 0.013 ± 0.011 |
| FinalH2O | 0.004 ± 0.016 | 0.005 ± 0.003 |
| DHLc: (CD3)2SO | Lc: (CD3)2SO | |||
|---|---|---|---|---|
| Position | 1H (mult, J Hz) | 13C | 1H (mult, J Hz) | 13C |
| 1 | - | 132.0 | - | 132.3 |
| 2 | - | 194.9 | - | 194.8 |
| 3 | 6.27 (s) | 132.5 | 6.28 (d, 1.0) | 132.7 |
| 4 | - | 175.8 | - | 169.2 |
| 5 | 3.76–3.64 (m) | 48.4 | 3.83–3.67 (m) | 48.3 |
| 6 | 3.76–3.64 (m) | 80.9 | 3.83–3.67 (m) | 81.1 |
| 7 | 2.17–2.05 (m) | 60.5 | 3.12–3.03 (m) | 56.8 |
| 8 | 3.61–3.49 (m) | 68.4 | 3.83–3.67 (m) | 66.9 |
| 9 | 9: 2.74–2.55 (m) | 48.7 | 9: 2.74–2.55 (m) | 48.8 |
| 9’: 2.24 (dd, 13.6, 1.8) | 9’: 2.24 (dd, 13.6, 1.8) | |||
| 10 | - | 147.5 | - | 147.0 |
| 11 | 2.74–2.55 (m) | 40.9 | - | 138.4 |
| 12 | - | 178.3 | - | 175.4 |
| 13 | 1.25 (d, 7.0) | 15.7 | 13: 6.13 (dd, 3.0, 1.5) | 121.9 |
| 13’: 6.01 (dd, 3.2, 1.5) | ||||
| 14 | 2.33 (s) | 21.5 | 2.33 (s) | 21.5 |
| 15 | 15: 4.64 (ddd, 18.8, 5.8, 1.9) | 61.8 | 15: 4.66 (d, 18.7) | 61.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruggieri, F.; Hance, P.; Gioia, B.; Biela, A.; Roussel, P.; Hilbert, J.-L.; Willand, N. A Three-Step Process to Isolate Large Quantities of Bioactive Sesquiterpene Lactones from Cichorium intybus L. Roots and Semisynthesis of Chicory STLs Standards. Pharmaceuticals 2023, 16, 771. https://doi.org/10.3390/ph16050771
Ruggieri F, Hance P, Gioia B, Biela A, Roussel P, Hilbert J-L, Willand N. A Three-Step Process to Isolate Large Quantities of Bioactive Sesquiterpene Lactones from Cichorium intybus L. Roots and Semisynthesis of Chicory STLs Standards. Pharmaceuticals. 2023; 16(5):771. https://doi.org/10.3390/ph16050771
Chicago/Turabian StyleRuggieri, Francesca, Philippe Hance, Bruna Gioia, Alexandre Biela, Pascal Roussel, Jean-Louis Hilbert, and Nicolas Willand. 2023. "A Three-Step Process to Isolate Large Quantities of Bioactive Sesquiterpene Lactones from Cichorium intybus L. Roots and Semisynthesis of Chicory STLs Standards" Pharmaceuticals 16, no. 5: 771. https://doi.org/10.3390/ph16050771