

Assessment of Aptamer as a Potential Drug Targeted Delivery for Retinal Angiogenesis Inhibition
 , , , , and
, , , , and
Abstract
:1. Introduction
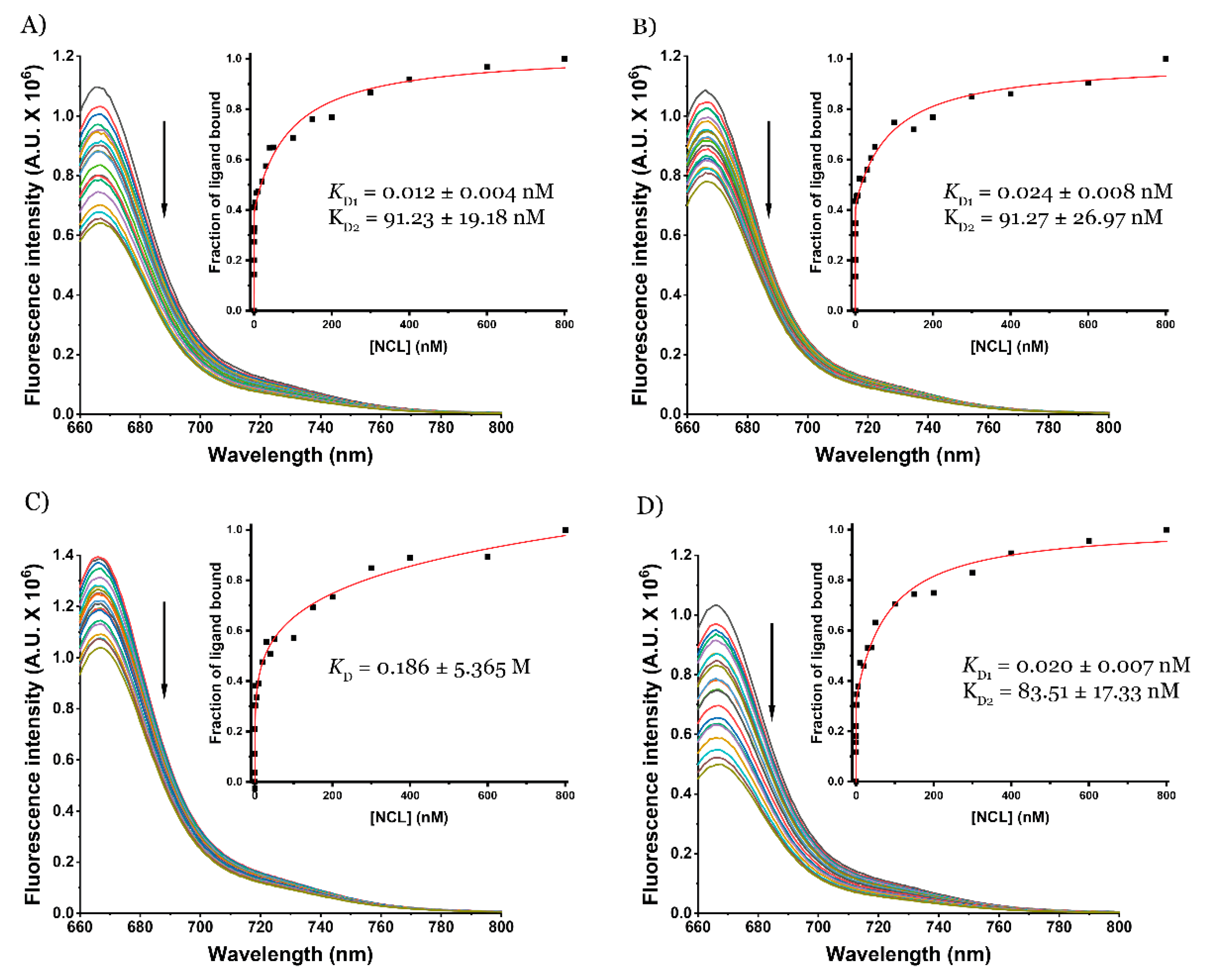
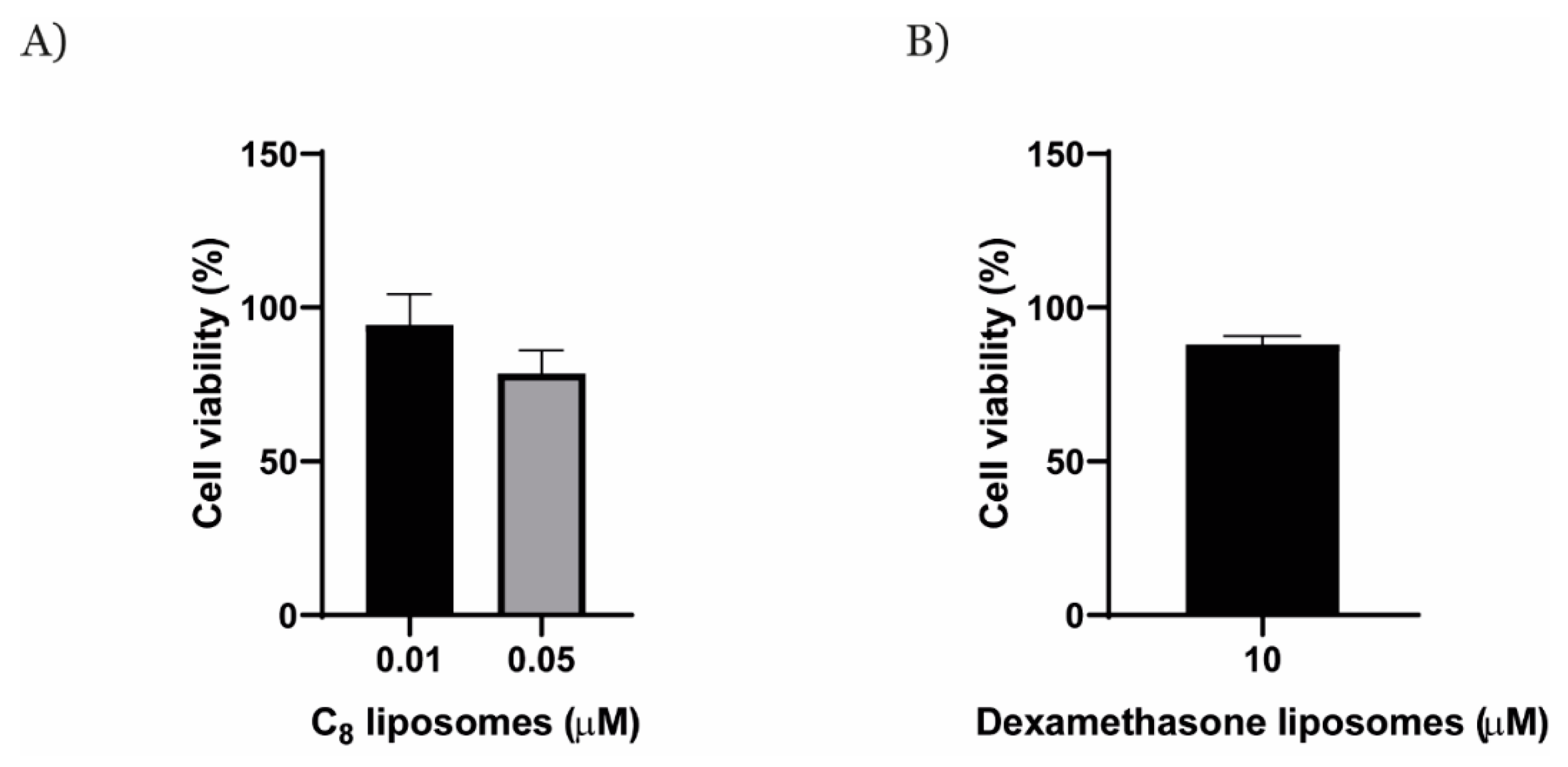
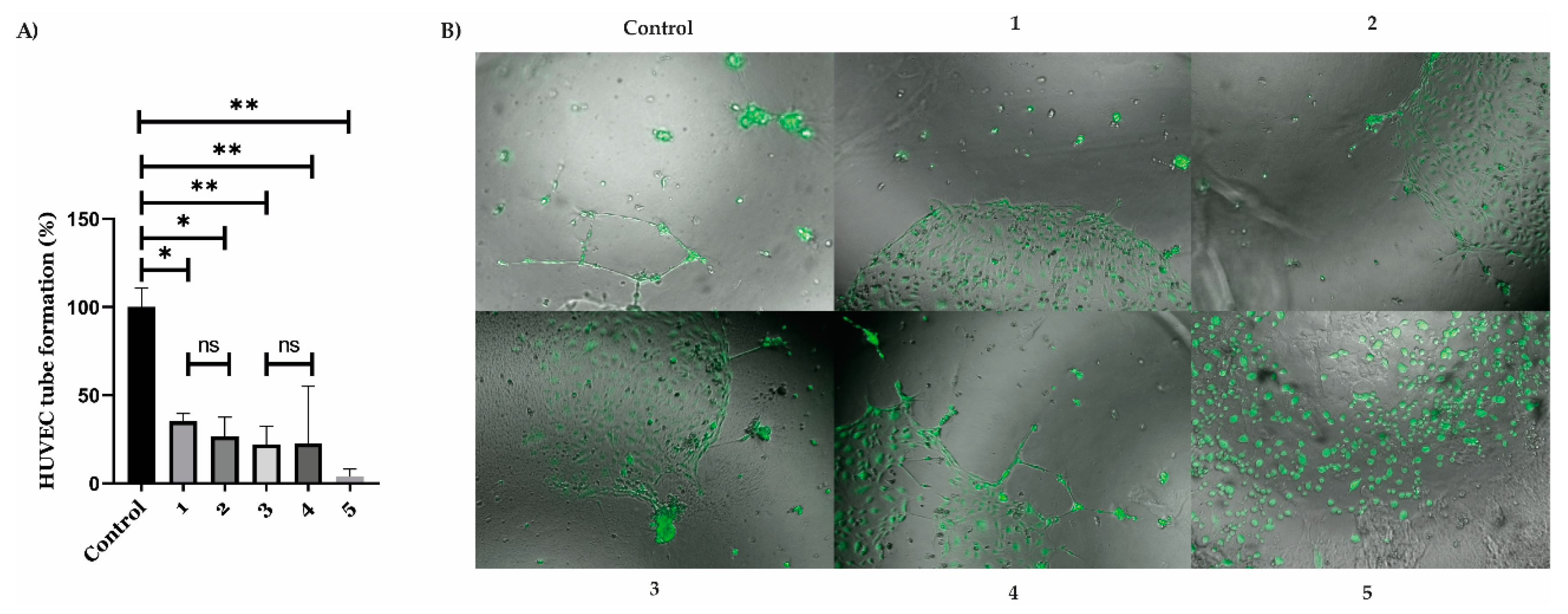
2. Results
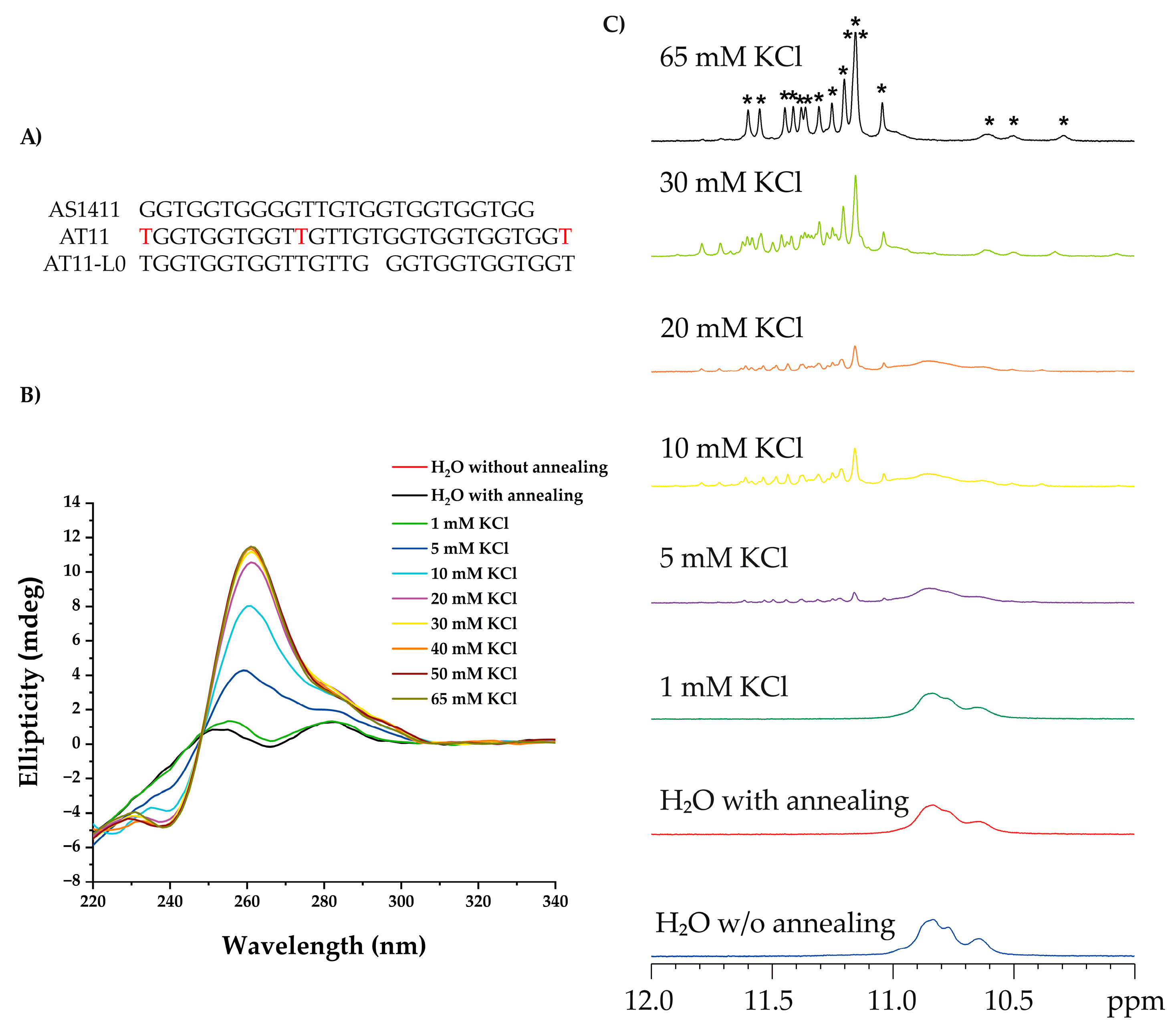
CD Spectroscopy Measurements
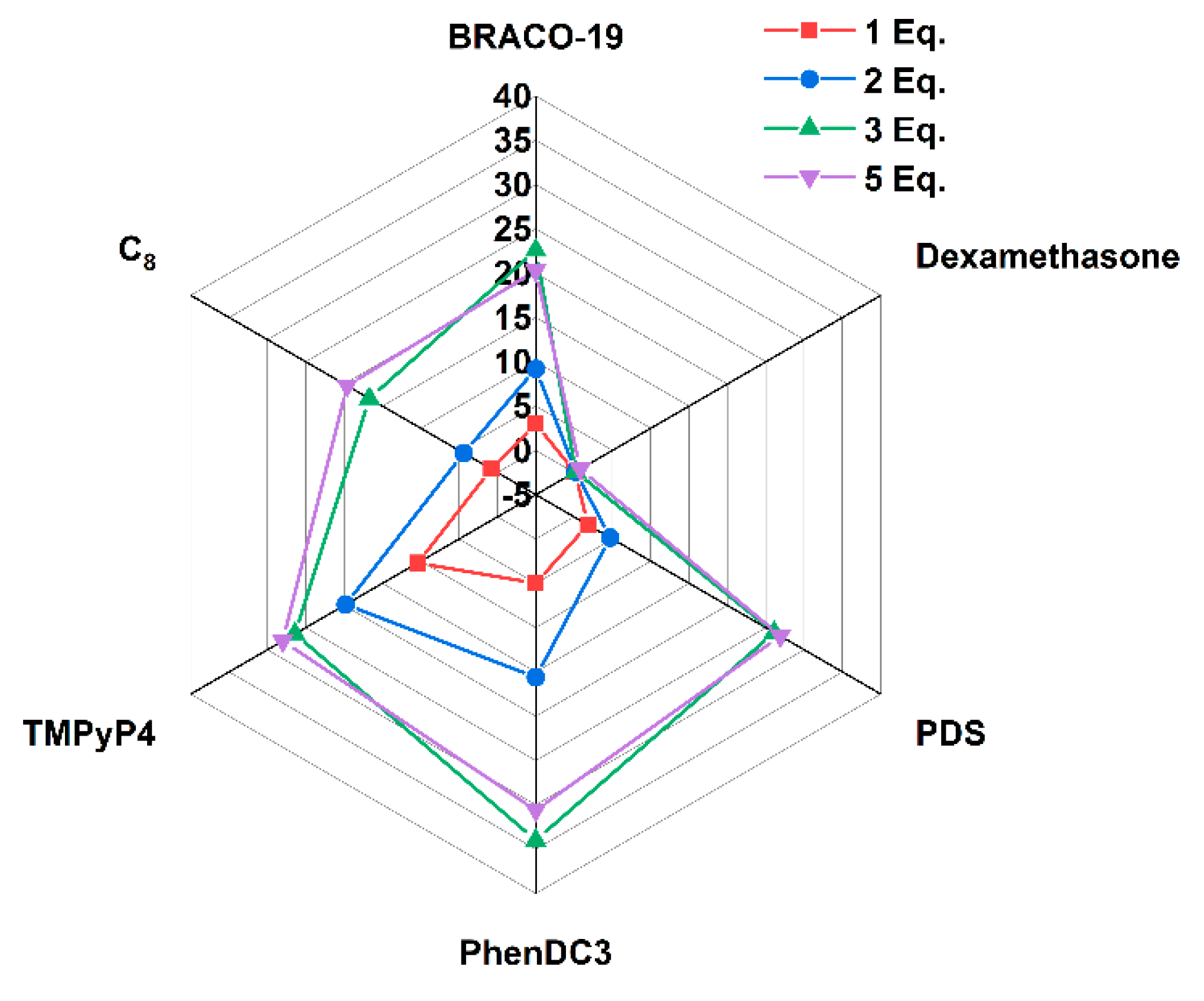
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Circular Dichroism
4.3. NMR Spectroscopy
4.4. Förster Resonance Energy Transfer (FRET)-Melting Experiments
4.5. Fluorescence Spectroscopy
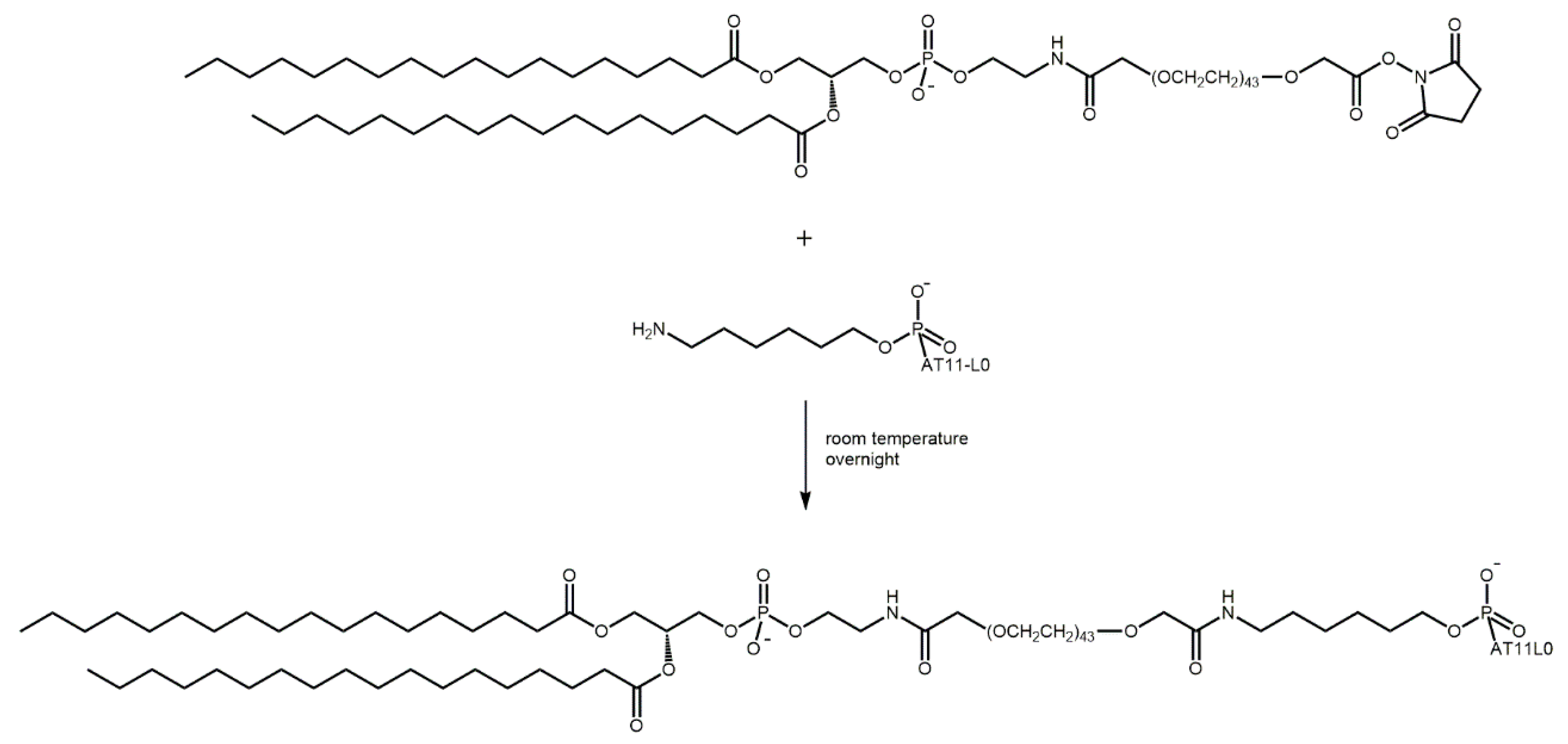
4.6. Synthesis of Liposomes Functionalized with AT11-L0 and Ligands
4.7. Structural Characterization of Liposomes
4.8. Confocal Fluorescence Microscopy
4.9. MTT Assay
4.10. Angiogenesis Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Usui, Y.; Wetenskow, P.D.; Murinello, S.; Dorrell, M.I.; Scheppke, L.; Bucher, F.; Sakimoto, S.; Paris, L.P.; Aguilar, E.; Friedlander, M. Angiogenesis and Eye Disease. Annu. Rev. Vis. Sci. 2015, 1, 155–184. [Google Scholar] [CrossRef] [PubMed]
- Gariano, R.F.; Gardner, T.W. Retinal Angiogenesis in Development and Disease. Nature 2005, 438, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Parachuri, N.; Kumar, N.; Bandello, F.; Kuppermann, B.D.; Loewenstein, A.; Regillo, C.D.; Chakravarthy, U. Terms Non-exudative and Non-neovascular: Awaiting Entry at the Doors of AMD Reclassification. Graefe’s Arch. Clin. Exp. Ophthalmol. 2021, 259, 1381–1383. [Google Scholar] [CrossRef]
- Rossino, M.G.; Monte, M.D.; Casini, G. Relationships Between Neurodegeneration and Vascular Damage in Diabetic Retinopathy. Front. Neurosci. 2019, 13, 1172. [Google Scholar] [CrossRef]
- McAllister, I.L. Central Retinal Vein Occlusion: A Review. Clin. Experiment. Ophthalmol. 2012, 40, 48–58. [Google Scholar] [CrossRef]
- Yorston, D. Retinal Diseases and VISION 2020. Community Eye Health 2003, 16, 19. [Google Scholar]
- Santos, F.M.; Ciordia, S.; Mesquita, J.; Cruz, C.; Sousa, J.P.C.; Passarinha, L.A.; Tomaz, C.T.; Paradela, A. Proteomics Profiling of Vitreous Humor Reveals Complement and Coagulation Components, Adhesion Factors, and Neurodegeneration Markers as Discriminatory Biomarkers of Vitreoretinal Eye Diseases. Front. Immunol. 2023, 14, 1107295. [Google Scholar] [CrossRef]
- Santos, F.M.; Mesquita, J.; Castro-de-Sousa, J.P.; Ciordia, S.; Paradela, A.; Tomaz, C.T. Vitreous Humor Proteome: Targeting Oxidative Stress, Inflammation, and Neurodegeneration in Vitreoretinal Diseases. Antioxidants 2022, 11, 505. [Google Scholar] [CrossRef]
- Jadeja, R.N.; Martin, P.M. Oxidative Stress and Inflammation in Retinal Degeneration. Antioxidants 2021, 10, 790. [Google Scholar] [CrossRef]
- Urias, E.A.; Urias, G.A.; Monickaraj, F.; McGuire, P.; Das, A. Novel Therapeutic Targets in Diabetic Macular Edema: Beyond VEGF. Vision Res. 2017, 139, 221–227. [Google Scholar] [CrossRef]
- Mesquita, J.; Castro Sousa, J.P.; Vaz-Pereira, S.; Neves, A.; Passarinha, L.; Tomaz, C.T. Evaluation of the Growth Factors VEGF-A and VEGF-B in the Vitreous and Serum of Patients with Macular and Retinal Vascular Diseases. Growth Factors 2018, 36, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Schoenberger, S.D.; Kim, S.J. Nonsteroidal Anti-Inflammatory Drugs for Retinal Disease. Int. J. Inflam. 2013, 2013, 281981. [Google Scholar] [CrossRef]
- Zou, L.; Lai, H.; Zhou, Q.; Xiao, F. Lasting Controversy on Ranibizumab and Bevacizumab. Theranostics 2012, 1, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Anguita, A.; Tasiopoulou, A.; Shahid, S.; Roth, J.; Sim, S.Y.; Patel, P.j. A Review of Aflibercept Treatment for Macular Disease Ophthalmol. Theranostics 2021, 10, 413–428. [Google Scholar] [CrossRef]
- Heier, J.S.; Khanani, A.M.; Ruiz, C.Q.; Basu, K.; Ferrone, P.J.; Brittain, C.; Figueroa, M.S.; Lin, H.; Holz, F.G.; Patel, V.; et al. Efficacy, Durability, and Safety of Intravitreal Faricimab Up to Every 16 Weeks for Neovascular Age-related Macular Degeneration (Tenaya and Lucerne): Two Randomised, Double-masked, Phase 3, Non-inferiority Trials. Lancet 2022, 399, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Figueira, J.; Henriques, J.; Carneiro, A.; Marques-Neves, C.; Flores, R.; Castro-Sousa, J.P.; Meireles, A.; Gomes, N.; Nascimento, J.; Amaro, M.; et al. Guidelines for the Management of Center-Involving Diabetic Macular Edema: Treatment Options and Patient Monitorization. Clin. Ophthalmol. 2021, 15, 3221–3230. [Google Scholar] [CrossRef] [PubMed]
- Iturriaga-Goyon, E.; Buentello-Volante, B.; Magaña-Guerrero, F.S.; Garfias, Y. Future Perspectives of Therapeutic, Diagnostic and Prognostic Aptamers in Eye Pathological Angiogenesis. Cells 2021, 10, 1455. [Google Scholar] [CrossRef]
- Drolet, D.W.; Green, L.S.; Gold, L.; Janjic, N. Fit for the Eye: Aptamers in Ocular Disorders. Nucleic Acid Ther. 2016, 26, 127–146. [Google Scholar] [CrossRef]
- Ismail, S.I.; Alshaer, W. Therapeutic Aptamers in Discovery, Preclinical and Clinical Stages. Adv. Drug Deliv. Rev. 2018, 134, 51–64. [Google Scholar] [CrossRef]
- Ng, E.W.M.; Shima, D.T.; Calias, P.; Cunningham, E.T.; Guyer, D.R.; Adamis, A.P. Pegaptanib, a Targeted Anti-VEGF Aptamer for Ocular Vascular Disease. Nat. Rev. Drug Discov. 2006, 5, 123–132. [Google Scholar] [CrossRef]
- Phase 2A Open Label Safety Study of Fovista® (Anti-PDGF BB) Regimen Administered in Combination With Anti-VEGF Therapy to Study Sub-Retinal Fibrosis in Neovascular AMD-Full Text View-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02214628?term=Fovista&phase=01&draw=2&rank=1 (accessed on 27 April 2022).
- Jaffe, G.J.; Eliott, D.; Wells, J.A.; Prenner, J.L.; Papp, A.; Patel, S. A Phase 1 Study of Intravitreous E10030 in Combination with Ranibizumab in Neovascular Age-Related Macular Degeneration. Ophthalmology 2016, 123, 78–85. [Google Scholar] [CrossRef] [PubMed]
- A Phase 3 Safety and Efficacy Study of Fovista® (E10030) Intravitreous Administration in Combination With Lucentis® Compared to Lucentis® Monotherapy-Study Results-ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/results/NCT01940900 (accessed on 27 April 2022).
- Jia, W.; Yao, Z.; Zhao, J.; Guan, Q.; Gao, L. New Perspectives of Physiological and Pathological Functions of Nucleolin (NCL). Life Sci. 2017, 186, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Shi, H.; Zhou, H.; Song, X.; Yuan, S.; Luo, Y. The Angiogenic Function of Nucleolin is Mediated by Vascular Endothelial Growth Factor and Nonmuscle Myosin. Blood 2006, 107, 3564–3571. [Google Scholar] [CrossRef]
- Romano, S.; Fonseca, N.; Simões, S.; Gonçalves, J.; Moreira, J.N. Nucleolin-based Targeting Strategies for Cancer Therapy: From Targeted Drug Delivery to Cytotoxic Ligands. Drug Discov. Today 2019, 24, 1985–2001. [Google Scholar] [CrossRef] [PubMed]
- Leaderer, D.; Cashman, S.M.; Kumar-Singh, R. Topical Application of a G-Quartet Aptamer Targeting Nucleolin Attenuates Choroidal Neovascularization in a model of age-related macular degeneration. Exp. Eye Res. 2015, 140, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Iturriaga-Goyon, E.; Vivanco-Rojas, O.; Magaña-Guerrero, F.S.; Buentello-Volante, B.; Castro-Salas, I.; Aguayo-Flores, J.S.; Gracia-Mora, I.; Rivera-Huerta, M.; Sánchez-Bartés, F.; Garfias, Y. As1411 Nucleolin-specific Binding Aptamers Reduce Pathological Angiogenesis Through Inhibition of Nucleolin Phosphorylation. Int. J. Mol. Sci. 2021, 22, 13150. [Google Scholar] [CrossRef]
- Miranda, A.; Santos, T.; Largy, E.; Cruz, C. Locking up the AS1411 Aptamer with a Flanking Duplex: Towards an Improved Nucleolin-Targeting. Pharmaceuticals 2021, 14, 121. [Google Scholar] [CrossRef] [PubMed]
- Santos, T.; Miranda, A.; Campello, M.P.C.; Paulo, A.; Salgado, G.; Cabrita, E.J.; Cruz, C. Recognition of Nucleolin Through Interaction with RNA G-quadruplex. Biochem. Pharmacol. 2021, 189, 114208. [Google Scholar] [CrossRef]
- Do, N.Q.; Chung, W.J.; Truong, T.H.A.; Heddi, B.; Phan, A.T. “G-quadruplex Structure of an Anti-proliferative DNA Sequence,”. Nucleic Acids Res. 2017, 45, 7487–7493. [Google Scholar] [CrossRef]
- Moreira, D.; Leitão, D.; Lopes-Nunes, J.; Santos, T.; Figueiredo, J.; Miranda, A.; Alexandre, D.; Tomaz, C.; Mergny, J.L.; Cruz, C. G-Quadruplex Aptamer-Ligand Characterization. Molecules 2022, 27, 6781. [Google Scholar] [CrossRef]
- Carvalho, J.; Lopes-Nunes, J.; Lopes, A.C.; Campello, M.P.C.; Paulo, A.; Queiroz, J.A.; Cruz, C. Aptamer-Guided Acridine Derivatives for Cervical Cancer. Org. Biomol. Chem. 2019, 17, 2992–3002. [Google Scholar] [CrossRef] [PubMed]
- Iglicki, M.; Busch, C.; Zur, D.; Okada, M.M.; Mariussi, M.; Chhablani, J.K.; Cebeci, Z.; Fraser-Bell, S.; Chaikitmongkol, V.; Couturier, A.; et al. Dexamethasone Implant for Diabetic Macular Edema in Naive Compared with Refractory Eyes: The International Retina Group Real-life 24-month Multicenter Study. The Irgrel-dex Study. Retina 2019, 39, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Rache, A.D.; Mergny, J.L. Assessment of Selectivity of G-quadruplex Ligands Via an Optimised FRET Melting Assay. Biochimie 2015, 115, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Gueddouda, N.M.; Hurtado, M.R.; Moreau, S.; Ronga, L.; Das, R.N.; Savrimoutou, S.; Rubio, S.; Marchand, A.; Mendonza, A.; Marchivie, M.; et al. Design, Synthesis, and Evaluation of 2,9-Bis[(substituted-aaminomethyl)phenyl]-1,10-phenanthroline Derivatives as G-Quadruplex Ligands. ChemMedChem 2017, 12, 146–160. [Google Scholar] [CrossRef]
- Suzuki, N.; Kobayashi, M.; Sakata, K.; Suzuki, T.; Hosoya, T. Synergistic Stimulatory Effect of Glucocorticoid, EGF and Insulin on the Synthesis of Ribosomal RNA and Phosphorylation of Nucleolin in Primary Cultured Rat Hepatocytes. BBA Mol. Cell Res. 1991, 1092, 367–375. [Google Scholar] [CrossRef]
- Xing, H.; Tang, L.; Yang, X.; Hwang, K.; Wang, W.; Yin, Q.; Wong, N.Y.; Dobrucki, L.W.; Yasui, N.; Katzenellenbogen, J.A.; et al. Selective Delivery of an Anticancer Drug with Aptamer-functionalized Liposomes to Breast Cancer Cells In Vitro and In Vivo. J. Mater. Chem. B 2013, 1, 5288–5297. [Google Scholar] [CrossRef]
- Park, J.W.; Hong, K.; Kirpotin, D.B.; Colbern, G.; Shalaby, R.; Baselga, J.; Shao, Y.; Nielsen, U.B.; Marks, J.D.; Moore, D.; et al. Anti-HER2 Immunoliposomes: Enhanced Efficacy an Attributable to Targeted Delivery. Clin. Cancer Res. 2002, 8, 1172–1181. [Google Scholar]
- Guimarães, D.; Cavaco-Paulo, A.; Nogueira, E. Design of liposomes as drug delivery system for therapeutic applications. Int. J. Pharm. 2021, 601, 120571. [Google Scholar] [CrossRef]
- Gilbert, D.E.; Feigon, J. Multistranded DNA Structures. Curr. Opin. Struct. Biol. 1999, 9, 305–314. [Google Scholar] [CrossRef]
- Adrian, M.; Heddi, B.; Phan, A.T. NMR Spectroscopy of G-quadruplexes. Methods 2012, 57, 11–24. [Google Scholar] [CrossRef]
- Wang, Y.; Patel, D.J. Guanine Residues in d(T2AG3) and d(T2G4) Form Parallel-Stranded Potassium Cation Stabilized G-quadruplexes with Anti Glycosidic Torsion Angles in Solution. Biochemistry 1992, 35, 8112–8119. [Google Scholar] [CrossRef]
- Lombardo, D.; Kiselev, M.A. Methods of Liposomes Preparation: Formation and Control Factors of Versatile Nanocarriers for Biomedical and Nanomedicine Application. Pharmaceutics 2022, 14, 543. [Google Scholar] [CrossRef] [PubMed]
- Perrie, Y.; Mohammed, A.U.R.; Vangala, A.; McNeil, S.E. Environmental Scanning Electron Microscopy Offers Real-Time Morphological Analysis of Liposomes and Niosomes. J. Liposome Res. 2007, 17, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Bibi, S.; Kaur, R.; Henriksen-Lacey, M.; McNeil, S.E.; Wilkhu, J.; Lattmann, E.; Christensen, D.; Mohammed, A.R.; Perrie, Y. Microscopy imaging of liposomes: From coverslips to environmental SEM. Int. J. Pharm. 2011, 417, 138–150. [Google Scholar] [CrossRef]
- Khalil, M.; Hashmi, U.; Riaz, R.; Abbas, S.R. Chitosan Coated Liposomes (CCL) Containing Triamcinolone Acetonide for Sustained Delivery: A Potential Topical Treatment for Posterior Segment Diseases. Int. J. Biol. Macromol. 2020, 143, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.; Wei, Y.; Wu, Q.; Zhou, K.; Liu, T.; Zhang, Y.; Jiang, N.; Xiao, W.; Chen, J.; Liu, Q.; et al. Liposomes for Effective Drug Delivery to the Ocular Posterior Chamber. J. Nanobiotechnol. 2019, 17, 64. [Google Scholar] [CrossRef] [PubMed]
- Sercombe, L.; Veerati, T.; Moheimani, F.; Wu, S.Y.; Sood, A.K.; Hua, S. Advances and Challenges of Liposome Assisted Drug Delivery. Front. Pharmacol. 2015, 6, 286. [Google Scholar] [CrossRef]
- Joo, E.J.; Yang, H.; Park, Y.; Park, N.Y.; Toida, T.; Linhardt, R.J.; Kim, Y.S. Induction of nucleolin translocation by acharan sulfate in A549 human lung adenocarcinoma. J. Cell. Biochem. 2010, 110, 1272–1278. [Google Scholar] [CrossRef] [PubMed]
- Bolte, S.; Cordelières, F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006, 224, 213–232. [Google Scholar] [CrossRef]
- Carvalho, J.; Lopes-Nunes, J.; Vialet, B.; Rosado, T.; Gallardo, E.; Vale, J.; Eloy, C.; Ferreira, S.; Palmeira-de-Oliveira, R.; Campello, M.; et al. Nanoaggregate-forming lipid-conjugated AS1411 aptamer as a promising tumor-targeted delivery system of anticancer agents in vitro. Nanomed. Nanotechnol. Biol. Med. 2021, 36, 102429. [Google Scholar] [CrossRef]
- Lopes-Nunes, J.; Agonia, A.S.; Rosado, T.; Gallardo, E.; Palmeira-de-Oliveira, R.; Palmeira-de-Oliveira, A.; Martinez-de-Oliveira, J.; Fonseca-Moutinho, J.; Campello, M.P.C.; Paiva, A.; et al. Aptamer-Functionalized Gold Nanoparticles for Drug Delivery to Gynecological Carcinoma Cells. Cancers 2021, 13, 4038. [Google Scholar] [CrossRef] [PubMed]
- Pereira, E.; Quental, L.; Palma, E.; Oliveira, M.C.; Mendes, F.; Raposinho, P.; Correia, I.; Lavrado, J.; Di Maria, S.; Belchior, A.; et al. Evaluation of Acridine Orange Derivatives as DNA-Targeted Radiopharmaceuticals for Auger Therapy: Influence of the Radionuclide and Distance to DNA. Sci. Rep. 2017, 7, 42544. [Google Scholar] [CrossRef] [PubMed]
- Aravind, A.; Jeyamohan, P.; Nair, R.; Veeranarayanan, S.; Nagaoka, Y.; Yoshida, Y.; Maekawa, T.; Kumar, D.S. AS1411 Aptamer Tagged PLGA-Lecithin-PEG Nanoparticles for Tumor Cell Targeting and Drug Delivery. Biotechnol. Bioeng. 2012, 109, 2920–2931. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S Factor | ||
|---|---|---|
| Ligand | 15 Molar eq. ds26 | 50 Molar eq. ds26 |
| BRACO-19 | 0.66 | 0.45 |
| C8 | 0.47 | 0.38 |
| PDS | 0.83 | 0.73 |
| PhenDC3 | 0.94 | 0.89 |
| TMPyP4 | 0.51 | 0.33 |
| Name | Molar Extinction Coefficient (L·mol−1·cm−1) |
|---|---|
| AT11-L0 | 256.1 |
| FAM-AT11-L0-TAMRA | 317.1 |
| Cy5-AT11-L0 | 266.1 |
| NH2-AT11-L0 | 256.1 |
| ds26 | 253.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreira, D.; Lopes-Nunes, J.; Santos, F.M.; Campello, M.P.C.; Oliveira, M.C.; Paulo, A.; Tomaz, C.; Cruz, C. Assessment of Aptamer as a Potential Drug Targeted Delivery for Retinal Angiogenesis Inhibition. Pharmaceuticals 2023, 16, 751. https://doi.org/10.3390/ph16050751
Moreira D, Lopes-Nunes J, Santos FM, Campello MPC, Oliveira MC, Paulo A, Tomaz C, Cruz C. Assessment of Aptamer as a Potential Drug Targeted Delivery for Retinal Angiogenesis Inhibition. Pharmaceuticals. 2023; 16(5):751. https://doi.org/10.3390/ph16050751
Chicago/Turabian StyleMoreira, David, Jéssica Lopes-Nunes, Fátima Milhano Santos, Maria Paula Cabral Campello, Maria Cristina Oliveira, António Paulo, Cândida Tomaz, and Carla Cruz. 2023. "Assessment of Aptamer as a Potential Drug Targeted Delivery for Retinal Angiogenesis Inhibition" Pharmaceuticals 16, no. 5: 751. https://doi.org/10.3390/ph16050751