
Peptide Radioligands in Cancer Theranostics: Agonists and Antagonists
 , , , and
, , , and
Abstract
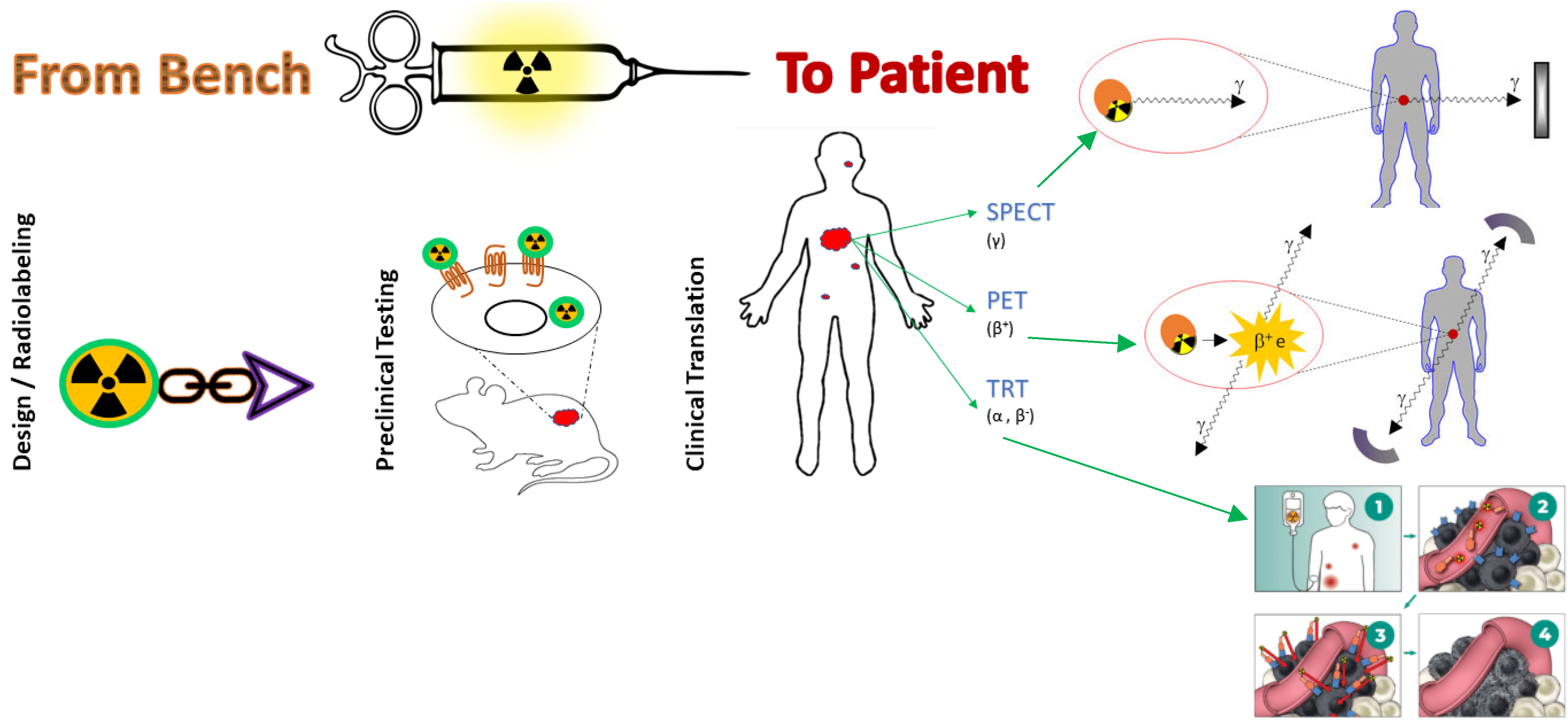
:1. Introduction
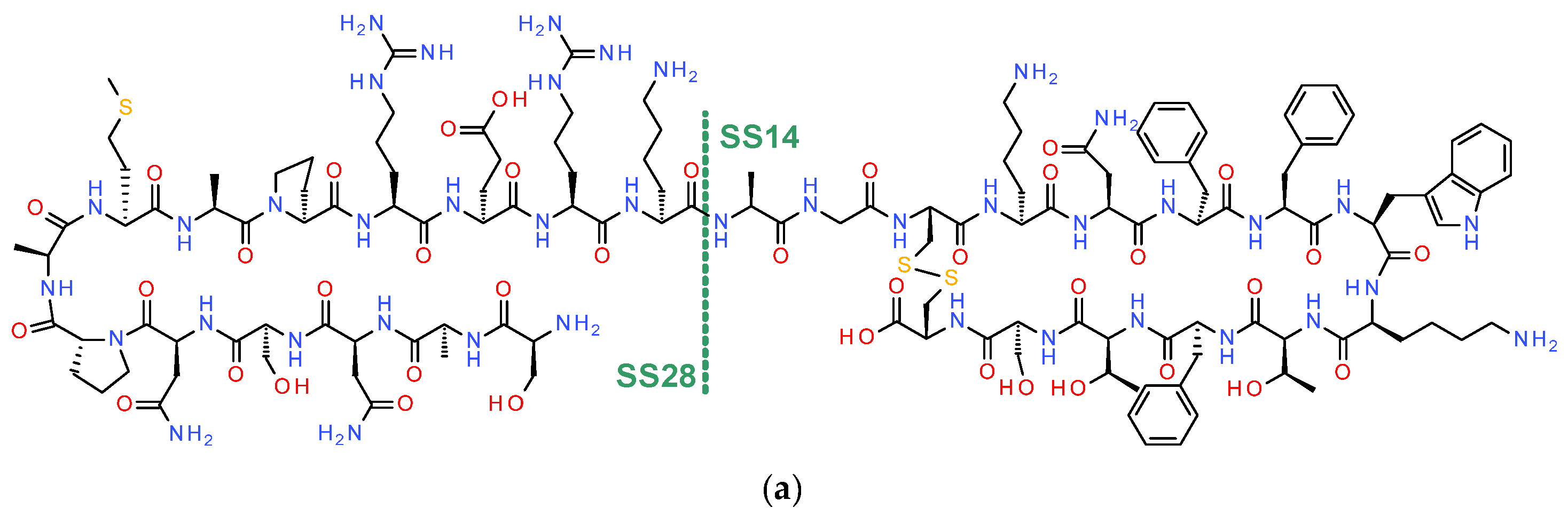
2. Somatostatin
2.1. Somatostatin Its Receptors and NETs
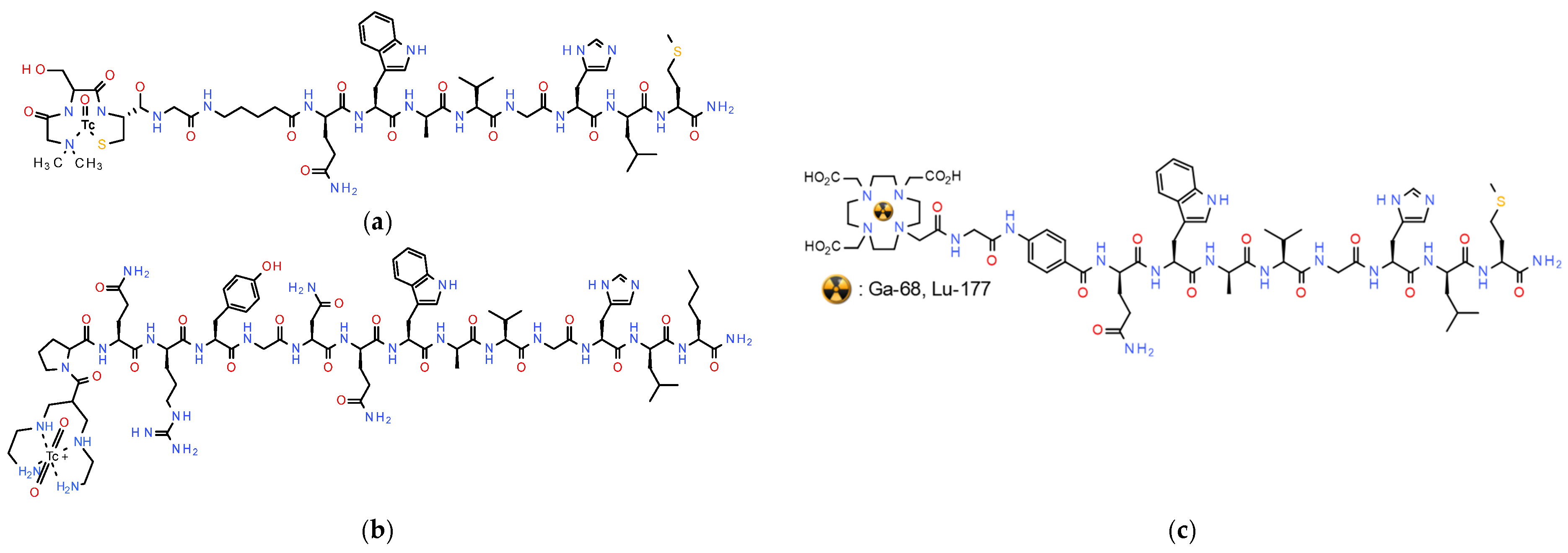
2.2. Radiolabeled Somatostatin in Theranostics of NETs—Clinical Impact
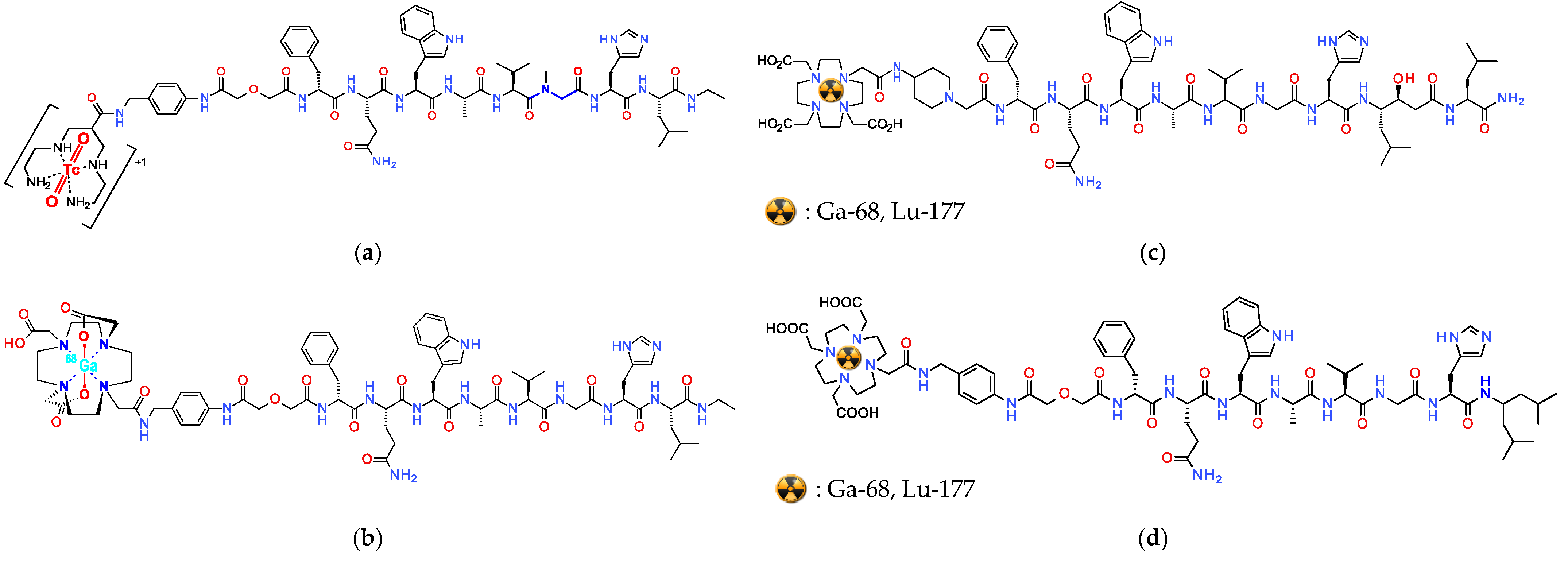
2.3. Shift in Paradigm: From Radiolabeled SST2R-Agonists to Antagonists
2.4. Conclusions
3. Bombesin—Gastrin-Releasing Peptide
3.1. Bombesin/Gastrin-Releasing Peptide and Their Receptors in Human Tumors
3.2. Radiolabeled BBN Analogs in Cancer Theranostics: Limitations in Clinical Translation
3.3. Biosafety Concerns: Switching to Radiolabeled GRPR-Antagonists
3.4. Conclusions
4. Gastrin-Cholecystokinin
4.1. Gastrin-Cholecystokinin and CCK2R Expression in Cancer
4.2. Radiolabeled Gastrin/CCK Analogs in Cancer Theranostics: Major Breakthroughs
4.3. Biosafety Problems: Development of Radiolabeled CCK2R-Antagonists Today
4.4. Conclusions
5. Exendin—Glucagon-like Peptide 1 Receptor
5.1. Exendin and Glucagon-like Peptide 1 Receptor in Insulinoma
5.2. Radiolabeled Exendin Analogs in Theranostics: Problems in Clinical Translation
5.2.1. PRRT
5.2.2. PDT
5.3. From Radiolabeled GLP-1R-Agonists to Antagonists
5.4. Conclusions
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Reubi, J.C. Peptide receptors as molecular targets for cancer diagnosis and therapy. Endocr. Rev. 2003, 24, 389–427. [Google Scholar] [CrossRef] [PubMed]
- Sgouros, G.; Bodei, L.; McDevitt, M.R.; Nedrow, J.R. Radiopharmaceutical therapy in cancer: Clinical advances and challenges. Nat. Rev. Drug Discov. 2020, 19, 589–608. [Google Scholar] [CrossRef] [PubMed]
- Notni, J.; Wester, H.J. Re-thinking the role of radiometal isotopes: Towards a future concept for theranostic radiopharmaceuticals. J. Labelled Comp. Radiopharm. 2018, 61, 141–153. [Google Scholar] [CrossRef] [PubMed]
- Witney, T.H.; Blower, P.J. The chemical tool-kit for molecular imaging with radionuclides in the age of targeted and immune therapy. Cancer Imaging 2021, 21, 18. [Google Scholar] [CrossRef]
- De Jong, M.; Breeman, W.A.; Kwekkeboom, D.J.; Valkema, R.; Krenning, E.P. Tumor imaging and therapy using radiolabeled somatostatin analogues. Acc. Chem. Res. 2009, 42, 873–880. [Google Scholar] [CrossRef]
- Levine, R.; Krenning, E.P. Clinical history of the theranostic radionuclide approach to neuroendocrine tumors and other types of cancer: Historical review based on an interview of Eric P. Krenning by Rachel Levine. J. Nucl. Med. 2017, 58 (Suppl. S2), 3S–9S. [Google Scholar] [CrossRef]
- Reubi, J.C. Regulatory peptide receptors as molecular targets for cancer diagnosis and therapy. Q. J. Nucl. Med. 1997, 41, 63–70. [Google Scholar]
- Fani, M.; Nicolàs, G.P.; Wild, D. Somatostatin receptor antagonists for imaging and therapy. J. Nucl. Med. 2017, 58 (Suppl. S2), 61S–66S. [Google Scholar] [CrossRef]
- Fani, M.; Mansi, R.; Nicolàs, G.P.; Wild, D. Radiolabeled somatostatin analogs-a continuously evolving class of radiopharmaceuticals. Cancers 2022, 14, 1172. [Google Scholar] [CrossRef]
- Reubi, J.C.; Waser, B.; Mäcke, H.; Rivier, J. Highly increased 125I-JR11 antagonist binding in vitro reveals novel indications for SST2 targeting in human cancers. J. Nucl. Med. 2017, 58, 300–306. [Google Scholar] [CrossRef]
- Ginj, M.; Zhang, H.; Waser, B.; Cescato, R.; Wild, D.; Wang, X.; Erchegyi, J.; Rivier, J.; Mäcke, H.R.; Reubi, J.C. Radiolabeled somatostatin receptor antagonists are preferable to agonists for in vivo peptide receptor targeting of tumors. Proc. Natl. Acad. Sci. USA 2006, 103, 16436–16441. [Google Scholar] [CrossRef] [PubMed]
- Cescato, R.; Maina, T.; Nock, B.; Nikolopoulou, A.; Charalambidis, D.; Piccand, V.; Reubi, J.C. Bombesin receptor antagonists may be preferable to agonists for tumor targeting. J. Nucl. Med. 2008, 49, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Mansi, R.; Nock, B.A.; Dalm, S.U.; Busstra, M.B.; van Weerden, W.M.; Maina, T. Radiolabeled bombesin analogs. Cancers 2021, 13, 5766. [Google Scholar] [CrossRef] [PubMed]
- Kaloudi, A.; Kanellopoulos, P.; Radolf, T.; Chepurny, O.G.; Rouchota, M.; Loudos, G.; Andreae, F.; Holz, G.G.; Nock, B.A.; Maina, T. [99mTc]Tc-DGA1, a promising CCK2R-antagonist-based tracer for tumor diagnosis with single-photon emission computed tomography. Mol. Pharm. 2020, 17, 3116–3128. [Google Scholar] [CrossRef]
- Waser, B.; Reubi, J.C. Value of the radiolabelled GLP-1 receptor antagonist exendin(9-39) for targeting of GLP-1 receptor-expressing pancreatic tissues in mice and humans. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 1054–1058. [Google Scholar] [CrossRef]
- Ambrosini, V.; Kunikowska, J.; Baudin, E.; Bodei, L.; Bouvier, C.; Capdevila, J.; Cremonesi, M.; de Herder, W.W.; Dromain, C.; Falconi, M.; et al. Consensus on molecular imaging and theranostics in neuroendocrine neoplasms. Eur. J. Cancer 2021, 146, 56–73. [Google Scholar] [CrossRef]
- Barbieri, F.; Bajetto, A.; Pattarozzi, A.; Gatti, M.; Wurth, R.; Thellung, S.; Corsaro, A.; Villa, V.; Nizzari, M.; Florio, T. Peptide receptor targeting in cancer: The somatostatin paradigm. Int. J. Pept. 2013, 2013, 926295. [Google Scholar] [CrossRef]
- Reubi, J.C.; Waser, B.; Schaer, J.C.; Laissue, J.A. Somatostatin receptor SST1–SST5 expression in normal and neoplastic human tissues using receptor autoradiography with subtype-selective ligands. Eur. J. Nucl. Med. 2001, 28, 836–846. [Google Scholar] [CrossRef]
- Gunther, T.; Tulipano, G.; Dournaud, P.; Bousquet, C.; Csaba, Z.; Kreienkamp, H.J.; Lupp, A.; Korbonits, M.; Castano, J.P.; Wester, H.J.; et al. International union of basic and clinical pharmacology. CV. Somatostatin receptors: Structure, function, ligands, and new nomenclature. Pharmacol. Rev. 2018, 70, 763–835. [Google Scholar] [CrossRef]
- Reubi, J.C.; Schaer, J.C.; Waser, B.; Mengod, G. Expression and localization of somatostatin receptor SSTR1, SSTR2, and SSTR3 messenger RNAs in primary human tumors using in situ hybridization. Cancer Res. 1994, 54, 3455–3459. [Google Scholar]
- Patel, Y.C.; Wheatley, T. In vivo and in vitro plasma disappearance and metabolism of somatostatin-28 and somatostatin-14 in the rat. Endocrinology 1983, 112, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Lamberts, S.W.J.; Hofland, L.J. Anniversary review: Octreotide, 40 years later. Eur. J. Endocrinol. 2019, 181, R173–R183. [Google Scholar] [CrossRef] [PubMed]
- Wolin, E.M. The expanding role of somatostatin analogs in the management of neuroendocrine tumors. Gastrointest. Cancer Res. 2012, 5, 161–168. [Google Scholar]
- Dasari, A.; Shen, C.; Halperin, D.; Zhao, B.; Zhou, S.; Xu, Y.; Shih, T.; Yao, J.C. Trends in the incidence, prevalence, and survival outcomes in patients with neuroendocrine tumors in the United States. JAMA Oncol. 2017, 3, 1335–1342. [Google Scholar] [CrossRef] [PubMed]
- Van Essen, M.; Krenning, E.P.; de Jong, M.; Valkema, R.; Kwekkeboom, D.J. Peptide receptor radionuclide therapy with radiolabelled somatostatin analogues in patients with somatostatin receptor positive tumours. Acta Oncol. 2007, 46, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Hicks, R.J.; Kwekkeboom, D.J.; Krenning, E.; Bodei, L.; Grozinsky-Glasberg, S.; Arnold, R.; Borbath, I.; Cwikla, J.; Toumpanakis, C.; Kaltsas, G.; et al. ENETS consensus guidelines for the standards of care in neuroendocrine neoplasia: Peptide receptor radionuclide therapy with radiolabeled somatostatin analogues. Neuroendocrinology 2017, 105, 295–309. [Google Scholar] [CrossRef] [PubMed]
- Krenning, E.P.; Bakker, W.H.; Breeman, W.A.; Koper, J.W.; Kooij, P.P.; Ausema, L.; Lameris, J.S.; Reubi, J.C.; Lamberts, S.W. Localisation of endocrine-related tumours with radioiodinated analogue of somatostatin. Lancet 1989, 1, 242–244. [Google Scholar] [CrossRef]
- Decristoforo, C.; Mather, S.J.; Cholewinski, W.; Donnemiller, E.; Riccabona, G.; Moncayo, R. 99mTc-EDDA/HYNIC-TOC: A new 99mTc-labelled radiopharmaceutical for imaging somatostatin receptor-positive tumours; first clinical results and intra-patient comparison with 111In-labelled octreotide derivatives. Eur. J. Nucl. Med. 2000, 27, 1318–1325. [Google Scholar] [CrossRef]
- Decristoforo, C.; Maina, T.; Nock, B.; Gabriel, M.; Cordopatis, P.; Moncayo, R. 99mTc-Demotate 1: First data in tumour patients-results of a pilot/phase i study. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 1211–1219. [Google Scholar] [CrossRef]
- Gabriel, M.; Decristoforo, C.; Maina, T.; Nock, B.; von Guggenberg, E.; Cordopatis, P.; Moncayo, R. 99mTc-N4-[Tyr3]octreotate versus 99mTc-EDDA/HYNIC-[Tyr3]octreotide: An intrapatient comparison of two novel technetium-99m labeled tracers for somatostatin receptor scintigraphy. Cancer Biother. Radiopharm. 2004, 19, 73–79. [Google Scholar] [CrossRef]
- Eychenne, R.; Bouvry, C.; Bourgeois, M.; Loyer, P.; Benoist, E.; Lepareur, N. Overview of radiolabeled somatostatin analogs for cancer imaging and therapy. Molecules 2020, 25, 4012. [Google Scholar] [CrossRef] [PubMed]
- Hennrich, U.; Kopka, K. Lutathera®: The first FDA- and EMA-approved radiopharmaceutical for peptide receptor radionuclide therapy. Pharmaceuticals 2019, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Barrio, M.; Czernin, J.; Fanti, S.; Ambrosini, V.; Binse, I.; Du, L.; Eiber, M.; Herrmann, K.; Fendler, W.P. The impact of SSTR-directed PET/CT on the management of patients with neuroendocrine tumor: A systematic review and meta-analysis. J. Nucl. Med. 2017, 58, 756–761. [Google Scholar] [CrossRef] [PubMed]
- Yadav, D.; Ballal, S.; Yadav, M.P.; Tripathi, M.; Roesch, F.; Bal, C. Evaluation of [68Ga]Ga-DATA-TOC for imaging of neuroendocrine tumours: Comparison with [68Ga]Ga-DOTA-NOC PET/CT. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Paterson, B.M.; Roselt, P.; Denoyer, D.; Cullinane, C.; Binns, D.; Noonan, W.; Jeffery, C.M.; Price, R.I.; White, J.M.; Hicks, R.J.; et al. PET imaging of tumours with a 64Cu labeled macrobicyclic cage amine ligand tethered to Tyr3-octreotate. Dalton Trans. 2014, 43, 1386–1396. [Google Scholar] [CrossRef] [PubMed]
- Hicks, R.J.; Jackson, P.; Kong, G.; Ware, R.E.; Hofman, M.S.; Pattison, D.A.; Akhurst, T.A.; Drummond, E.; Roselt, P.; Callahan, J.; et al. 64Cu-SARTATE PET imaging of patients with neuroendocrine tumors demonstrates high tumor uptake and retention, potentially allowing prospective dosimetry for peptide receptor radionuclide therapy. J. Nucl. Med. 2019, 60, 777–785. [Google Scholar] [CrossRef]
- Pauwels, E.; Cleeren, F.; Tshibangu, T.; Koole, M.; Serdons, K.; Boeckxstaens, L.; Dekervel, J.; Vandamme, T.; Lybaert, W.; van den Broeck, B.; et al. 18F-AlF-NOTA-octreotide outperforms 68Ga-DOTA-TATE/-NOC PET in neuroendocrine tumor patients: Results from a prospective, multicenter study. J. Nucl. Med. 2022, 64, 632–638. [Google Scholar] [CrossRef]
- Hofman, M.S.; Michael, M.; Hicks, R.J. 177Lu-DOTATATE for midgut neuroendocrine tumors. N. Engl. J. Med. 2017, 376, 1390–1391. [Google Scholar] [CrossRef]
- Strosberg, J.; Wolin, E.; Chasen, B.; Kulke, M.; Bushnell, D.; Caplin, M.; Baum, R.P.; Kunz, P.; Hobday, T.; Hendifar, A.; et al. Health-related quality of life in patients with progressive midgut neuroendocrine tumors treated with 177Lu-DOTATATE in the Phase III NETTER-1 trial. J. Clin. Oncol. 2018, 36, 2578–2584. [Google Scholar] [CrossRef]
- Strosberg, J.R.; Caplin, M.E.; Kunz, P.L.; Ruszniewski, P.B.; Bodei, L.; Hendifar, A.; Mittra, E.; Wolin, E.M.; Yao, J.C.; Pavel, M.E.; et al. 177Lu-DOTATATE plus long-acting octreotide versus high-dose long-acting octreotide in patients with midgut neuroendocrine tumours (NETTER-1): Final overall survival and long-term safety results from an open-label, randomised, controlled, Phase 3 trial. Lancet Oncol. 2021, 22, 1752–1763. [Google Scholar] [CrossRef]
- Saravana-Bawan, B.; Bajwa, A.; Paterson, J.; McEwan, A.J.B.; McMullen, T.P.W. Efficacy of 177Lu peptide receptor radionuclide therapy for the treatment of neuroendocrine tumors: A meta-analysis. Clin. Nucl. Med. 2019, 44, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Clement, D.; Navalkissoor, S.; Srirajaskanthan, R.; Courbon, F.; Dierickx, L.; Eccles, A.; Lewington, V.; Mitjavila, M.; Percovich, J.C.; Lequoy, B.; et al. Efficacy and safety of 177Lu-DOTATATE in patients with advanced pancreatic neuroendocrine tumours: Data from the NETTER-R international, retrospective study. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 3529–3537. [Google Scholar] [CrossRef]
- Kratochwil, C.; Giesel, F.L.; Bruchertseifer, F.; Mier, W.; Apostolidis, C.; Boll, R.; Murphy, K.; Haberkorn, U.; Morgenstern, A. 213Bi-DOTATOC receptor-targeted alpha-radionuclide therapy induces remission in neuroendocrine tumours refractory to beta radiation: A first-in-human experience. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 2106–2119. [Google Scholar] [CrossRef] [PubMed]
- Kratochwil, C.; Apostolidis, L.; Rathke, H.; Apostolidis, C.; Bicu, F.; Bruchertseifer, F.; Choyke, P.L.; Haberkorn, U.; Giesel, F.L.; Morgenstern, A. Dosing 225Ac-DOTATOC in patients with somatostatin-receptor-positive solid tumors: 5-year follow-up of hematological and renal toxicity. Eur. J. Nucl. Med. Mol. Imaging 2021, 49, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Ballal, S.; Yadav, M.P.; Bal, C.; Sahoo, R.K.; Tripathi, M. Broadening horizons with 225Ac-DOTATATE targeted alpha therapy for gastroenteropancreatic neuroendocrine tumour patients stable or refractory to 177Lu-DOTATATE PRRT: First clinical experience on the efficacy and safety. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 934–946. [Google Scholar] [CrossRef]
- Delpassand, E.S.; Tworowska, I.; Esfandiari, R.; Torgue, J.; Hurt, J.; Shafie, A.; Nunez, R. Targeted alpha-emitter therapy with 212Pb-DOTAMTATE for the treatment of metastatic SSTR-expressing neuroendocrine tumors: First-in-humans dose-escalation clinical trial. J. Nucl. Med. 2022, 63, 1326–1333. [Google Scholar] [CrossRef]
- Perrin, M.H.; Sutton, S.W.; Cervini, L.A.; Rivier, J.E.; Vale, W.W. Comparison of an agonist, urocortin, and an antagonist, astressin, as radioligands for characterization of corticotropin-releasing factor receptors. J. Pharmacol. Exp. Ther. 1999, 288, 729–734. [Google Scholar]
- Bass, R.T.; Buckwalter, B.L.; Patel, B.P.; Pausch, M.H.; Price, L.A.; Strnad, J.; Hadcock, J.R. Identification and characterization of novel somatostatin antagonists. Mol. Pharmacol. 1996, 50, 709–715. [Google Scholar]
- Cescato, R.; Waser, B.; Fani, M.; Reubi, J.C. Evaluation of 177Lu-DOTA-SST2 antagonist versus 177Lu-DOTA-SST2 agonist binding in human cancers in vitro. J. Nucl. Med. 2011, 52, 1886–1890. [Google Scholar] [CrossRef]
- Wang, X.; Fani, M.; Schulz, S.; Rivier, J.; Reubi, J.C.; Maecke, H.R. Comprehensive evaluation of a somatostatin-based radiolabelled antagonist for diagnostic imaging and radionuclide therapy. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 1876–1885. [Google Scholar] [CrossRef]
- Wild, D.; Fani, M.; Béhé, M.; Brink, I.; Rivier, J.E.; Reubi, J.C.; Maecke, H.R.; Weber, W.A. First clinical evidence that imaging with somatostatin receptor antagonists is feasible. J. Nucl. Med. 2011, 52, 1412–1417. [Google Scholar] [CrossRef] [PubMed]
- Cescato, R.; Erchegyi, J.; Waser, B.; Piccand, V.; Maecke, H.R.; Rivier, J.E.; Reubi, J.C. Design and in vitro characterization of highly SST2-selective somatostatin antagonists suitable for radiotargeting. J. Med. Chem. 2008, 51, 4030–4037. [Google Scholar] [CrossRef] [PubMed]
- Fani, M.; Del Pozzo, L.; Abiraj, K.; Mansi, R.; Tamma, M.L.; Cescato, R.; Waser, B.; Weber, W.A.; Reubi, J.C.; Maecke, H.R. PET of somatostatin receptor-positive tumors using 64Cu- and 68Ga-somatostatin antagonists: The chelate makes the difference. J. Nucl. Med. 2011, 52, 1110–1118. [Google Scholar] [CrossRef] [PubMed]
- Fani, M.; Braun, F.; Waser, B.; Beetschen, K.; Cescato, R.; Erchegyi, J.; Rivier, J.E.; Weber, W.A.; Maecke, H.R.; Reubi, J.C. Unexpected sensitivity of SST2 antagonists to N-terminal radiometal modifications. J. Nucl. Med. 2012, 53, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
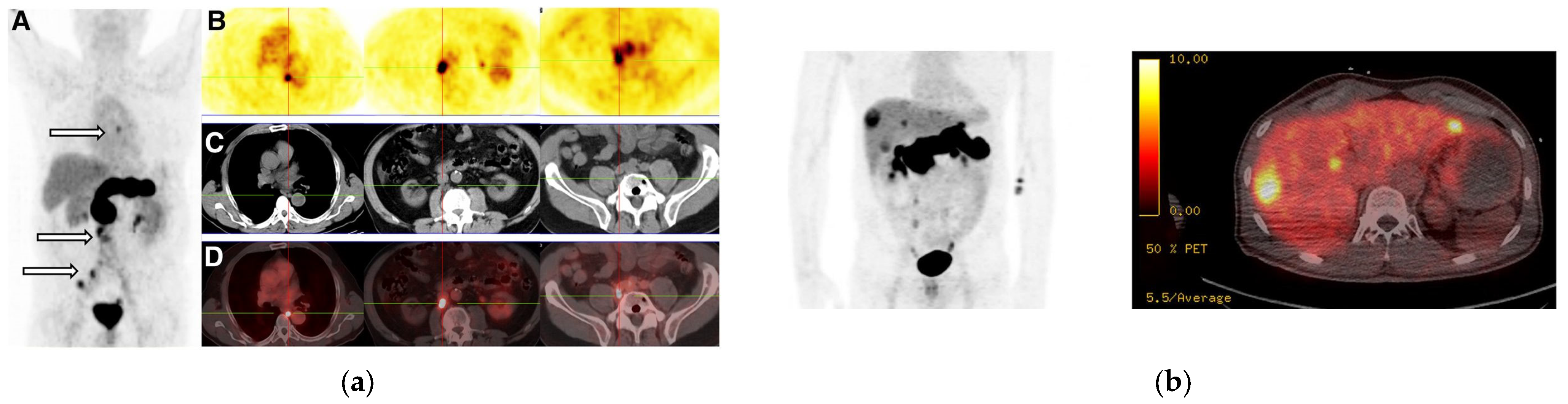
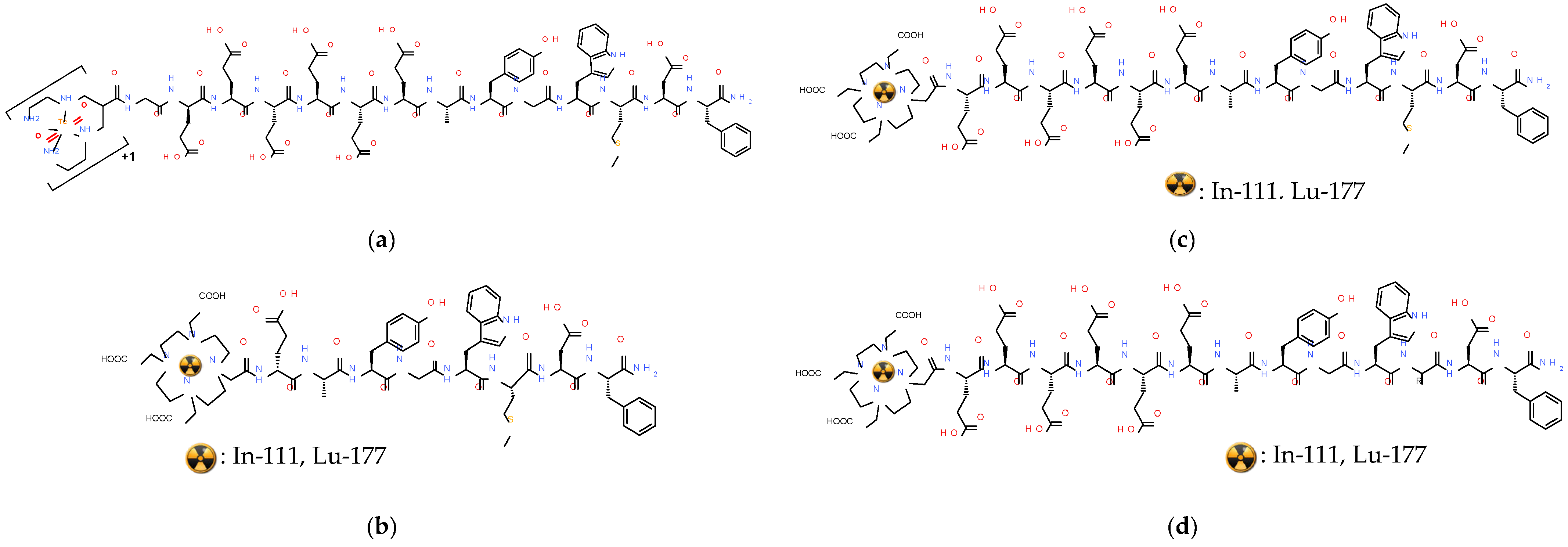
- Kanellopoulos, P.; Nock, B.A.; Greifenstein, L.; Baum, R.P.; Roesch, F.; Maina, T. [68Ga]Ga-DATA5m-LM4, a PET radiotracer in the diagnosis of SST2R-positive tumors: Preclinical and first clinical results. Int. J. Mol. Sci. 2022, 23, 4590. [Google Scholar] [CrossRef]
- Nock, B.A.; Kanellopoulos, P.; Moon, E.S.; Rouchota, M.; Loudos, G.; Ballal, S.; Yadav, M.P.; Bal, C.; Mishra, P.; Sheokand, P.; et al. [111In]In/[177Lu]Lu-AAZTA5-LM4 SST2R-antagonists in cancer theranostics: From preclinical testing to first patient results. Pharmaceutics 2023, 15, 776. [Google Scholar] [CrossRef]
- Nicolàs, G.P.; Beykan, S.; Bouterfa, H.; Kaufmann, J.; Bauman, A.; Lassmann, M.; Reubi, J.C.; Rivier, J.E.F.; Maecke, H.R.; Fani, M.; et al. Safety, biodistribution, and radiation dosimetry of 68Ga-OPS202 in patients with gastroenteropancreatic neuroendocrine tumors: A prospective Phase I imaging study. J. Nucl. Med. 2018, 59, 909–914. [Google Scholar] [CrossRef]
- Nicolàs, G.P.; Schreiter, N.; Kaul, F.; Uiters, J.; Bouterfa, H.; Kaufmann, J.; Erlanger, T.E.; Cathomas, R.; Christ, E.; Fani, M.; et al. Sensitivity comparison of 68Ga-OPS202 and 68Ga-DOTATOC PET/CT in patients with gastroenteropancreatic neuroendocrine tumors: A prospective Phase II imaging study. J. Nucl. Med. 2018, 59, 915–921. [Google Scholar] [CrossRef]
- Wild, D.; Fani, M.; Fischer, R.; Del Pozzo, L.; Kaul, F.; Krebs, S.; Fischer, R.; Rivier, J.E.; Reubi, J.C.; Maecke, H.R.; et al. Comparison of somatostatin receptor agonist and antagonist for peptide receptor radionuclide therapy: A pilot study. J. Nucl. Med. 2014, 55, 1248–1252. [Google Scholar] [CrossRef]
- Reidy-Lagunes, D.; Pandit-Taskar, N.; O’Donoghue, J.A.; Krebs, S.; Staton, K.D.; Lyashchenko, S.K.; Lewis, J.S.; Raj, N.; Gonen, M.; Lohrmann, C.; et al. Phase I trial of well-differentiated neuroendocrine tumors (NETs) with radiolabeled somatostatin antagonist 177Lu-Satoreotide Tetraxetan. Clin. Cancer Res. 2019, 25, 6939–6947. [Google Scholar] [CrossRef]
- Nicolàs, G.P.; Mansi, R.; McDougall, L.; Kaufmann, J.; Bouterfa, H.; Wild, D.; Fani, M. Biodistribution, pharmacokinetics, and dosimetry of 177Lu-, 90Y-, and 111In-labeled somatostatin receptor antagonist OPS201 in comparison to the agonist 177Lu-DOTATATE: The mass effect. J. Nucl. Med. 2017, 58, 1435–1441. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Cheng, Y.; Jia, R.; Zhao, H.; Bai, C.; Xu, J.; Yao, S.; Huo, L. A prospective, randomized, double-blind study to evaluate the safety, biodistribution, and dosimetry of 68Ga-NODAGA-LM3 and 68Ga-DOTA-LM3 in patients with well-differentiated neuroendocrine tumors. J. Nucl. Med. 2021, 62, 1398–1405. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Jia, R.; Yang, Q.; Cheng, Y.; Zhao, H.; Bai, C.; Xu, J.; Yao, S.; Huo, L. A prospective randomized, double-blind study to evaluate the diagnostic efficacy of 68Ga-NODAGA-LM3 and 68Ga-DOTA-LM3 in patients with well-differentiated neuroendocrine tumors: Compared with 68Ga-DOTATATE. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 1613–1622. [Google Scholar] [CrossRef] [PubMed]
- Baum, R.P.; Zhang, J.; Schuchardt, C.; Müller, D.; Mäcke, H. First-in-humans study of the SSTR antagonist 177Lu-DOTA-LM3 for peptide receptor radionuclide therapy in patients with metastatic neuroendocrine neoplasms: Dosimetry, safety, and efficacy. J. Nucl. Med. 2021, 62, 1571–1581. [Google Scholar] [CrossRef] [PubMed]
- Borgna, F.; Barritt, P.; Grundler, P.V.; Talip, Z.; Cohrs, S.; Zeevaart, J.R.; Koster, U.; Schibli, R.; van der Meulen, N.P.; Müller, C. Simultaneous visualization of 161Tb- and 177Lu-labeled somatostatin analogues using dual-isotope spect imaging. Pharmaceutics 2021, 13, 536. [Google Scholar] [CrossRef]
- Borgna, F.; Haller, S.; Rodriguez, J.M.M.; Ginj, M.; Grundler, P.V.; Zeevaart, J.R.; Koster, U.; Schibli, R.; van der Meulen, N.P.; Müller, C. Combination of terbium-161 with somatostatin receptor antagonists-A potential paradigm shift for the treatment of neuroendocrine neoplasms. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 1113–1126. [Google Scholar] [CrossRef]
- Feijtel, D.; de Jong, M.; Nonnekens, J. Peptide receptor radionuclide therapy: Looking back, looking forward. Curr. Top. Med. Chem. 2020, 20, 2959–2969. [Google Scholar] [CrossRef]
- Reubi, J.C.; Maecke, H.R. Approaches to multireceptor targeting: Hybrid radioligands, radioligand cocktails, and sequential radioligand applications. J. Nucl. Med. 2017, 58 (Suppl. S2), 10S–16S. [Google Scholar] [CrossRef]
- Minczeles, N.S.; Hofland, J.; de Herder, W.W.; Brabander, T. Strategies towards improving clinical outcomes of peptide receptor radionuclide therapy. Curr. Oncol. Rep. 2021, 23, 46. [Google Scholar] [CrossRef]
- Cullinane, C.; Waldeck, K.; Kirby, L.; Rogers, B.E.; Eu, P.; Tothill, R.W.; Hicks, R.J. Enhancing the anti-tumour activity of 177Lu-DOTA-octreotate radionuclide therapy in somatostatin receptor-2 expressing tumour models by targeting PARP. Sci. Rep. 2020, 10, 10196. [Google Scholar] [CrossRef]
- Klomp, M.J.; Dalm, S.U.; de Jong, M.; Feelders, R.A.; Hofland, J.; Hofland, L.J. Epigenetic regulation of somatostatin and somatostatin receptors in neuroendocrine tumors and other types of cancer. Rev. Endocr. Metab. Disord. 2021, 22, 495–510. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.T.; Battey, J.F.; Spindel, E.R.; Benya, R.V. International union of pharmacology. LXVIII. Mammalian bombesin receptors: Nomenclature, distribution, pharmacology, signaling, and functions in normal and disease states. Pharmacol. Rev. 2008, 60, 1–42. [Google Scholar] [CrossRef] [PubMed]
- Moreno, P.; Ramos-Alvarez, I.; Moody, T.W.; Jensen, R.T. Bombesin related peptides/receptors and their promising therapeutic roles in cancer imaging, targeting and treatment. Expert. Opin. Ther. Targets 2016, 20, 1055–1073. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C.; Wenger, S.; Schmuckli-Maurer, J.; Schaer, J.C.; Gugger, M. Bombesin receptor subtypes in human cancers: Detection with the universal radioligand 125I-[D-Tyr6,beta-Ala11,Phe13,Nle14]bombesin(6-14). Clin. Cancer Res. 2002, 8, 1139–1146. [Google Scholar]
- Körner, M.; Waser, B.; Rehmann, R.; Reubi, J.C. Early over-expression of GRP receptors in prostatic carcinogenesis. Prostate 2014, 74, 217–224. [Google Scholar] [CrossRef]
- Markwalder, R.; Reubi, J.C. Gastrin-releasing peptide receptors in the human prostate: Relation to neoplastic transformation. Cancer Res. 1999, 59, 1152–1159. [Google Scholar]
- Beer, M.; Montani, M.; Gerhardt, J.; Wild, P.J.; Hany, T.F.; Hermanns, T.; Muntener, M.; Kristiansen, G. Profiling gastrin-releasing peptide receptor in prostate tissues: Clinical implications and molecular correlates. Prostate 2012, 72, 318–325. [Google Scholar] [CrossRef]
- Gugger, M.; Waser, B.; Kappeler, A.; Schönbrunn, A.; Reubi, J.C. Immunohistochemical localization of somatostatin receptor SST2A in human gut and lung tissue: Possible implications for physiology and carcinogenesis. Ann. N. Y. Acad. Sci. 2004, 1014, 132–136. [Google Scholar] [CrossRef]
- Morgat, C.; MacGrogan, G.; Brouste, V.; Velasco, V.; Sevenet, N.; Bonnefoi, H.; Fernandez, P.; Debled, M.; Hindie, E. Expression of gastrin-releasing peptide receptor in breast cancer and its association with pathologic, biologic, and clinical parameters: A study of 1,432 primary tumors. J. Nucl. Med. 2017, 58, 1401–1407. [Google Scholar] [CrossRef]
- Dalm, S.U.; Martens, J.W.; Sieuwerts, A.M.; van Deurzen, C.H.; Koelewijn, S.J.; de Blois, E.; Maina, T.; Nock, B.A.; Brunel, L.; Fehrentz, J.A.; et al. In vitro and in vivo application of radiolabeled gastrin-releasing peptide receptor ligands in breast cancer. J. Nucl. Med. 2015, 56, 752–757. [Google Scholar] [CrossRef]
- Reubi, C.; Gugger, M.; Waser, B. Co-expressed peptide receptors in breast cancer as a molecular basis for in vivo multireceptor tumour targeting. Eur. J. Nucl. Med. Mol. Imaging 2002, 29, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Mattei, J.; Achcar, R.D.; Cano, C.H.; Macedo, B.R.; Meurer, L.; Batlle, B.S.; Groshong, S.D.; Kulczynski, J.M.; Roesler, R.; Dal Lago, L.; et al. Gastrin-releasing peptide receptor expression in lung cancer. Arch. Pathol. Lab. Med. 2014, 138, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C.; Körner, M.; Waser, B.; Mazzucchelli, L.; Guillou, L. High expression of peptide receptors as a novel target in gastrointestinal stromal tumours. Eur. J. Nucl. Med. Mol. Imaging 2004, 31, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C. Peptide receptor expression in GEP-NET. Virchows Archiv. 2007, 451, S47–S50. [Google Scholar] [CrossRef]
- Maina, T.; Nock, B.A. From bench to bed: New gastrin-releasing peptide receptor-directed radioligands and their use in prostate cancer. PET Clin. 2017, 12, 205–217. [Google Scholar] [CrossRef]
- Li, X.; Cai, H.; Wu, X.; Li, L.; Wu, H.; Tian, R. New frontiers in molecular imaging using peptide-based radiopharmaceuticals for prostate cancer. Front. Chem. 2020, 8, 583309. [Google Scholar] [CrossRef]
- Maina, T.; Nock, B.A.; Kulkarni, H.; Singh, A.; Baum, R.P. Theranostic prospects of gastrin-releasing peptide receptor-radioantagonists in oncology. PET Clin. 2017, 12, 297–309. [Google Scholar] [CrossRef]
- Bodei, L.; Ferrari, M.; Nunn, A.; Llull, J.; Cremonesi, M.; Martano, L.; Laurora, G.; Scardino, E.; Tiberini, S.; Bufi, G.; et al. 177Lu-AMBA bombesin analogue in hormone refractory prostate cancer patients: A Phase I escalation study with single-cycle administrations. Eur. J. Nucl. Med. Mol. Imaging 2007, 34 (Suppl. S2), S221. [Google Scholar]
- Rozengurt, E.; Sinnett-Smith, J. Bombesin stimulation of fibroblast mitogenesis: Specific receptors, signal transduction and early events. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1990, 327, 209–221. [Google Scholar] [CrossRef]
- Delle Fave, G.; Annibale, B.; de Magistris, L.; Severi, C.; Bruzzone, R.; Puoti, M.; Melchiorri, P.; Torsoli, A.; Erspamer, V. Bombesin effects on human GI functions. Peptides 1985, 6 (Suppl. S3), 113–116. [Google Scholar] [CrossRef]
- Bruzzone, R.; Tamburrano, G.; Lala, A.; Mauceri, M.; Annibale, B.; Severi, C.; de Magistris, L.; Leonetti, F.; Delle Fave, G. Effect of bombesin on plasma insulin, pancreatic glucagon, and gut glucagon in man. J. Clin. Endocrinol. Metab. 1983, 56, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Linder, K.E.; Metcalfe, E.; Arunachalam, T.; Chen, J.; Eaton, S.M.; Feng, W.; Fan, H.; Raju, N.; Cagnolini, A.; Lantry, L.E.; et al. In vitro and in vivo metabolism of Lu-AMBA, a GRP-receptor binding compound, and the synthesis and characterization of its metabolites. Bioconjug. Chem. 2009, 20, 1171–1178. [Google Scholar] [CrossRef] [PubMed]
- Lymperis, E.; Kaloudi, A.; Sallegger, W.; Bakker, I.L.; Krenning, E.P.; de Jong, M.; Maina, T.; Nock, B.A. Radiometal-dependent biological profile of the radiolabeled gastrin-releasing peptide receptor antagonist SB3 in cancer theranostics: Metabolic and biodistribution patterns defined by neprilysin. Bioconjug. Chem. 2018, 29, 1774–1784. [Google Scholar] [CrossRef] [PubMed]
- Marsouvanidis, P.J.; Maina, T.; Sallegger, W.; Krenning, E.P.; de Jong, M.; Nock, B.A. Tumor diagnosis with new 111In-radioligands based on truncated human gastrin releasing peptide sequences: Synthesis and preclinical comparison. J. Med. Chem. 2013, 56, 8579–8587. [Google Scholar] [CrossRef] [PubMed]
- Marsouvanidis, P.J.; Maina, T.; Sallegger, W.; Krenning, E.P.; de Jong, M.; Nock, B.A. 99mTc radiotracers based on human GRP(18-27): Synthesis and comparative evaluation. J. Nucl. Med. 2013, 54, 1797–1803. [Google Scholar] [CrossRef] [PubMed]
- Chatalic, K.L.; Kwekkeboom, D.J.; de Jong, M. Radiopeptides for imaging and therapy: A radiant future. J. Nucl. Med. 2015, 56, 1809–1812. [Google Scholar] [CrossRef] [PubMed]
- Van de Wiele, C.; Dumont, F.; Vanden Broecke, R.; Oosterlinck, W.; Cocquyt, V.; Serreyn, R.; Peers, S.; Thornback, J.; Slegers, G.; Dierckx, R.A. Technetium-99m RP527, a GRP analogue for visualisation of GRP receptor-expressing malignancies: A feasibility study. Eur. J. Nucl. Med. 2000, 27, 1694–1699. [Google Scholar] [CrossRef]
- Nock, B.A.; Nikolopoulou, A.; Galanis, A.; Cordopatis, P.; Waser, B.; Reubi, J.C.; Maina, T. Potent bombesin-like peptides for GRP-receptor targeting of tumors with 99mTc: A preclinical study. J. Med. Chem. 2005, 48, 100–110. [Google Scholar] [CrossRef]
- Lantry, L.E.; Cappelletti, E.; Maddalena, M.E.; Fox, J.S.; Feng, W.; Chen, J.; Thomas, R.; Eaton, S.M.; Bogdan, N.J.; Arunachalam, T.; et al. 177Lu-AMBA: Synthesis and characterization of a selective 177Lu-labeled GRP-R agonist for systemic radiotherapy of prostate cancer. J. Nucl. Med. 2006, 47, 1144–1152. [Google Scholar]
- Nock, B.A.; Maina, T.; Krenning, E.P.; de Jong, M. "To serve and protect": Enzyme inhibitors as radiopeptide escorts promote tumor targeting. J. Nucl. Med. 2014, 55, 121–127. [Google Scholar] [CrossRef]
- Kanellopoulos, P.; Kaloudi, A.; Rouchota, M.; Loudos, G.; de Jong, M.; Krenning, E.P.; Nock, B.A.; Maina, T. One step closer to clinical translation: Enhanced tumor targeting of [99mTc]Tc-DB4 and [111In]In-SG4 in mice treated with Entresto. Pharmaceutics 2020, 12, 1145. [Google Scholar] [CrossRef] [PubMed]
- Mather, S.J.; Nock, B.A.; Maina, T.; Gibson, V.; Ellison, D.; Murray, I.; Sobnack, R.; Colebrook, S.; Wan, S.; Halberrt, G.; et al. GRP receptor imaging of prostate cancer using [Tc-99m]Demobesin 4: A first-in-man study. Mol. Imaging Biol. 2014, 16, 888–895. [Google Scholar] [CrossRef] [PubMed]
- Baum, R.P.V.; Mutloka, N.; Frischknecht, M.; Maecke, H.; Reubi, J.C. Molecular imaging of bombesin receptors in various tumors by Ga-68 AMBA PET/CT: First results. J. Nucl. Med. 2007, 48 (Suppl. S2). [Google Scholar]
- De Castiglione, R.; Gozzini, L. Bombesin receptor antagonists. Crit. Rev. Oncol. Hematol. 1996, 24, 117–151. [Google Scholar] [CrossRef] [PubMed]
- Azay, J.; Nagain, C.; Llinares, M.; Devin, C.; Fehrentz, J.A.; Bernad, N.; Roze, C.; Martinez, J. Comparative study of in vitro and in vivo activities of bombesin pseudopeptide analogs modified on the C-terminal dipeptide fragment. Peptides 1998, 19, 57–63. [Google Scholar] [CrossRef]
- Wang, L.H.; Coy, D.H.; Taylor, J.E.; Jiang, N.Y.; Moreau, J.P.; Huang, S.C.; Frucht, H.; Haffar, B.M.; Jensen, R.T. Des-Met carboxyl-terminally modified analogues of bombesin function as potent bombesin receptor antagonists, partial agonists, or agonists. J. Biol. Chem. 1990, 265, 15695–15703. [Google Scholar] [CrossRef] [PubMed]
- Heimbrook, D.C.; Saari, W.S.; Balishin, N.L.; Fisher, T.W.; Friedman, A.; Kiefer, D.M.; Rotberg, N.S.; Wallen, J.W.; Oliff, A. Gastrin releasing peptide antagonists with improved potency and stability. J. Med. Chem. 1991, 34, 2102–2107. [Google Scholar] [CrossRef]
- Nock, B.A.; Kaloudi, A.; Kanellopoulos, P.; Janota, B.; Bromińska, B.; Iżycki, D.; Mikołajczak, R.; Czepczynski, R.; Maina, T. [99mTc]Tc-DB15 in GRPR-targeted tumor imaging with SPECT: From preclinical evaluation to the first clinical outcomes. Cancers 2021, 13, 5093. [Google Scholar] [CrossRef]
- Maina, T.; Bergsma, H.; Kulkarni, H.R.; Mueller, D.; Charalambidis, D.; Krenning, E.P.; Nock, B.A.; de Jong, M.; Baum, R.P. Preclinical and first clinical experience with the gastrin-releasing peptide receptor-antagonist [68Ga]SB3 and PET/CT. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 964–973. [Google Scholar] [CrossRef]
- Bakker, I.L.; Fröberg, A.C.; Busstra, M.B.; Verzijlbergen, J.F.; Konijnenberg, M.; van Leenders, G.; Schoots, I.G.; de Blois, E.; van Weerden, W.M.; Dalm, S.U.; et al. GRPR antagonist 68Ga-SB3 PET/CT-imaging of primary prostate cancer in therapy-naive patients. J. Nucl. Med. 2021, 62, 1517–1523. [Google Scholar] [CrossRef]
- Stoykow, C.; Erbes, T.; Maecke, H.R.; Bulla, S.; Bartholoma, M.; Mayer, S.; Drendel, V.; Bronsert, P.; Werner, M.; Gitsch, G.; et al. Gastrin-releasing peptide receptor imaging in breast cancer using the receptor antagonist 68Ga-RM2 and PET. Theranostics 2016, 6, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Kähkonen, E.; Jambor, I.; Kemppainen, J.; Lehtio, K.; Gronroos, T.J.; Kuisma, A.; Luoto, P.; Sipila, H.J.; Tolvanen, T.; Alanen, K.; et al. In vivo imaging of prostate cancer using [68Ga]-labeled bombesin analog BAY86-7548. Clin. Cancer Res. 2013, 19, 5434–5443. [Google Scholar] [CrossRef] [PubMed]
- Kurth, J.; Krause, B.J.; Schwarzenbock, S.M.; Bergner, C.; Hakenberg, O.W.; Heuschkel, M. First-in-human dosimetry of gastrin-releasing peptide receptor antagonist [177Lu]Lu-RM2: A radiopharmaceutical for the treatment of metastatic castration-resistant prostate cancer. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Nock, B.A.; Kaloudi, A.; Lymperis, E.; Giarika, A.; Kulkarni, H.R.; Klette, I.; Singh, A.; Krenning, E.P.; de Jong, M.; Maina, T.; et al. Theranostic perspectives in prostate cancer with the gastrin-releasing peptide receptor antagonist NeoBOMB1: Preclinical and first clinical results. J. Nucl. Med. 2017, 58, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Kaloudi, A.; Lymperis, E.; Giarika, A.; Dalm, S.; Orlandi, F.; Barbato, D.; Tedesco, M.; Maina, T.; de Jong, M.; Nock, B.A. NeoBOMB1, a GRPR-antagonist for breast cancer theragnostics: First results of a preclinical study with [67Ga]NeoBOMB1 in T-47D cells and tumor-bearing mice. Molecules 2017, 22, 1950. [Google Scholar] [CrossRef]
- Dalm, S.U.; Bakker, I.L.; de Blois, E.; Doeswijk, G.N.; Konijnenberg, M.W.; Orlandi, F.; Barbato, D.; Tedesco, M.; Maina, T.; Nock, B.A.; et al. Ga-68/Lu-177-NeoBOMB1, a novel radiolabeled GRPR antagonist for theranostic use in oncology. J. Nucl. Med. 2017, 58, 293–299. [Google Scholar] [CrossRef]
- Gruber, L.; Jimenez-Franco, L.D.; Decristoforo, C.; Uprimny, C.; Glatting, G.; Hohenberger, P.; Schoenberg, S.O.; Reindl, W.; Orlandi, F.; Mariani, M.; et al. MITIGATE-NeoBOMB1, a Phase I/IIA study to evaluate safety, pharmacokinetics and preliminary imaging of 68Ga-NeoBOMB1, a gastrin-releasing peptide receptor antagonist, in GIST patients. J. Nucl. Med. 2020, 61, 1749–1755. [Google Scholar] [CrossRef]
- Gruber, L.; Decristoforo, C.; Uprimny, C.; Hohenberger, P.; Schoenberg, S.O.; Orlandi, F.; Mariani, M.F.; Manzl, C.; Kasseroler, M.T.; Tilg, H.; et al. Imaging properties and tumor targeting of 68Ga-NeoBOMB1, a gastrin-releasing peptide receptor antagonist, in GIST patients. Biomedicines 2022, 10, 2899. [Google Scholar] [CrossRef]
- Ruigrok, E.A.M.; Verhoeven, M.; Konijnenberg, M.W.; de Blois, E.; de Ridder, C.M.A.; Stuurman, D.C.; Bertarione, L.; Rolfo, K.; de Jong, M.; Dalm, S.U. Safety of [177Lu]Lu-NeoBOMB1 treatment: A preclinical study characterizing absorbed dose and acute, early, and late organ toxicity. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 4440–4451. [Google Scholar] [CrossRef]
- Minamimoto, R.; Sonni, I.; Hancock, S.; Vasanawala, S.; Loening, A.; Gambhir, S.S.; Iagaru, A. Prospective evaluation of 68Ga-RM2 PET/MRI in patients with biochemical recurrence of prostate cancer and negative findings on conventional imaging. J. Nucl. Med. 2018, 59, 803–808. [Google Scholar] [CrossRef]
- Rozengurt, E.; Walsh, J.H. Gastrin, CCK, signaling, and cancer. Annu. Rev. Physiol. 2001, 63, 49–76. [Google Scholar] [CrossRef]
- Townsend, C.M., Jr.; Singh, P.; Thompson, J.C. Gastrointestinal hormones and gastrointestinal and pancreatic carcinomas. Gastroenterology 1986, 91, 1002–1006. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Ou, L.; Wang, W.; Guo, D.Y. Gastrin, cholecystokinin, signaling, and biological activities in cellular processes. Front. Endocrinol. 2020, 11, 112. [Google Scholar] [CrossRef]
- Béhé, M.; Behr, T.M. Cholecystokinin-B (CCK-B)/gastrin receptor targeting peptides for staging and therapy of medullary thyroid cancer and other CCK-B receptor expressing malignancies. Biopolymers 2002, 66, 399–418. [Google Scholar] [CrossRef] [PubMed]
- Dufresne, M.; Seva, C.; Fourmy, D. Cholecystokinin and gastrin receptors. Physiol. Rev. 2006, 86, 805–847. [Google Scholar] [CrossRef] [PubMed]
- Wank, S.A. Cholecystokinin receptors. Am. J. Physiol. 1995, 269 Pt 1, G628–G646. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, P.M.; Humphrey, P.P.; Spedding, M.X. International union of pharmacology recommendations for nomenclature of new receptor subtypes. Pharmacol. Rev. 1996, 48, 1–2. [Google Scholar]
- Hellmich, M.R.; Rui, X.L.; Hellmich, H.L.; Fleming, R.Y.; Evers, B.M.; Townsend, C.M., Jr. Human colorectal cancers express a constitutively active cholecystokinin-B/gastrin receptor that stimulates cell growth. J. Biol. Chem. 2000, 275, 32122–32128. [Google Scholar] [CrossRef]
- Reubi, J.C.; Schaer, J.C.; Waser, B. Cholecystokinin(CCK)-A and CCK-B/gastrin receptors in human tumors. Cancer Res. 1997, 57, 1377–1386. [Google Scholar]
- Reubi, J.C.; Waser, B. Unexpected high incidence of cholecystokinin-B/gastrin receptors in human medullary thyroid carcinomas. Int. J. Cancer 1996, 67, 644–647. [Google Scholar] [CrossRef]
- Camby, I.; Salmon, I.; Danguy, A.; Pasteels, J.L.; Brotchi, J.; Martinez, J.; Kiss, R. Influence of gastrin on human astrocytic tumor cell proliferation. J. Natl. Cancer Inst. 1996, 88, 594–600. [Google Scholar] [CrossRef] [PubMed]
- Matsumori, Y.; Katakami, N.; Ito, M.; Taniguchi, T.; Iwata, N.; Takaishi, T.; Chihara, K.; Matsui, T. Cholecystokinin-B/gastrin receptor: A novel molecular probe for human small cell lung cancer. Cancer Res. 1995, 55, 276–279. [Google Scholar] [PubMed]
- Upp, J.R., Jr.; Singh, P.; Townsend, C.M., Jr.; Thompson, J.C. Clinical significance of gastrin receptors in human colon cancers. Cancer Res. 1989, 49, 488–492. [Google Scholar] [PubMed]
- Kaloudi, A.; Nock, B.A.; Krenning, E.P.; Maina, T.; de Jong, M. Radiolabeled gastrin/CCK analogs in tumor diagnosis: Towards higher stability and improved tumor targeting. Q. J. Nucl. Med. Mol. Imaging 2015, 59, 287–302. [Google Scholar]
- Von Guggenberg, E.; Kolenc, P.; Rottenburger, C.; Mikolajczak, R.; Hubalewska-Dydejczyk, A. Update on preclinical development and clinical translation of cholecystokinin-2 receptor targeting radiopharmaceuticals. Cancers 2021, 13, 5776. [Google Scholar] [CrossRef]
- Behr, T.M.; Jenner, N.; Radetzky, S.; Béhé, M.; Gratz, S.; Yucekent, S.; Raue, F.; Becker, W. Targeting of cholecystokinin-B/gastrin receptors in vivo: Preclinical and initial clinical evaluation of the diagnostic and therapeutic potential of radiolabelled gastrin. Eur. J. Nucl. Med. 1998, 25, 424–430. [Google Scholar] [CrossRef]
- Kosowicz, J.; Mikolajczak, R.; Czepczynski, R.; Ziemnicka, K.; Gryczynska, M.; Sowinski, J. Two peptide receptor ligands 99mTc-EDDA/HYNIC-Tyr3-octreotide and 99mTc-EDDA/HYNIC-DGlu-octagastrin for scintigraphy of medullary thyroid carcinoma. Cancer Biother. Radiopharm. 2007, 22, 613–628. [Google Scholar] [CrossRef]
- Nock, B.A.; Maina, T.; Béhé, M.; Nikolopoulou, A.; Gotthardt, M.; Schmitt, J.S.; Behr, T.M.; Mäcke, H.R. CCK-2/gastrin receptor-targeted tumor imaging with 99mTc-labeled minigastrin analogs. J. Nucl. Med. 2005, 46, 1727–1736. [Google Scholar]
- Fröberg, A.C.; de Jong, M.; Nock, B.A.; Breeman, W.A.; Erion, J.L.; Maina, T.; Verdijsseldonck, M.; de Herder, W.W.; van der Lugt, A.; Kooij, P.P.; et al. Comparison of three radiolabelled peptide analogues for CCK-2 receptor scintigraphy in medullary thyroid carcinoma. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 1265–1272. [Google Scholar] [CrossRef]
- Breeman, W.A.; Fröberg, A.C.; de Blois, E.; van Gameren, A.; Melis, M.; de Jong, M.; Maina, T.; Nock, B.A.; Erion, J.L.; Mäcke, H.R.; et al. Optimised labeling, preclinical and initial clinical aspects of CCK-2 receptor-targeting with 3 radiolabeled peptides. Nucl. Med. Biol. 2008, 35, 839–849. [Google Scholar] [CrossRef]
- Béhé, M.; Becker, W.; Gotthardt, M.; Angerstein, C.; Behr, T.M. Improved kinetic stability of DTPA- DGlu as compared with conventional monofunctional DTPA in chelating indium and yttrium: Preclinical and initial clinical evaluation of radiometal labelled minigastrin derivatives. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 1140–1146. [Google Scholar] [CrossRef] [PubMed]
- Gotthardt, M.; Béhé, M.P.; Beuter, D.; Battmann, A.; Bauhofer, A.; Schurrat, T.; Schipper, M.; Pollum, H.; Oyen, W.J.; Behr, T.M. Improved tumour detection by gastrin receptor scintigraphy in patients with metastasised medullary thyroid carcinoma. Eur. J. Nucl. Med. Mol. Imaging 2006, 33, 1273–1279. [Google Scholar] [CrossRef] [PubMed]
- Laverman, P.; Roosenburg, S.; Gotthardt, M.; Park, J.; Oyen, W.J.; de Jong, M.; Hellmich, M.R.; Rutjes, F.P.; van Delft, F.L.; Boerman, O.C. Targeting of a CCK2 receptor splice variant with 111In-labelled cholecystokinin-8 (CCK8) and 111In-labelled minigastrin. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 386–392. [Google Scholar] [CrossRef]
- Kaloudi, A.; Nock, B.A.; Lymperis, E.; Sallegger, W.; Krenning, E.P.; de Jong, M.; Maina, T. In vivo inhibition of neutral endopeptidase enhances the diagnostic potential of truncated gastrin 111In-radioligands. Nucl. Med. Biol. 2015, 42, 824–832. [Google Scholar] [CrossRef] [PubMed]
- Kaloudi, A.; Nock, B.A.; Lymperis, E.; Valkema, R.; Krenning, E.P.; de Jong, M.; Maina, T. Impact of clinically tested NEP/ACE inhibitors on tumor uptake of [111In-DOTA]MG11-first estimates for clinical translation. EJNMMI Res. 2016, 6, 15. [Google Scholar] [CrossRef] [PubMed]
- Valkema, R.; Schonebaum, E.L.; Fröberg, A.C.; Maina, T.; Nock, B.A.; de Blois, E.; Konijnenberg, M.W.; Koolen, S.L.W.; Peeters, R.P.; Visser, W.E.; et al. PepProtect: Improved detection of cancer and metastases by peptide scanning under the protection of enzyme inhibitors. Eur. J. Nucl. Med. Mol. Imaging 2022, 49 (Suppl. S1), S81. [Google Scholar]
- Pawlak, D.; Rangger, C.; Kolenc Peitl, P.; Garnuszek, P.; Maurin, M.; Ihli, L.; Kroselj, M.; Maina, T.; Maecke, H.; Erba, P.; et al. From preclinical development to clinical application: Kit formulation for radiolabelling the minigastrin analogue CP04 with In-111 for a first-in-human clinical trial. Eur. J. Pharm. Sci. 2016, 85, 1–9. [Google Scholar] [CrossRef]
- Maina, T.; Konijnenberg, M.W.; Kolenc-Peitl, P.; Garnuszek, P.; Nock, B.A.; Kaloudi, A.; Kroselj, M.; Zaletel, K.; Maecke, H.; Mansi, R.; et al. Preclinical pharmacokinetics, biodistribution, radiation dosimetry and toxicity studies required for regulatory approval of a Phase I clinical trial with 111In-CP04 in medullary thyroid carcinoma patients. Eur. J. Pharm. Sci. 2016, 91, 236–242. [Google Scholar] [CrossRef]
- Lezaic, L.; Erba, P.A.; Decristoforo, C.; Zaletel, K.; Mikolajczak, R.; Maecke, H.; Maina, T.; Konijnenberg, M.; Kolenc, P.; Trofimiuk-Muldner, M.; et al. [111In]In-CP04 as a novel cholecystokinin-2 receptor ligand with theranostic potential in patients with progressive or metastatic medullary thyroid cancer: Final results of a GRAN-T-MTC Phase I clinical trial. Eur. J. Nucl. Med. Mol. Imaging 2023, 50, 892–907. [Google Scholar] [CrossRef]
- Rottenburger, C.; Nicolàs, G.P.; McDougall, L.; Kaul, F.; Cachovan, M.; Vija, A.H.; Schibli, R.; Geistlich, S.; Schumann, A.; Rau, T.; et al. Cholecystokinin 2 receptor agonist 177Lu-PP-F11N for radionuclide therapy of medullary thyroid carcinoma: Results of the LUMED Phase 0a study. J. Nucl. Med. 2020, 61, 520–526. [Google Scholar] [CrossRef]
- Von Guggenberg, E.; Uprimny, C.; Klingler, M.; Warwitz, B.; Sviridenko, A.; Bayerschmidt, S.; Di Santo, G.; Virgolini, I.J. Preliminary clinical experience of cholecystokinin-2 receptor PET/CT imaging using the 68Ga-labeled minigastrin analog DOTA-MGS5 in patients with medullary thyroid cancer. J. Nucl. Med. 2023. [Google Scholar] [CrossRef] [PubMed]
- Ubl, P.; Gincu, T.; Keilani, M.; Ponhold, L.; Crevenna, R.; Niederle, B.; Hacker, M.; Li, S. Comparison of side effects of pentagastrin test and calcium stimulation test in patients with increased basal calcitonin concentration: The gender-specific differences. Endocrine 2014, 46, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Novak, D.; Anderluh, M.; Kolenc Peitl, P. CCK2R antagonists: From SAR to clinical trials. Drug. Discov. Today 2020, 25, 1322–1336. [Google Scholar] [CrossRef] [PubMed]
- Ukawa, H.; Miura, N.; Morita, H.; Hori, Y.; Ueki, S.; Yoneta, T.; Kurimoto, T.; Itoh, Z. Effect of Z-360, a selective CCKB/gastrin receptor antagonist, on chronic acid reflux esophagitis in rats. Gastroenterology 2002, 122, A194. [Google Scholar]
- Ueno, M.; Li, C.P.; Ikeda, M.; Ishii, H.; Mizuno, N.; Yamaguchi, T.; Ioka, T.; Oh, D.Y.; Ichikawa, W.; Okusaka, T.; et al. A randomized Phase II study of gemcitabine plus Z-360, a CCK2 receptor-selective antagonist, in patients with metastatic pancreatic cancer as compared with gemcitabine plus placebo. Cancer Chemother. Pharmacol. 2017, 80, 307–315. [Google Scholar] [CrossRef]
- Wayua, C.; Low, P.S. Evaluation of a nonpeptidic ligand for imaging of cholecystokinin 2 receptor-expressing cancers. J. Nucl. Med. 2015, 56, 113–119. [Google Scholar] [CrossRef]
- Nock, B.A.; Kanellopoulos, P.; Chepurny, O.G.; Rouchota, M.; Loudos, G.; Holz, G.G.; Krenning, E.P.; Maina, T. Nonpeptidic Z360-analogs tagged with trivalent radiometals as anti-CCK2R cancer theranostic agents: A preclinical study. Pharmaceutics 2022, 14, 666. [Google Scholar] [CrossRef]
- Novak, D.; Tomasic, T.; Kroselj, M.; Javornik, U.; Plavec, J.; Anderluh, M.; Kolenc Peitl, P. Radiolabelled CCK2 R antagonists containing PEG linkers: Design, synthesis and evaluation. ChemMedChem 2021, 16, 155–163. [Google Scholar] [CrossRef]
- Verona, M.; Rubagotti, S.; Croci, S.; Sarpaki, S.; Borgna, F.; Tosato, M.; Vettorato, E.; Marzaro, G.; Mastrotto, F.; Asti, M. Preliminary study of a 1,5-benzodiazepine-derivative labelled with indium-111 for CCK-2 receptor targeting. Molecules 2021, 26, 918. [Google Scholar] [CrossRef]
- Grzmil, M.; Qin, Y.; Schleuniger, C.; Frank, S.; Imobersteg, S.; Blanc, A.; Spillmann, M.; Berger, P.; Schibli, R.; Béhé, M. Pharmacological inhibition of mTORC1 increases CCKBR-specific tumor uptake of radiolabeled minigastrin analogue [177Lu]Lu-PP-F11N. Theranostics 2020, 10, 10861–10873. [Google Scholar] [CrossRef]
- Qin, Y.; Imobersteg, S.; Blanc, A.; Frank, S.; Schibli, R.; Béhé, M.P.; Grzmil, M. Evaluation of actinium-225 labeled minigastrin analogue [225Ac]Ac-DOTA-PP-F11N for targeted alpha particle therapy. Pharmaceutics 2020, 12, 1088. [Google Scholar] [CrossRef] [PubMed]
- Körner, M.; Stöckli, M.; Waser, B.; Reubi, J.C. GLP-1 receptor expression in human tumors and human normal tissues: Potential for in vivo targeting. J. Nucl. Med. 2007, 48, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C.; Waser, B. Concomitant expression of several peptide receptors in neuroendocrine tumours: Molecular basis for in vivo multireceptor tumour targeting. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 781–793. [Google Scholar] [CrossRef] [PubMed]
- Gotthardt, M.; Fischer, M.; Naeher, I.; Holz, J.B.; Jungclas, H.; Fritsch, H.W.; Béhé, M.; Goke, B.; Joseph, K.; Behr, T.M. Use of the incretin hormone glucagon-like peptide-1 (GLP-1) for the detection of insulinomas: Initial experimental results. Eur. J. Nucl. Med. Mol. Imaging 2002, 29, 597–606. [Google Scholar] [CrossRef]
- Mehrabi, A.; Fischer, L.; Hafezi, M.; Dirlewanger, A.; Grenacher, L.; Diener, M.K.; Fonouni, H.; Golriz, M.; Garoussi, C.; Fard, N.; et al. A systematic review of localization, surgical treatment options, and outcome of insulinoma. Pancreas 2014, 43, 675–686. [Google Scholar] [CrossRef]
- Wild, D.; Christ, E.; Caplin, M.E.; Kurzawinski, T.R.; Forrer, F.; Brandle, M.; Seufert, J.; Weber, W.A.; Bomanji, J.; Perren, A.; et al. Glucagon-like peptide-1 versus somatostatin receptor targeting reveals 2 distinct forms of malignant insulinomas. J. Nucl. Med. 2011, 52, 1073–1078. [Google Scholar] [CrossRef]
- Calabro, D.; Argalia, G.; Ambrosini, V. Role of PET/CT and therapy management of pancreatic neuroendocrine tumors. Diagnostics 2020, 10, 1059. [Google Scholar] [CrossRef]
- Eng, J.; Kleinman, W.A.; Singh, L.; Singh, G.; Raufman, J.P. Isolation and characterization of exendin-4, an exendin-3 analogue, from heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. J. Biol. Chem. 1992, 267, 7402–7405. [Google Scholar] [CrossRef]
- Antwi, K.; Fani, M.; Heye, T.; Nicolàs, G.; Rottenburger, C.; Kaul, F.; Merkle, E.; Zech, C.J.; Boll, D.; Vogt, D.R.; et al. Comparison of glucagon-like peptide-1 receptor (GLP-1R) PET/CT, SPECT/CT and 3T MRI for the localisation of occult insulinomas: Evaluation of diagnostic accuracy in a prospective crossover imaging study. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 2318–2327. [Google Scholar] [CrossRef]
- De Carbonnieres, A.; Challine, A.; Cottereau, A.S.; Coriat, R.; Soyer, P.; Abou Ali, E.; Prat, F.; Terris, B.; Bertherat, J.; Dousset, B.; et al. Surgical management of insulinoma over three decades. HPB 2021, 23, 1799–1806. [Google Scholar] [CrossRef]
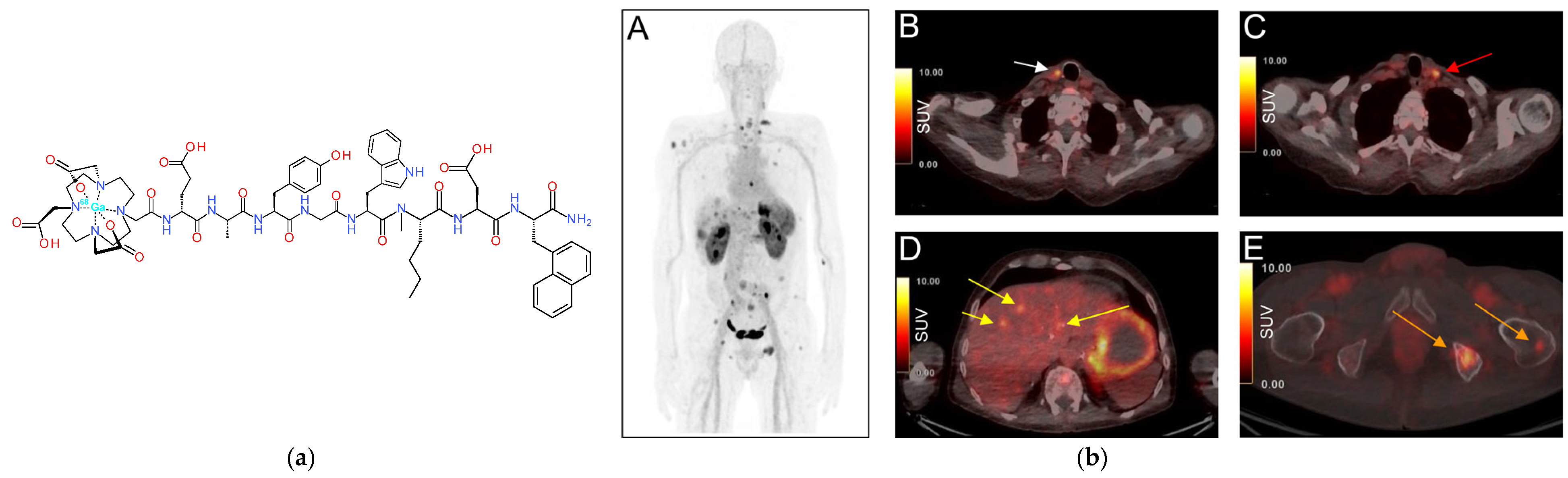
- Eriksson, O.; Velikyan, I.; Selvaraju, R.K.; Kandeel, F.; Johansson, L.; Antoni, G.; Eriksson, B.; Sorensen, J.; Korsgren, O. Detection of metastatic insulinoma by positron emission tomography with [68Ga]exendin-4-a case report. J. Clin. Endocrinol. Metab. 2014, 99, 1519–1524. [Google Scholar] [CrossRef] [PubMed]
- Richards, M.L.; Gauger, P.G.; Thompson, N.W.; Kloos, R.G.; Giordano, T.J. Pitfalls in the surgical treatment of insulinoma. Surgery 2002, 132, 1040–1049, discussion 1049. [Google Scholar] [CrossRef] [PubMed]
- Wicki, A.; Wild, D.; Storch, D.; Seemayer, C.; Gotthardt, M.; Béhé, M.; Kneifel, S.; Mihatsch, M.J.; Reubi, J.C.; Mäcke, H.R.; et al. [Lys40(Ahx-DTPA-111In)NH2]-exendin-4 is a highly efficient radiotherapeutic for glucagon-like peptide-1 receptor-targeted therapy for insulinoma. Clin. Cancer Res. 2007, 13, 3696–3705. [Google Scholar] [CrossRef]
- Velikyan, I.; Bulenga, T.N.; Selvaraju, R.; Lubberink, M.; Espes, D.; Rosenstrom, U.; Eriksson, O. Dosimetry of [177Lu]-DO3A-VS-Cys40-exendin-4—impact on the feasibility of insulinoma internal radiotherapy. Am. J. Nucl. Med. Mol. Imaging 2015, 5, 109–126. [Google Scholar] [PubMed]
- Zhang, M.; Jacobson, O.; Kiesewetter, D.O.; Ma, Y.; Wang, Z.; Lang, L.; Tang, L.; Kang, F.; Deng, H.; Yang, W.; et al. Improving the theranostic potential of exendin 4 by reducing the renal radioactivity through brush border membrane enzyme-mediated degradation. Bioconjug. Chem. 2019, 30, 1745–1753. [Google Scholar] [CrossRef]
- Kaeppeli, S.A.M.; Jodal, A.; Gotthardt, M.; Schibli, R.; Béhé, M. Exendin-4 derivatives with an albumin-binding moiety show decreased renal retention and improved GLP-1 receptor targeting. Mol. Pharm. 2019, 16, 3760–3769. [Google Scholar] [CrossRef]
- Iikuni, S.; Kamei, I.; Ohara, T.; Watanabe, H.; Ono, M. Development of an 111In-labeled glucagon-like peptide-1 receptor-targeting exendin-4 derivative that exhibits reduced renal uptake. Mol. Pharm. 2022, 19, 1019–1027. [Google Scholar] [CrossRef]
- Buitinga, M.; Jansen, T.; van der Kroon, I.; Woliner-van der Weg, W.; Boss, M.; Janssen, M.; Aarntzen, E.; Béhé, M.; Wild, D.; Visser, E.; et al. Succinylated gelatin improves the theranostic potential of radiolabeled exendin-4 in insulinoma patients. J. Nucl. Med. 2019, 60, 812–816. [Google Scholar] [CrossRef]
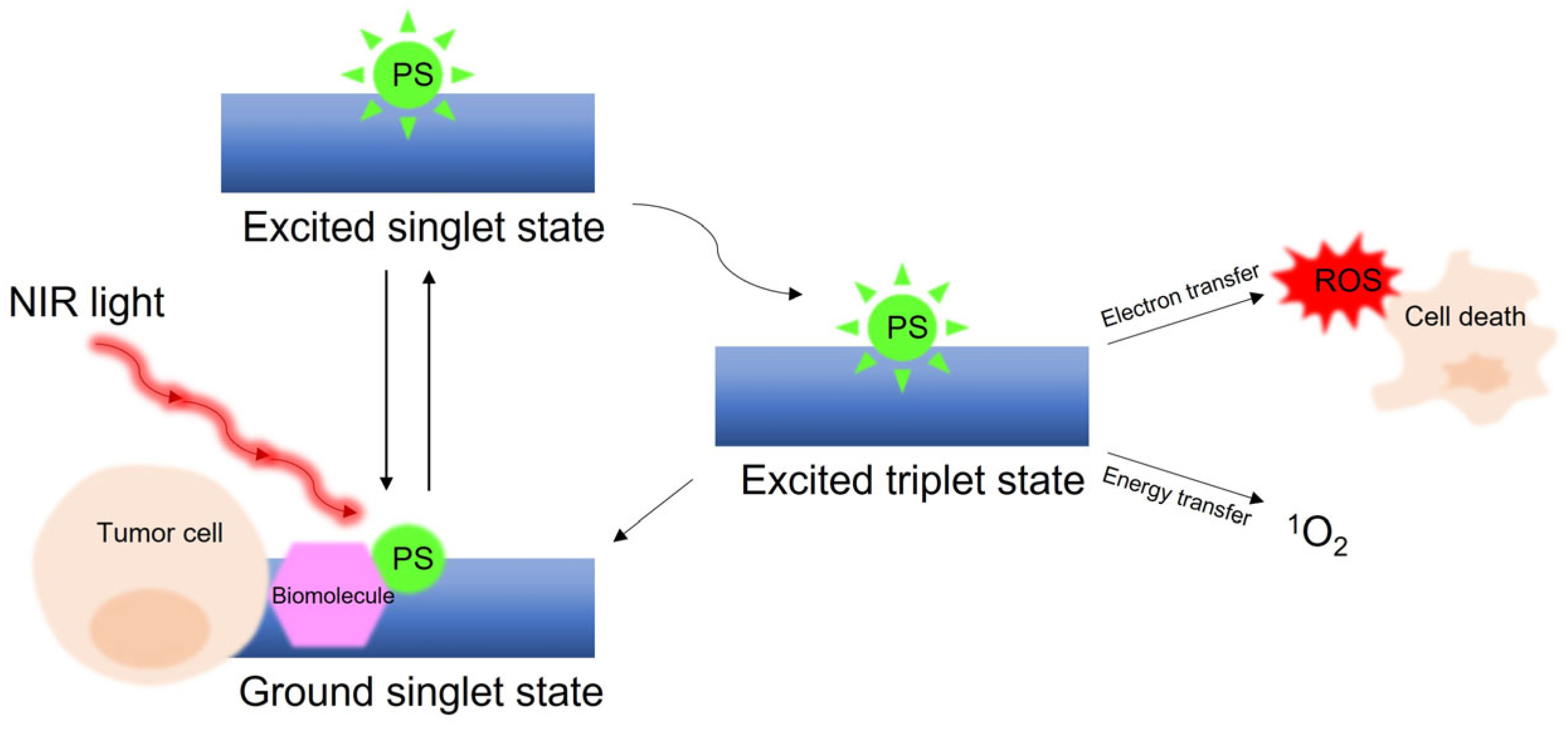
- Dougherty, T.J.; Gomer, C.J.; Henderson, B.W.; Jori, G.; Kessel, D.; Korbelik, M.; Moan, J.; Peng, Q. Photodynamic therapy. J. Natl. Cancer Inst. 1998, 90, 889–905. [Google Scholar] [CrossRef]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic therapy of cancer: An update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef]
- Wang, X.; Luo, D.; Basilion, J.P. Photodynamic therapy: Targeting cancer biomarkers for the treatment of cancers. Cancers 2021, 13, 2992. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Yang, X.; Zheng, L.; Yang, G.; Sun, L.; Bao, Y.; Wu, Y.; Huang, Y.; Yu, C.; Yang, S.N.; et al. Photoactivation of hypericin decreases the viability of RINM5F insulinoma cells through reduction in JNK/ERK phosphorylation and elevation of caspase-9/caspase-3 cleavage and BAX-to-BCL-2 ratio. Biosci. Rep. 2015, 35, e00195. [Google Scholar] [CrossRef] [PubMed]
- Karimnia, V.; Slack, F.J.; Celli, J.P. Photodynamic therapy for pancreatic ductal adenocarcinoma. Cancers 2021, 13, 4354. [Google Scholar] [CrossRef] [PubMed]
- Boss, M.; Bos, D.; Frielink, C.; Sandker, G.; Bronkhorst, P.; van Lith, S.A.M.; Brom, M.; Buitinga, M.; Gotthardt, M. Receptor-targeted photodynamic therapy of glucagon-like peptide 1 receptor-positive lesions. J. Nucl. Med. 2020, 61, 1588–1593. [Google Scholar] [CrossRef]
- Capozza, M.; Stefania, R.; Dinatale, V.; Bitonto, V.; Conti, L.; Grange, C.; Skovronova, R.; Terreno, E. A novel PSMA-targeted probe for NIRF-guided surgery and photodynamic therapy: Synthesis and preclinical validation. Int. J. Mol. Sci. 2022, 23, 2878. [Google Scholar] [CrossRef] [PubMed]
- Christ, E.; Antwi, K.; Fani, M.; Wild, D. Innovative imaging of insulinoma: The end of sampling? A review. Endocr. Relat. Cancer 2020, 27, R79–R92. [Google Scholar] [CrossRef] [PubMed]
- Thorens, B.; Porret, A.; Buhler, L.; Deng, S.P.; Morel, P.; Widmann, C. Cloning and functional expression of the human islet GLP-1 receptor. Demonstration that exendin-4 is an agonist and exendin-(9-39) an antagonist of the receptor. Diabetes 1993, 42, 1678–1682. [Google Scholar] [CrossRef] [PubMed]
- Goke, R.; Fehmann, H.C.; Linn, T.; Schmidt, H.; Krause, M.; Eng, J.; Goke, B. Exendin-4 is a high potency agonist and truncated exendin-(9-39)-amide an antagonist at the glucagon-like peptide 1-(7-36)-amide receptor of insulin-secreting beta-cells. J. Biol. Chem. 1993, 268, 19650–19655. [Google Scholar] [CrossRef] [PubMed]
- Brom, M.; Joosten, L.; Oyen, W.J.; Gotthardt, M.; Boerman, O.C. Radiolabelled GLP-1 analogues for in vivo targeting of insulinomas. Contrast Media Mol. Imaging 2012, 7, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Rylova, S.N.; Waser, B.; Del Pozzo, L.; Tonnesmann, R.; Mansi, R.; Meyer, P.T.; Reubi, J.C.; Maecke, H.R. Approaches to improve the pharmacokinetics of radiolabeled glucagon-like peptide-1 receptor ligands using antagonistic tracers. J. Nucl. Med. 2016, 57, 1282–1288. [Google Scholar] [CrossRef]
- Kimura, H.; Matsuda, H.; Ogawa, Y.; Fujimoto, H.; Toyoda, K.; Fujita, N.; Arimitsu, K.; Hamamatsu, K.; Yagi, Y.; Ono, M.; et al. Development of 111In-labeled exendin(9-39) derivatives for single-photon emission computed tomography imaging of insulinoma. Bioorg. Med. Chem. 2017, 25, 1406–1412. [Google Scholar] [CrossRef] [PubMed]
- Lappchen, T.; Tonnesmann, R.; Eersels, J.; Meyer, P.T.; Maecke, H.R.; Rylova, S.N. Radioiodinated exendin-4 is superior to the radiometal-labelled glucagon-like peptide-1 receptor probes overcoming their high kidney uptake. PLoS ONE 2017, 12, e0170435. [Google Scholar] [CrossRef] [PubMed]
- Waser, B.; Reubi, J.C. Radiolabelled GLP-1 receptor antagonist binds to GLP-1 receptor-expressing human tissues. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 1166–1171. [Google Scholar] [CrossRef] [PubMed]
- Collado Camps, E.; van Lith, S.A.M.; Kip, A.; Frielink, C.; Joosten, L.; Brock, R.; Gotthardt, M. Conjugation to a cell-penetrating peptide drives the tumour accumulation of the GLP1R antagonist exendin(9-39). Eur. J. Nucl. Med. Mol. Imaging 2023, 50, 996–1004. [Google Scholar] [CrossRef] [PubMed]
- Cherry, S.R.; Jones, T.; Karp, J.S.; Qi, J.; Moses, W.W.; Badawi, R.D. Total-body PET: Maximizing sensitivity to create new opportunities for clinical research and patient care. J. Nucl. Med. 2018, 59, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Targeted Alpha Therapy Working Group; Parker, C.; Lewington, V.; Shore, N.; Kratochwil, C.; Levy, M.; Linden, O.; Noordzij, W.; Park, J.; Saad, F. Targeted alpha therapy, an emerging class of cancer agents: A review. JAMA Oncol. 2018, 4, 1765–1772. [Google Scholar] [CrossRef]
- Ku, A.; Facca, V.J.; Cai, Z.; Reilly, R.M. Auger electrons for cancer therapy—A review. EJNMMI Radiopharm. Chem. 2019, 4, 27. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Receptor | Tumor Expression |
|---|---|---|
| Somatostatin | SST1R, SST2R, SST3R, SST4R, SST5R | NET, NHL, melanoma, BC, MTC, SCLC |
| Bombesin/GRP | BB1R/NMBR, BB2R/GRPR, BB3R | PC, BC, SCLC, colorectal cancer, glioblastoma, gastrinoma, GIST |
| CCK/gastrin | CCK1R, CCK2R | MTC, SCLC, astrocytoma, stromal ovarian cancer, GIST |
| GLP/Exendin | GLP-1R | insulinoma |
| Neurotensin | NTS1R, NTS2R, NTS3R | SCLC, PDAC, Ewing sarcoma, meningioma, astrocytoma |
| Substance P | NK1R, NK2R, NK3R | Glioblastoma, astrocytoma, SCLC, MTC, BC |
| NPY | NPY1R, NPY2R, NPY4, NPY5 | PC, renal cell carcinoma, ovarian adenocarcinoma, neuroblastoma, paraganglioma, GIST |
| VIP | VPAC1R, VPAC2R | SCLC, colorectal cancer, BC, gastrinoma, PC |
| α-MSH | MC1-5R | Melanoma |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nock, B.A.; Kanellopoulos, P.; Joosten, L.; Mansi, R.; Maina, T. Peptide Radioligands in Cancer Theranostics: Agonists and Antagonists. Pharmaceuticals 2023, 16, 674. https://doi.org/10.3390/ph16050674
Nock BA, Kanellopoulos P, Joosten L, Mansi R, Maina T. Peptide Radioligands in Cancer Theranostics: Agonists and Antagonists. Pharmaceuticals. 2023; 16(5):674. https://doi.org/10.3390/ph16050674
Chicago/Turabian StyleNock, Berthold A., Panagiotis Kanellopoulos, Lieke Joosten, Rosalba Mansi, and Theodosia Maina. 2023. "Peptide Radioligands in Cancer Theranostics: Agonists and Antagonists" Pharmaceuticals 16, no. 5: 674. https://doi.org/10.3390/ph16050674