
Disruption of Proteostasis by Natural Products and Synthetic Compounds That Induce Pervasive Unfolding of Proteins: Therapeutic Implications
 , , ,
, , ,
Abstract
:1. Introduction
2. Results
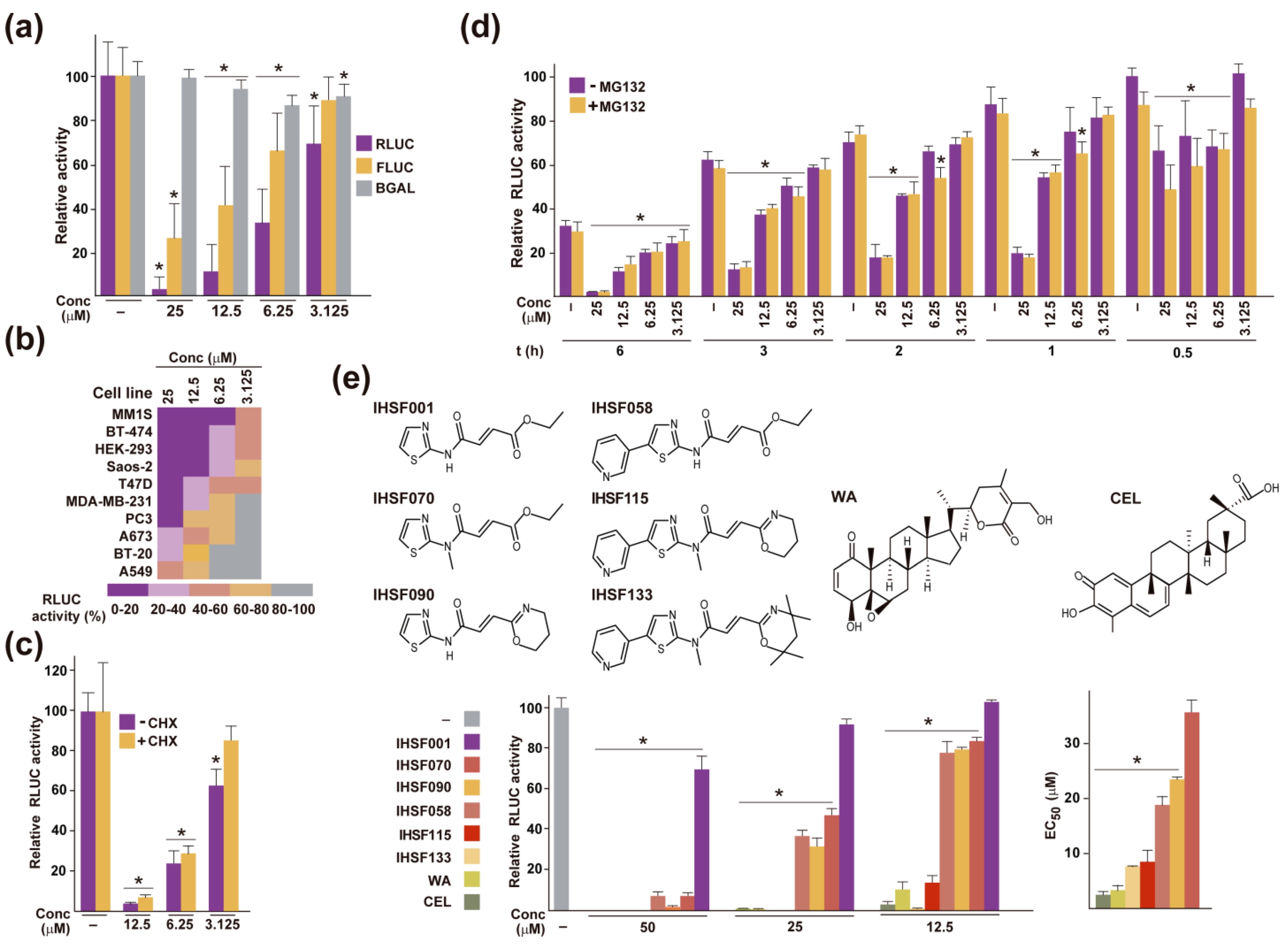
2.1. Inactivation of RLUC by IHSF Series Compounds, CEL and WA
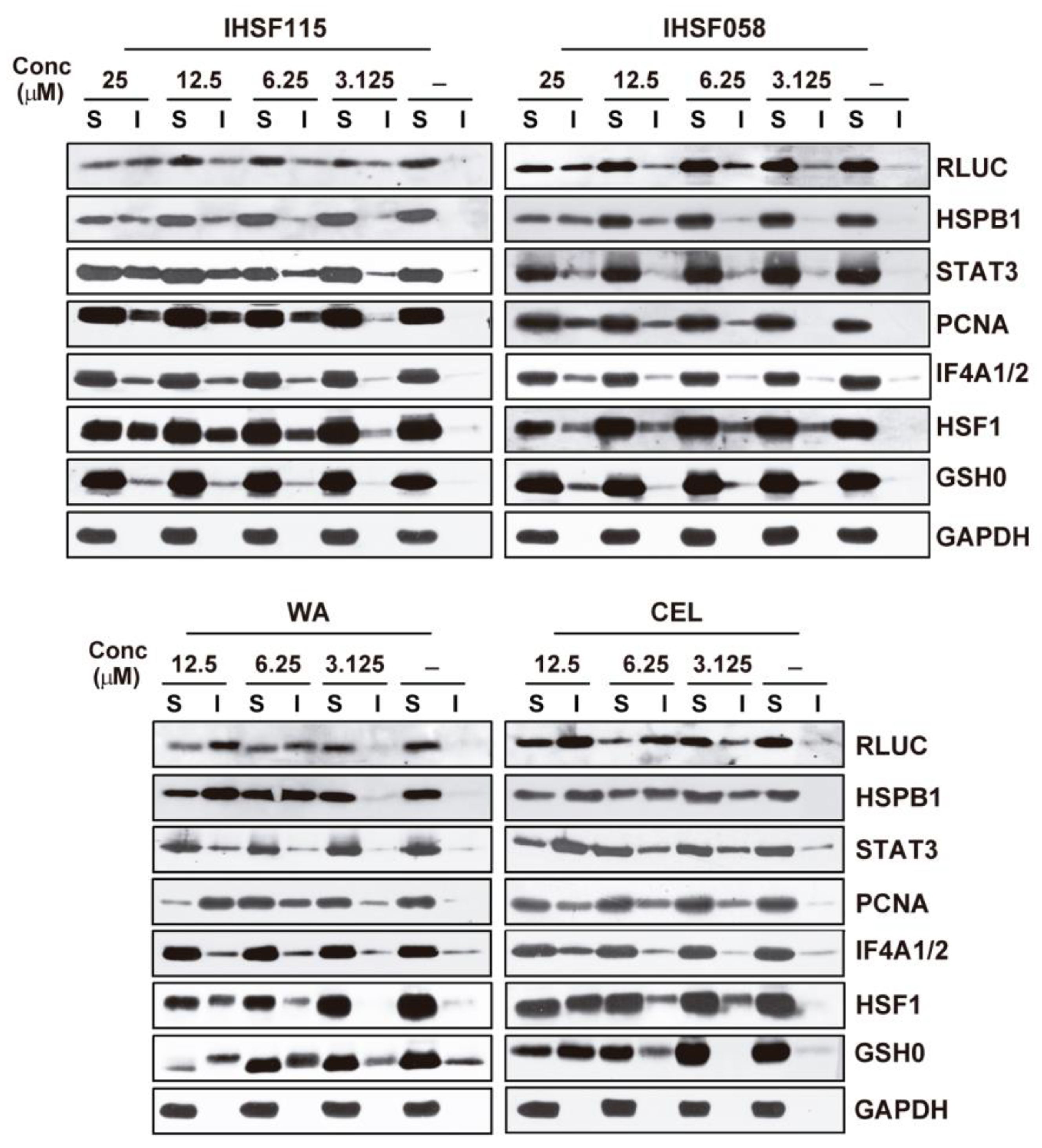
2.2. IHSF Series Compounds, CEL and WA Cause RLUC and Cellular Proteins to Unfold
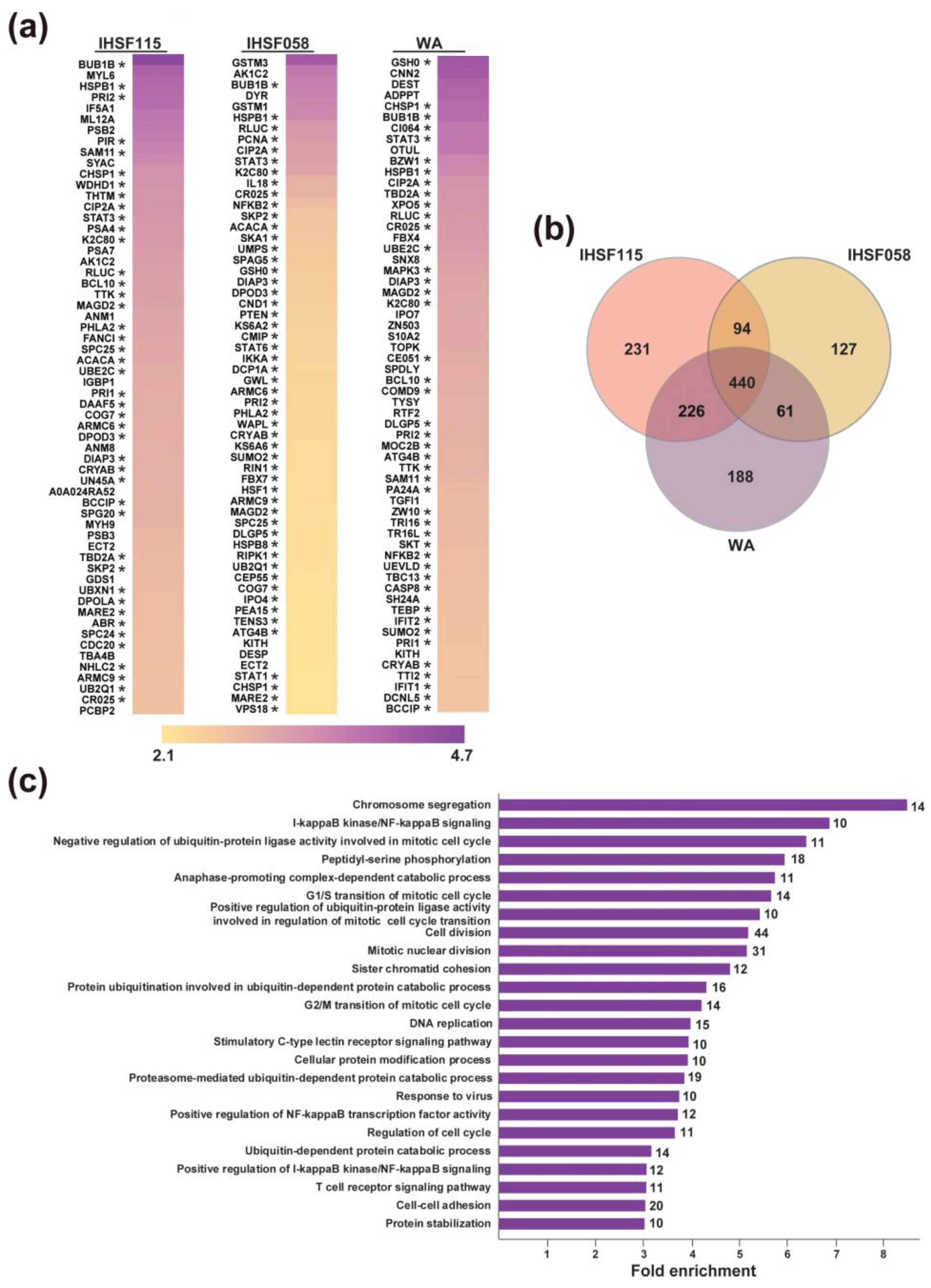
2.3. Proteomic Analyses of Induced Protein Unfolding
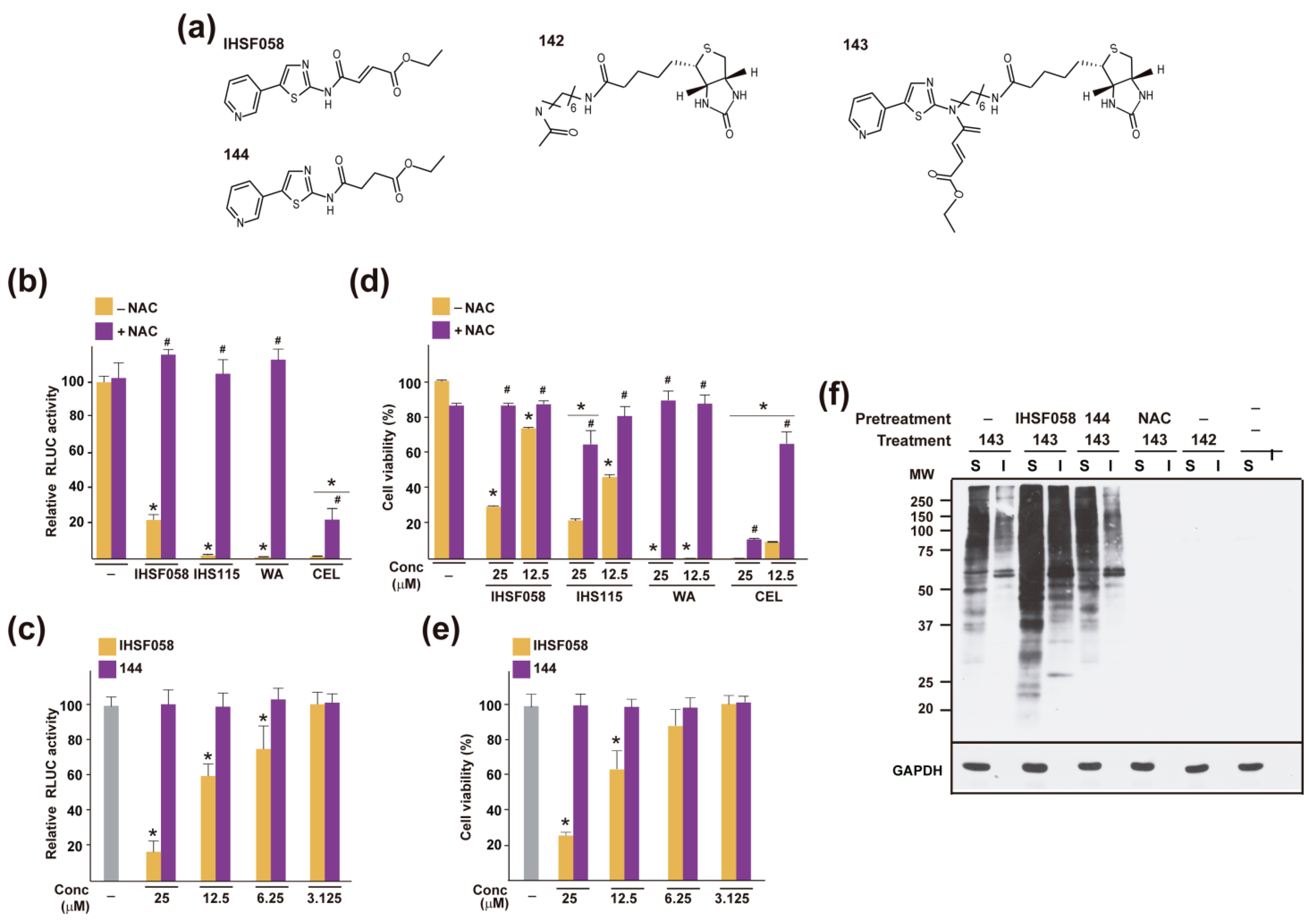
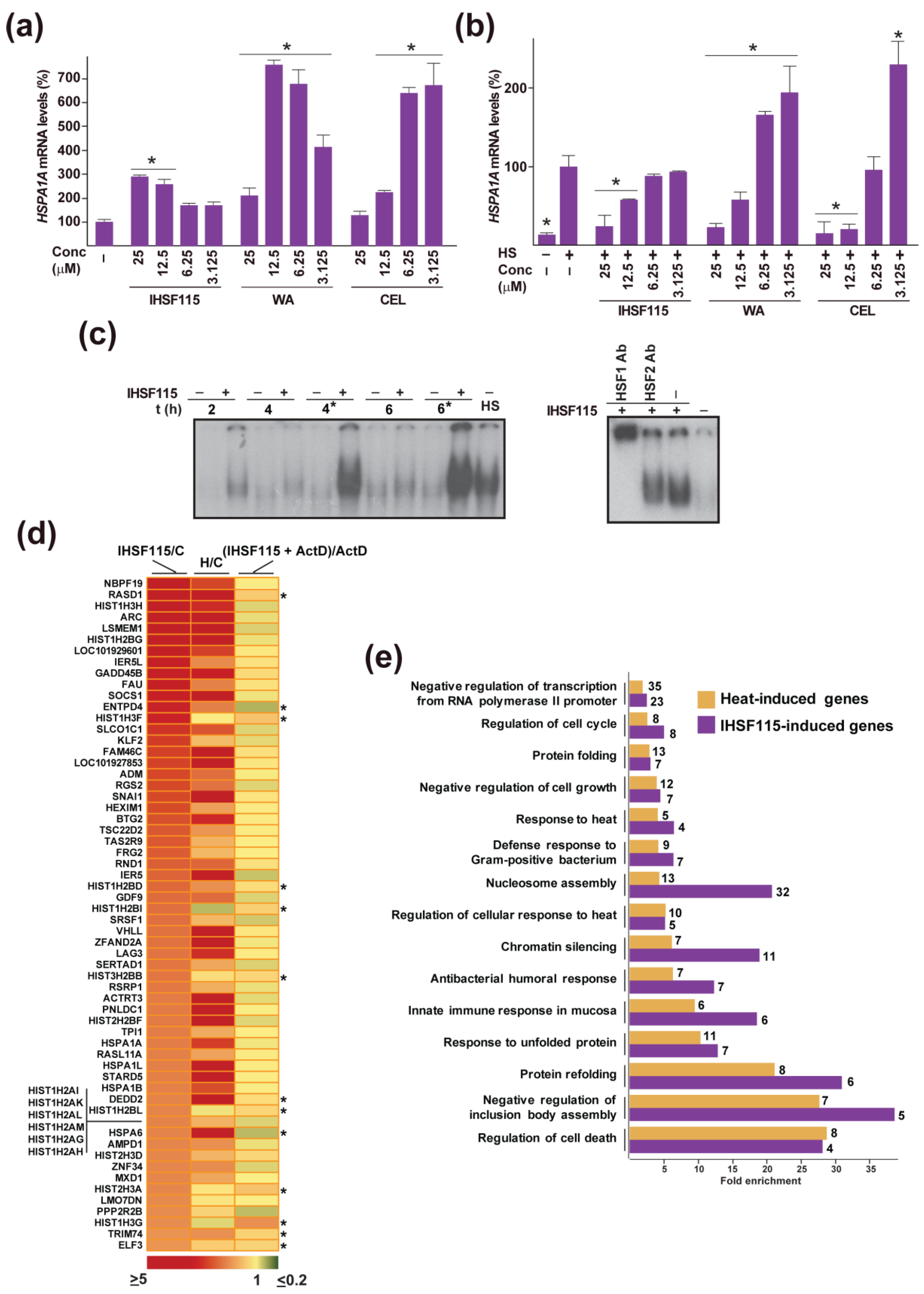
2.4. Other Shared Properties of CEL, WA and IHSF Series Compounds
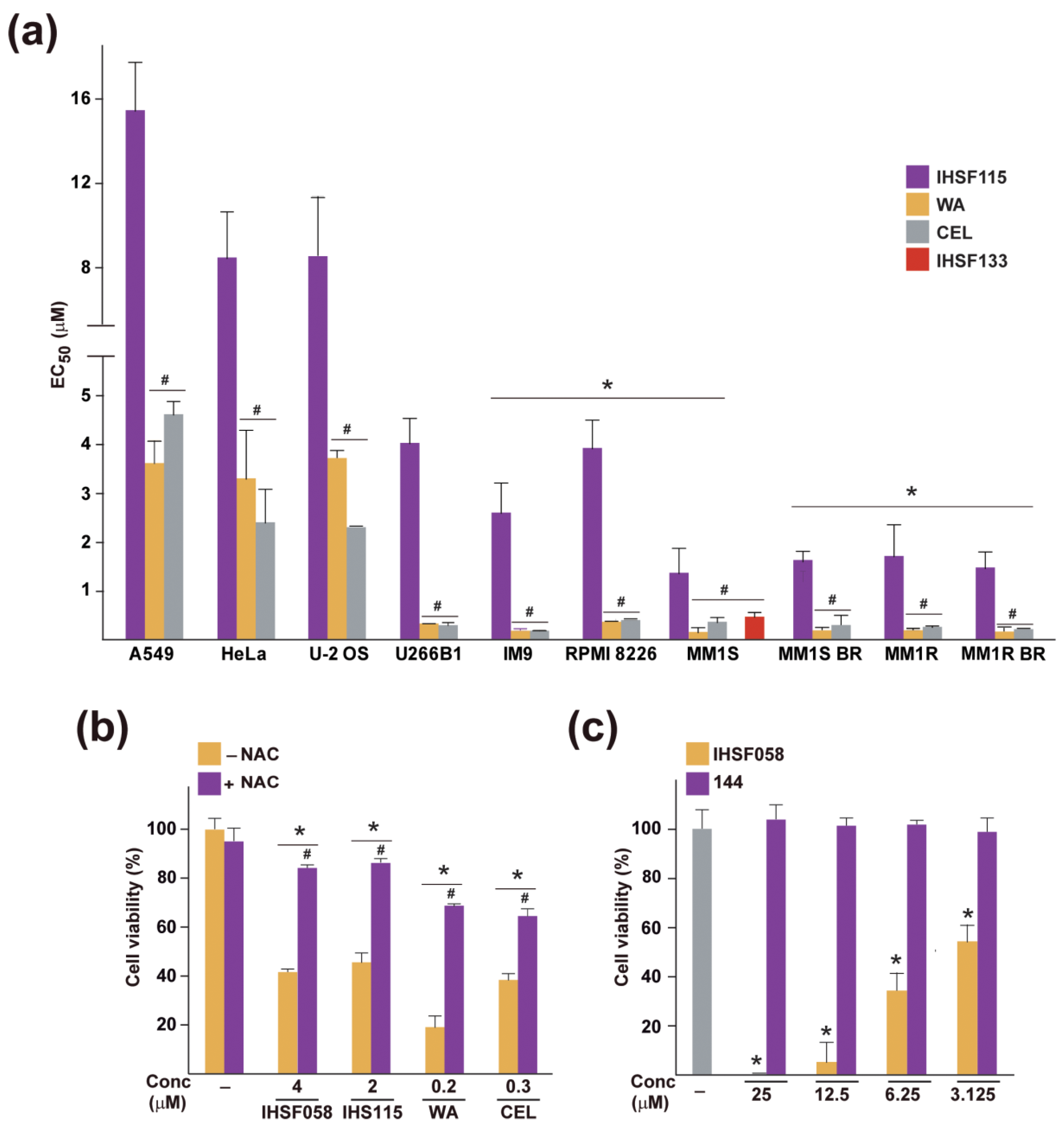
2.5. Elevated Sensitivity of MM Cells to CEL, WA and IHSF
2.6. Synergistic Interactions of CEL, WA and IHSF115 with Proteasome Inhibitor Bortezomib in MM Cells
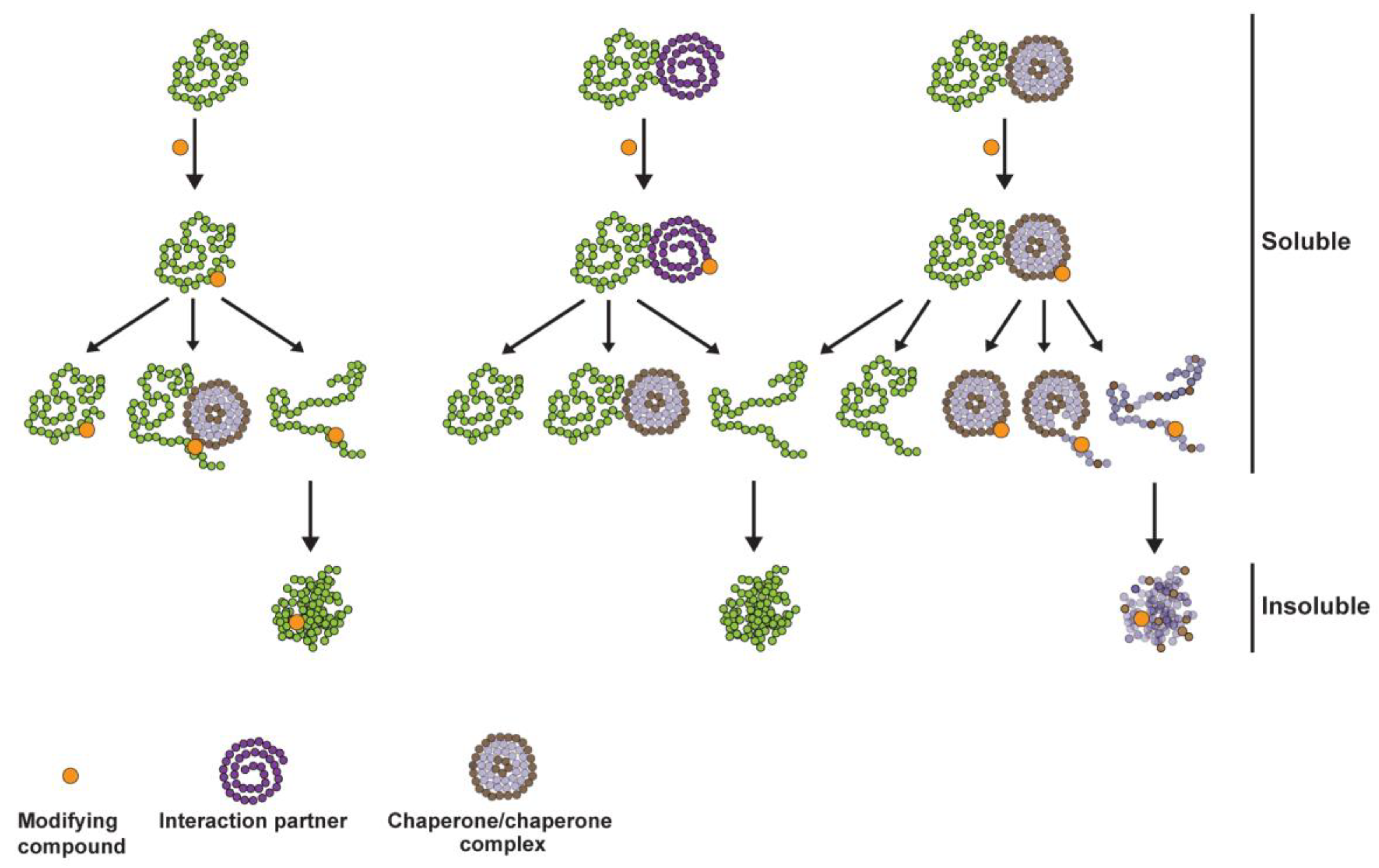
3. Discussion
4. Materials and Methods
4.1. Compounds, PLASMIDS and Viruses
4.2. Cell Culture, Cell Viability and Reporter Assays
4.3. Detergent-Insoluble and Detergent-Soluble Extract Fractions
4.4. Western Blot (WB)
4.5. Tandem Mass Tag (TMT) Mass Spectrometry
4.6. Analysis of Covalent Protein Modification
4.7. Electrophoretic Mobility Shift Assay (EMSA), RT-qPCR and Transcriptomic Assays
4.8. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Labbadia, J.; Morimoto, R.I. The biology of proteostasis in aging and disease. Ann. Rev. Biochem. 2015, 84, 435–464. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, S.L.; Kelly, J.W. Chemical and biological approaches for adapting proteostasis to ameliorate protein misfolding and aggregation diseases: Progress and prognosis. Cold Spring Harb. Perspect. Biol. 2011, 3, a004507. [Google Scholar] [CrossRef] [PubMed]
- Klaips, C.L.; Jayaraj, C.G.; Hartl, F.U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 2017, 217, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Gandolfi, S.; Laubach, J.P.; Hideshima, T.; Chauha, D.; Anderson, K.C.; Richardson, P.G. The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev. 2017, 36, 561–584. [Google Scholar] [CrossRef]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef]
- Meister, S.; Schubert, U.; Neubert, K.; Herrmann, K.; Burger, R.; Gramatzki, M.; Hahn, S.; Schreiber, S.; Wilhelm, S.; Hermann, M.; et al. Excessive immunoglobulin production sensitizes myeloma cells for proteasome inhibition. Cancer Res. 2007, 67, 1783–1792. [Google Scholar] [CrossRef]
- Sidera, K.; Patsavoudi, E. HSP90 inhibitors: Current development and potential in cancer therapy. Recent Pat. Anti-Cancer Drug Discov. 2014, 9, 1–20. [Google Scholar] [CrossRef]
- Jaeger, A.M.; Whitesell, L. HSP90: Enabler of cancer adaptation. Annu. Rev. Cancer Biol. 2019, 3, 275–297. [Google Scholar] [CrossRef]
- Whitesell, L.; Lindquist, S. Inhibiting the transcription factor HSF1 as an anticancer strategy. Expert Opin. Ther. Targets 2009, 13, 69–478. [Google Scholar] [CrossRef]
- Piper, P.W.; Millson, S.H. Mechanisms of resistance to Hsp90 inhibitor drugs: A complex mosaic emerges. Pharmaceuticals 2011, 4, 1400–1422. [Google Scholar] [CrossRef]
- Samarasinghe, B.; Wales, C.T.K.; Taylor, F.R.; Jacobs, A.T. Heat shock factor 1 confers resistance to HSP90 inhibitors through p62/SQSRM1 expression and promotion of autophagic flux. Biochem. Pharmacol. 2014, 87, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Leu, J.I.; Pimkina, J.; Frank, A.; Murphy, M.E.; George, D.L. A small molecule inhibitor of inducible heat shock protein 70. Mol. Cell 2009, 36, 15–27. [Google Scholar] [CrossRef]
- Galluzzi, L.; Giordanetto, F.; Kroemer, G. Targeting HSP70 for cancer therapy. Mol. Cell. 2009, 36, 176–177. [Google Scholar] [CrossRef] [PubMed]
- McMillan, D.R.; Xiao, X.; Shao, L.; Graves, K.; Benjamin, I.J. Targeted disruption of heat shock transcription factor 1 abolishes thermotolerance and protection against heat-inducible apoptosis. J. Biol. Chem. 1998, 273, 7523–7528. [Google Scholar] [CrossRef] [PubMed]
- Akerfelt, M.; Morimoto, R.I.; Sistonen, L. Heat shock factors: Integrators of cell stress, development and life span. Nat. Rev. Mol. Cell Biol. 2010, 11, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Whitesell, L.; Rogers, A.B.; Lindquist, S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 2007, 130, 1005–1018. [Google Scholar] [CrossRef] [PubMed]
- Mendillo, M.L.; Santagata, S.; Koeva, M.; Bell, G.W.; Hu, R.; Tamimi, R.M.; Fraenkel, E.; Ince, T.A.; Whitesell, L.; Lindquist, S. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell 2012, 150, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Vilaboa, N.; Boré, A.; Martin-Saavedra, F.; Bayford, M.; Winfield, N.; Firth-Clark, S.; Kirton, S.B.; Voellmy, R. New inhibitor targeting human transcription factor HSF1: Effects on the heat shock response and tumor cell survival. Nucleic Acids Res. 2017, 45, 5797–5817. [Google Scholar] [CrossRef]
- International Patent Application Publication WO 2018/050656. Available online: https://worldwide.espacenet.com/patent/search/family/061619347/publication/WO2018050656A2?q=WO%202018%2F050656 (accessed on 22 February 2023).
- Chen, S.-R.; Dai, Y.; Zhao, J.; Lin, L.; Wang, Y.; Wang, Y. A mechanistic overview of triptolide and celastrol, natural products from Tripterygium wilfordii Hook F. Front. Pharmacol. 2018, 9, 104. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.W.; Chana, Y.; Chellappanb, D.K.; Madheswaranc, T.; Zeeshanc, F.; Chan, Y.-L.; Collet, T.; Gupta, G.; Oliver, B.G.; Wark, P.; et al. Molecular modulators of celastrol as the keystones for its diverse pharmacological activities. Biomed. Pharmacother. 2019, 109, 1785–1792. [Google Scholar] [CrossRef]
- Cascao, R.; Fonseca, J.E.; Moita, L.F. Celastrol: A spectrum of treatment opportunities in chronic diseases. Front. Med. 2017, 4, 69. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Cheng, S.; Liang, G.; Honggang, L.; Wu, H. Celastrol inhibits cancer metastasis by suppressing M2-like polarization of macrophages. Biochem. Biophys. Res. Commun. 2018, 503, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Ahn, K.S.; Lee, J.H.; Kannaiyan, R.; Mustafa, N.; Manu, K.A.; Siveen, K.S.; Sethi, G.; Chng, W.J.; Kumar, A.P. Celastrol attenuates the invasion and migration and augments the anticancer effects of bortezomib in a xenograft mouse model of multiple myeloma. Front. Pharmacol. 2018, 9, 365. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ma, L.; Wen, Z.S.; Hu, Z.; Wu, F.-Q.; Li, W.; Liu, J.; Zhou, G.-B. Cancerous inhibitor of PP2A is targeted by natural compound celastrol for degradation in non small-cell lung cancer. Carcinogenesis 2014, 35, 905–914. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Jang, K.S.; Choi, H.J.; Jo, A.; Cheong, J.H.; Chun, K.H. Celastrol inhibits gastric cancer growth by induction of apoptosis and autophagy. BMB Rep. 2014, 47, 697–702. [Google Scholar] [CrossRef]
- Li, H.Y.; Zhang, J.; Sun, L.L.; Li, B.H.; Gao, H.L.; Xie, T.; Zhang, N.; Ye, Z.-M. Celastrol induces apoptosis and autophagy via the ROS/JNK signaling pathway in human osteosarcoma cells: An in vitro and in vivo study. Cell Death Dis. 2015, 6, e1604. [Google Scholar] [CrossRef]
- Lee, J.H.; Won, Y.S.; Park, K.H.; Lee, M.K.; Tachibana, H.; Yamada, K.; Seo, K.-I. Celastrol inhibits growth and induces apoptotic cell death in melanoma cells via the activation ROS-dependent mitochondrial pathway and the suppression of PI3K/AKT signaling. Apoptosis 2012, 17, 1275–1286. [Google Scholar] [CrossRef]
- Zhang, T.; Hamza, A.; Cao, X.; Wang, B.; Yu, S.; Zhan, C.G.; Sun, D. A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol. Cancer Ther. 2008, 7, 162–170. [Google Scholar] [CrossRef]
- Lin, L.; Sun, Y.; Wang, D.; Zheng, S.; Zhang, J.; Zheng, C. Celastrol ameliorates ulcerative colitis-related colorectal cancer in mice via suppressing inflammatory responses and epithelial-mesenchymal transition. Front. Pharmacol. 2015, 6, 320. [Google Scholar] [CrossRef]
- Ni, H.; Zhao, W.; Kong, X.; Li, H.; Ouyang, J. NF-kappa B modulation is involved in celastrol induced human multiple myeloma cell apoptosis. PLoS ONE 2014, 9, e95846. [Google Scholar] [CrossRef]
- Kannaijan, R.; Hay, S.H.; Rajendran, P.; Li, F.; Shanmugan, M.K.; Vali, S.; Abbasi, T.; Kapoor, S.; Sharma, A.; Kumar, A.P.; et al. Celastrol inhibits proliferation and induces chemosensitization through down-regulation of NF-kB and STAT3 regulated gene products in multiple myeloma cells. Br. J. Pharmacol. 2011, 164, 1506–1521. [Google Scholar] [CrossRef] [PubMed]
- Chadli, A.; Felts, S.J.; Wang, Q.; Sullivan, W.P.; Botuyan, M.V.; Fauq, A.; Ramirez-Alvarado, M.; Mer, G. Celastrol inhibits Hsp90 chaperoning of steroid receptors by inducing fibrillization of the co-chaperone p23. J. Biol. Chem. 2010, 285, 4224–4231. [Google Scholar] [CrossRef] [PubMed]
- Klaic, L.; Morimoto, R.I.; Silverman, R.B. Celastrol analogs as inducers of the heat shock response. Design and Synthesis of Affinity Probes for the Identification of Protein Targets. ACS Chem. Biol. 2012, 7, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, W.; Wang, M.; Zhang, X.; Zhang, H.; Tong, X.; Xiao, Y. Competitive profiling of celastrol targets in human cervical cancer HeLa cells via quantitative chemical proteomics. Mol. Biosyst. 2017, 13, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.-C.; Choi, B.Y. Withaferin A—A natural anticancer agent with pleiotropic mechanisms of action. Int. J. Mol. Sci. 2016, 17, 290. [Google Scholar] [CrossRef] [PubMed]
- Behl, T.; Sharma, A.; Sharma, L.; Sehgal, A.; Zengin, G.; Brata, R.; Fratila, O.; Bungau, S. Exploring the Multifaceted Therapeutic Potential of Withaferin A and Its Derivatives. Biomedicines 2020, 8, 571. [Google Scholar] [CrossRef]
- Yang, H.; Shi, G.; Dou, Q.P. The tumor proteasome is a primary target for the natural anticancer compound withaferin A isolated from “Indian winter cherry”. Mol. Pharmacol. 2007, 71, 428–437. [Google Scholar] [CrossRef]
- Thaiparambil, J.T.; Bender, L.; Ganesh, T.; Kline, E.; Patel, P.; Liu, Y.; Tighiouart, M.; Vertino, P.M.; Harvey, R.D.; Garcia, A.; et al. Withaferin-A inhibits breast cancer invasion and metastasis at sub-cytotoxic doses by inducing vimentin disassembly and serine 56 phosphorylation. Int. J. Cancer 2011, 129, 2744–2755. [Google Scholar] [CrossRef]
- Kakar, S.S.; Ratajczak, M.Z.; Powell, K.S.; Moghadamfalahi, M.; Miller, D.M.; Batra, S.K.; Singh, S.K. Withaferin-A alone and in combination with cisplatin suppresses growth and metastasis of ovarian cancer by targeting putative cancer stem cells. PLoS ONE 2014, 9, e107596. [Google Scholar] [CrossRef]
- Devi, P.U.; Kamath, R.; Rao, B.S. Radiosensitization of a mouse melanoma by withaferin A: In vivo studies. Indian J. Exp. Biol. 2000, 38, 432–437. [Google Scholar]
- Yu, Y.; Hamza, A.; Zhang, T.; Gu, M.; Zou, P.; Newman, B.; Li, Y.; Gunatilaka, A.A.L.; Zhan, C.-G.; Suh, D. Withaferin-A targets heat shock protein 90 in pancreatic cancer cells. Biochem. Pharmacol. 2010, 79, 542–551. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Kim, B.W. Withaferin-A inhibits colon cancer cell growth by blocking STAT3 transcriptional activity. J. Cancer Prev. 2015, 20, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.; Pohling, C.; Natarajan, A.; Witney, T.H.; Kaur, J.; Xu, L.; Gowrishankar, G.; D’Souza, A.L.; Murty, S.; Schick, S.; et al. AshwaMAX and withaferin-A inhibits gliomas in cellular and murine orthotopic models. J. Neurooncol. 2016, 126, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Santagata, S.; Xu, Y.; Kithsiri Wijeratne, E.M.; Kontnik, R.; Rooney, C.; Perley, C.C.; Kwon, H.; Clardy, J.; Kesari, S.; Whitesell, L.; et al. Using the heat shock response to discover anticancer compounds that target protein homeostasis. ACS Chem. Biol. 2012, 7, 340–349. [Google Scholar] [CrossRef]
- Stan, S.D.; Hahm, E.R.; Warin, R.; Singh, S.V. Withaferin A causes FOXO3a- and Bim-dependent apoptosis and inhibits growth of human breast cancer cells in vivo. Cancer Res. 2008, 68, 7661–7669. [Google Scholar] [CrossRef]
- Grover, A.; Shandilya, A.; Bisaria, V.S.; Sundar, D. Probing the anticancer mechanism of prospective herbal drug withaferin A on mammals: A case study on human and bovine proteasomes. BMC Genom. 2010, 11 (Suppl. S4), S15. [Google Scholar] [CrossRef]
- Falsey, R.R.; Marron, M.T.; Gunaherath, G.M.; Shirahatti, N.; Mahadevan, D.; Gunatilaka, A.A.; Whitesell, L. Actin microfilament aggregation induced by withaferin A is mediated by annexin II. Nat. Chem. Biol. 2006, 2, 33–38. [Google Scholar] [CrossRef]
- Zhang, X.; Timmermann, B.; Samadi, A.K.; Cohen, M.S. Withaferin A induces proteasome-dependent degradation of breast cancer susceptibility gene 1 and heat shock factor 1 proteins in breast cancer cells. ISRN Biochem. 2012, 2012, 707586. [Google Scholar] [CrossRef]
- Yco, L.P.; Mocz, G.; Opoku-Ansah, J.; Bachmann, A.S. Withaferin A Inhibits STAT3 and induces tumor cell death in neuroblastoma and multiple myeloma. Biochem. Insights 2014, 7, BCI-S18863. [Google Scholar] [CrossRef]
- Dom, M.; Vanden Berghe, W.; Van Ostade, X. Broad-spectrum antitumor properties of Withaferin A: A proteomic perspective. RSC Med. Chem. 2020, 11, 30. [Google Scholar] [CrossRef]
- Dom, M.; Offner, F.; Vanden Berghe, W.; Van Ostade, X. Proteomic characterization of Withaferin A-targeted protein networks for the treatment of monoclonal myeloma gammopathies. J. Proteom. 2018, 179, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Grossman, E.A.; Ward, C.C.; Spradlin, J.N.; Bateman, L.A.; Huffman, T.R.; Miyamoto, D.K.; Kleinman, J.I.; Nomura, D.K. Covalent ligand discovery against druggable hotspots targeted by anti-cancer natural products. Cell Chem. Biol. 2017, 24, 1368–1376. [Google Scholar] [CrossRef] [PubMed]
- Westerheide, S.D.; Bosman, J.D.; Mbadugha, N.A.; Kawahara, T.L.A.; Matsumoto, G.; Kim, S.; Gu, W.; Devlin, J.P.; Silverman, R.B.; Morimoto, R.I. Celastrols as inducers of the heat shock response and cytoprotection. J. Biol. Chem. 2004, 279, 56053–56060. [Google Scholar] [CrossRef] [PubMed]
- Trott, A.; West, G.D.; Klaic, L.; Westerheide, S.D.; Silverman, R.B.; Morimoto, R.I.; Morano, K.A. Activation of heat shock and antioxidant responses by the natural product celastrol: Transcriptional signatures of a thiol-targeted molecule. Mol. Biol. Cell 2008, 19, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- Meierhofer, D.; Wang, X.; Huang, L.; Kaiser, P. Quantitative analysis of global ubiquitination in HeLa cells by mass spectrometry. J. Proteome Res. 2008, 7, 4566–4576. [Google Scholar] [CrossRef]
- Xia, W.; Vilaboa, N.; Martin, J.L.; Mestril, R.; Guo, Y.; Voellmy, R. Modulation of tolerance by mutant heat shock transcription factors. Cell Stress Chaperones 1999, 4, 8–18. [Google Scholar] [CrossRef]
- Martín-Saavedra, F.M.; Ruíz-Hernández, E.; Boré, A.; Arcos, D.; Vallet-Regí, M.; Vilaboa, N. Magnetic mesoporous silica spheres for hyperthermia therapy. Acta Biomater. 2010, 6, 4522–4531. [Google Scholar] [CrossRef]
- Cardona, M.; López, J.A.; Serafín, A.; Rongvaux, A.; Inserte, J.; García-Dorado, D.; Flawell, R.; Llovera, M.; Canas, X.; Vazquez, J.; et al. Executioner caspase-3 and 7 deficiency reduces myocyte number in the developing mouse heart. PLoS ONE 2015, 10, e0131411. [Google Scholar] [CrossRef]
- Martínez-Bartolomé, S.; Navarro, P.; Martín-Maroto, F.; López-Ferrer, D.; Ramos-Fernández, A.; Villar, M.; García-Ruiz, J.P.; Vázquez, J. Properties of average score distributions of SEQUEST: The probability ratio method. Mol. Cell. Proteom. 2008, 7, 1135–1145. [Google Scholar] [CrossRef]
- Navarro, P.; Vázquez, J. A refined method to calculate false discovery rates for peptide identification using decoy databases. J. Proteome Res. 2009, 8, 1792–1796. [Google Scholar] [CrossRef]
- Bonzon-Kulichenko, E.; Garcia-Marques, F.; Trevisan-Herraz, M.; Vázquez, J. Revisiting peptide identification by high-accuracy mass spectrometry: Problems associated with the use of narrow mass precursor windows. J. Proteome Res. 2015, 14, 700–710. [Google Scholar] [CrossRef] [PubMed]
- Navarro, P.; Trevisan-Herraz, M.; Bonzon-Kulichenko, E.; Núñez, E.; Martínez-Acedo, P.; Pérez-Hernández, D.; Jorge, I.; Mesa, R.; Calvo, E.; Carrascal, M.; et al. General statistical framework for quantitative proteomics by stable isotope labeling. J. Proteome Res. 2014, 13, 1234–1247. [Google Scholar] [CrossRef] [PubMed]
- Zuo, J.; Baler, R.; Dahl, G.; Voellmy, R. Activation of the DNA-binding ability of human heat shock transcription factor 1 may involve the transition from an intramolecular to an intermolecular triple-stranded coiled-coil structure. Mol. Cell. Biol. 1994, 14, 7557–7568. [Google Scholar] [PubMed]
- Zuo, J.; Rungger, D.; Voellmy, R. Multiple layers of regulation of human heat shock transcription factor 1. Mol. Cell. Biol. 1995, 15, 4319–4330. [Google Scholar] [CrossRef] [PubMed]
- Vilaboa, N.E.; Calle, C.; Pérez, C.; de Blas, E.; García-Bermejo, L.; Aller, P. cAMP increasing agents prevent the stimulation of heat-shock protein 70 (HSP70) gene expression by cadmium chloride in human myeloid cell lines. J. Cell Sci. 1995, 108, 2877–2883. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using David bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Cell Line | Ratio Compound/Bortezomib | Combination Index Values (CI) at Different Effect Levels (ED) * | ||
|---|---|---|---|---|---|
| ED50 | ED75 | ED90 | |||
| Celastrol | MM1S | 200 | 0.60 | 0.60 | 0.60 |
| 160 | 0.74 | 0.67 | 0.14 | ||
| IM9 | 75 | 1.12 | 0.95 | 0.81 | |
| 67 | 0.77 | 0.56 | 0.42 | ||
| RPMI 8226 | 100 | 1.29 | 0.77 | 0.50 | |
| 100 | 1.25 | 0.98 | 0.77 | ||
| Withaferin A | MM1S | 50 | 1.20 | 0.86 | 0.63 |
| 40 | 1.38 | 0.99 | 0.75 | ||
| IM9 | 40 | 1.08 | 0.82 | 0.66 | |
| 33 | 0.99 | 0.79 | 0.67 | ||
| RPMI 8226 | 175 | 0.95 | 0.56 | 0.34 | |
| 150 | 0.80 | 0.50 | 0.31 | ||
| IHSF115 | MM1S | 1200 | 1.33 | 1.00 | 0.78 |
| 1300 | 1.26 | 0.95 | 0.73 | ||
| IM9 | 700 | 1.29 | 0.91 | 0.65 | |
| 600 | 1.25 | 0.98 | 0.78 | ||
| RPMI 8226 | 1000 | 1.40 | 0.99 | 0.71 | |
| 800 | 1.63 | 1.16 | 0.86 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vilaboa, N.; Lopez, J.A.; de Mesa, M.; Escudero-Duch, C.; Winfield, N.; Bayford, M.; Voellmy, R. Disruption of Proteostasis by Natural Products and Synthetic Compounds That Induce Pervasive Unfolding of Proteins: Therapeutic Implications. Pharmaceuticals 2023, 16, 616. https://doi.org/10.3390/ph16040616
Vilaboa N, Lopez JA, de Mesa M, Escudero-Duch C, Winfield N, Bayford M, Voellmy R. Disruption of Proteostasis by Natural Products and Synthetic Compounds That Induce Pervasive Unfolding of Proteins: Therapeutic Implications. Pharmaceuticals. 2023; 16(4):616. https://doi.org/10.3390/ph16040616
Chicago/Turabian StyleVilaboa, Nuria, Juan Antonio Lopez, Marco de Mesa, Clara Escudero-Duch, Natalie Winfield, Melanie Bayford, and Richard Voellmy. 2023. "Disruption of Proteostasis by Natural Products and Synthetic Compounds That Induce Pervasive Unfolding of Proteins: Therapeutic Implications" Pharmaceuticals 16, no. 4: 616. https://doi.org/10.3390/ph16040616