Author Contributions
Conceptualization, M.O.; methodology, M.O. and I.H.; formal analysis, M.O., I.H. and D.C.; resources, D.C. and A.E.A.; writing—original draft preparation, M.O. and I.H.; writing—review and editing, C.D., A.S., D.C. and. A.E.A.; supervision, D.C. and A.E.A. All authors have read and agreed to the published version of the manuscript.
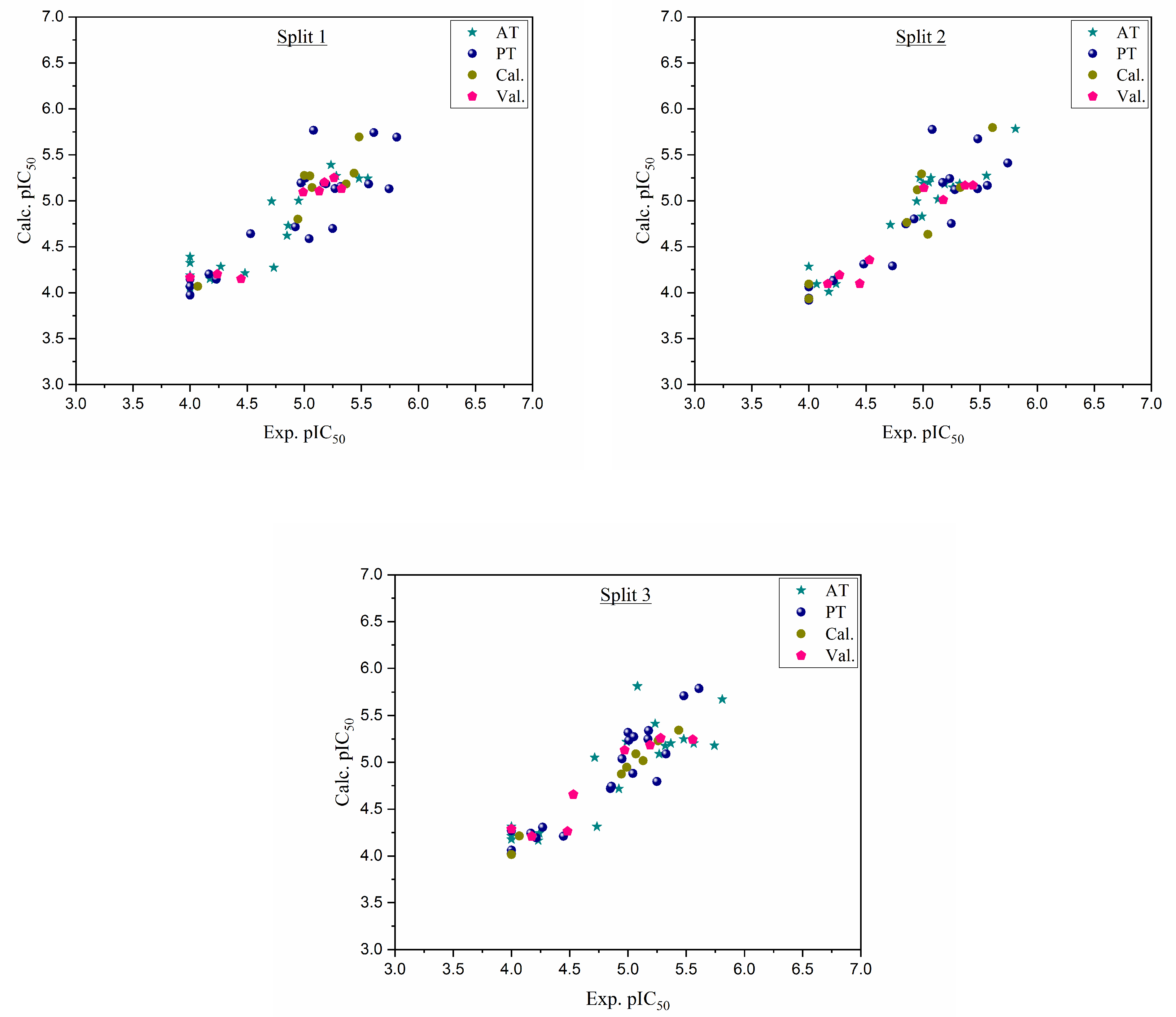
Figure 1.
Graphical representation of experimental pIC50 versus calculated pIC50 for three splits.
Figure 1.
Graphical representation of experimental pIC50 versus calculated pIC50 for three splits.
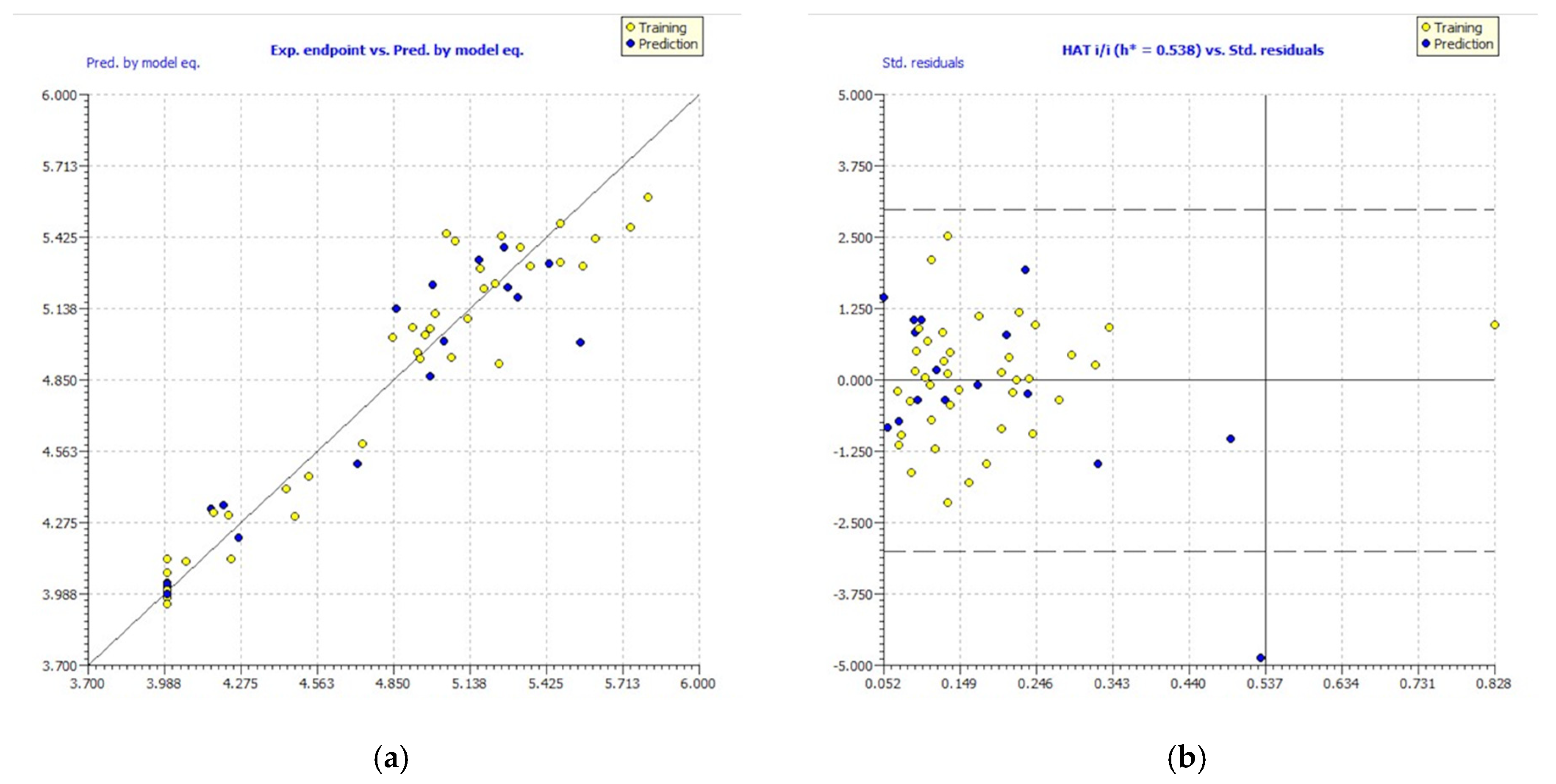
Figure 2.
Experimental vs. predicted pIC50 values computed by GA-MLR (a). William’s plot (b).
Figure 2.
Experimental vs. predicted pIC50 values computed by GA-MLR (a). William’s plot (b).
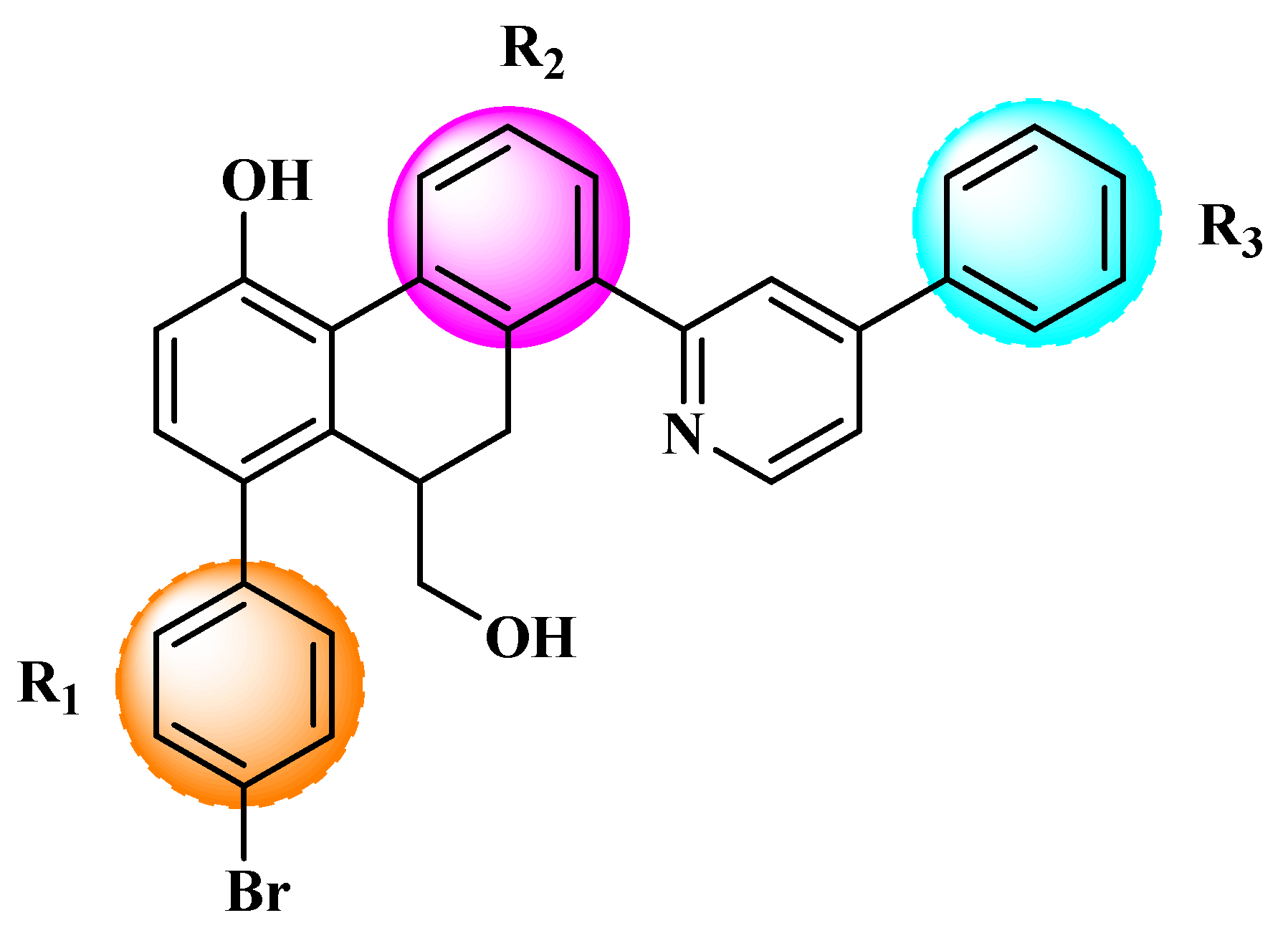
Figure 3.
Chemical structures of the newly designed compounds.
Figure 3.
Chemical structures of the newly designed compounds.
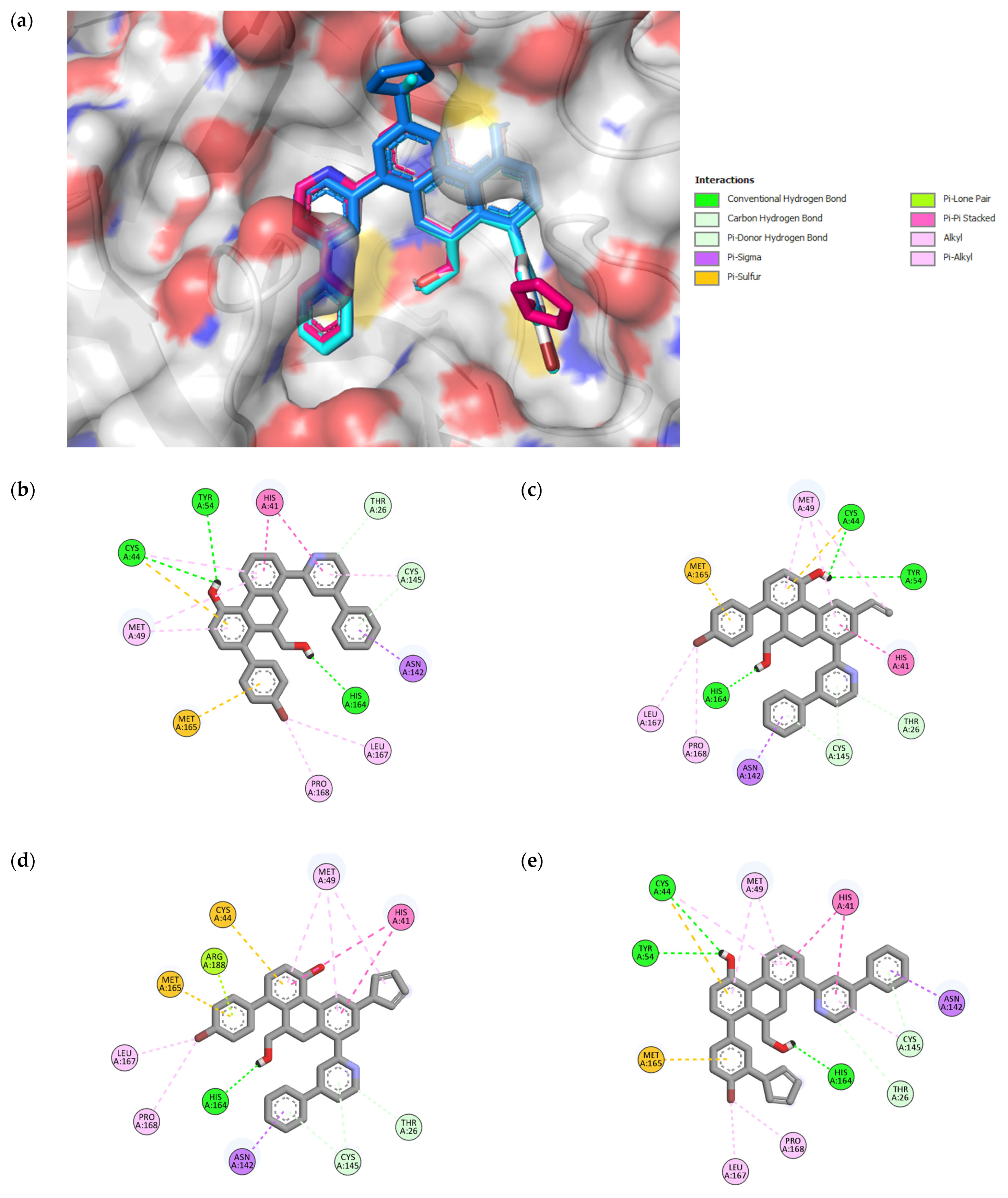
Figure 4.
3D representation of superimposition of the binding poses within the CS of SARS-CoV-2 Mpro of 49, 49n, 49p, and 49x molecules in white, cyan, magenta and blue color respectively (a); 2D representation of interactions within the CS of SARS-CoV-2 Mpro of 49 (b), 49n (c), 49p (d), and 49x (e) molecules.
Figure 4.
3D representation of superimposition of the binding poses within the CS of SARS-CoV-2 Mpro of 49, 49n, 49p, and 49x molecules in white, cyan, magenta and blue color respectively (a); 2D representation of interactions within the CS of SARS-CoV-2 Mpro of 49 (b), 49n (c), 49p (d), and 49x (e) molecules.
Figure 5.
3D representation of superimposition of the binding poses within the DIS of SARS-CoV-2 of 49, 49n, 49p, and 49x molecules in white, cyan, magenta and blue color respectively (a); 2D representation of interactions within the DIS of SARS-CoV-2 Mpro of 49 (b), 49n (c), 49p (d), and 49x (e) molecules.
Figure 5.
3D representation of superimposition of the binding poses within the DIS of SARS-CoV-2 of 49, 49n, 49p, and 49x molecules in white, cyan, magenta and blue color respectively (a); 2D representation of interactions within the DIS of SARS-CoV-2 Mpro of 49 (b), 49n (c), 49p (d), and 49x (e) molecules.
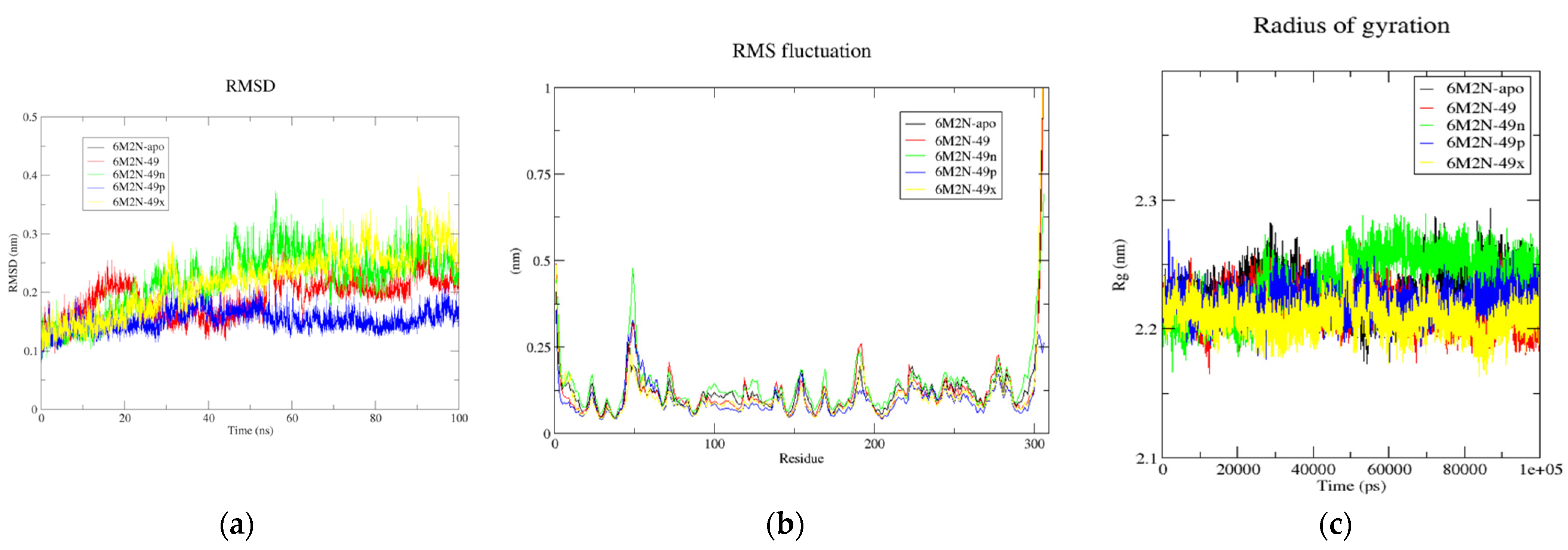
Figure 6.
(a) Time-dependent RMSD of c-α backbone of the Mpro-apo, 6M2N-49, 6M2N-49n, 6M2N-49p, and 6M2N-49x. (b) The RMSF for c-α atoms of Mpro-apo, 6M2N-49, 6M2N-49n, 6M2N-49p, and 6M2N-49x. (c) Plot of Rg vs. time for Mpro-apo, 6M2N-49, 6M2N-49n, 6M2N-49p, and 6M2N-49x.
Figure 6.
(a) Time-dependent RMSD of c-α backbone of the Mpro-apo, 6M2N-49, 6M2N-49n, 6M2N-49p, and 6M2N-49x. (b) The RMSF for c-α atoms of Mpro-apo, 6M2N-49, 6M2N-49n, 6M2N-49p, and 6M2N-49x. (c) Plot of Rg vs. time for Mpro-apo, 6M2N-49, 6M2N-49n, 6M2N-49p, and 6M2N-49x.
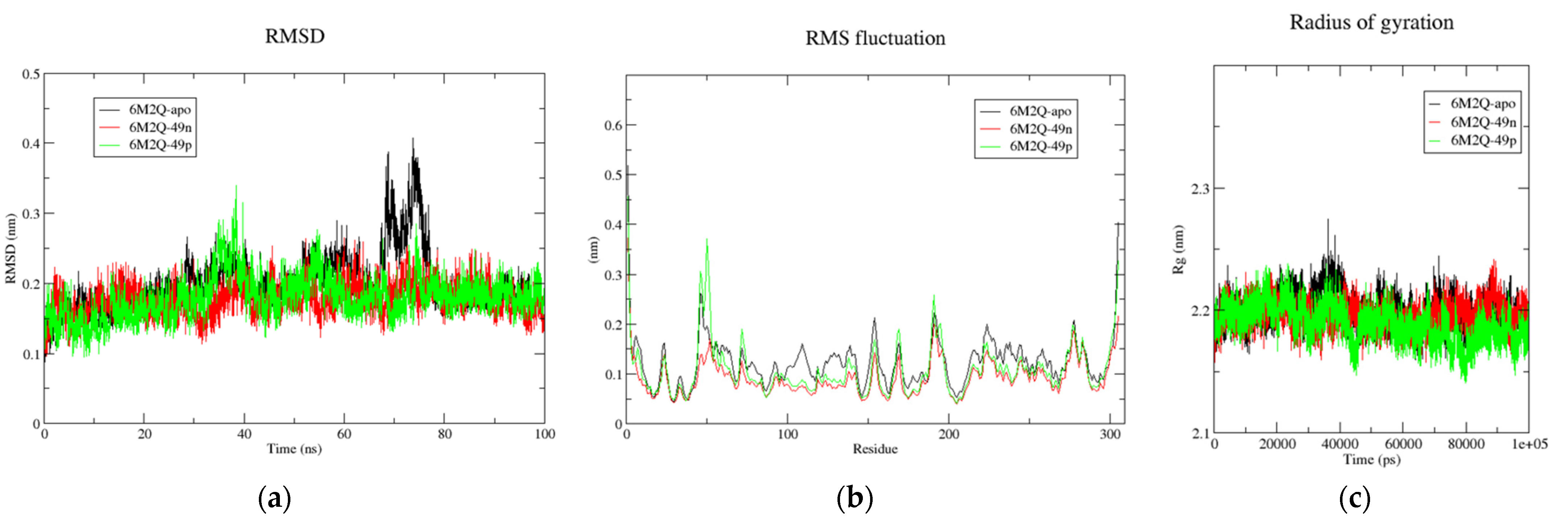
Figure 7.
(a) Time-dependent RMSD of c-α backbone of the Mpro-apo, 6M2N-49n, and 6M2N-49p. (b) The RMSF for c-α atoms of Mpro-apo, 6M2N-49n, and 6M2N-49p. (c) Plot of Rg vs. time for Mpro-apo, 6M2N-49n, and 6M2N-49p.
Figure 7.
(a) Time-dependent RMSD of c-α backbone of the Mpro-apo, 6M2N-49n, and 6M2N-49p. (b) The RMSF for c-α atoms of Mpro-apo, 6M2N-49n, and 6M2N-49p. (c) Plot of Rg vs. time for Mpro-apo, 6M2N-49n, and 6M2N-49p.
Table 1.
Statistical parameters of built QSAR models and their corresponding equations.
Table 1.
Statistical parameters of built QSAR models and their corresponding equations.
| Split | TF | Set | n | R2 | CCC | IIC | Q2 | Q2F1 | Q2F2 | Q2F3 | Rm2av | ΔRm2 | Crp2 | Equation |
|---|
| 1 | TF1 | AT | 19 | 0.8031 | 0.8908 | 0.6518 | 0.7605 | | | | | | 0.7855 | pIC50 = 1.8172593 (+− 0.0736101) + 0.0553060 (+− 0.0014479) × DCW(1,1) |
| PT | 20 | 0.7237 | 0.8152 | 0.4176 | 0.6364 | | | | | | 0.6964 |
| Cal | 8 | 0.8085 | 0.8965 | 0.7468 | 0.5238 | 0.8468 | 0.7854 | 0.8854 | 0.7270 | 0.0438 | 0.7259 |
| Val | 8 | 0.7579 | 0.8490 | 0.6372 | 0.5803 | | | | 0.6545 | 0.1869 | |
| TF2 | AT | 19 | 0.8145 | 0.8978 | 0.8123 | 0.7734 | | | | | | 0.7868 | pIC50 = 2.1090187 (+− 0.0660490) + 0.0635121 (+− 0.0016442) × DCW(5,14) |
| PT | 20 | 0.7510 | 0.8619 | 0.6401 | 0.6889 | | | | | | 0.7309 |
| Cal | 8 | 0.8536 | 0.9182 | 0.9239 | 0.6657 | 0.8743 | 0.8239 | 0.9060 | 0.7874 | 0.1008 | 0.7687 |
| Val | 8 | 0.9161 | 0.9549 | 0.8433 | 0.8274 | | | | 0.8771 | 0.0040 | |
| 2 | TF1 | AT | 20 | 0.9314 | 0.9645 | 0.6434 | 0.9187 | | | | | | 0.8879 | pIC50 = 2.0227798 (+− 0.0329913) + 0.0786046 (+− 0.0009506) × DCW(7,5) |
| PT | 19 | 0.8821 | 0.9245 | 0.4217 | 0.8428 | | | | | | 0.8595 |
| Cal | 8 | 0.8039 | 0.8857 | 0.6191 | 0.6788 | 0.7331 | 0.7329 | 0.7594 | 0.6830 | 0.1830 | 0.6787 |
| Val | 8 | 0.8506 | 0.8826 | 0.4703 | 0.7229 | | | | 0.7827 | 0.1187 | |
| TF2 | AT | 20 | 0.9191 | 0.9579 | 0.7844 | 0.9023 | | | | | | 0.9029 | pIC50 = 1.7915330 (+− 0.0454500) + 0.0672532 (+− 0.0010114) × DCW(3,17) |
| PT | 19 | 0.8283 | 0.9001 | 0.7511 | 0.7652 | | | | | | 0.8001 |
| Cal | 8 | 0.8612 | 0.9251 | 0.9277 | 0.7845 | 0.8374 | 0.8373 | 0.8535 | 0.7932 | 0.1285 | 0.8266 |
| Val | 8 | 0.9203 | 0.9157 | 0.6768 | 0.8508 | | | | 0.8825 | 0.0647 | |
| 3 | TF1 | AT | 19 | 0.7424 | 0.8521 | 0.7754 | 0.6545 | | | | | | 0.7102 | pIC50 = 1.9879653 (+− 0.1287947) + 0.0650533 (+− 0.0030713) × DCW(3,1) |
| PT | 20 | 0.8129 | 0.8983 | 0.5442 | 0.7805 | | | | | | 0.8008 |
| Cal | 9 | 0.9736 | 0.9712 | 0.4888 | 0.9615 | 0.9492 | 0.9483 | 0.9622 | 0.8647 | 0.0379 | 0.9167 |
| Val | 7 | 0.8589 | 0.9068 | 0.7634 | 0.7030 | | | | 0.6779 | 0.1627 | |
| TF2 | AT | 19 | 0.7473 | 0.8554 | 0.7780 | 0.6645 | | | | | | 0.6957 | pIC50 = 2.0784826 (+− 0.1186544) + 0.0690012 (+− 0.0031186) × DCW(10,7) |
| PT | 20 | 0.8210 | 0.9039 | 0.5496 | 0.7899 | | | | | | 0.7929 |
| Cal | 9 | 0.9860 | 0.9864 | 0.9925 | 0.9700 | 0.9758 | 0.9754 | 0.9820 | 0.8849 | 0.0241 | 0.8731 |
| Val | 7 | 0.8728 | 0.9197 | 0.7740 | 0.7444 | | | | 0.7169 | 0.1417 | |
Table 2.
Molecular descriptors and their description.
Table 2.
Molecular descriptors and their description.
| Molecular Descriptor | Description |
|---|
| Eig09_AEA(dm) | Eigenvalue n. 9 from augmented edge adjacency matrix weighted by dipole moment |
| P_VSA_MR_5 | P_VSA-like on Molar Refractivity, bin 5 |
| s3_numSharedNeighbors | Number of shared neighbours in substituent 3 with other substituents |
| s3_relPathLength_2 | Maximum path length of the substituent 3 normed by s3_size |
| SpMin2_Bh(m) | Smallest eigenvalue n. 2 of Burden matrix weighted by mass |
| GATS5m | Geary autocorrelation of lag 5 weighted by mass |
Table 3.
Promoters of increase and decrease of pIC50 endpoint value from split 2 and their description.
Table 3.
Promoters of increase and decrease of pIC50 endpoint value from split 2 and their description.
| | CWs Probe 1 | CWs Probe 2 | CWs Probe 3 | NAT a | NPT b | NCal c | Defect [SAk] d | Comment |
|---|
| Promoters of increase |
| (........... | 0.21284 | 0.24591 | 0.24281 | 20 | 19 | 8 | 0.0000 | Branching |
| (...O...(... | 1.23889 | 1.32719 | 1.06767 | 20 | 19 | 8 | 0.0000 | Two-sided branching of oxygen |
| ++++N---O=== | 0.92838 | 1.33202 | 1.79720 | 20 | 19 | 8 | 0.0000 | Presence of nitrogen with oxygen |
| 1........... | 0.30243 | 0.03065 | 0.22419 | 20 | 19 | 8 | 0.0000 | Presence of one ring |
| C........... | 0.01768 | 0.10610 | 0.24270 | 20 | 19 | 8 | 0.0000 | Presence of sp3 carbon |
| c...(....... | 0.10123 | 0.12192 | 0.30233 | 20 | 19 | 8 | 0.0000 | Sp2 carbon with branching |
| c...(...c... | 0.14109 | 0.27861 | 0.30707 | 20 | 19 | 8 | 0.0000 | Branching between two sp2 carbons |
| c........... | 0.57756 | 0.61985 | 0.68283 | 20 | 19 | 8 | 0.0000 | Presence of a sp2 carbon |
| c...2....... | 0.03199 | 0.01794 | 0.13095 | 20 | 19 | 8 | 0.0000 | Presence of at least two aromatic rings/Presence of sp2 carbon with two rings |
| c...2...c... | 0.64083 | 0.33025 | 0.27204 | 20 | 19 | 8 | 0.0000 | Aromatic ring surrounded by two sp2 carbons |
| c...c...(... | 0.45761 | 0.46821 | 0.64020 | 20 | 19 | 8 | 0.0000 | Presence of two sp2 carbons with branching |
| c...c....... | 0.09429 | 0.44519 | 0.11101 | 20 | 19 | 8 | 0.0000 | Presence of two sp2 carbons |
| c...c...c... | 0.28956 | 0.77633 | 0.66046 | 20 | 19 | 8 | 0.0000 | Presence of three sp2carbons |
| C...C...(... | 0.40471 | 0.84999 | 1.11520 | 19 | 19 | 8 | 0.0019 | Presence of two sp3 carbons with branching |
| Promoters of decrease |
| O........... | −0.96058 | −1.30101 | −1.49847 | 20 | 19 | 8 | 0.0000 | Presence of oxygen |
| n........… | −1.27893 | −1.65511 | −1.62009 | 20 | 19 | 8 | 0.0000 | Presence of sp2 nitrogen |
| n...c....... | −0.11779 | −0.15247 | −0.22729 | 20 | 19 | 7 | 0.0046 | Presence of sp2 nitrogen with sp2 carbon |
| =………. | −0.52134 | −0.04031 | −0.07066 | 15 | 19 | 7 | 0.0057 | Presence of double covalent bond |
| c...C....... | −0.28402 | −0.70453 | −0.44302 | 9 | 12 | 4 | 0.0038 | Combination of sp2 carbon and sp3 carbon |
| c...(...C... | −0.24438 | −0.23144 | −0.63654 | 8 | 12 | 4 | 0.0083 | Branching between sp2 carbon and sp3 carbon |
| C...O...C... | −0.19900 | −0.49944 | −0.58811 | 7 | 7 | 3 | 0.0025 | Sp3 oxygen surrounded by two sp3 carbons |
Table 4.
The newly designed compounds and their predicted pIC50 using the Monte Carlo optimization and the GA-MLR models.
Table 5.
The calculated average parameters for all the systems throughout 100 ns MD simulation run.
Table 5.
The calculated average parameters for all the systems throughout 100 ns MD simulation run.
| Systems | 6M2N-Apo | 6M2N-49 | 6M2N-49n | 6M2N-49p | 6M2N-49x |
|---|
| RMSD (nm) | 0.220 | 0.191 | 0.221 | 0.152 | 0.241 |
| RMSF (nm) | 0.115 | 0.113 | 0.131 | 0.092 | 0.103 |
| Rg (nm) | 2.223 | 2.209 | 2.231 | 2.213 | 2.203 |
Table 6.
Detailed binding free energy calculated by MM/GBSA for all complexes. All the values are given in kcal/mol.
Table 6.
Detailed binding free energy calculated by MM/GBSA for all complexes. All the values are given in kcal/mol.
| Systems | 6M2N-49 | 6M2N-49n | 6M2N-49p | 6M2N-49x |
|---|
| ΔEvdw | −31.98 | −33.54 | −40.95 | −51.68 |
| ΔEele | −11.93 | −8.38 | −16.3 | −18.1 |
| ΔEGB | 31.1 | 26.48 | 42.76 | 45.97 |
| ΔEsurf | −4.26 | −4.19 | −5.28 | −6.5 |
| ΔGgas | −43.91 | −41.92 | −57.25 | −69.78 |
| ΔGsolv | 26.84 | 22.29 | 37.48 | 39.48 |
| Δtotal | −17.07 | −19.63 | −19.77 | −30.3 |
Table 7.
The calculated average parameters for all the systems after 100 ns MD simulation run.
Table 7.
The calculated average parameters for all the systems after 100 ns MD simulation run.
| Systems | 6M2Q-Apo | 6M2Q-49n | 6M2Q-49p |
|---|
| RMSD (nm) | 0.198 | 0.176 | 0.178 |
| RMSF (nm) | 0.116 | 0.087 | 0.102 |
| Rg (nm) | 2.194 | 2.190 | 2.183 |
Table 8.
ADMET properties for the designed compounds and the reference compound.
Table 8.
ADMET properties for the designed compounds and the reference compound.
| Compounds | 49 | 49n | 49p | 49x |
|---|
| Pharmacokinetic and ADME properties |
| HIA (%) | 97.575 | 97.494 | 97.534 | 97.873 |
| BBB (Log BB) | −0.428 | −0.249 | −0.214 | −0.218 |
| P-glycoprotein substrate | Yes | No | No | No |
| CYP2C19 inhibitor | No | No | No | No |
| CYP2C9 inhibitor | No | No | No | No |
| CYP2D6 inhibitor | No | No | No | No |
| CYP3A4 inhibitor | No | No | No | No |
| Toxicological properties |
| Mutagenic | No risk | No risk | No risk | No risk |
| Tumorigenic | No risk | No risk | No risk | No risk |
| Irritant | No risk | No risk | No risk | No risk |
| Reproductive effect | No risk | No risk | No risk | No risk |
Table 9.
Percentage of the identity of the three splits.
Table 9.
Percentage of the identity of the three splits.
| | Split 1 (%) | Split 2 (%) | Split 3 (%) |
|---|
| Set | Total | AT | PT | Cal | Val | Total | AT | PT | Cal | Val | Total | AT | PT | Cal | Val |
|---|
| Split 1 (%) | 100 | 100 | 100 | 100 | 100 | 25.5 | 25.6 | 35.9 | 0.0 | 25.0 | 32.7 | 36.8 | 35.0 | 50.0 | 0.0 |
| Split 2 (%) | | | | | | 100 | 100 | 100 | 100 | 100 | 30.9 | 35.9 | 41.0 | 12.5 | 12.5 |
| Split 3 (%) | | | | | | | | | | | 100 | 100 | 100 | 100 | 100 |
Table 10.
The detailed description of SMILES attributes.
Table 10.
The detailed description of SMILES attributes.
| SMILES Attributes | Description |
|---|
| Sk | One symbol or two symbols that cannot be examined separately |
| SSk | Combination of two SMILES-atomes |
| SSSk | Combination of three SMILES-atomes |
| PAIR | Alliance of two descriptors NOSP a and BOND b |
| HARD | Existence of some chemical element |
| Cmax | Number of rings |
| Omax | Number of oxygen atoms |
| Nmax | Number of nitrogen atoms |
Table 11.
List of key residues of SARS-CoV-2 Mpro.
Table 11.
List of key residues of SARS-CoV-2 Mpro.
| Role in the SARS-CoV-2 Mpro | Residues |
|---|
| Substrate binding | HIS41, MET49, GLY143, SER144, HIS163, HIS164, MET165, GLU166, LEU167, ASN187, ARG188, GLN189, THR190, ALA191, GLN192 |
| Dimerization | ARG4, SER10, GLY11, GLU14, ASN28, SER139, PHE140, SER147, GLU290, ARG298 |
| Catalytic dyad | HIS41, CYS145 |

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



