
Suramin Affects the Renal VEGF-A/VEGFR Axis in Short-Term Streptozotocin-Induced Diabetes
 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Results
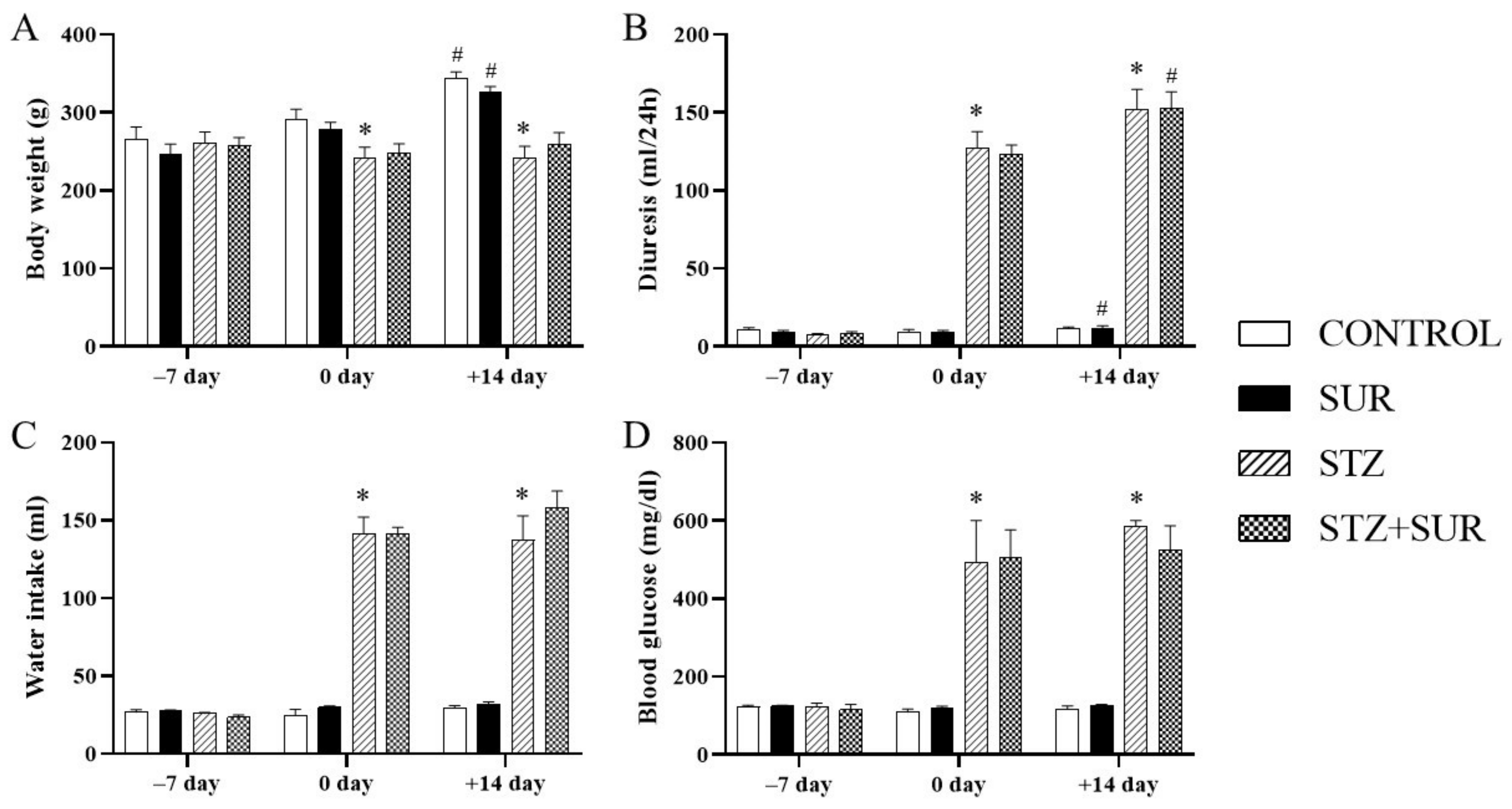
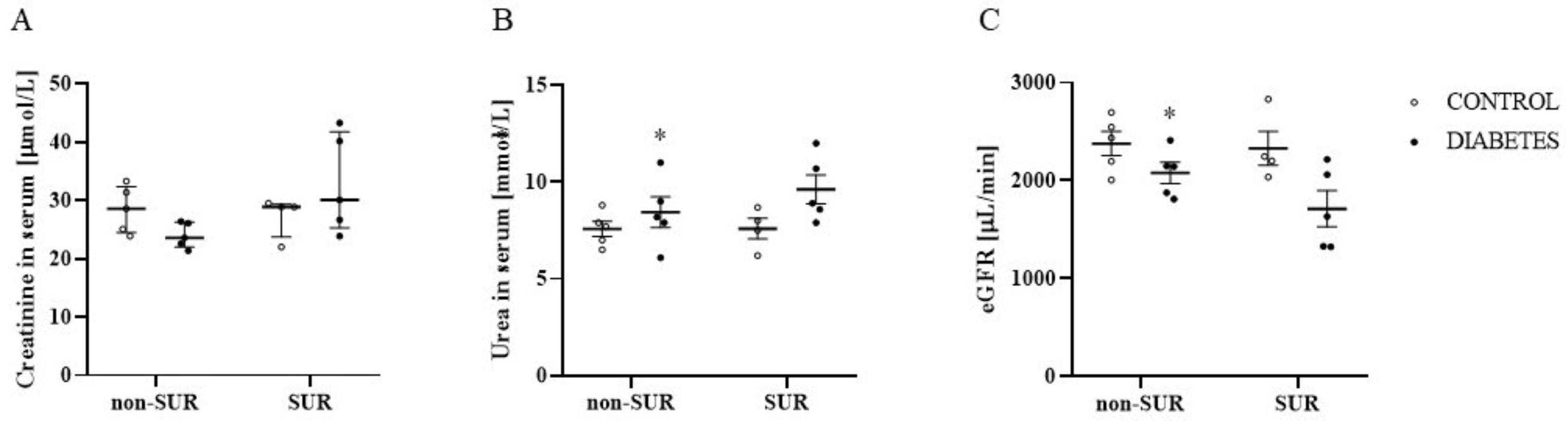
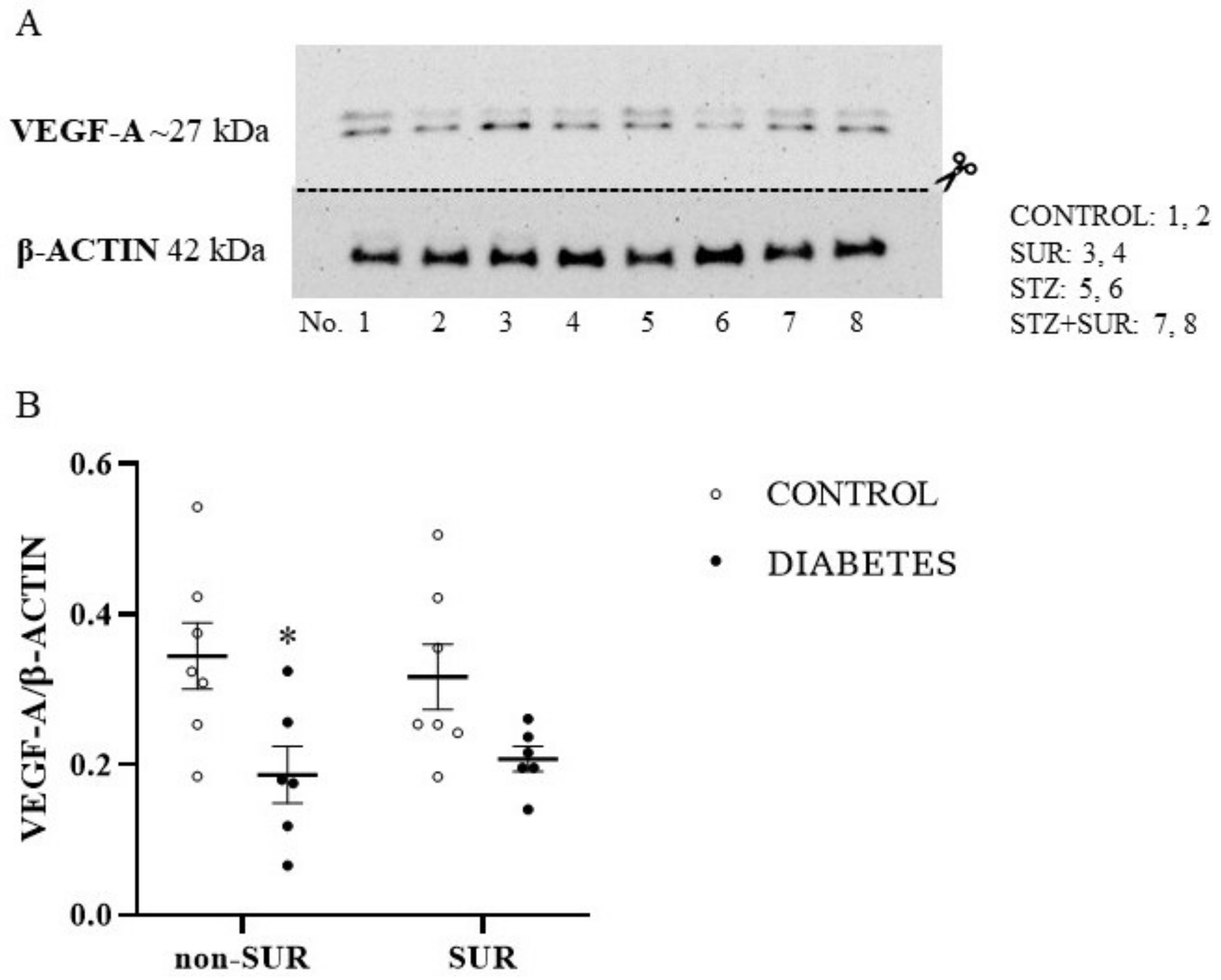
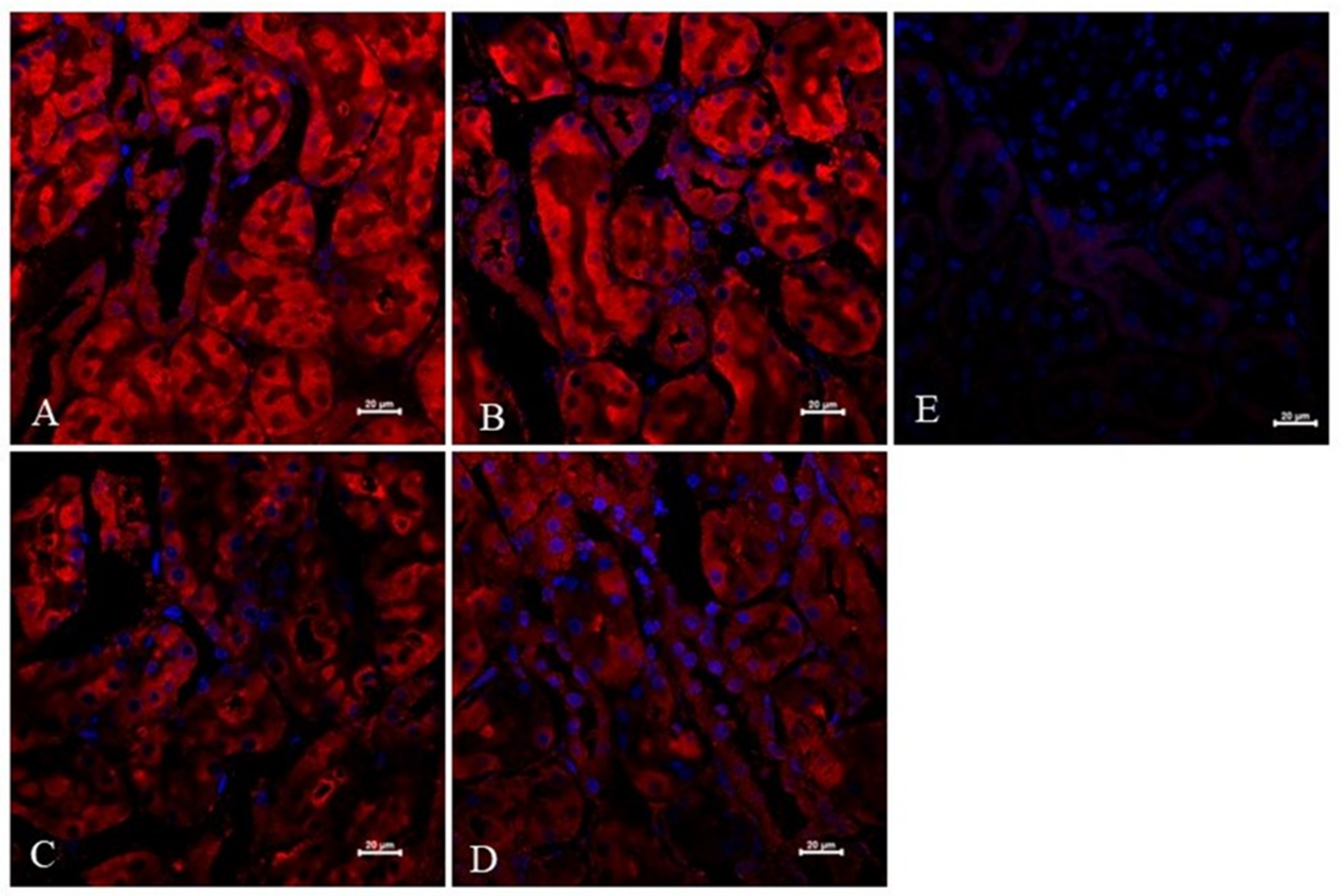
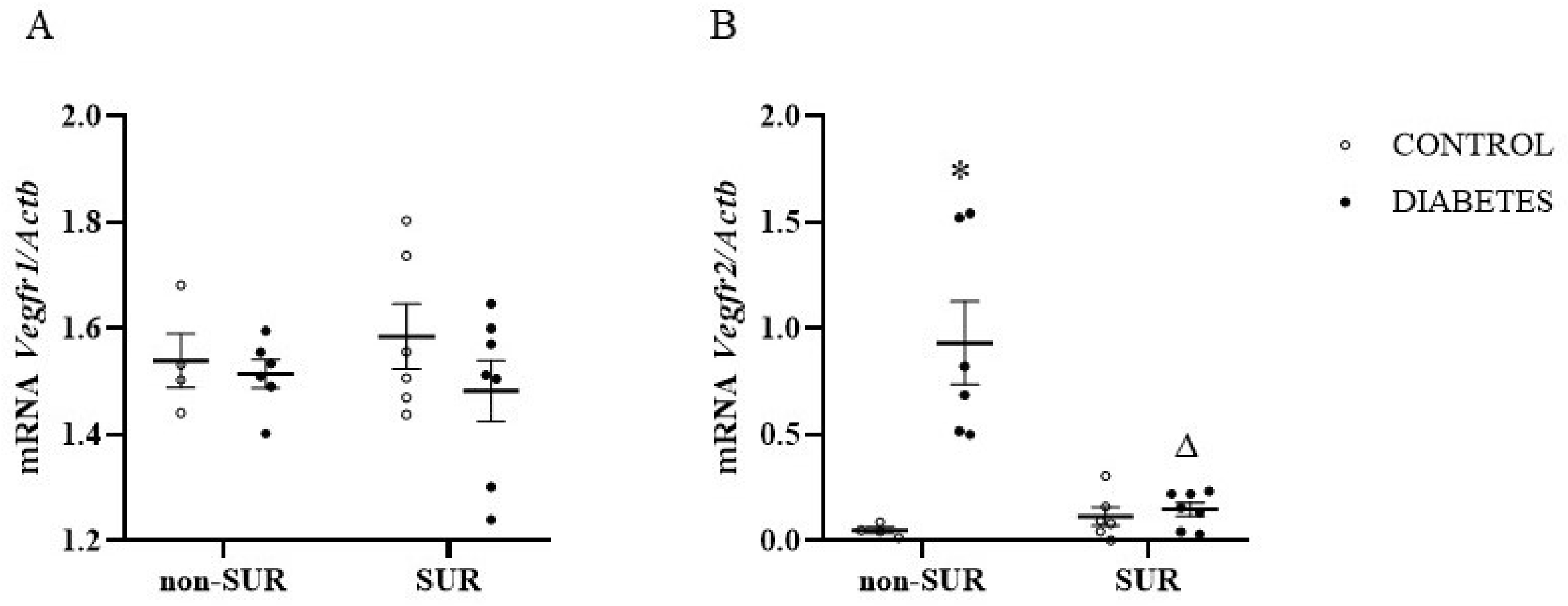
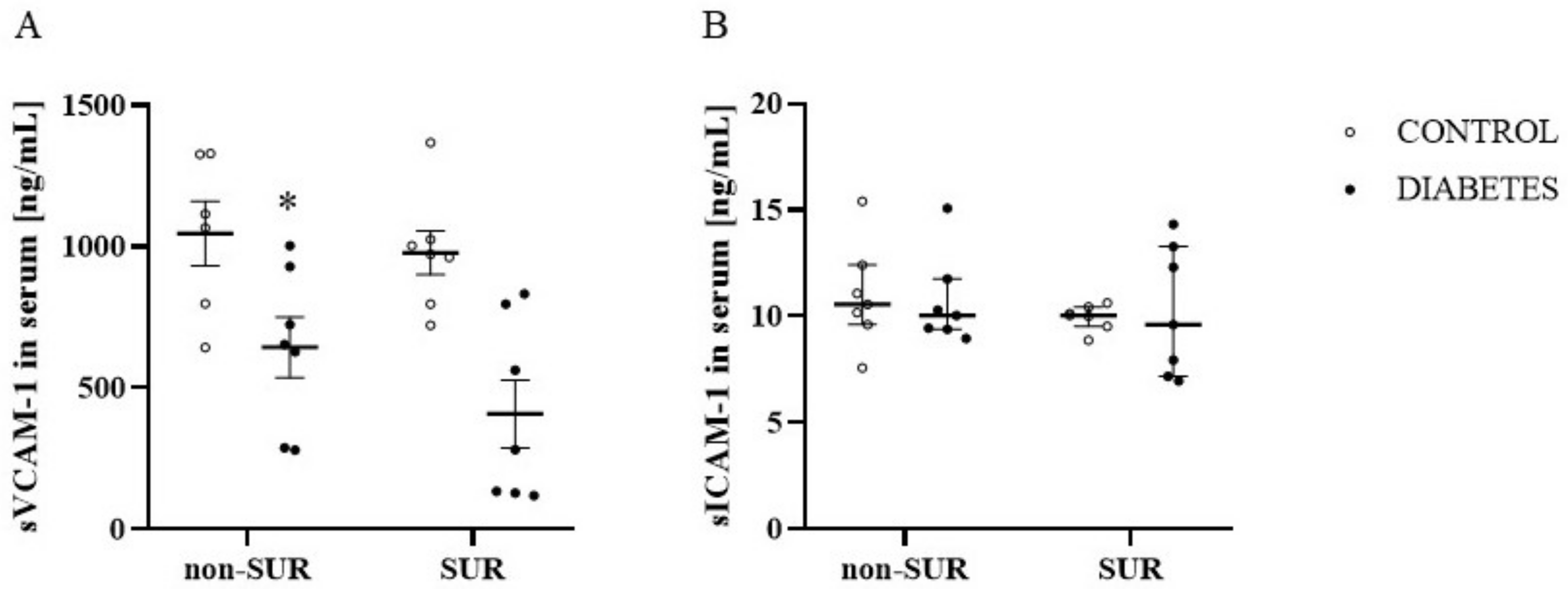
2.1. Experiments In Vivo
2.2. Experiments In Vitro
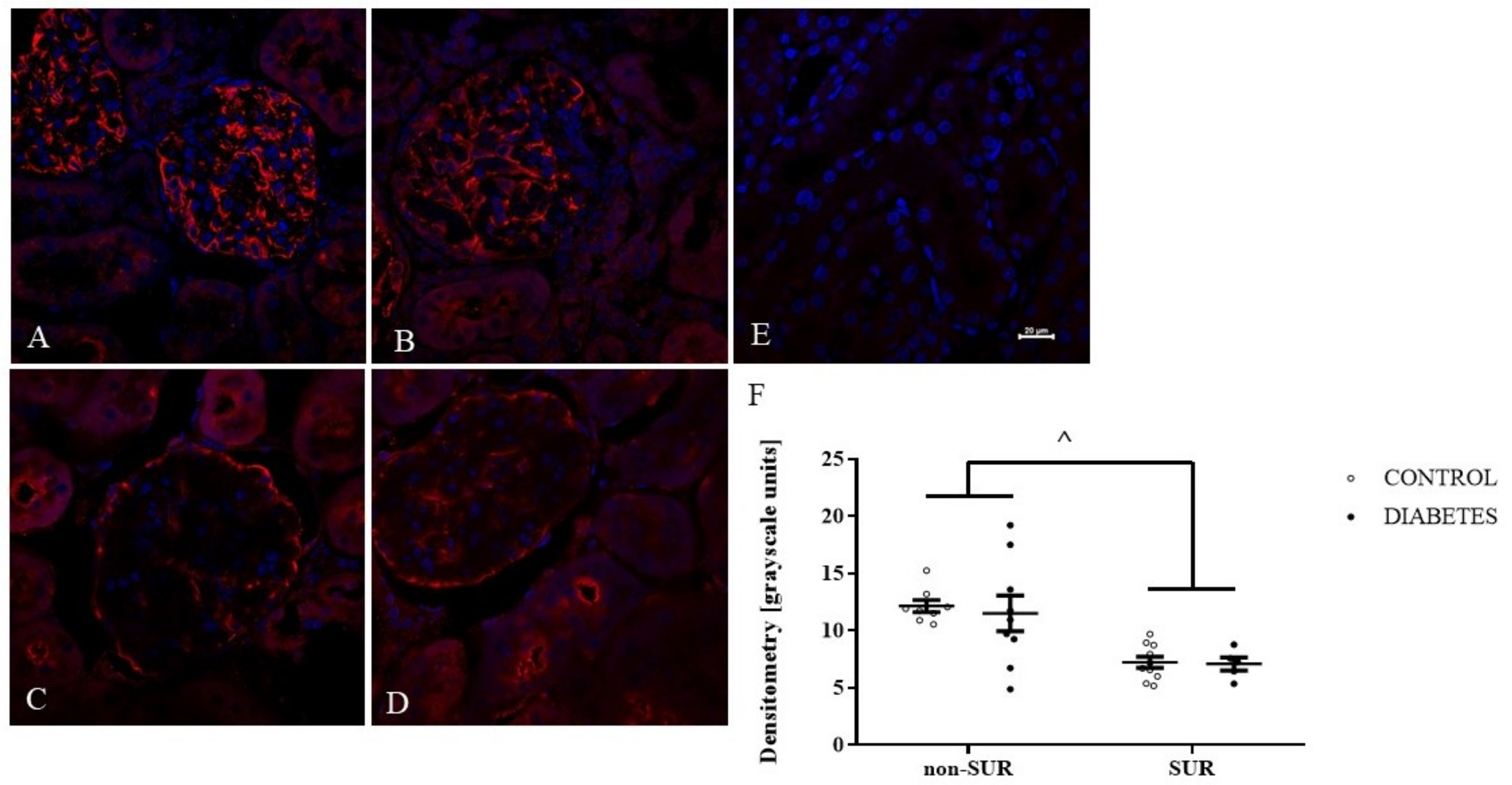
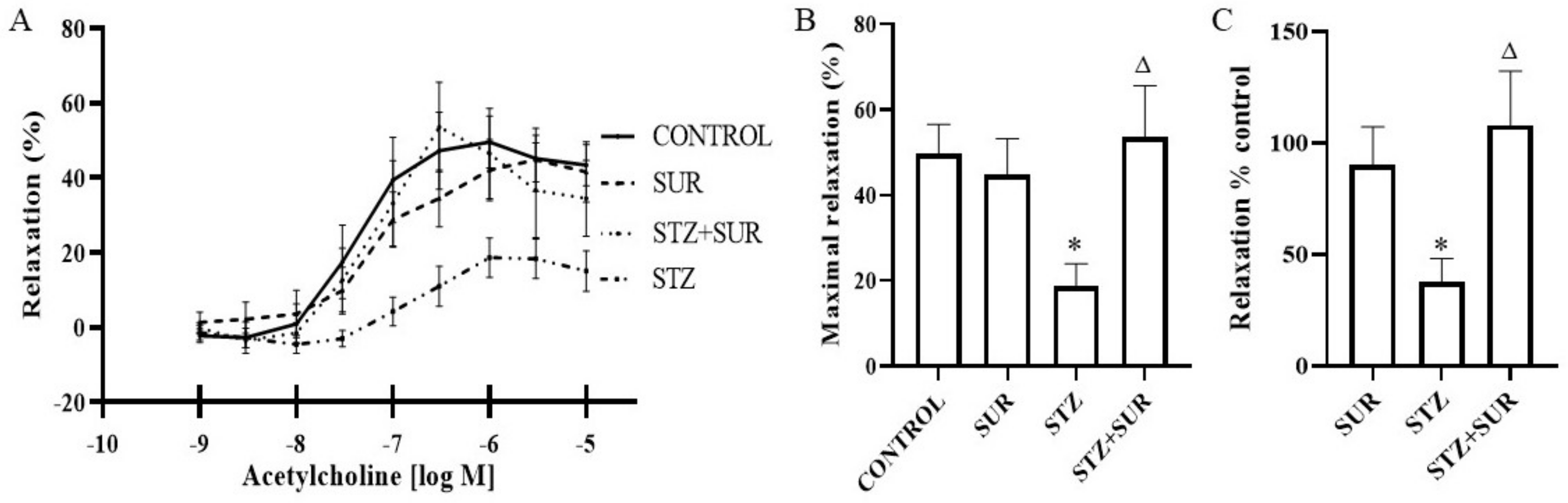
2.3. Experiments Ex Vivo
3. Discussion
4. Materials and Methods
4.1. Ethical Approval
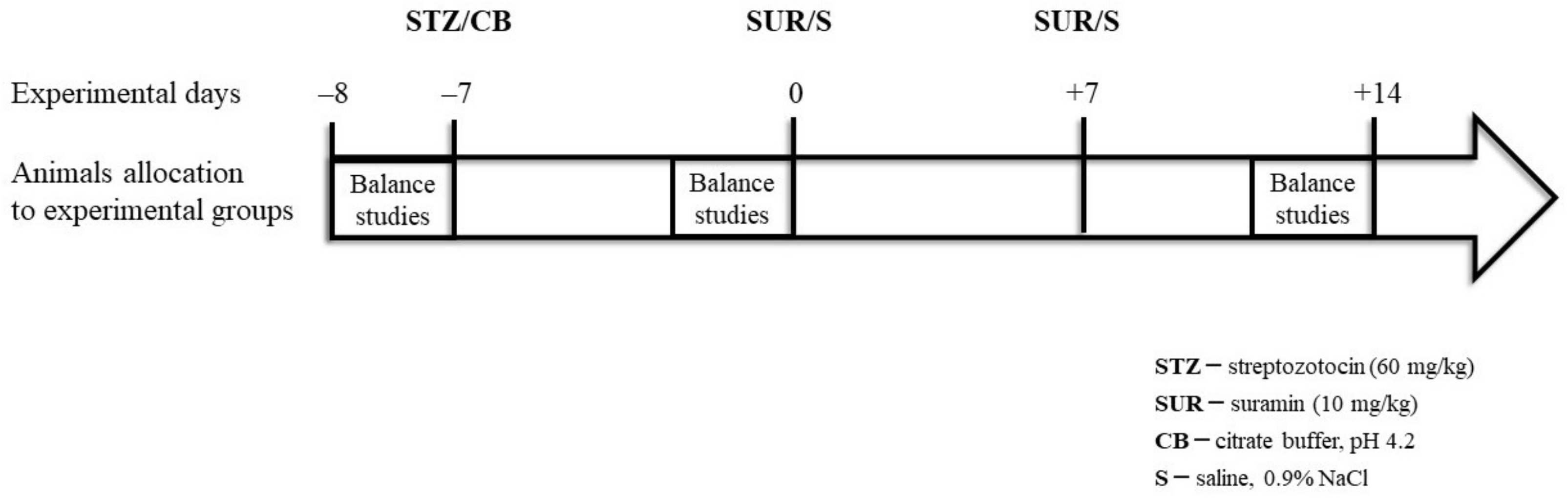
4.2. Animals
- Non-diabetes group—control group (CON): citrate buffer injected on day –7, saline injected on days 0 and +7.
- Suramin-treated non-diabetes group (SUR): suramin (10 mg/kg, ip) injected on days 0 and +7 after the citrate buffer injection on day –7.
- Diabetes group (STZ): streptozotocin (60 mg/kg, ip) injected on day –7, saline injected on days 0 and +7.
- Suramin-treated diabetes group (STZ+SUR): streptozotocin (60 mg/kg, ip) injected on day –7 and suramin (10 mg/kg, ip) injected on days 0 and +7.
4.3. Preparation of Renal Interlobar Arteries
4.4. Measurements of Vascular Responses
4.5. Relative Quantitative Real-Time RT-PCR Analysis
4.6. Western Blotting Analysis
4.7. Immunofluorescence Analysis and Confocal Microscopy of Kidney Sections
4.8. Analytical Methods
4.9. eGFR Analysis
4.10. Nutrition Variable Analysis
4.11. Statistical Analysis
4.12. Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, Regional and Country-Level Diabetes Prevalence Estimates for 2021 and Projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef]
- Day, R.T.; de Cássia Cavaglieri, R.; Tabatabaimir, H.; Mantravadi, V.; Lee, M.J.; Barnes, J.L.; Kasinath, B.S.; Feliers, D. Acute Hyperglycemia Rapidly Stimulates VEGF MRNA Translation in the Kidney. Role of Angiotensin Type 2 Receptor (AT2). Cell Signal. 2010, 22, 1849–1857. [Google Scholar] [CrossRef] [Green Version]
- Tervaert, T.W.C.; Mooyaart, A.L.; Amann, K.; Cohen, A.H.; TerenceCook, H.; Drachenberg, C.B.; Ferrario, F.; Fogo, A.B.; Haas, M.; de Heer, E.; et al. Pathologic Classification of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2010, 21, 556–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Persson, F.; Rossing, P. Diagnosis of Diabetic Kidney Disease: State of the Art and Future Perspective. Kidney Int. Suppl. 2018, 8, 2–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hempel, A.; Maasch, C.; Heintze, U.; Lindschau, C.; Dietz, R.; Luft, F.C.; Haller, H. High Glucose Concentrations Increase Endothelial Cell Permeability via Activation of Protein Kinase Cα. Circ. Res. 1997, 81, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Varma, S.; Lal, B.K.; Zheng, R.; Breslin, J.W.; Saito, S.; Pappas, P.J.; Hobson, R.W.; Durán, W.N. Hyperglycemia Alters PI3k and Akt Signaling and Leads to Endothelial Cell Proliferative Dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H1744–H1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lassén, E.; Daehn, I.S. Molecular Mechanisms in Early Diabetic Kidney Disease: Glomerular Endothelial Cell Dysfunction. Int. J. Mol. Sci. 2020, 21, 9456. [Google Scholar] [CrossRef]
- Vempati, P.; Popel, A.S.; mac Gabhann, F. Extracellular Regulation of VEGF: Isoforms, Proteolysis, and Vascular Patterning. Cytokine Growth Factor Rev. 2014, 25, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Chen, T.T.; Barber, C.L.; Jordan, M.C.; Murdock, J.; Desai, S.; Ferrara, N.; Nagy, A.; Roos, K.P.; Iruela-Arispe, M.L. Autocrine VEGF Signaling Is Required for Vascular Homeostasis. Cell 2007, 130, 691–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamba, T.; Tam, B.Y.Y.; Hashizume, H.; Haskell, A.; Sennino, B.; Mancuso, M.R.; Norberg, S.M.; O’Brien, S.M.; Davis, R.B.; Gowen, L.C.; et al. VEGF-Dependent Plasticity of Fenestrated Capillaries in the Normal Adult Microvasculature. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H560–H576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, I.; Moon, S.O.; Kim, S.H.; Kim, H.J.; Koh, Y.S.; Koh, G.Y. Vascular Endothelial Growth Factor Expression of Intercellular Adhesion Molecule 1 (ICAM-1), Vascular Cell Adhesion Molecule 1 (VCAM-1), and E-Selectin through Nuclear Factor-ΚB Activation in Endothelial Cells. J. Biol. Chem. 2001, 276, 7614–7620. [Google Scholar] [CrossRef] [Green Version]
- Nakagawa, T. Uncoupling of the VEGF-Endothelial Nitric Oxide Axis in Diabetic Nephropathy: An Explanation for the Paradoxical Effects of VEGF in Renal Disease. Am. J. Physiol. Renal. Physiol. 2007, 292, F1665–F1672. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, Q.; Ma, B.; Wang, X.; Bove, A.M.; Simone, G. Molecular Bases of VEGFR-2-Mediated Physiological Function and Pathological Role. Front. Cell Dev. Biol. 2020, 8, 599281. [Google Scholar] [CrossRef]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The Biology of VEGF and Its Receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Bussolati, B.; Dunk, C.; Grohman, M.; Kontos, C.D.; Mason, J.; Ahmed, A. Vascular Endothelial Growth Factor Receptor-1 Modulates Vascular Endothelial Growth Factor-Mediated Angiogenesis via Nitric Oxide. Am. J. Pathol. 2001, 159, 993–1008. [Google Scholar] [CrossRef] [Green Version]
- Sison, K.; Eremina, V.; Baelde, H.; Min, W.; Hirashima, M.; Fantus, I.G.; Quaggin, S.E. Glomerular Structure and Function Require Paracrine, Not Autocrine, VEGF-VEGFR-2 Signaling. J. Am. Soc. Nephrol. 2010, 21, 1691–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, M.; Röckl, W.; Hornig, C.; Gröne, E.F.; Theis, H.; Weich, H.A.; Fuchs, E.; Yayon, A.; Gröne, H.J. Receptors of Vascular Endothelial Growth Factor/Vascular Permeability Factor (VEGF/VPF) in Fetal and Adult Human Kidney: Localization and [125I]VEGF Binding Sites. J. Am. Soc. Nephrol. 1998, 9, 1032–1044. [Google Scholar] [CrossRef]
- Bartlett, C.S.; Jeansson, M.; Quaggin, S.E. Vascular Growth Factors and Glomerular Disease. Annu. Rev. Physiol. 2016, 78, 437–461. [Google Scholar] [CrossRef]
- Cooper, M.E.; Vranes, D.; Youssef, S.; Stacker, S.A.; Cox, A.J.; Rizkalla, B.; Casley, D.J.; Bach, L.A.; Kelly, D.J.; Gilbert, R.E. Increased Renal Expression of Vascular Endothelial Growth Factor (VEGF) and Its Receptor VEGFR-2 in Experimental Diabetes. Diabetes 1999, 48, 2229–2239. [Google Scholar] [CrossRef]
- Zafar, M.I.; Mills, K.; Ye, X.; Blakely, B.; Min, J.; Kong, W.; Zhang, N.; Gou, L.; Regmi, A.; Hu, S.Q.; et al. Association between the Expression of Vascular Endothelial Growth Factors and Metabolic Syndrome or Its Components: A Systematic Review and Meta-Analysis. Diabetol. Metab. Syndr. 2018, 10, 62. [Google Scholar] [CrossRef]
- Majumder, S.; Advani, A. VEGF and the Diabetic Kidney: More than Too Much of a Good Thing. J. Diabetes Complicat. 2017, 31, 273–279. [Google Scholar] [CrossRef]
- Eremina, V.; Sood, M.; Haigh, J.; Nagy, A.; Lajoie, G.; Ferrara, N.; Gerber, H.P.; Kikkawa, Y.; Miner, J.H.; Quaggin, S.E. Glomerular-Specific Alterations of VEGF-A Expression Lead to Distinct Congenital and Acquired Renal Diseases. J. Clin. Investig. 2003, 111, 707–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivaskandarajah, G.A.; Jeansson, M.; Maezawa, Y.; Eremina, V.; Baelde, H.J.; Quaggin, S.E. Vegfa Protects the Glomerular Microvasculature in Diabetes. Diabetes 2012, 61, 2958–2966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veron, D.; Aggarwal, P.K.; Li, Q.; Moeckel, G.; Kashgarian, M.; Tufro, A. Podocyte VEGF-A Knockdown Induces Diffuse Glomerulosclerosis in Diabetic and in ENOS Knockout Mice. Front. Pharm. 2022, 12, 788886. [Google Scholar] [CrossRef]
- Oltean, S.; Qiu, Y.; Ferguson, J.K.; Stevens, M.; Neal, C.; Russell, A.; Kaura, A.; Arkill, K.P.; Harris, K.; Symonds, C.; et al. Vascular Endothelial Growth Factor-A165b Is Protective and Restores Endothelial Glycocalyx in Diabetic Nephropathy. J. Am. Soc. Nephrol. 2015, 26, 1889–1904. [Google Scholar] [CrossRef] [Green Version]
- Lindenmeyer, M.T.; Kretzler, M.; Boucherot, A.; Berra, S.; Yasuda, Y.; Henger, A.; Eichinger, F.; Gaiser, S.; Schmid, H.; Rastaldi, M.P.; et al. Interstitial Vascular Rarefaction and Reduced VEGF-A Expression in Human Diabetic Nephropathy. J. Am. Soc. Nephrol. 2007, 18, 1765–1776. [Google Scholar] [CrossRef] [Green Version]
- Hohenstein, B.; Hausknecht, B.; Boehmer, K.; Riess, R.; Brekken, R.A.; Hugo, C.P.M. Local VEGF Activity but Not VEGF Expression Is Tightly Regulated during Diabetic Nephropathy in Man. Kidney Int. 2006, 69, 1654–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanabe, K.; Maeshima, Y.; Sato, Y.; Wada, J. Antiangiogenic Therapy for Diabetic Nephropathy. Biomed. Res. Int. 2017, 2017, 5724069. [Google Scholar] [CrossRef] [Green Version]
- Chyła, G.; Sałaga-Zaleska, K.; Dąbkowski, K.; Kuchta, A.; Jankowski, M. Suramin Enhances the Urinary Excretion of VEGF-A in Normoglycemic and Streptozotocin-Induced Diabetic Rats. Pharmacol. Rep. 2021, 73, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Wiedemar, N.; Hauser, D.A.; Mäser, P. 100 Years of Suramin. Antimicrob. Agents Chemother. 2020, 64, e01168-19. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Tolbert, E.; Pang, M.; Ponnusamy, M.; Yan, H.; Zhuang, S. Suramin Inhibits Renal Fibrosis in Chronic Kidney Disease. J. Am. Soc. Nephrol. 2011, 22, 1064–1075. [Google Scholar] [CrossRef] [Green Version]
- Korrapati, M.C.; Shaner, B.E.; Neely, B.A.; Alge, J.L.; Arthur, J.M.; Schnellmann, R.G. Diabetes-Induced Renal Injury in Rats Is Attenuated by Suramin. J. Pharmacol. Exp. Ther. 2012, 343, 34–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korrapati, M.C.; Howell, L.H.; Shaner, B.E.; Megyesi, J.K.; Siskind, L.J.; Schnellmann, R.G. Suramin: A Potential Therapy for Diabetic Nephropathy. PLoS ONE 2013, 8, e73655. [Google Scholar] [CrossRef]
- Waltenberger, J.; Mayr, U.; Frank, H.; Hombach, V. Suramin Is a Potent Inhibitor of Vascular Endothelial Growth Factor. A Contribution to the Molecular Basis of Its Antiangiogenic Action. J. Mol. Cell Cardiol. 1996, 28, 1523–1529. [Google Scholar] [CrossRef]
- McNally, W.P.; DeHart, P.D.; Lathia, C.; Whitfield, L.R. Distribution of [14C]Suramin in Tissues of Male Rats Following a Single Intravenous Dose. Life Sci. 2000, 67, 1847–1857. [Google Scholar] [CrossRef]
- Gagliardi, A.R.T.; Taylor, M.F.; Collins, D.C. Uptake of Suramin by Human Microvascular Endothelial Cells. Cancer Lett. 1998, 125, 97–102. [Google Scholar] [CrossRef]
- Singh, A.K.; Gudehithlu, K.P.; Pegoraro, A.A.; Singh, G.K.; Basheerudin, K.; Robey, R.B.; Arruda, J.A.L.; Dunea, G. Vascular Factors Altered in Glucose-Treated Mesangial Cells and Diabetic Glomeruli. Changes in Vascular Factors Impair Endothelial Cell Growth and Matrix. Lab. Investig. 2004, 84, 597–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, E.; Bottomley, M.J.; Westwell, S.; Pringle, J.H.; Furness, P.N.; Fechally, J.; Brenchley, P.E.C.; Harper, S.J. Vascular Endothelial Growth Factor MRNA Expression in Minimal Change, Membranous, and Diabetic Nephropathy Demonstrated by Non-Isotopic in Situ Hybridisation. J. Clin. Pathol. 1999, 52, 735–738. [Google Scholar] [CrossRef] [Green Version]
- Ichinose, K.; Maeshima, Y.; Yamamoto, Y.; Kitayama, H.; Takazawa, Y.; Hirokoshi, K.; Sugiyama, H.; Yamasaki, Y.; Eguchi, K.; Makino, H. Antiangiogenic Endostatin Peptide Ameliorates Renal Alterations in the Early of a Type 1 Diabetic Nephropathy Model. Diabetes 2005, 54, 2891–2903. [Google Scholar] [CrossRef] [Green Version]
- Shulman, K.; Rosen, S.; Tognazzi, K.; Manseau, E.J.; Brown, L.F. Expression of Vascular Permeability Factor (VPF/VEGF) Is Altered in Many Glomerular Diseases. J. Am. Soc. Nephrol. 1996, 7, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Lin, J.-H.; Hammes, H.-P.; Zhang, C. Cellular Phenotypic Transitions in Diabetic Nephropathy: An Update. Front. Pharmacol. 2022, 13, 1038073. [Google Scholar] [CrossRef] [PubMed]
- Sato, W.; Tanabe, K.; Kosugi, T.; Hudkins, K.; Lanaspa, M.A.; Zhang, L.; Campbell-Thompson, M.; Li, Q.; Long, D.A.; Alpers, C.E.; et al. Selective Stimulation of VEGFR2 Accelerates Progressive Renal Disease. Am. J. Pathol. 2011, 179, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Sung, S.H.; Ziyadeh, F.N.; Wang, A.; Pyagay, P.E.; Kanwar, Y.S.; Chen, S. Blockade of Vascular Endothelial Growth Factor Signaling Ameliorates Diabetic Albuminuria in Mice. J. Am. Soc. Nephrol. 2006, 17, 3093–3104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oates, J.C.; Russell, D.L.; van Beusecum, J.P. Endothelial Cells: Potential Novel Regulators of Renal Inflammation. Am. J. Physiol. Renal. Physiol. 2022, 322, F309–F321. [Google Scholar] [CrossRef] [PubMed]
- Marfella, R.; Esposito, K.; Giunta, R.; Coppola, G.; de Angelis, L.; Farzati, B.; Paolisso, G.; Giugliano, D. Circulating Adhesion Molecules in Humans: Role of Hyperglycemia and Hyperinsalinemia. Circulation 2000, 101, 2247–2251. [Google Scholar] [CrossRef] [Green Version]
- Gu, H.F.; Ma, J.; Gu, K.T.; Brismar, K. Association of Intercellular Adhesion Molecule 1 (ICAM1) with Diabetes and Diabetic Nephropathy. Front. Endocrinol. 2013, 3, 179. [Google Scholar] [CrossRef] [Green Version]
- Leeuwenberg, J.F.; Smeets, E.F.; Neefjes, J.J.; Shaffer, M.A.; Cinek, T.; Jeunhomme, T.M.; Ahern, T.J.; Buurman, W.A. E-Selectin and Intercellular Adhesion Molecule-1 Are Released by Activated Human Endothelial Cells in Vitro. Immunology 1992, 77, 543–549. [Google Scholar]
- Altannavch, T.; Roubalová, K.; Kučera, P.; Anděl, M. Effect of High Glucose Concentrations on Expression of ELAM-1, VCAM-1 and ICAM-1 in HUVEC with and without Cytokine Activation. Physiol. Res. 2004, 53, 77–82. [Google Scholar] [CrossRef]
- Güler, S.; Cakir, B.; Demirbas, B.; Yönem, A.; Odabasi, E.; Önde, U.; Aykut, Ö.; Gürsoy, G. Plasma Soluble Intercellular Adhesion Molecule 1 Levels Are Increased in Type 2 Diabetic Patients with Nephropathy. Horm. Res. 2002, 58, 67–70. [Google Scholar] [CrossRef]
- Clausen, P.; Jacobsen, P.; Rossing, K.; Jensen, J.S.; Parving, H.H.; Feldt-Rasmussen, B. Plasma Concentrations of VCAM-1 and ICAM-1 Are Elevated in Patients with Type 1 Diabetes Mellitus with Microalbuminuria and Overt Nephropathy. Diabet. Med. 2000, 17, 644–649. [Google Scholar] [CrossRef] [PubMed]
- Maric-Bilkan, C.; Flynn, E.R.; Chade, A.R. Microvascular Disease Precedes the Decline in Renal Function in the Streptozotocin-Induced Diabetic Rat. Am. J. Physiol. Ren. Physiol. 2012, 302, F308–F315. [Google Scholar] [CrossRef] [Green Version]
- Edgley, A.J.; Tare, M.; Evans, R.G.; Skordilis, C.; Parkington, H.C. In Vivo Regulation of Endothelium-Dependent Vasodilation in the Rat Renal Circulation and the Effect of Streptozotocin-Induced Diabetes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R829–R839. [Google Scholar] [CrossRef] [Green Version]
- Johnstone, M.T.; Creager, S.J.; Scales, K.M.; Cusco, J.A.; Lee, B.K.; Creager, M.A. Impaired Endothelium-Dependent Vasodilation in Patients with Insulin- Dependent Diabetes Mellitus. Circulation 1993, 88, 2510–2516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamata, K.; Miyata, N.; Kasuya, Y. Impairment of Endothelium-Dependent Relaxation and Changes in Levels of Cyclic GMP in Aorta from Streptozotocin-Induced Diabetic Rats. Br. J. Pharm. 1989, 97, 614–618. [Google Scholar] [CrossRef] [Green Version]
- Jourde-Chiche, N.; Fakhouri, F.; Dou, L.; Bellien, J.; Burtey, S.; Frimat, M.; Jarrot, P.A.; Kaplanski, G.; le Quintrec, M.; Pernin, V.; et al. Endothelium Structure and Function in Kidney Health and Disease. Nat. Rev. Nephrol. 2019, 15, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Bardal, S.; Misurski, D.; Qiu, X.; Desai, K.; McNeill, J.R. Chronic Treatment with Vascular Endothelial Growth Factor Preserves Agonist-Evoked Vascular Responses in the Streptozotocin-Induced Diabetic Rat. Diabetologia 2006, 49, 811–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chomczynski, P.; Sacchi, N. Single-Step Method of RNA Isolation by Acid Guanidinium Thiocyanate-Phenol-Chloroform Extraction. Anal. Biochem. 1987, 162, 156–159. [Google Scholar] [CrossRef]
- Besseling, P.J.; Pieters, T.T.; Nguyen, I.T.N.; de Bree, P.M.; Willekes, N.; Dijk, A.H.; Bovee, D.M.; Hoorn, E.J.; Rookmaaker, M.B.; Gerritsen, K.G.; et al. A Plasma Creatinine- And Urea-Based Equation to Estimate Glomerular Filtration Rate in Rats. Am. J. Physiol. Ren. Physiol. 2021, 320, F518–F524. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Day | Experimental Groups | |||
|---|---|---|---|---|---|
| CON | SUR | STZ | STZ+SUR | ||
| Food intake, (g) | 0 | 20 ± 2 | 23 ± 1 | 29 ± 1 * | 29 ± 1 |
| +14 | 23 ± 1 | 23 ± 0.4 | 35 ± 2 #,* | 35 ± 2 # | |
| Specific rate of body mass gain, (g/kg) | +14 | 184 ± 31 | 180 ± 26 | −0.51 ± 22.2 & | 43 ± 17 |
| Feed efficiency ratio | +14 | 17 ± 1 | 15 ± 2 | −0.06 ± 1.2 & | 2 ± 1 |
| Efficiency of food utilization for body weight, (g/cal) | +14 | 62 ± 5 | 56 ± 6 | −0.2 ± 4.4 & | 9 ± 4 |
| Gene Transcript | Accession No. | Oligonucleotide Sequence 5′-3′ | Universal Probe Library Probe |
|---|---|---|---|
| rFlt1 (Vegfr1) | D28498 | (F) cagtttccaagtggccagag | #22 |
| (R) aggtcgcgatgaatgcac | |||
| rFlk1 (Vegfr2) | U93306 | (F) gagacccgcgttttcaga | #65 |
| (R) aagaacaatatagtctttgccatcc | |||
| Actb | Universal ProbeLibrary Rat Actb Gene Assay (Roche, Cat #05046203001) | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chyła-Danił, G.; Sałaga-Zaleska, K.; Kreft, E.; Krzesińska, A.; Herman, S.; Kuchta, A.; Sakowicz-Burkiewicz, M.; Lenartowicz, M.; Jankowski, M. Suramin Affects the Renal VEGF-A/VEGFR Axis in Short-Term Streptozotocin-Induced Diabetes. Pharmaceuticals 2023, 16, 470. https://doi.org/10.3390/ph16030470
Chyła-Danił G, Sałaga-Zaleska K, Kreft E, Krzesińska A, Herman S, Kuchta A, Sakowicz-Burkiewicz M, Lenartowicz M, Jankowski M. Suramin Affects the Renal VEGF-A/VEGFR Axis in Short-Term Streptozotocin-Induced Diabetes. Pharmaceuticals. 2023; 16(3):470. https://doi.org/10.3390/ph16030470
Chicago/Turabian StyleChyła-Danił, Gabriela, Kornelia Sałaga-Zaleska, Ewelina Kreft, Aleksandra Krzesińska, Sylwia Herman, Agnieszka Kuchta, Monika Sakowicz-Burkiewicz, Małgorzata Lenartowicz, and Maciej Jankowski. 2023. "Suramin Affects the Renal VEGF-A/VEGFR Axis in Short-Term Streptozotocin-Induced Diabetes" Pharmaceuticals 16, no. 3: 470. https://doi.org/10.3390/ph16030470