

Amphiphilic Cell-Penetrating Peptides Containing Arginine and Hydrophobic Residues as Protein Delivery Agents
 ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
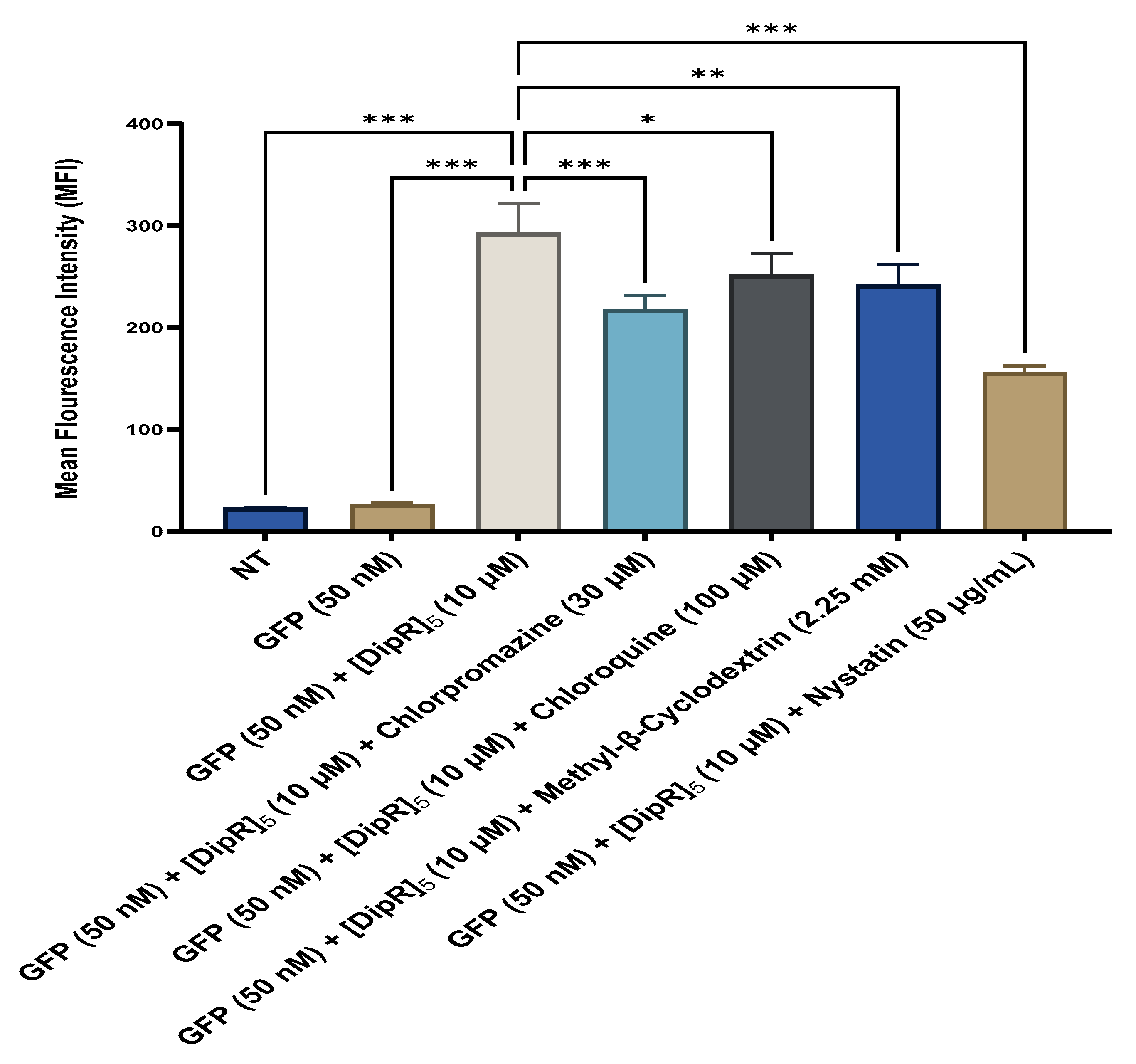{kind=link}
Abstract
:1. Introduction
2. Results and Discussions
2.1. Chemistry
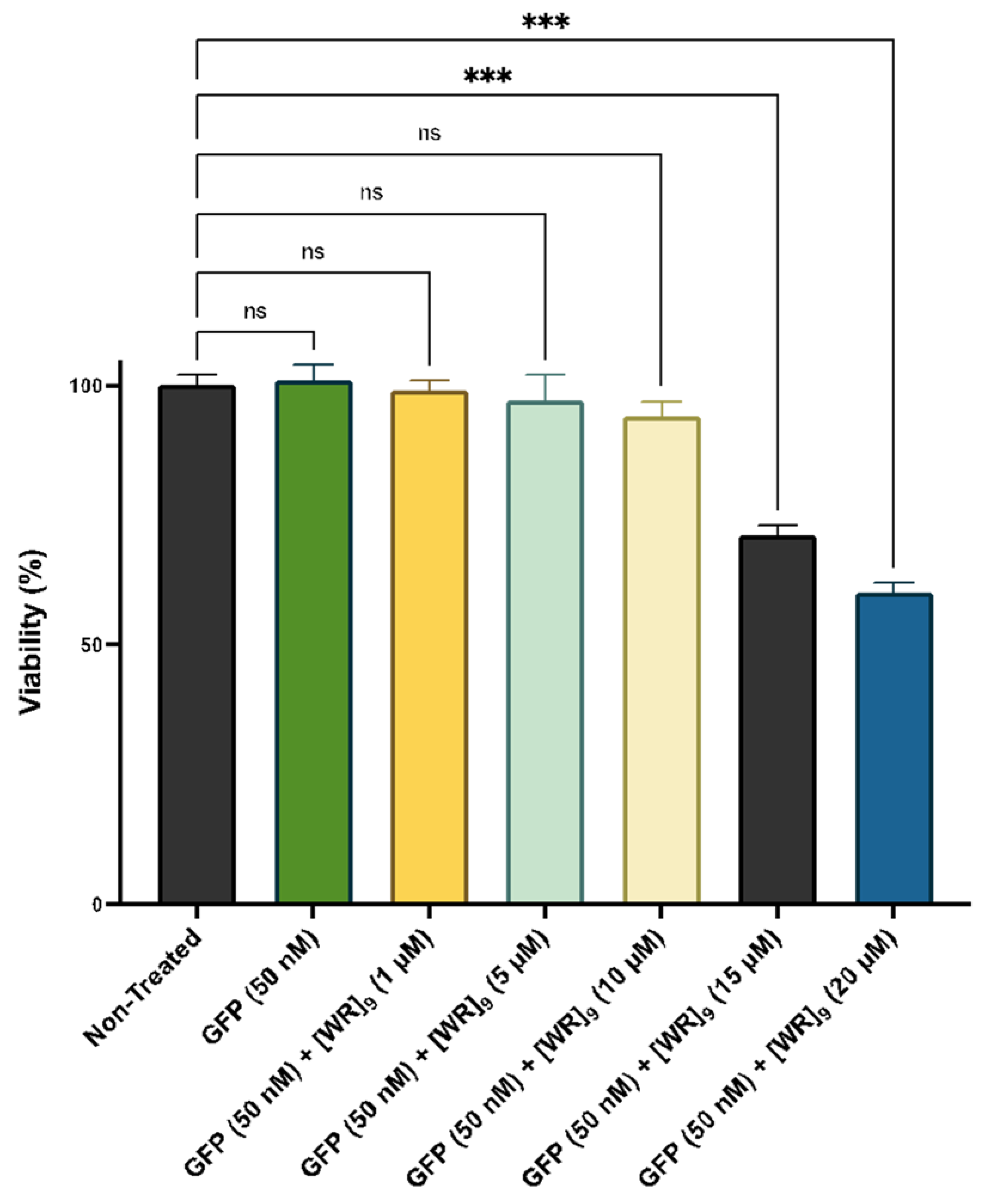
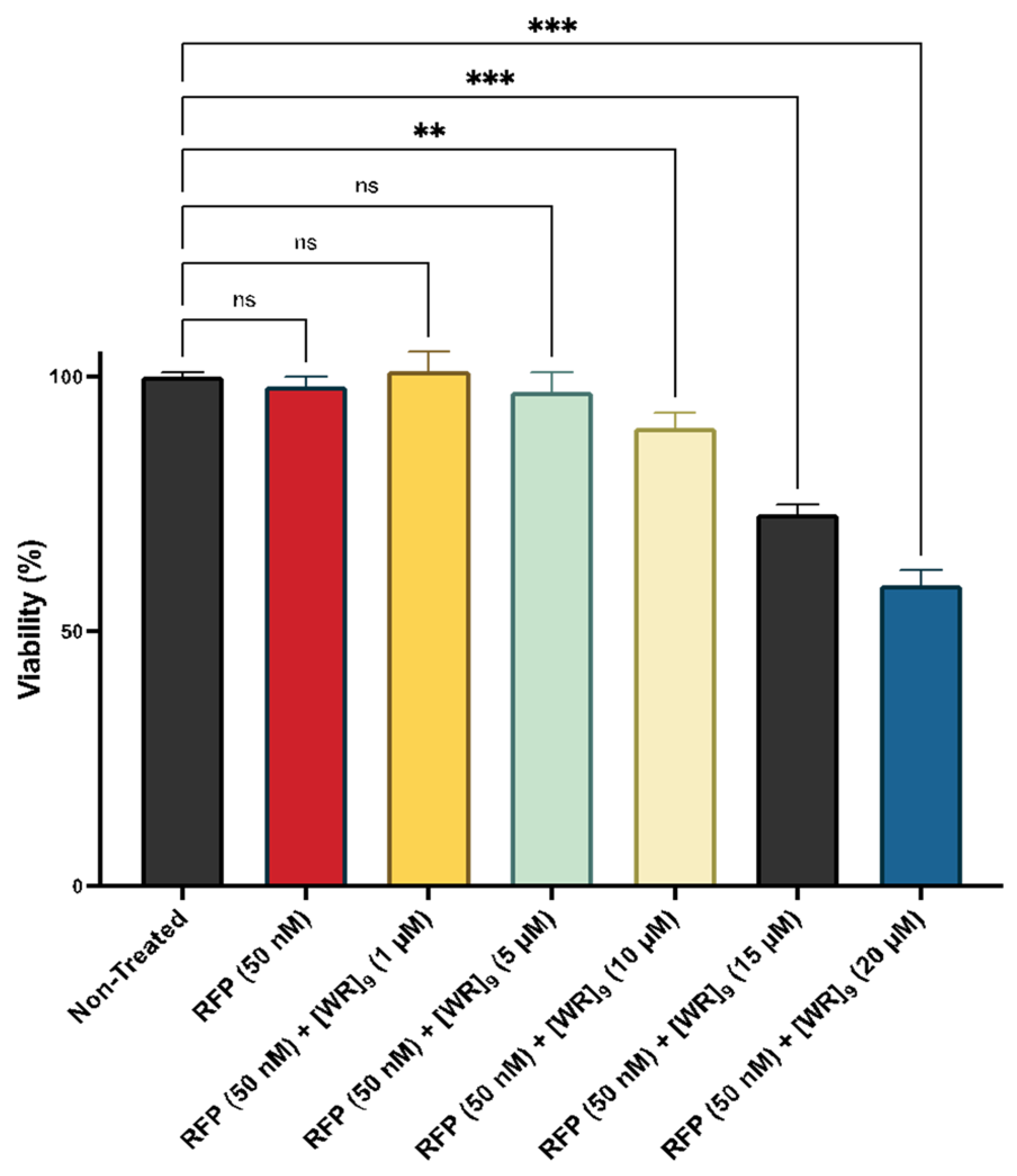
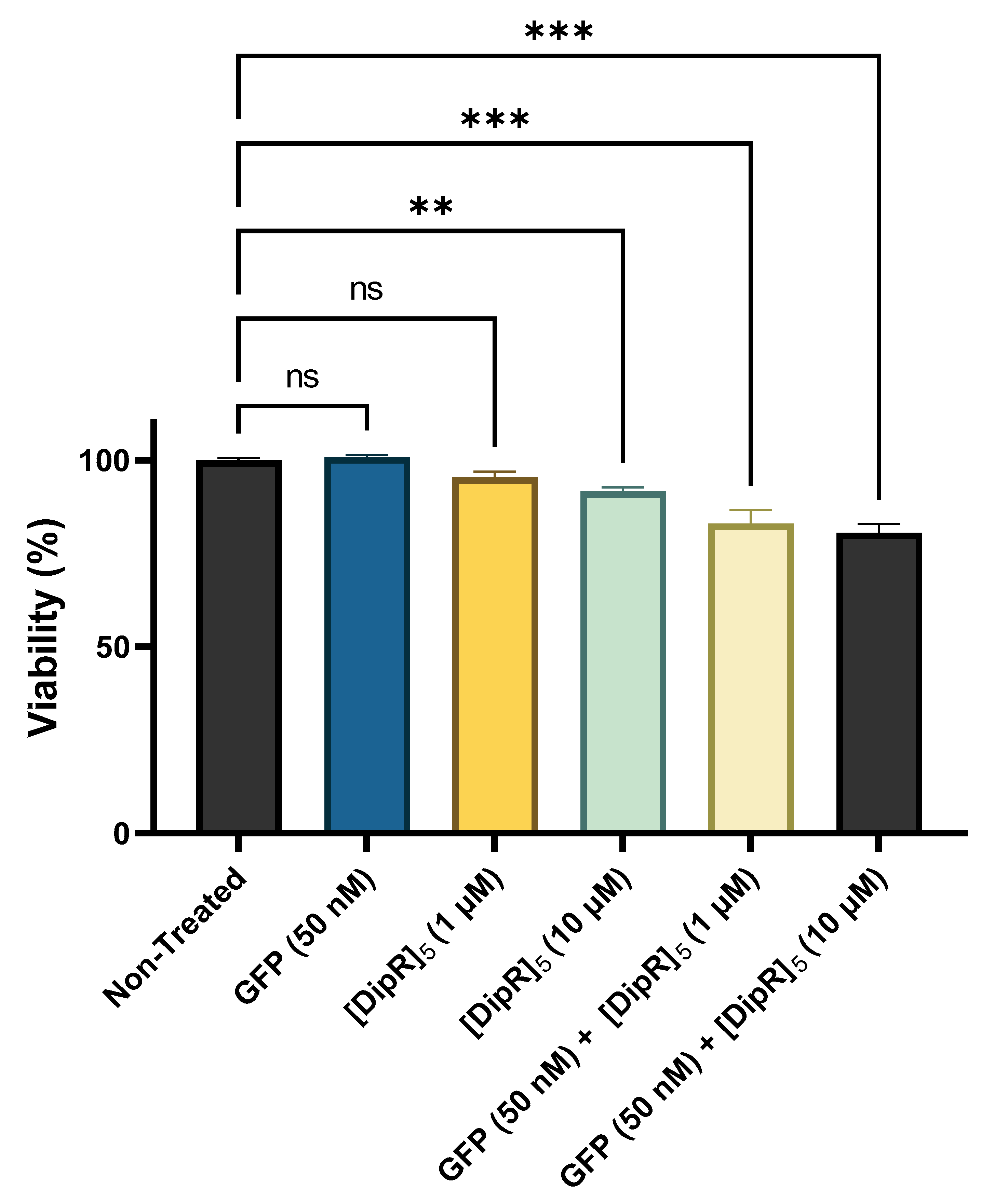
2.2. Cytotoxicity of Peptide-Protein Physical Mixture
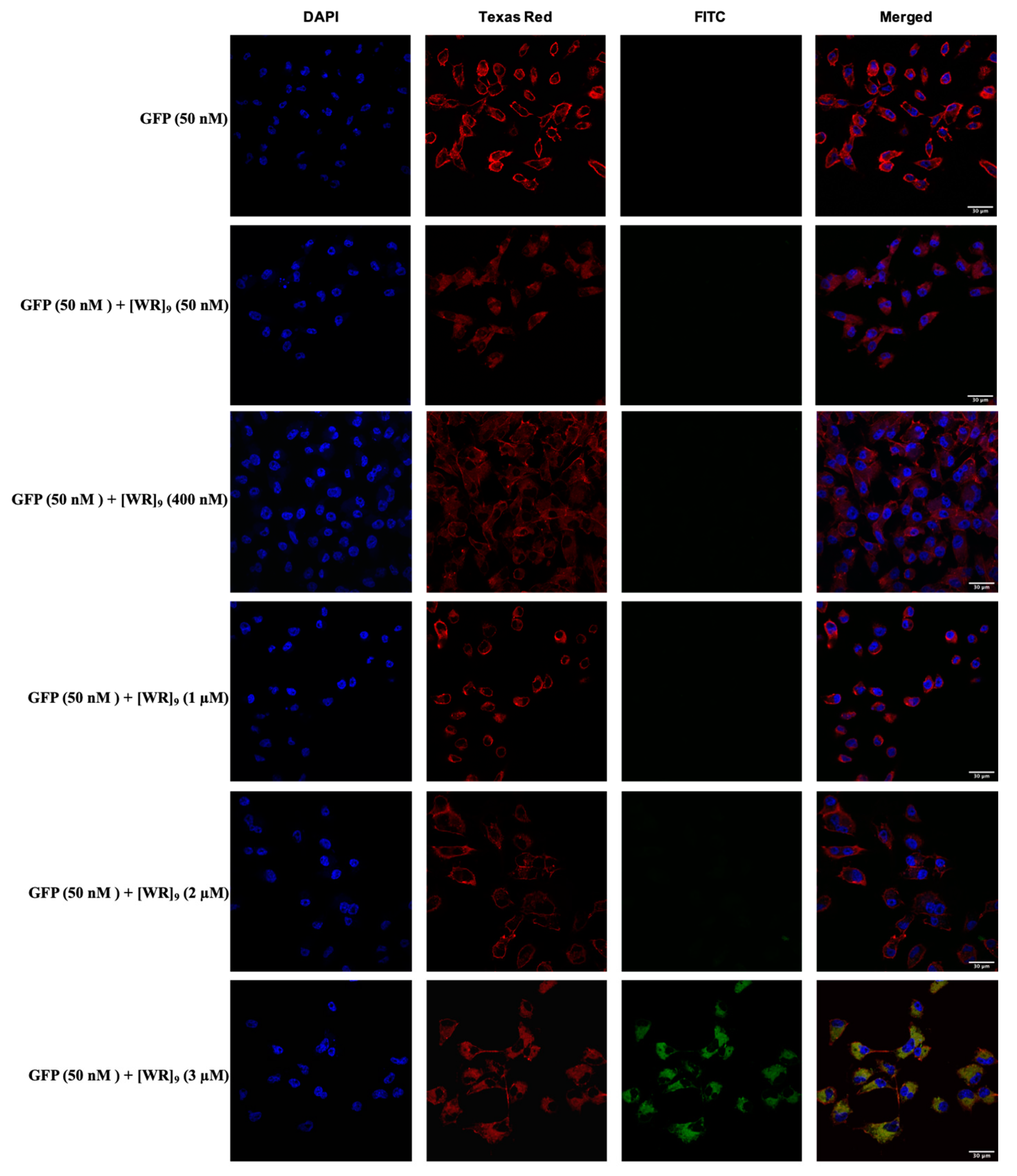
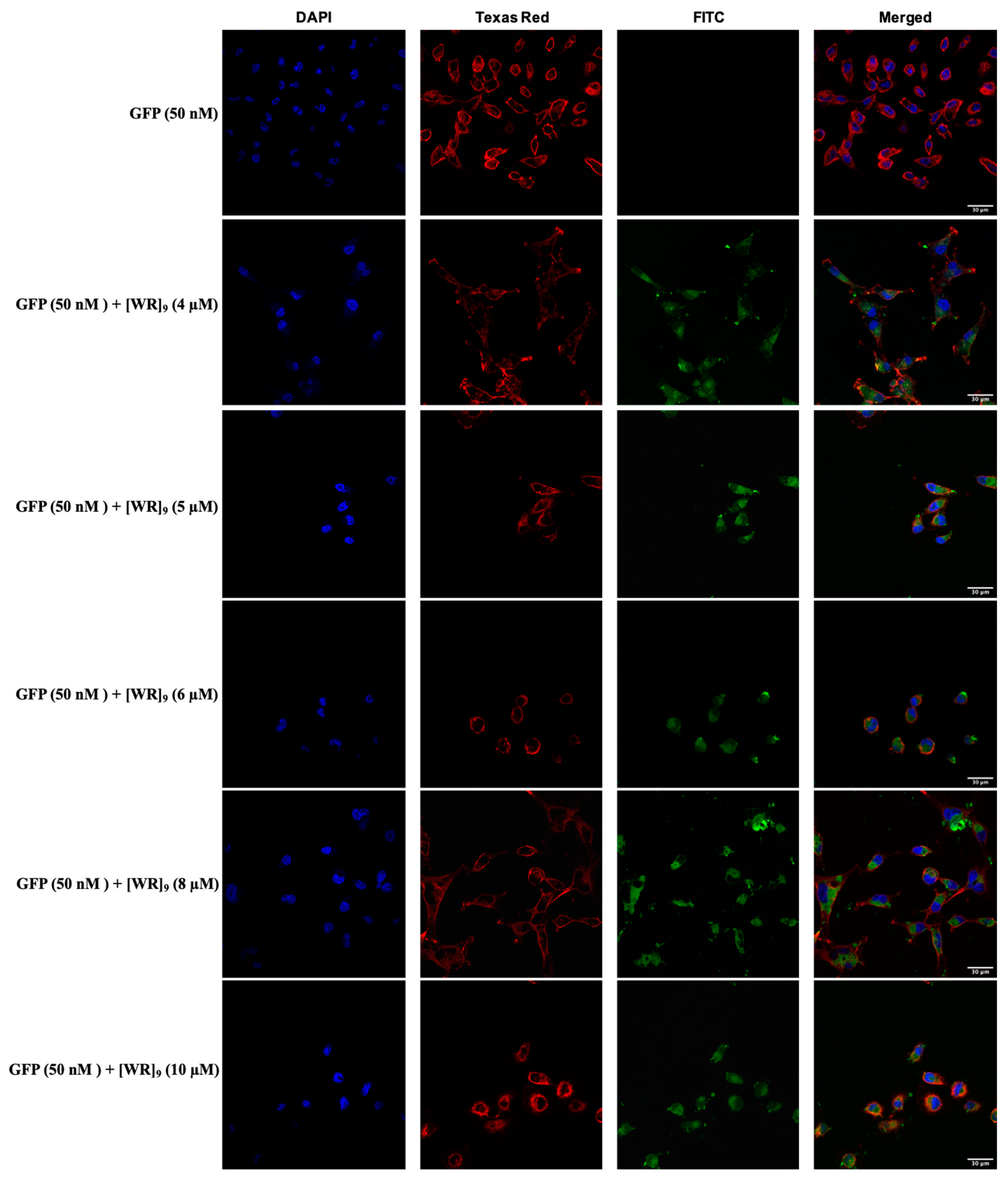
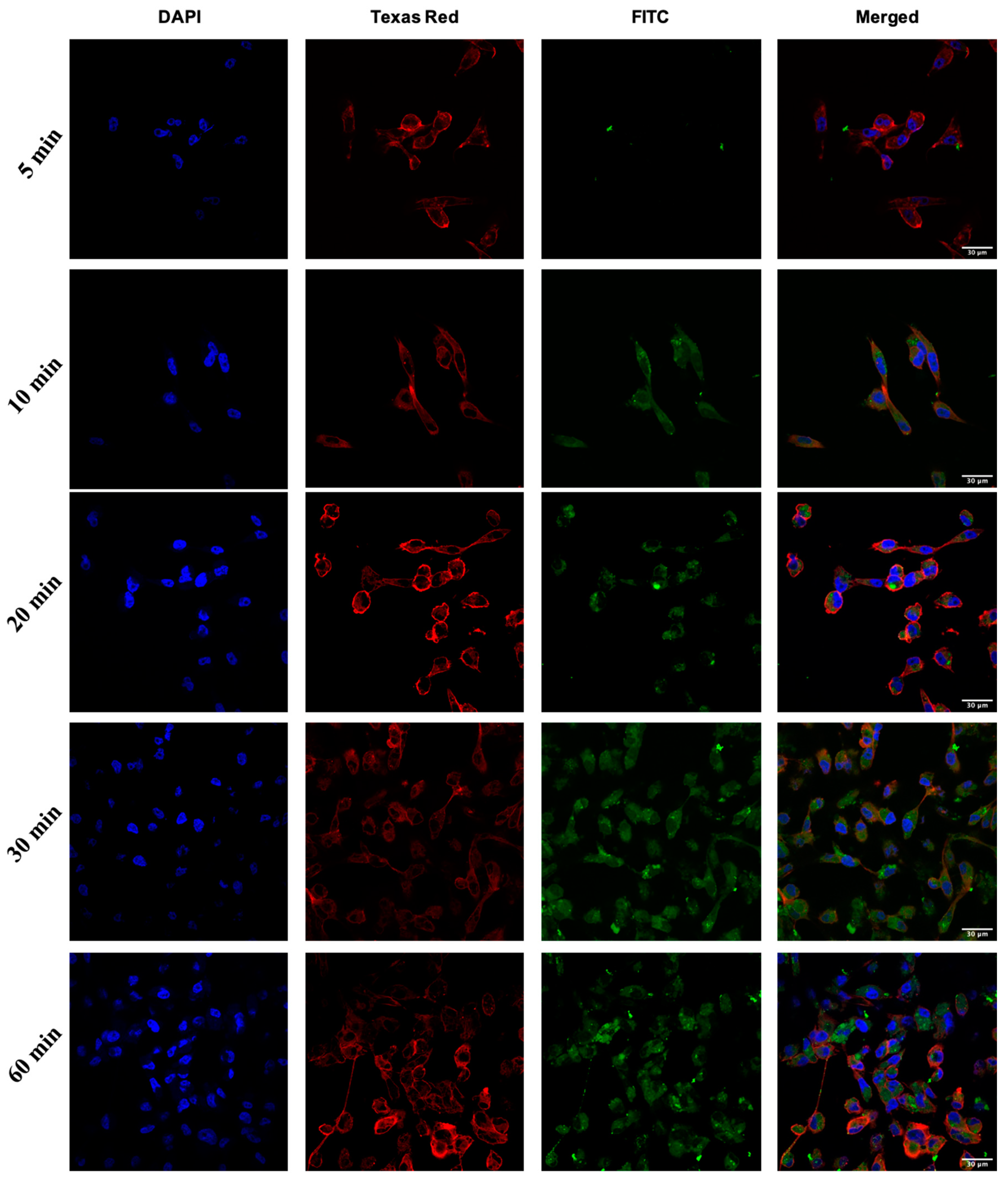
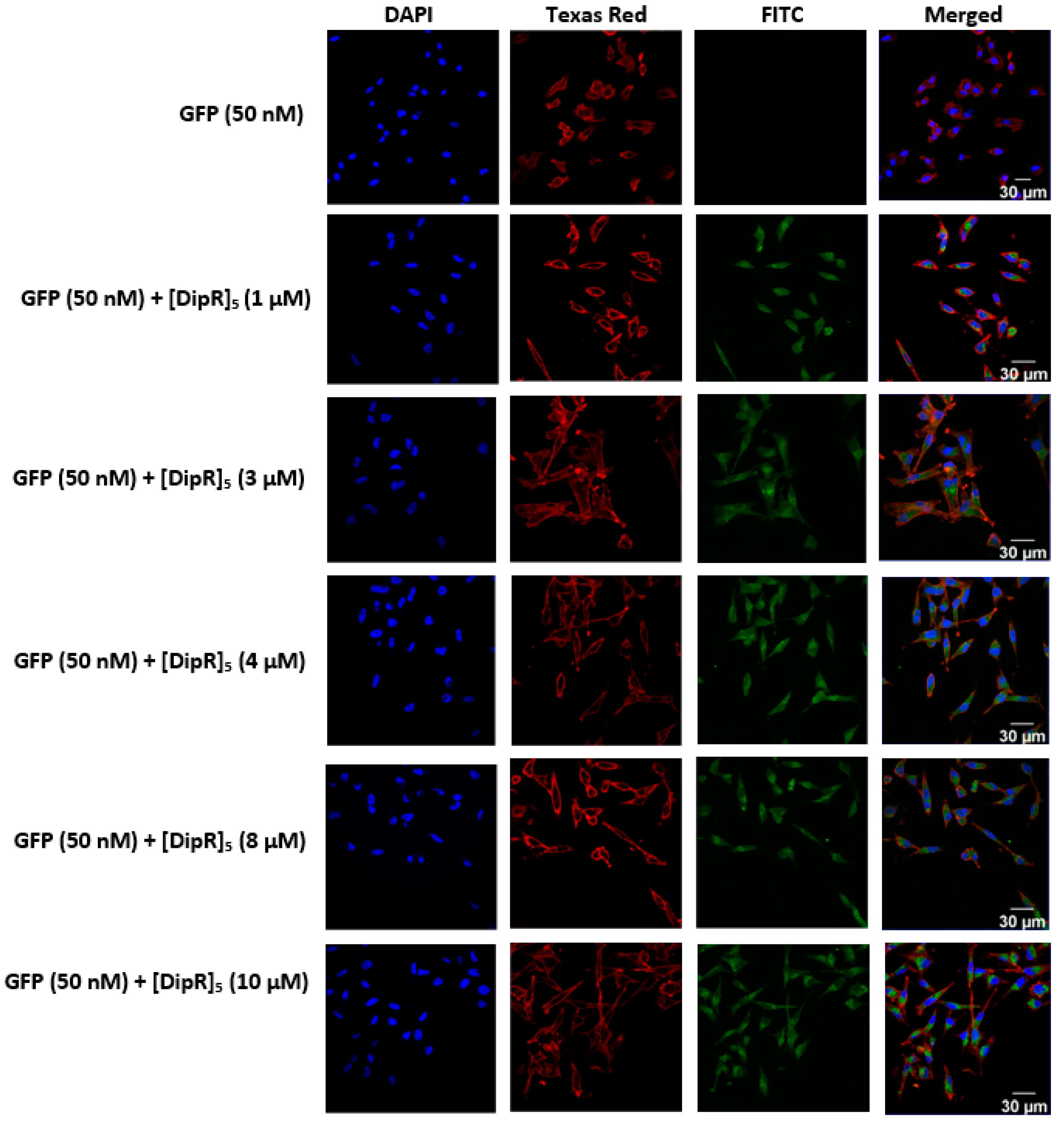
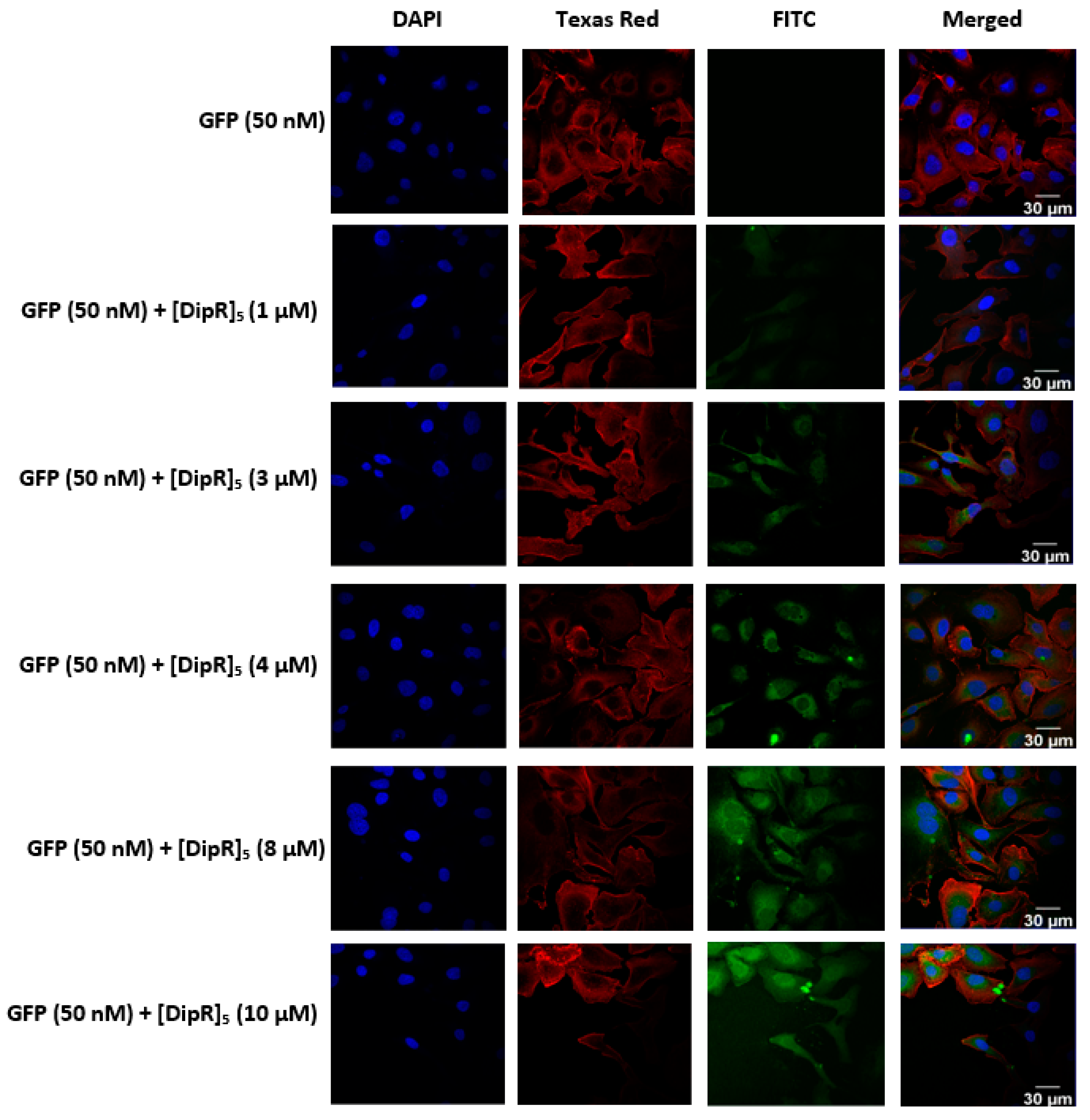
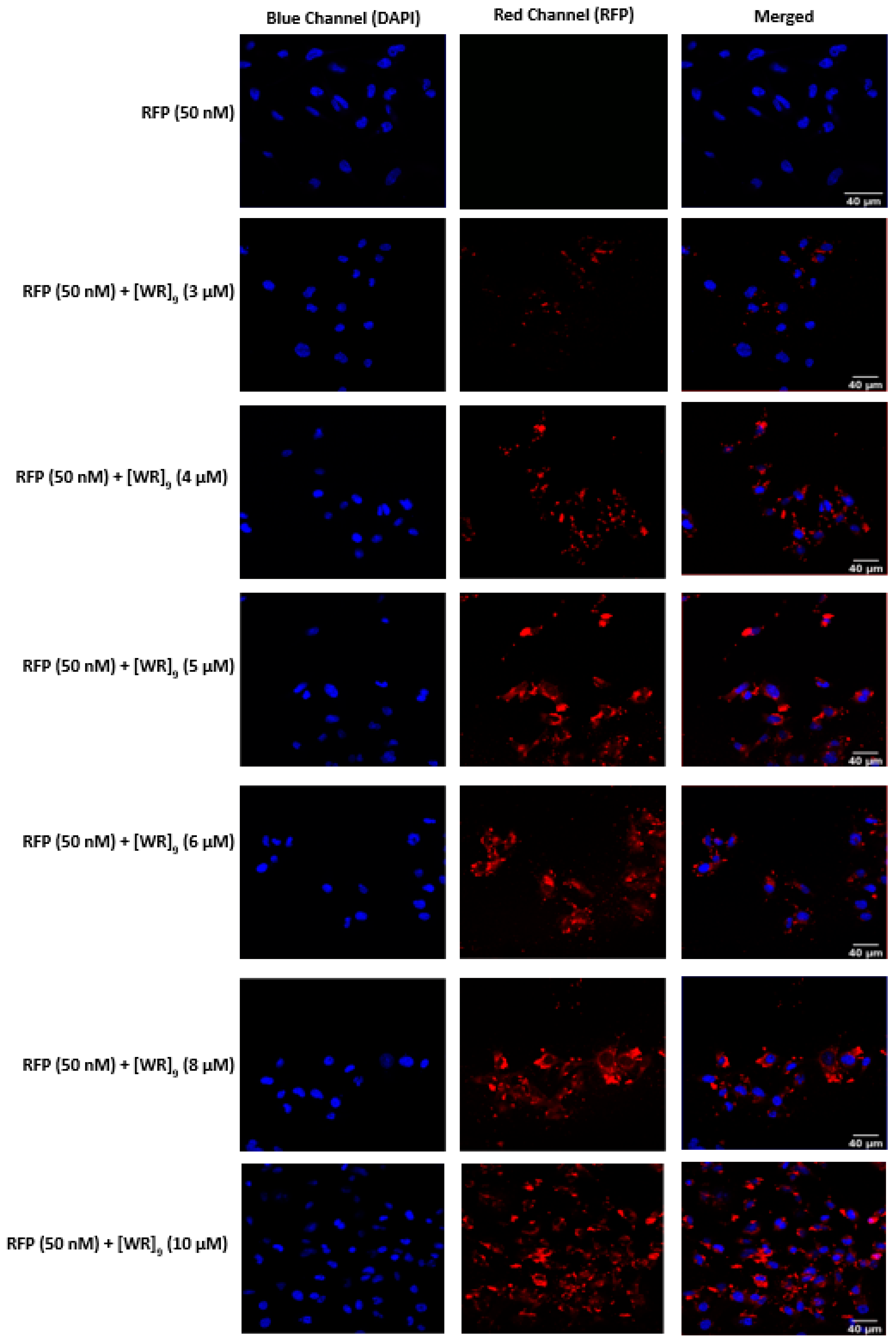
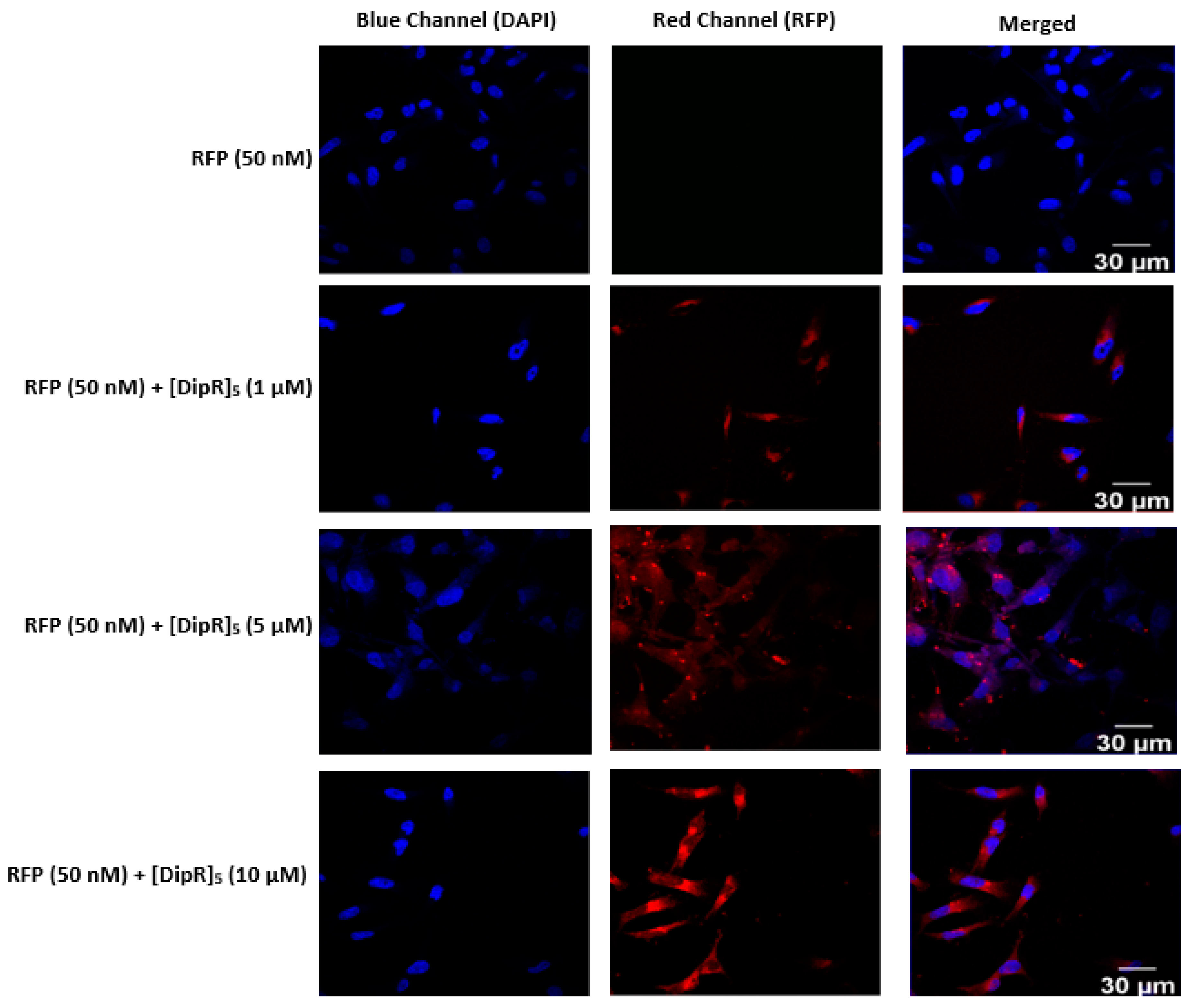
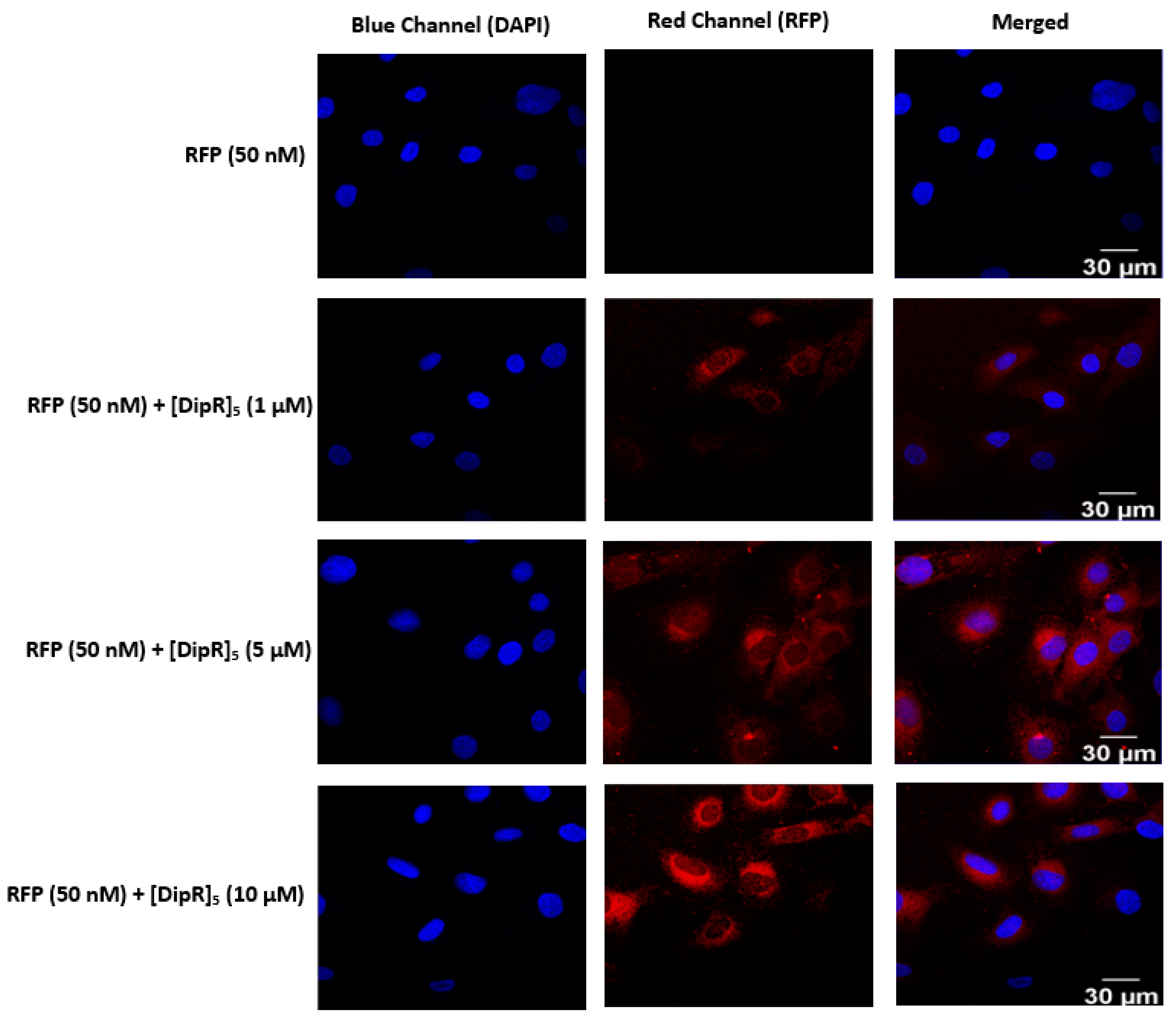
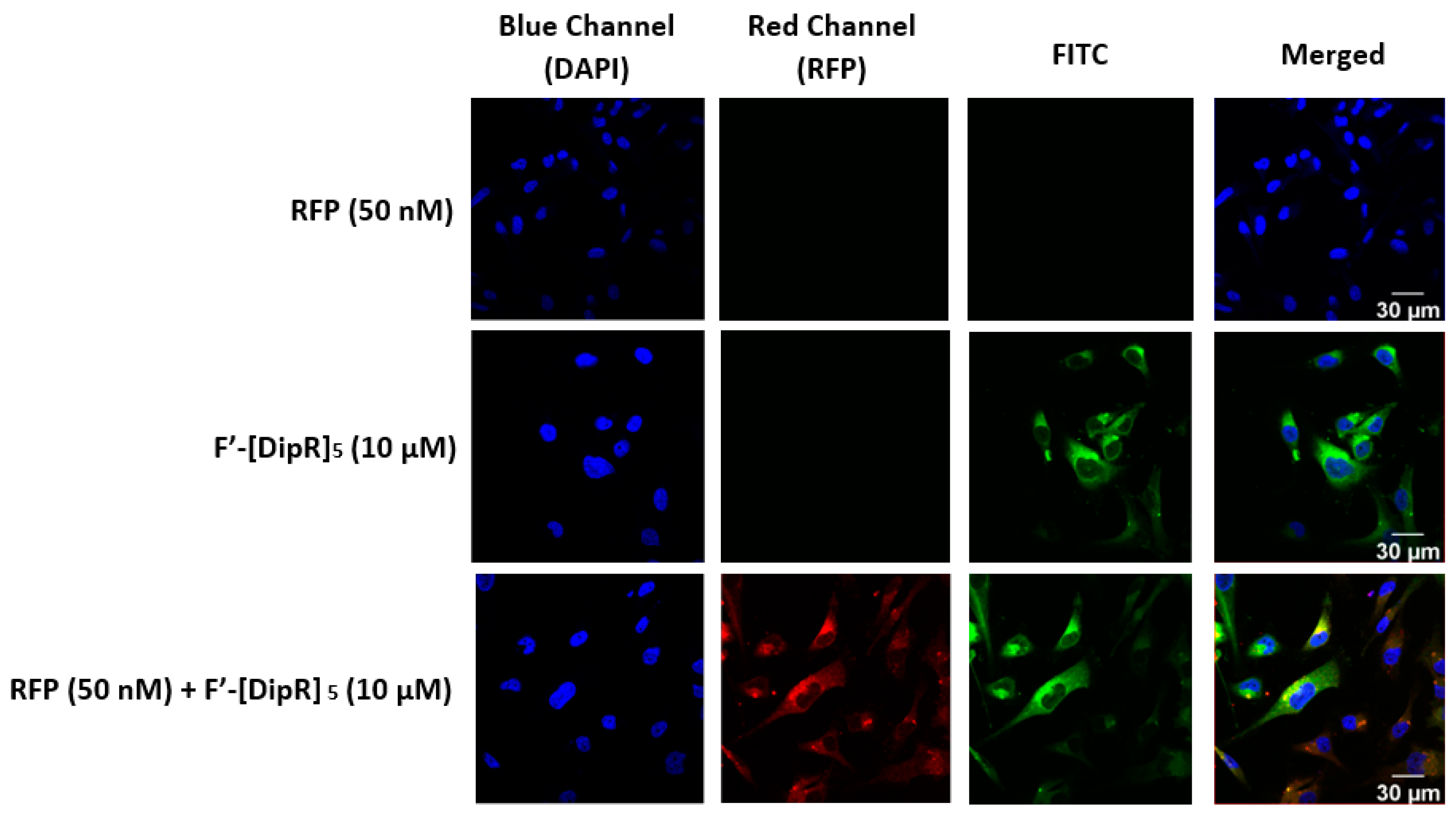
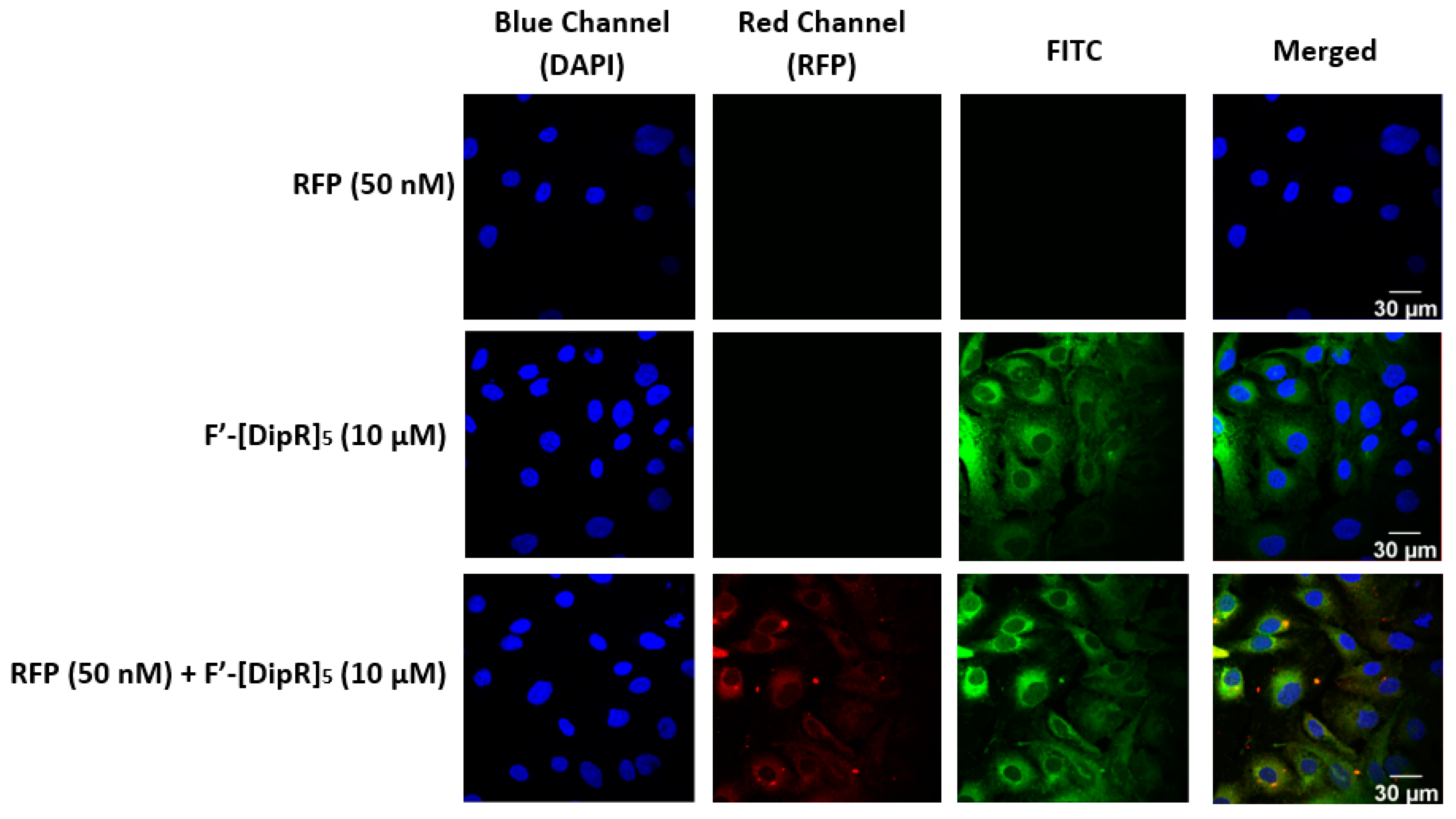
2.3. Cellular Internalization Using Confocal Microscopy
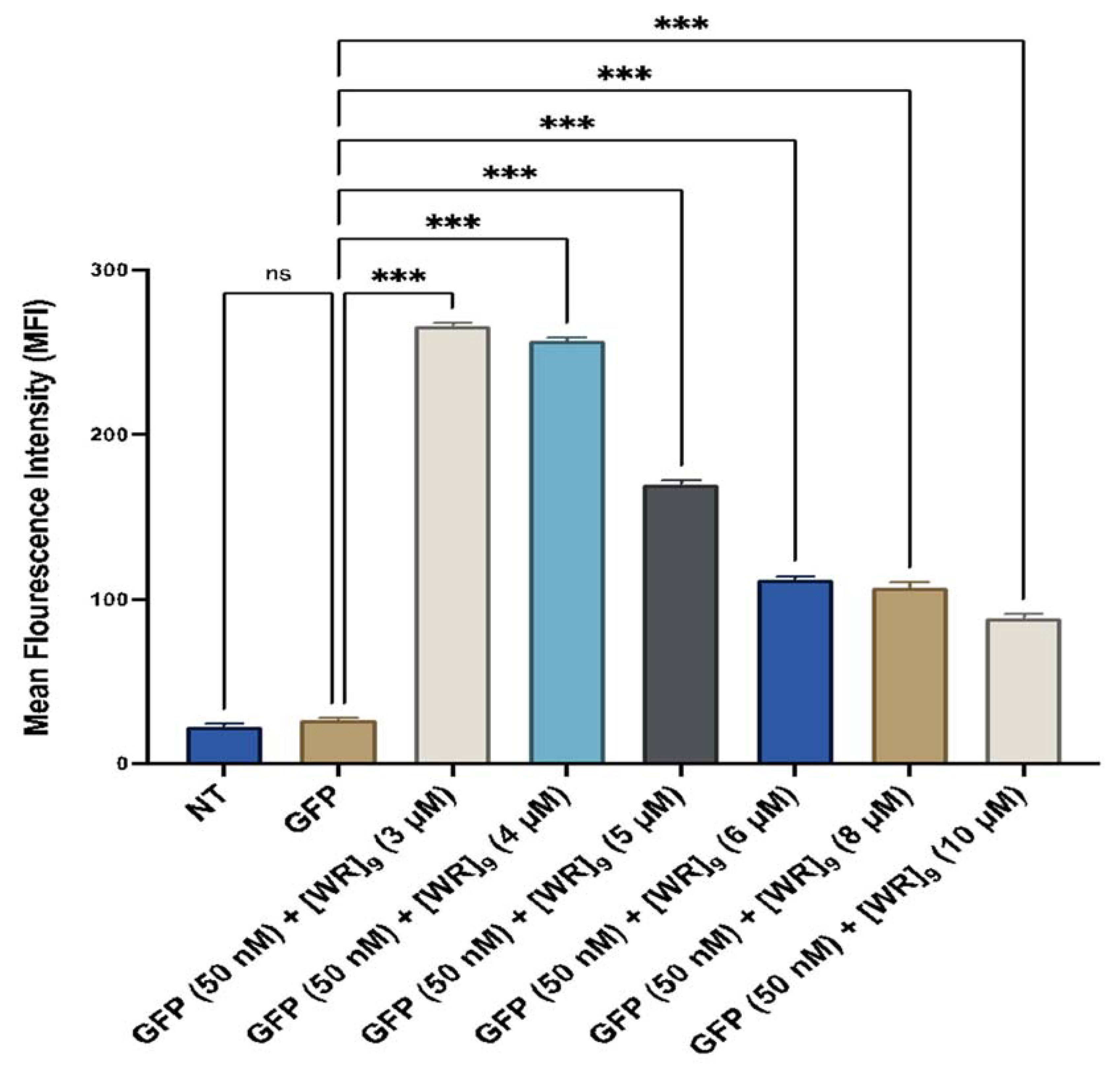
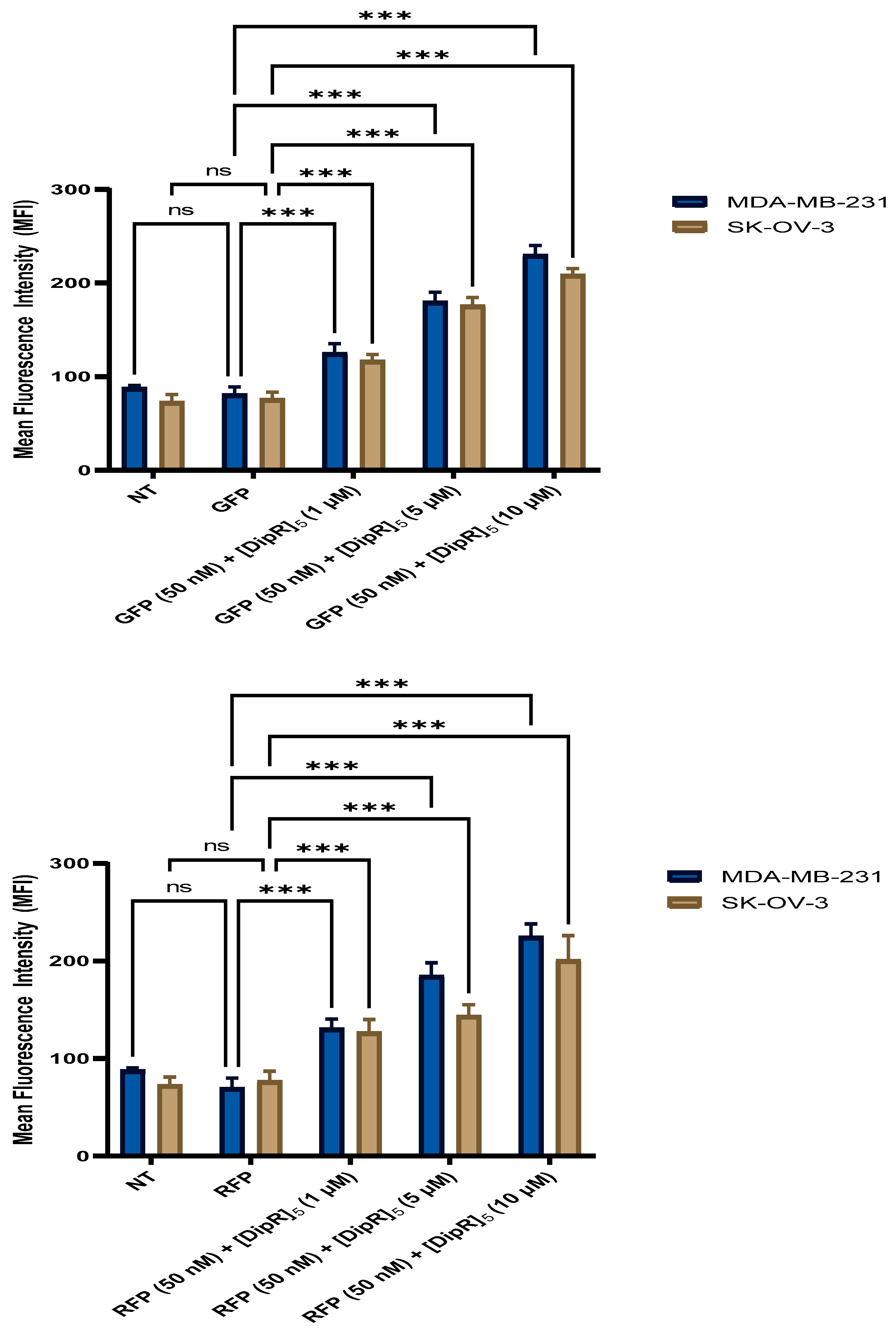
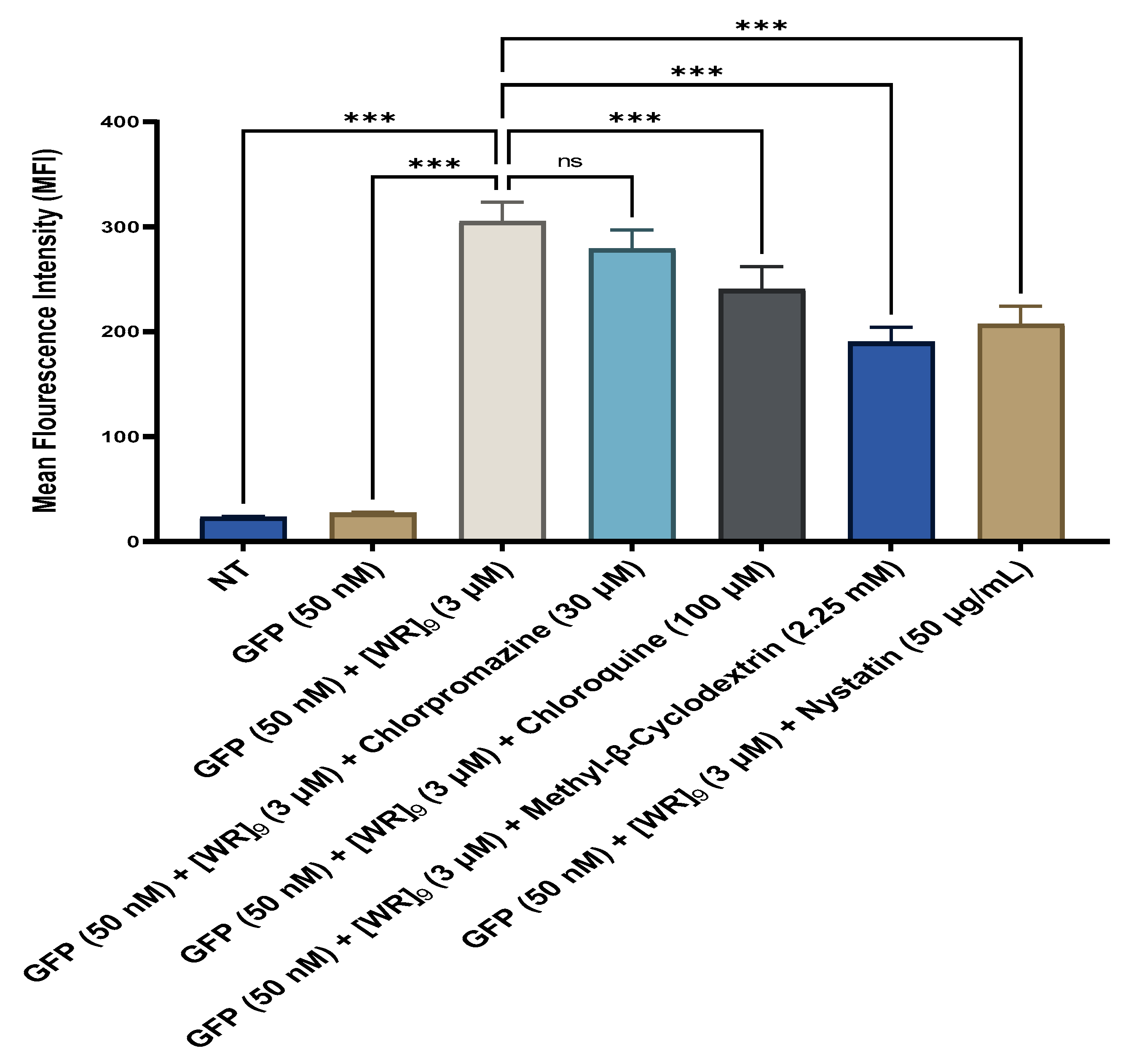
2.4. Fluorescent-Assisted Cell Sorting (FACS)
3. Materials and Methods
3.1. Materials
3.2. Synthesis of Peptides
3.3. Cell Culture and Cytotoxicity Assay of [WR]9-Protein Physical Mixture and [DipR]5-Protein Physical Mixture
3.4. Confocal Microscopy
3.5. Cellular Internalization and Mechanistic Studies (Flow Cytometry)
3.6. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pisal, D.S.; Kosloski, M.P.; Balu-Iyer, S.V. Delivery of therapeutic proteins. J. Pharm. Sci. 2010, 99, 2557–2575. [Google Scholar] [CrossRef] [Green Version]
- Leader, B.; Baca, Q.J.; Golan, D.E. Protein therapeutics: A summary and pharmacological classification. Nat. Rev. Drug Discov. 2008, 7, 21–39. [Google Scholar] [CrossRef]
- Mohanty, S.; Panda, S.; Devadharshini, U.; Paul, S. Proteins and their functionalization for finding therapeutic avenues in cancer: Current status and future prospective. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 188862. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Shumate, A.; Pertea, G.; Varabyou, A.; Breitwieser, F.P.; Chang, Y.C.; Madugundu, A.K.; Pandey, A.; Salzberg, S.L. CHESS: A new human gene catalog curated from thousands of large-scale RNA sequencing experiments reveals extensive transcriptional noise. Genome Biol. 2018, 19, 208. [Google Scholar] [CrossRef]
- Piovesan, A.; Antonaros, F.; Vitale, L.; Strippoli, P.; Pelleri, M.C.; Caracausi, M. Human protein-coding genes and gene feature statistics in 2019. BMC Res. Notes 2019, 12, 315. [Google Scholar] [CrossRef] [Green Version]
- Park, B.H.; Levitt, M. The complexity and accuracy of discrete state models of protein structure. J. Mol. Biol. 1995, 249, 493–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berkowitz, S.A.; Houde, D.J. The complexity of protein structure and the challenges it poses in developing biopharmaceuticals. In Biophysical Characterization of Proteins in Developing Biopharmaceuticals; Elsevier: Amsterdam, The Netherlands, 2020; Volume 361, pp. 3–26. [Google Scholar]
- Lagassé, H.A.; Alexaki, A.; Simhadri, V.L.; Katagiri, N.H.; Jankowski, W.; Sauna, Z.E.; Kimchi-Sarfaty, C. Recent advances in (therapeutic protein) drug development. F1000Research 2017, 6, 113. [Google Scholar] [CrossRef] [Green Version]
- Krejsa, C.; Rogge, M.; Sadee, W. Protein therapeutics: New applications for pharmacogenetics. Nat. Rev. Drug Discov. 2006, 5, 507–521. [Google Scholar] [CrossRef] [PubMed]
- De Groot, A.S.; Scott, D.W. Immunogenicity of protein therapeutics. Trends Immunol. 2007, 28, 482–490. [Google Scholar] [CrossRef]
- Chirino, A.J.; Ary, M.L.; Marshall, S.A. Minimizing the immunogenicity of protein therapeutics. Drug Discov. Today 2004, 9, 82–90. [Google Scholar] [CrossRef]
- Baker, M.; Reynolds, H.M.; Lumicisi, B.; Bryson, C.J. Immunogenicity of protein therapeutics: The key causes, consequences and challenges. Self/No Self 2010, 1, 314–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.R.; Kundu, S.K.; Nam, J.S.; Sharma, G.; Priya Doss, C.G.; Lee, S.S.; Chakraborty, C. Next generation delivery system for proteins and genes of therapeutic purpose: Why and how? Biomed. Res. Int. 2014, 2014, 327950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usmani, S.S.; Bedi, G.; Samuel, J.S.; Singh, S.; Kalra, S.; Kumar, P.; Ahuja, A.A.; Sharma, M.; Gautam, A.; Raghava, G.P.S. THPdb: Database of FDA-approved peptide and protein therapeutics. PLoS ONE 2017, 12, e0181748. [Google Scholar] [CrossRef] [Green Version]
- Mahmood, I.; Green, M.D. Pharmacokinetic and pharmacodynamic considerations in the development of therapeutic proteins. Clin. Pharmacokinet. 2005, 44, 331–347. [Google Scholar] [CrossRef]
- Vugmeyster, Y.; Xu, X.; Theil, F.P.; Khawli, L.A.; Leach, M.W. Pharmacokinetics and toxicology of therapeutic proteins: Advances and challenges. World J. Biol. Chem. 2012, 3, 73. [Google Scholar] [CrossRef]
- Conner, K.P.; Devanaboyina, S.C.; Thomas, V.A.; Rock, D.A. The biodistribution of therapeutic proteins: Mechanism, implications for pharmacokinetics, and methods of evaluation. Pharmacol. Ther. 2020, 212, 107574. [Google Scholar] [CrossRef] [PubMed]
- Solá, R.J.; Griebenow, K. Glycosylation of therapeutic proteins. BioDrugs 2010, 24, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Zaman, R.; Islam, R.A.; Ibnat, N.; Othman, I.; Zaini, A.; Lee, C.Y.; Chowdhury, E.H. Current strategies in extending half–lives of therapeutic proteins. J. Control. Release 2019, 301, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Ishii-Watabe, A.; Tada, M.; Kobayashi, T.; Kanayasu-Toyoda, T.; Kawanishi, T.; Yamaguchi, T. Importance of neonatal FcR in regulating the serum half-life of therapeutic proteins containing the Fc domain of human IgG1: A comparative study of the affinity of monoclonal antibodies and Fc-fusion proteins to human neonatal FcR. J. Immunol. 2010, 184, 1968–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jevševar, S.; Kunstelj, M.; Porekar, V.G. PEGylation of therapeutic proteins. Biotechnol. J. Healthc. Nutr. Technol. 2010, 5, 113–128. [Google Scholar]
- Wang, F.; Wang, Y.; Zhang, X.; Zhang, W.; Guo, S.; Jin, F. Recent progress of cell-penetrating peptides as new carriers for intracellular cargo delivery. J. Control. Release 2014, 174, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.D.; Luo, L.J.; Yang, C.J.; Lai, J.Y. Highly retina-permeating and long-acting resveratrol/metformin nanotherapeutics for enhanced treatment of macular degeneration. ACS Nano. 2023, 17, 168–183. [Google Scholar] [CrossRef] [PubMed]
- Park, S.E.; Sajid, M.I.; Parang, K.; Tiwari, R.K. Cyclic cell-penetrating peptides as efficient intracellular drug delivery tools. Mol Pharm. 2019, 16, 3727–3743. [Google Scholar] [CrossRef] [PubMed]
- Böhmová, E.; Machová, D.; Pechar, M.; Pola, R.; Venclíková, K.; Janoušková, O.; Etrych, T. Cell-penetrating peptides: A useful tool for the delivery of various cargoes into cells. Physiol. Res. 2018, 67, S267–S279. [Google Scholar] [CrossRef]
- El-Andaloussi, S.; Holm, T.; Langel, U. Cell-penetrating peptides: Mechanisms and applications. Curr. Pharm. Des. 2005, 11, 3597–3611. [Google Scholar] [CrossRef] [PubMed]
- Drin, G.; Cottin, S.; Blanc, E.; Rees, A.R.; Temsamani, J. Studies on the internalization mechanism of cationic cell-penetrating peptides. J. Biol. Chem. 2003, 278, 31192–31201. [Google Scholar] [CrossRef] [Green Version]
- Patel, L.N.; Zaro, J.L.; Shen, W.C. Cell penetrating peptides: Intracellular pathways and pharmaceutical perspectives. Pharm. Res. 2007, 24, 1977–1992. [Google Scholar] [CrossRef]
- Lundberg, P.; Langel, Ü. A brief introduction to cell-penetrating peptides. J. Mol. Recognit. 2003, 16, 227–233. [Google Scholar] [CrossRef]
- Sandvig, K.; Pust, S.; Skotland, T.; van Deurs, B. Clathrin-independent endocytosis: Mechanisms and function. Curr. Opin. Cell Biol. 2011, 23, 413–420. [Google Scholar] [CrossRef]
- Hansen, C.G.; Nichols, B.J. Molecular mechanisms of clathrin-independent endocytosis. J. Cell Sci. 2009, 122, 1713–1721. [Google Scholar] [CrossRef] [Green Version]
- Daly, C.; Sugimori, M.; Moreira, J.E.; Ziff, E.B.; Llinás, R. Synaptophysin regulates clathrin-independent endocytosis of synaptic vesicles. Proc. Natl. Acad. Sci. USA 2000, 97, 6120–6125. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.G.; Sayers, E.J.; He, L.; Narayan, R.; Williams, T.L.; Mills, E.M.; Tsai, Y.H. Cell-penetrating peptide sequence and modification dependent uptake and subcellular distribution of green fluorescent protein in different cell lines. Sci. Rep. 2019, 9, 6298. [Google Scholar] [CrossRef] [Green Version]
- Nischan, N.; Herce, H.D.; Natale, F.; Bohlke, N.; Budisa, N.; Cardoso, M.C.; Hackenberger, C.P. Covalent attachment of cyclic TAT peptides to GFP results in protein delivery into live cells with immediate bioavailability. Angew. Chem. Int. Ed. 2015, 54, 1950–1953. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Nanasato, Y.; Omura, K.; Endoh, K.; Kawano, T.; Iwasaki, T. Direct protein delivery into intact plant cells using polyhistidine peptides. Biosci. Biotechnol. Biochem. 2021, 85, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.F.L.; Wallabregue, A.L.D.; Franz, L.; Hackenberger, C.P.R. Targeted subcellular protein delivery using cleavable cyclic cell-penetrating peptides. Bioconjug. Chem. 2019, 30, 400–404. [Google Scholar] [CrossRef] [PubMed]
- Nasrolahi Shirazi, A.; Tiwari, R.K.; Oh, D.; Banerjee, A.; Yadav, A.; Parang, K. Efficient delivery of cell impermeable phosphopeptides by a cyclic peptide amphiphile containing tryptophan and arginine. Mol. Pharm. 2013, 10, 2008–2020. [Google Scholar] [CrossRef] [Green Version]
- Mandal, D.; Nasrolahi Shirazi, A.; Parang, K. Cell-penetrating homochiral cyclic peptides as nuclear-targeting molecular transporters. Angew. Chem. Int. Ed. 2011, 50, 9633–9637. [Google Scholar] [CrossRef]
- Hanna, S.E.; Mozaffari, S.; Tiwari, R.K.; Parang, K. Comparative molecular transporter efficiency of cyclic peptides containing tryptophan and arginine residues. ACS Omega 2018, 3, 16281–16291. [Google Scholar] [CrossRef]
- El-Sayed, A.; Futaki, S.; Harashima, H. Delivery of macromolecules using arginine-rich cell-penetrating peptides: Ways to overcome endosomal entrapment. AAPS J. 2009, 11, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Wender, P.A.; Galliher, W.C.; Goun, E.A.; Jones, L.R.; Pillow, T.H. The design of guanidinium-rich transporters and their internalization mechanisms. Adv. Drug Deliv. Rev. 2008, 60, 452–472. [Google Scholar] [CrossRef] [Green Version]
- Wadia, J.S.; Dowdy, S.F. Transmembrane delivery of protein and peptide drugs by TAT-mediated transduction in the treatment of cancer. Adv. Drug Deliv. Rev. 2005, 57, 579–596. [Google Scholar] [CrossRef]
- Mitchell, D.J.; Steinman, L.; Kim, D.T.; Fathman, C.G.; Rothbard, J.B. Polyarginine enters cells more efficiently than other polycationic homopolymers. J. Pept. Res. 2000, 56, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.; Schwarze, S.R.; Mermelstein, S.J.; Waksman, G.; Dowdy, S.F. Synthetic protein transduction domains: Enhanced transduction potential in vitro and in vivo. Cancer Res. 2001, 61, 474–477. [Google Scholar] [PubMed]
- Kim, L.; Lohan, S.; Moreno, J.; Zoghebi, K.; Tiwari, R.K.; Parang, K. Cyclic and linear peptides containing alternate WW and RR residues as molecular cargo delivery tools. Mol. Pharm. 2023, 20, 341–356. [Google Scholar] [CrossRef]
- Khayyatnejad Shoushtari, S.; Zoghebi, K.; Sajid, M.I.; Tiwari, R.; Parang, K. Hybrid cyclic-linear cell-penetrating peptides containing alternative positively charged and hydrophobic residues as molecular transporters. Mol. Pharm. 2021, 18, 3909–3919. [Google Scholar] [CrossRef] [PubMed]
- Mozaffari, S.; Salehi, D.; Mahdipoor, P.; Beuttler, R.; Tiwari, R.; Montazeri Aliabadi, H.; Parang, K. Design and application of hybrid cyclic-linear peptide-doxorubicin conjugates as a strategy to overcome doxorubicin resistance and toxicity. Eur. J. Med. Chem. 2021, 226, 113836. [Google Scholar] [CrossRef]
- Mozaffari, S.; Bousoik, E.; Amirrad, F.; Lamboy, R.; Coyle, M.; Hall, R.; Alasmari, A.; Mahdipoor, P.; Parang, K.; Montazeri Aliabadi, H. Amphiphilic peptides for efficient siRNA delivery. Polymers 2019, 11, 703. [Google Scholar] [CrossRef] [Green Version]
- Salehi, D.; Mozaffari, S.; Lohan, S.; Mandal, D.; Zoghebi, K.; Tiwari, R.K.; Parang, K. Amphiphilic cell-penetrating peptides containing natural and unnatural amino acids as drug delivery agents. Cells 2022, 11, 1156. [Google Scholar] [CrossRef]
- Gambotto, A.; Dworacki, G.; Cicinnati, V.; Kenniston, T.; Steitz, J.; Tüting, T.; DeLeo, A.B. Immunogenicity of enhanced green fluorescent protein (EGFP) in BALB/c mice: Identification of an H2-Kd-restricted CTL epitope. Gene Ther. 2000, 7, 2036–2040. [Google Scholar] [CrossRef] [Green Version]
- Stripecke, R.; del Carmen Villacres, M.; Skelton, D.C.; Satake, N.; Halene, S.; Kohn, D.B. Immune response to green fluorescent protein: Implications for gene therapy. Gene Ther. 1999, 6, 1305–1312. [Google Scholar] [CrossRef]
- Zoghebi, K.; Aliabadi, H.M.; Tiwari, R.K.; Parang, K. [(WR)8WKβA]-doxorubicin conjugate: A delivery system to overcome multi-drug resistance against doxorubicin. Cells 2022, 11, 301. [Google Scholar] [CrossRef]
- Downs, J.A.; Lowndes, N.F.; Jackson, S.P. A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature 2000, 408, 1001–1004. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, L.; Erdjument-Bromage, H.; Vidal, M.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature 2004, 431, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Jiao, C.; Chen, Y.; Chen, L.; Li, X.; Liu, X.; Yang, D.; Zhao, J. Characteristic and antibacterial effect of a histone H2A and its preliminary roles in extracellular traps in manila clam Ruditapes philippinarum. Fish Shellfish Immunol. 2022, 131, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, J.M.; Kemp, G.D.; Molle, M.G.; Smith, V.J. Anti-microbial properties of histone H2A from skin secretions of rainbow trout, Oncorhynchus mykiss. Biochem. J. 2002, 368, 611–620. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, H.; Iwamuro, S. Potential roles of histones in host defense as antimicrobial agents. Infect. Disord. Drug Targets (Former. Curr. Drug Targets-Infect. Disord.) 2008, 8, 195–205. [Google Scholar] [CrossRef]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Preissner, K.T. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: A predominant role of histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef]
- Song, Y.; Kadiyala, U.; Weerappuli, P.; Valdez, J.J.; Yalavarthi, S.; Louttit, C.; Takayama, S. Antimicrobial Microwebs of DNA–Histone Inspired from Neutrophil Extracellular Traps. Adv. Mater. 2019, 31, 1807436. [Google Scholar] [CrossRef]
- Doolin, T.; Amir, H.M.; Duong, L.; Rosenzweig, R.; Urban, L.A.; Bosch, M.; Siryaporn, A. Mammalian histones facilitate antimicrobial synergy by disrupting the bacterial proton gradient and chromosome organization. Nat. Commun. 2020, 11, 3888. [Google Scholar] [CrossRef]
- Darwish, S.; Mozaffari, S.; Parang, K.; Tiwari, R. Cyclic peptide conjugate of curcumin and doxorubicin as an anticancer agent. Tetrahedron Lett. 2017, 58, 4617–4622. [Google Scholar] [CrossRef] [Green Version]
- Darwish, S.; Sadeghiani, N.; Fong, S.; Mozaffari, S.; Hamidi, P.; Withana, T.; Yang, S.; Tiwari, R.K.; Parang, K. Synthesis and antiproliferative activities of doxorubicin thiol conjugates and doxorubicin-SS-cyclic peptide. Eur. J. Med. Chem. 2019, 161, 594–606. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, N.S.; Shirazi, A.N.; Sajid, M.I.; Park, S.E.; Parang, K.; Tiwari, R.K. Synthesis and antiproliferative activities of conjugates of paclitaxel and camptothecin with a cyclic cell-penetrating peptide. Molecules 2019, 24, 1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]



















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno, J.; Zoghebi, K.; Salehi, D.; Kim, L.; Shoushtari, S.K.; Tiwari, R.K.; Parang, K. Amphiphilic Cell-Penetrating Peptides Containing Arginine and Hydrophobic Residues as Protein Delivery Agents. Pharmaceuticals 2023, 16, 469. https://doi.org/10.3390/ph16030469
Moreno J, Zoghebi K, Salehi D, Kim L, Shoushtari SK, Tiwari RK, Parang K. Amphiphilic Cell-Penetrating Peptides Containing Arginine and Hydrophobic Residues as Protein Delivery Agents. Pharmaceuticals. 2023; 16(3):469. https://doi.org/10.3390/ph16030469
Chicago/Turabian StyleMoreno, Jonathan, Khalid Zoghebi, David Salehi, Lois Kim, Sorour Khayyatnejad Shoushtari, Rakesh K. Tiwari, and Keykavous Parang. 2023. "Amphiphilic Cell-Penetrating Peptides Containing Arginine and Hydrophobic Residues as Protein Delivery Agents" Pharmaceuticals 16, no. 3: 469. https://doi.org/10.3390/ph16030469