The reagents and solvents used in this article were chemically pure or analytically pure, and could be used directly without purification. The silica gel used for flash column chromatography and thin layer chromatography was purchased from Qingdao Yumingyuan Silicone Chemical Factory (Qingdao, China). The NMR spectra was recorded by a Bruker AV-400/600 nuclear magnetic resonance instrument, in which TMS was used as the internal standard, the chemical shift is expressed in ppm(δ), and the coupling constant (J) is expressed in Hertz (Hz). The HRMS spectrum was determined by an Agilent Accurate Mass Q-TOF 6530 mass spectrometer (Agilent, Santa Clara, CA, USA).
5.1. Chemistry
General Procedure A was used for the synthesis of 11a–l.
The synthesis of N-(5-bromo-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide (4). To a solution of 5-bromo-2-methoxypyridin-3-amine (3) (4.04 g, 20 mmol) in pyridine (anhydrous, 50 mL), 2,4-difluorobenzenesulfonyl chloride (3.22 mL, 24 mmol) was added dropwise. Then it was stirred at room temperature for 24 h. The solvent was evaporated, followed by addition of H2O (100 mL), and stirring for 1 h. The precipitate was filtered, washed with hexane, and dried to give a brown yellow solid (4) (6.91 g, 91%).
The synthesis of 2,4-difluoro-N-(2-methoxy-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-3-yl)benzenesulfonamide (5). PdCl2(dppf)•DCM (204 mg, 0.25 mmol) was added to a mixture of bromo 4 (1.99 g, 5 mmol), KOAc (1.23 g, 7.5 mmol), bis(pinacolato)diborane (1.65 g, 4.5 mmol), and 1,4-dioxane (anhydrous). It was refluxed for 4 h under argon. The solvent was evaporated, diluted with DCM, and washed with H2O. The separated organic layer was evaporated in vacuum and the residue was purified by flash column chromatography (Hexane: ethyl acetate = 3:1) to obtain an off-white solid (5) (2.00 g, 94%). 1H NMR (400 MHz, DMSO-d6) δ 10.15 (s, 1H), 8.20 (d, J = 1.8 Hz, 1H), 7.71 (d, J = 1.8 Hz, 1H), 7.68 (dd, J = 8.6, 6.3 Hz, 1H), 7.61–7.49 (m, 1H), 7.29–7.11 (m, 1H), 3.62 (s, 3H), 1.29 (s, 12H).
The synthesis of ethyl 3-amino-6-bromobenzo[b]thiophene-2-carboxylate (7). Ethyl 2-mercaptoacetate (2.20 mL, 20 mmol) was added to a mixture of 4-bromo-2-fluorobenzonitrile (6) (4.00 g, 20 mmol), DIPEA (3.30 mL, 20 mmol), K2CO3 (2.80 g), and DMF (50 mL). The mixture was stirred at 80 °C for 20 h. It was cooled to room temperature and treated with ice water (50 mL) and stirred for 1 h. The formed precipitate was washed with H2O (20 mL) and filtered to give a faint yellow solid (7) (5.88 g, 98%).1H NMR (400 MHz, DMSO-d6) δ 8.13 (d, J = 1.8 Hz, 1H), 8.07 (d, J = 8.6 Hz, 1H), 7.57 (dd, J = 8.6, 1.8 Hz, 1H), 7.16 (s, 2H), 4.26 (q, J = 7.1 Hz, 2H), 1.29 (t, J = 7.1 Hz, 3H).
The synthesis of 7-bromobenzo[4,5]thieno[3,2-d]pyrimidin-4-ol (8). A mixture of the ester 7 (4.50 g, 15 mmol) and formamidine acetate (1.56 g 15 mmol) in formamide (50 mL) was stirred at 150 °C for 45 min. Then an additional portion of formamidine acetate (1.56 g, 15 mmol) was added every 45 min to the mixture. The total formamidine acetate required was 12.48 g, 120 mmol, and the mixture was stirred for 6 h. It was cooled, treated with ice water (100 mL), and stirred for 1 h. The precipitate was filtered, washed with H2O, and dried to obtain an ashy solid (3.99 g, 95%).1H NMR (400 MHz, DMSO-d6) δ 12.94 (s, 1H), 8.50 (d, J = 1.8 Hz, 1H), 8.35 (s, 1H), 8.15 (d, J = 8.5 Hz, 1H), 7.75 (dd, J = 8.5, 1.8 Hz, 1H).
The synthesis of 7-bromo-4-chlorobenzo[4,5]thieno[3,2-d]pyrimidine (9). A mixture of compound 8 (2.80 g, 10 mmol), phosphorous oxychloride (30 mL) and N,N-dimethylformamide (0.1 mL) was stirred under argon at 90 °C for 20 h. The phosphorous oxychloride was removed in vacuo, and the residue was diluted with DCM and treated with ice water (10 mL). Then it was carefully neutralized with NaHCO3, and the formed crude was filtered, washed with H2O, and purified with flash column chromatography (Hexane: ethyl acetate = 1:1) to give a yellow solid (2.47 g, 83%).
The synthesis of 4-(7-bromobenzo[4,5]thieno[3,2-d]pyrimidin-4-yl)morpholine (10a). Morpholine (146 μL, 1 mmol) was added to a mixture of chloride 9 (75 mg, 0.25 mmol) and Et3N (70 μL, 0.5 mmol) in THF (5 mL) under an ice cooling bath. Then, the mixture was gradually raised to room temperature, and stirred for additional 4 h. It was concentrated in vacuo and the residue was stirred in ethyl acetate (5 mL) and H2O (5 mL). The organic layer was separated and dried. The solvent was evaporated in vacuum to afford 10a as an ashy solid (89 mg, 88%), which was used without further purification.
10b−l were prepared by a similar procedure described for the synthesis of
10a. The synthesis of 2,4-difluoro-
N-(2-methoxy-5-(4-morpholinobenzo[4,5]thieno[3,2-
d]pyrimidin-7-yl)pyridin-3-yl)benzenesulfonamide (
11a). Under argon protection, PdCl
2(dppf)•DCM (21 mg, 0.025 mmol) was added to a mixture of bromo compound
10a (89 mg, 0.22 mmol), borate
5 (213 mg, 0.5 mmol), K
2CO
3 (52 mg, 0.375 mmol) in 1,4-dioxane (10 mL), and H
2O (2 mL). The mixture was refluxed for 4 h. Then it was concentrated and purified by flash column chromatography (hexane: ethyl acetate = 1:1) to obtain the coupled
11a as a white solid (94 mg, 75%).
1H NMR (400 MHz, DMSO-
d6) δ 10.35 (s, 1H), 8.69 (s, 1H), 8.47 (d, J = 2.3 Hz, 1H), 8.47–8.26 (m, 2H), 8.04 (d, J = 2.3 Hz, 1H), 7.85 (dd, J = 8.3, 1.7 Hz, 1H), 7.78 (td, J = 8.6, 6.3 Hz, 1H), 7.58 (ddd, J = 10.6, 9.2, 2.5 Hz, 1H), 7.21 (tt, J = 8.7, 1.6 Hz, 1H), 3.95 (dd, J = 5.7, 4.0 Hz, 4H), 3.79 (dd, J = 5.7, 3.9 Hz, 4H), 3.67 (s, 3H).
13C NMR (100 MHz, DMSO-
d6) δ 170.80, 164.35, 161.21, 158.65, 158.52, 158.41, 158.28, 157.18, 154.80, 143.31, 140.41, 138.47, 134.87, 132.99, 132.40, 132.30, 129.39, 125.64, 125.49, 124.58, 124.29, 120.91, 120.21, 114.30, 112.40, 112.21, 106.53, 106.28, 106.02, 66.46, 60.21, 53.94, 46.50, 21.22, 14.55.
1H and
13C NMR spectra were shown in
Figure S1. HRMS (ESI)
m/
z: [M + H]
+ calcd for C
26H
22F
2N
5O
4S
2, 570.1003; found, 570.1069.
11b−l were prepared by a similar procedure described for the synthesis of 11a.
N-(2-methoxy-5-(4-(4-methylpiperidin-1-yl)benzo[4,5]thieno[3,2-d]pyrimidin-7-yl)pyridin-3-yl) 2,4-difluorobenzenesulfonamide (
11b): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (99 mg, 68%).
1H NMR (400 MHz, DMSO-
d6) δ 10.34 (s, 1H), 8.63 (s, 1H), 8.47 (d,
J = 2.3 Hz, 1H), 8.40 (d,
J = 1.6 Hz, 1H), 8.38 (d,
J = 8.3 Hz, 1H), 8.03 (d,
J = 2.4 Hz, 1H), 7.83 (dd,
J = 8.3, 1.6 Hz, 1H), 7.81–7.74 (m, 1H), 7.58 (ddd,
J = 10.5, 9.2, 2.5 Hz, 1H), 7.21 (td,
J = 8.6, 2.7 Hz, 1H), 4.71 (d,
J = 13.3 Hz, 2H), 3.67 (s, 3H), 3.26–3.15 (m, 2H), 1.80 (d,
J = 11.7 Hz, 3H), 1.39–1.10 (m, 2H), 0.94 (d,
J = 6.1 Hz, 3H).
13C NMR (100 MHz, DMSO-
d6) δ 166.88, 166.76, 164.35, 164.24, 161.21, 161.07, 158.65, 158.51, 158.23, 157.87, 156.95, 154.95, 143.23, 140.26, 138.28, 134.77, 133.23, 132.41, 132.30, 129.44, 125.64, 125.61, 125.50, 125.46, 124.48, 124.23, 120.87, 120.23, 113.91, 112.44, 112.40, 112.22, 112.18, 106.53, 106.27, 106.02, 60.21, 53.94, 47.47, 46.60, 34.33, 33.34, 31.00, 22.03, 21.22, 14.55.
1H and
13C NMR spectra were shown in
Figure S2. HRMS (ESI)
m/
z: [M – H]
– calcd for C
28H
24F
2N
5O
3S
2, 580.1367; found, 580.1343.
N-(5-(4-((2-hydroxyethyl)amino)benzo[4,5]thieno[3,2-d]pyrimidin-7-yl)-2-methoxypyridin-3-yl) -2,4-difluoro-benzenesulfonamide (
11c): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (80 mg, 59%).
1H NMR (400 MHz, DMSO-
d6) δ 10.34 (s, 1H), 8.60 (s, 1H), 8.49 (d,
J = 2.4 Hz, 1H), 8.47 (d,
J = 1.5 Hz, 1H), 8.35 (d,
J = 8.3 Hz, 1H), 8.05 (d,
J = 2.3 Hz, 1H), 7.97–7.91 (m, 1H), 7.83 (dd,
J = 8.3, 1.6 Hz, 1H), 7.77 (td,
J = 8.6, 6.3 Hz, 1H), 7.58 (ddd,
J = 10.4, 9.2, 2.5 Hz, 1H), 7.21 (td,
J = 8.5, 2.5 Hz, 1H), 4.91–4.58 (m, 1H), 3.66 (s, 3H), 3.65–3.61 (m, 4H).
13C NMR (100 MHz, DMSO-
d6) δ 166.80 (d,
J = 12.1 Hz), 164.27 (d,
J = 11.7 Hz), 161.15 (d,
J = 13.4 Hz), 158.59 (d,
J = 13.6 Hz), 158.27, 157.64, 155.47, 154.90, 143.24, 140.83, 137.77, 135.03, 133.74, 132.34 (d,
J = 10.8 Hz), 129.51, 125.54, 124.12 (d,
J = 25.9 Hz), 121.78, 120.22, 115.22, 112.29 (d,
J = 22.4 Hz), 106.26 (t,
J = 26.1 Hz), 59.85, 53.92, 43.66.
1H and
13C NMR spectra were shown in
Figure S3. HRMS (ESI)
m/
z: [M + H]
+ calcd for C
24H
20F
2N
5O
4S
2, 544.0847; found, 544.0919.
N-(5-(4-(cyclopropylamino)benzo[4,5]thieno[3,2-d]pyrimidin-7-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide (
11d): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (105 mg, 78%).
1H NMR (400 MHz, DMSO-
d6) δ 10.33 (s, 1H), 8.63 (s, 1H), 8.49 (d,
J = 2.3 Hz, 1H), 8.45 (d,
J = 1.6 Hz, 1H), 8.36 (d,
J = 8.3 Hz, 1H), 8.09 (d,
J = 3.4 Hz, 1H), 8.05 (d,
J = 2.4 Hz, 1H), 7.83 (dd,
J = 8.3, 1.6 Hz, 1H), 7.77 (td,
J = 8.6, 6.3 Hz, 1H), 7.58 (ddd,
J = 10.5, 9.2, 2.5 Hz, 1H), 7.24–7.17 (m, 1H), 3.66 (s, 3H), 3.04 (tt,
J = 7.0, 3.6 Hz, 1H), 0.83 (td,
J = 7.0, 4.7 Hz, 2H), 0.71–0.65 (m, 2H).
13C NMR (100 MHz, DMSO-
d6) δ 166.80 (d,
J = 11.4 Hz), 164.28 (d,
J = 11.8 Hz), 161.15 (d,
J = 13.3 Hz), 158.92, 158.59 (d,
J = 13.1 Hz), 158.27, 155.48, 155.19, 143.26, 141.05, 137.84, 135.02, 133.55, 132.35 (d,
J = 11.0 Hz), 129.51, 125.59 (d,
J = 10.6 Hz), 124.12 (d,
J = 20.4 Hz), 121.62, 120.20, 114.85, 112.28 (d,
J = 18.8 Hz), 106.26 (t,
J = 26.1 Hz), 53.92.
1H and
13C NMR spectra were shown in
Figure S4. HRMS (ESI)
m/
z: [M – H]
– calcd for C
25H
18F
2N
5O
3S
2, 538.0897; found, 538.0857.
N-(2-methoxy-5-(4-((3-methoxypropyl)amino)benzo[4,5]thieno[3,2-d]pyrimidin-7-yl)pyridin-3-yl)-2,4-difluorobenzenesulfonamide (
11e): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (117 mg, 82%).
1H NMR (400 MHz, DMSO-
d6) δ 10.33 (s, 1H), 8.60 (s, 1H), 8.48 (d,
J = 2.4 Hz, 1H), 8.46 (d,
J = 1.6 Hz, 1H), 8.34 (d,
J = 8.3 Hz, 1H), 8.04 (d,
J = 2.4 Hz, 1H), 7.94 (t,
J = 5.5 Hz, 1H), 7.83 (dd,
J = 8.3, 1.6 Hz, 1H), 7.77 (td,
J = 8.6, 6.3 Hz, 1H), 7.57 (ddd,
J = 11.3, 9.2, 2.5 Hz, 1H), 7.24–7.17 (m, 1H), 3.66 (s, 3H), 3.63–3.56 (m, 2H), 3.43 (t,
J = 6.2 Hz, 2H), 3.26 (s, 3H), 1.89 (p,
J = 6.6 Hz, 2H).
13C NMR (100 MHz, DMSO-
d6) δ 166.72, 164.25 (d,
J = 11.7 Hz), 161.15 (d,
J = 13.6 Hz), 158.59 (d,
J = 13.3 Hz), 158.27, 157.52, 155.53, 154.86, 143.15, 140.78, 137.78, 134.93, 133.77, 132.34 (d,
J = 10.8 Hz), 129.51, 125.70, 124.11 (d,
J = 28.2 Hz), 121.76, 120.34, 115.15, 112.26 (d,
J = 19.4 Hz), 106.25 (t,
J = 26.0 Hz), 70.21, 58.40, 53.91.
1H and
13C NMR spectra were shown in
Figure S5. HRMS (ESI)
m/
z: [M – H]
– calcd for C
26H
22F
2N
5O
4S
2, 572.1160; found, 572.1221.
N-(5-(4-((2-aminoethyl)amino)benzo[4,5]thieno[3,2-d]pyrimidin-7-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide (
11f): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (87 mg, 64%).
1H NMR (400 MHz, DMSO-
d6) δ 8.62 (s, 1H), 8.31 (d,
J = 8.3 Hz, 1H), 8.24 (d,
J = 5.0 Hz, 1H), 8.22 (s, 1H), 7.94 (s, 1H), 7.90–7.78 (m, 1H), 7.68 (d,
J = 2.4 Hz, 1H), 7.63 (d,
J = 8.4 Hz, 1H), 7.29 (td,
J = 9.8, 2.6 Hz, 1H), 7.11 (td,
J = 8.5, 2.6 Hz, 1H), 3.82 (q,
J = 6.1 Hz, 2H), 3.77 (s, 3H), 3.11 (t,
J = 6.3 Hz, 2H), 1.89 (s, 2H).
13C NMR (100 MHz, DMSO-
d6) δ 160.46, 158.74 (d,
J = 12.9 Hz), 157.57 (d,
J = 20.6 Hz), 155.22 (d,
J = 9.3 Hz), 140.93, 139.89, 133.00, 131.98 (d,
J = 10.8 Hz), 129.03, 124.16, 123.93, 121.09, 115.38, 111.29 (d,
J = 21.5 Hz), 105.60 (t,
J = 26.5 Hz), 63.26, 60.22, 53.44, 49.01.
1H and
13C NMR spectra were shown in
Figure S6. HRMS (ESI)
m/
z: [M – H]
– calcd for C
24H
19F
2N
6O
3S
2, 543.1006; found, 543.1073.
N-(5-(4-(isopropylamino)benzo[4,5]thieno[3,2-
d]pyrimidin-7-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide (
11g): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (108 mg, 80%).
1H NMR (400 MHz, DMSO-
d6) δ 10.33 (s, 1H), 8.59 (s, 1H), 8.49 (d,
J = 2.4 Hz, 1H), 8.46 (d,
J = 1.6 Hz, 1H), 8.34 (d,
J = 8.3 Hz, 1H), 8.05 (d,
J = 2.4 Hz, 1H), 7.83 (dd,
J = 8.3, 1.6 Hz, 1H), 7.79–7.76 (m, 1H), 7.74 (d,
J = 7.7 Hz, 1H), 7.58 (ddd,
J = 10.6, 9.2, 2.5 Hz, 1H), 7.20 (td,
J = 8.6, 2.5 Hz, 1H), 4.50 (dq,
J = 13.4, 6.7 Hz, 1H), 3.66 (s, 3H), 1.28 (s, 3H), 1.26 (s, 3H).
13C NMR (100 MHz, DMSO-
d6) δ 166.80 (d,
J = 11.6 Hz), 164.27 (d,
J = 11.8 Hz), 161.16 (d,
J = 13.5 Hz), 158.60 (d,
J = 13.5 Hz), 158.28, 156.80, 155.50, 154.96, 143.26, 140.84, 137.73, 135.05, 133.79, 132.34 (d,
J = 10.7 Hz), 129.53, 126.31–125.16 (m), 124.07 (d,
J = 26.0 Hz), 121.73, 120.21, 115.07, 113.36–111.62 (m), 106.25 (t,
J = 26.0 Hz), 53.90, 42.50, 22.69.
1H and
13C NMR spectra were shown in
Figure S7. HRMS (ESI)
m/
z: [M + H]
+ calcd for C
25H
22F
2N
5O
3S
2, 542.1054; found, 542.1122.
N-(5-(4-((2-(dimethylamino)ethyl)amino)benzo[4,5]thieno[3,2-
d]pyrimidin-7-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide (
11h): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (100 mg, 70%).
1H NMR (400 MHz, DMSO-
d6) δ 8.61 (s, 1H), 8.34–8.25 (m, 2H), 8.17 (d,
J = 2.3 Hz, 1H), 7.89 (t,
J = 3.8 Hz, 2H), 7.79 (td,
J = 8.6, 6.5 Hz, 1H), 7.72 (dd,
J = 8.3, 1.6 Hz, 1H), 7.45 (td,
J = 9.8, 2.5 Hz, 1H), 7.16 (td,
J = 8.3, 2.5 Hz, 1H), 3.70 (s, 6H), 2.82 (t,
J = 6.4 Hz, 2H), 2.44 (s, 7H).
13C NMR (100 MHz, DMSO-
d6) δ 163.53 (d,
J = 11.9 Hz), 160.93, 158.51, 157.66 (d,
J = 40.9 Hz), 155.19 (d,
J = 34.8 Hz), 140.67, 139.58, 138.50, 133.37, 132.16 (d,
J = 10.4 Hz), 131.19, 128.99, 127.64, 124.76, 123.97 (d,
J = 27.1 Hz), 121.32, 115.05, 111.81 (d,
J = 24.9 Hz), 105.95 (t,
J = 26.2 Hz), 57.65, 53.65, 44.85, 38.13.
1H and
13C NMR spectra were shown in
Figure S8. HRMS (ESI)
m/
z: [M + H]
+ calcd for C
26H
25F
2N
6O
3S
2, 570.1319; found, 570.1388.
N-(5-(4-(butylamino)benzo[4,5]thieno[3,2-
d]pyrimidin-7-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide (
11i): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (80 mg, 58%).
1H NMR (400 MHz, DMSO-
d6) δ 10.39 (s, 1H), 8.59 (s, 1H), 8.49 (d,
J = 2.3 Hz, 1H), 8.46 (d,
J = 1.6 Hz, 1H), 8.34 (d,
J = 8.3 Hz, 1H), 8.05 (d,
J = 2.4 Hz, 1H), 7.93 (t,
J = 5.6 Hz, 1H), 7.85–7.81 (m, 1H), 7.77 (td,
J = 8.6, 6.3 Hz, 1H), 7.61–7.53 (m, 1H), 7.21 (td,
J = 8.6, 2.5 Hz, 1H), 3.66 (s, 3H), 3.58–3.50 (m, 2H), 1.63 (s, 2H), 1.38 (d,
J = 7.4 Hz, 2H), 0.93 (t,
J = 7.4 Hz, 3H).
13C NMR (100 MHz, DMSO-
d6) δ 166.80 (d,
J = 11.9 Hz), 164.27 (d,
J = 11.7 Hz), 161.15 (d,
J = 13.3 Hz), 158.59 (d,
J = 13.5 Hz), 158.26, 157.53, 155.55, 154.84, 143.22, 140.78, 137.73, 134.97, 133.81, 132.35 (d,
J = 10.9 Hz), 129.53, 126.66–125.11 (m), 124.23, 123.96, 121.76, 120.24, 115.08, 112.27 (dd,
J = 22.1, 3.5 Hz), 106.25 (t,
J = 26.1 Hz), 91.15, 73.99, 70.40, 65.65, 63.19, 60.21, 53.91, 31.45, 20.17, 14.24.
1H and
13C NMR spectra were shown in
Figure S9. HRMS (ESI)
m/
z: [M – H]
– calcd for C
26H
22F
2N
5O
3S
2, 554.1210; found, 554.1178.
N-(2-methoxy-5-(4-((3-morpholinopropyl)amino)benzo[4,5]thieno[3,2-
d]pyrimidin-7-yl)pyridin-3-yl)-2,4-difluorobenzenesulfonamide (
11j): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (92 mg, 59%).
1H NMR (400 MHz, DMSO-
d6) δ 10.38 (s, 1H), 8.60 (s, 1H), 8.46 (t,
J = 2.1 Hz, 2H), 8.34 (d,
J = 8.3 Hz, 1H), 8.03 (d,
J = 2.4 Hz, 1H), 7.97 (t,
J = 5.4 Hz, 1H), 7.82 (dd,
J = 8.4, 1.7 Hz, 1H), 7.80–7.73 (m, 1H), 7.56 (ddd,
J = 11.4, 9.3, 2.5 Hz, 1H), 7.20 (td,
J = 8.5, 2.5 Hz, 1H), 3.67 (s, 3H), 3.61 (s, 2H), 2.44 (d,
J = 7.2 Hz, 3H), 2.41 (d,
J = 4.3 Hz, 3H), 1.82 (s, 1H).
13C NMR (100 MHz, DMSO-
d6) δ 166.68 (d,
J = 11.7 Hz), 164.16 (d,
J = 11.6 Hz), 161.14 (d,
J = 13.6 Hz), 158.58 (d,
J = 13.5 Hz), 158.25, 157.49, 155.56, 154.86, 142.69, 140.70, 137.91, 134.46, 133.75, 132.32 (d,
J = 10.6 Hz), 129.47, 125.92 (d,
J = 14.3 Hz), 124.12 (d,
J = 29.1 Hz), 121.75, 120.93, 115.11, 112.22 (d,
J = 22.3 Hz), 106.21 (t,
J = 26.1 Hz), 67.48, 66.58, 56.62, 53.82 (d,
J = 10.4 Hz), 25.71 (d,
J = 23.9 Hz).
1H and
13C NMR spectra were shown in
Figure S10. HRMS (ESI)
m/
z: [M – H]
– calcd for C
29H
27F
2N
6O
4S
2, 625.1582; found, 625.1555.
N-(5-(4-(2-(dimethylamino)ethoxy)benzo[4,5]thieno[3,2-
d]pyrimidin-7-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide (
11k): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (104 mg, 73%).
1H NMR (400 MHz, DMSO-
d6) δ 8.90 (s, 1H), 8.42 (d,
J = 8.5 Hz, 2H), 8.32 (d,
J = 2.3 Hz, 1H), 7.96 (d,
J = 2.4 Hz, 1H), 7.83 (dd,
J = 8.2, 1.8 Hz, 1H), 7.81–7.76 (m, 1H), 7.51 (d,
J = 2.0 Hz, 1H), 7.19 (td,
J = 8.5, 2.5 Hz, 1H), 4.73 (t,
J = 5.6 Hz, 2H), 3.69 (s, 3H), 2.92 (t,
J = 5.6 Hz, 2H), 2.38 (s, 6H).
13C NMR (100 MHz, DMSO-
d6) δ 166.44 (d,
J = 11.5 Hz), 164.07, 163.93 (d,
J = 11.5 Hz), 161.15, 158.53 (d,
J = 13.1 Hz), 158.16 (d,
J = 5.1 Hz), 155.15, 141.59 (d,
J = 30.1 Hz), 139.17, 132.78 (d,
J = 40.9 Hz), 132.25 (d,
J = 10.7 Hz), 129.01, 126.59 (d,
J = 14.8 Hz), 124.49 (d,
J = 34.5 Hz), 122.59, 121.79, 117.52, 112.09 (d,
J = 21.6 Hz), 106.12 (t,
J = 26.2 Hz), 64.92, 57.37, 53.82, 45.52.
1H and
13C NMR spectra were shown in
Figure S11. HRMS (ESI)
m/
z: [M + H]
+ calcd for C
26H
24F
2N
5O
4S
2, 572.1160; found, 572.1229.
N-(2-methoxy-5-(4-(2-methoxyethoxy)benzo[4,5]thieno[3,2-
d]pyrimidin-7-yl)pyridin-3-yl)-2,4-difluorobenzenesulfonamide (
11l): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (102 mg, 73%).
1H NMR (400 MHz, DMSO-
d6) δ 10.35 (s, 1H), 8.91 (s, 1H), 8.52 (d,
J = 1.6 Hz, 1H), 8.49 (d,
J = 2.3 Hz, 1H), 8.45 (d,
J = 8.3 Hz, 1H), 8.06 (d,
J = 2.4 Hz, 1H), 7.90 (dd,
J = 8.3, 1.6 Hz, 1H), 7.78 (td,
J = 8.6, 6.3 Hz, 1H), 7.58 (ddd,
J = 10.5, 9.2, 2.5 Hz, 1H), 7.21 (td,
J = 8.6, 2.5 Hz, 1H), 4.82–4.66 (m, 2H), 3.87–3.74 (m, 2H), 3.67 (s, 3H), 3.35 (s, 3H).
13C NMR (100 MHz, DMSO-
d6) δ 166.80 (d,
J = 11.9 Hz), 164.34, 164.19, 161.15 (d,
J = 13.5 Hz), 158.58 (d,
J = 13.5 Hz), 158.38, 158.15, 155.20, 143.30, 141.76, 138.85, 134.91, 132.73, 132.34 (d,
J = 11.0 Hz), 129.21, 126.07–125.29 (m), 124.76, 124.40, 122.03, 120.33, 117.53, 112.30 (d,
J = 25.0 Hz), 106.27 (t,
J = 26.1 Hz), 70.28, 66.73, 58.72, 53.96.
1H and
13C NMR spectra were shown in
Figure S12. HRMS (ESI)
m/
z: [M – H]
– calcd for C
25H
19F
2N
4O
5S
2, 557.0543; found, 557.0820.
General Procedure B was used for the synthesis of 17a–l.
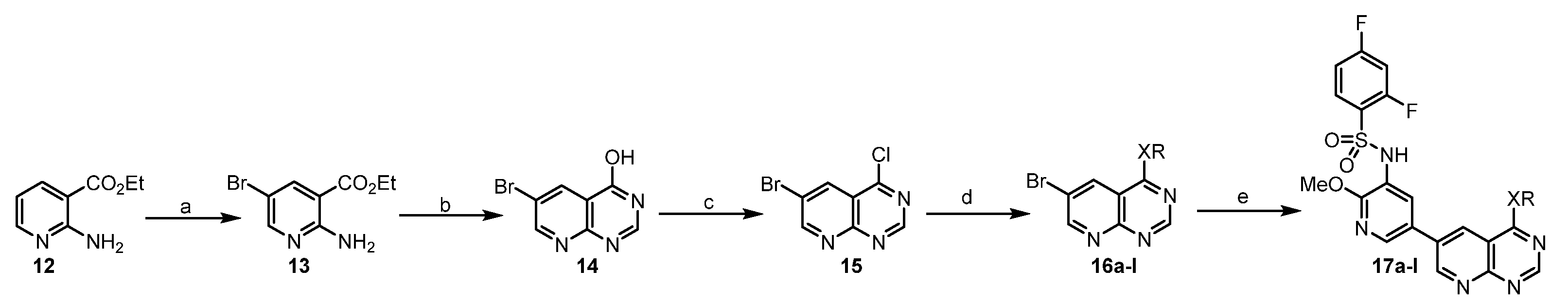
The synthesis of ethyl 2-amino-5-bromonicotinate (13). 1-bromo-2,5-pyrrolidinedione (13.8 g in 15 mL acetonitrile, 78 mmol) was added to a mixture of compound 12 (9.9 g, 60 mmol) in acetonitrile (50 mL) under ice cooling bath. The mixture was gradually raised to room temperature and stirred for an additional 1 h. The precipitate was filtered, washed with dilute ammonia, and dried to give a white solid (13) (14.1 g, 86%).
The synthesis of 6-bromopyrido[2,3-d]pyrimidin-4-ol (14). A mixture of compound 13 (14.1 g, 58 mmol) and formamide (80 mL) was stirred at 155 °C for 20 h. It was cooled down to room temperature and treated with ice water (400 mL). The mixture was stirred for 1 h, filtered and washed with ethyl acetate to give a white solid (14) (8.5 g, 65%). 1H NMR (400 MHz, DMSO-d6) δ 12.72 (s, 1H), 9.04 (d, J = 2.6 Hz, 1H), 8.61 (d, J = 2.6 Hz, 1H), 8.36 (s, 1H).
The synthesis of 6-bromo-4-chloropyrido[2,3-d]pyrimidine (15). A mixture of compound 14 (2.25 g 10 mmol), phosphorous oxychloride (30 mL), and N,N-dimethylformamide (0.1 mL) was stirred under argon at 90 °C for 20 h. The phosphorous oxychloride was removed in vacuo, and the residue was diluted with DCM and treated with ice water (10 mL). Then it was carefully neutralized with NaHCO3, the formed crude was filtered, washed with water, and purified by flash column chromatography (Hexane: ethyl acetate = 1:1) to give a yellow solid (15) (1.9 g, 77%).
The synthesis of 6-bromo-N-isopropylpyrido[2,3-d]pyrimidin-4-amine (16a). Isopropyl amine (86 μL, 1 mmol) was added to a mixture of 15 (75 mg, 0.25 mmol) and Et3N (70 μL, 0.5 mmol) in THF (5 mL) under ice cooling bath. Then, the mixture was gradually raised to room temperature, and stirred for an additional 4 h. It was concentrated in vacuo and the residue was stirred in ethyl acetate (5 mL) and H2O (5 mL). The organic layer was separated and dried. The solvent was evaporated in vacuum to afford 16a as an ashy solid (55 mg, 83.1%), which was used without further purification.
16b−l were prepared by a similar procedure described for the synthesis of 16a.
The synthesis of 2,4-difluoro-
N-(5-(4-(isopropylamino)pyrido[2,3-
d]pyrimidin-6-yl)-2-methoxypyridin-3-yl)benzenesulfonamide (
17a). Under argon protection, PdCl
2(dppf)•DCM (21 mg, 0.025 mmol) was added to a mixture of bromo compound
16a (55 mg, 0.21mmol), borate
5 (213 mg, 0.5 mmol), K
2CO
3 (52 mg, 0.375 mmol) in 1,4-dioxane (10 mL), and H
2O (2 mL). The mixture was refluxed for 4 h, then concentrated and purified by flash column chromatography (hexane: ethyl acetate = 1:1) to obtain the coupled
17a as a white solid (90 mg, 88%).
1H NMR (400 MHz, DMSO-
d6) δ 10.40 (s, 1H), 9.29 (d,
J = 2.4 Hz, 1H), 9.03 (d,
J = 2.5 Hz, 1H), 8.61 (s, 1H), 8.56 (d,
J = 2.4 Hz, 1H), 8.33 (d,
J = 7.5 Hz, 1H), 8.14 (d,
J = 2.4 Hz, 1H), 7.74 (td,
J = 8.6, 6.2 Hz, 1H), 7.58 (ddd,
J = 11.4, 9.3, 2.5 Hz, 1H), 7.20 (td,
J = 8.5, 2.5 Hz, 1H), 4.54 (hept,
J = 6.7 Hz, 1H), 3.63 (s, 3H), 1.31 (d,
J = 6.6 Hz, 6H).
13C NMR (100 MHz, DMSO-
d6) δ 166.80 (d,
J = 11.4 Hz), 164.27 (d,
J = 12.0 Hz), 161.18 (d,
J = 13.5 Hz), 160.41, 158.80 (d,
J = 13.0 Hz), 158.55, 158.02, 153.86, 143.54, 135.63, 132.29 (d,
J = 11.0 Hz), 129.89, 129.51, 126.69, 125.68 (d,
J = 10.8 Hz), 120.27, 113.22–111.50 (m), 109.78, 106.23 (t,
J = 26.2 Hz), 60.22, 53.88, 43.12, 22.45.
1H and
13C NMR spectra were shown in
Figure S13. HRMS (ESI)
m/
z: [M – H]
– calcd for C
22H
19F
2N
6O
3S, 485.1286; found, 485.1231.
17b–i were prepared by a similar procedure described for the synthesis of 17a.
N-(5-(4-(cyclopropylamino)pyrido[2,3-
d]pyrimidin-6-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide (
17b): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (80 mg, 65%).
1H NMR (400 MHz, DMSO-
d6) δ 10.37 (s, 1H), 9.31 (d, J = 2.4 Hz, 1H), 8.95 (d, J = 2.5 Hz, 1H), 8.67 (s, 1H), 8.64 (s, 1H), 8.54 (d, J = 2.4 Hz, 1H), 8.12 (d, J = 2.4 Hz, 1H), 7.74 (td, J = 8.6, 6.2 Hz, 1H), 7.57 (ddd, J = 10.5, 9.2, 2.5 Hz, 1H), 7.19 (td, J = 8.5, 2.5 Hz, 1H), 3.63 (s, 3H), 3.07 (dt, J = 7.7, 3.6 Hz, 1H), 0.86 (dt, J = 6.9, 3.4 Hz, 2H), 0.77–0.65 (m, 2H).
13C NMR (100 MHz, DMSO-
d6) δ 166.85, 166.74, 164.33, 164.21, 162.50, 161.24, 161.11, 158.88, 158.75, 158.68, 158.55, 157.77, 153.95, 143.47, 135.58, 132.33, 132.22, 129.77, 129.58, 126.63, 125.78, 125.74, 125.63, 125.60, 120.30, 112.34, 112.31, 112.12, 112.09, 109.75, 106.47, 106.21, 105.96, 60.21, 53.89, 24.95, 14.55, 6.72.
1H and
13C NMR spectra were shown in
Figure S14. HRMS (ESI)
m/
z: [M – H]
– calcd for C
22H
17F
2N
6O
3S, 483.1129; found, 483.1057.
N-(5-(4-(butylamino)pyrido[2,3-
d]pyrimidin-6-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide (
17c): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (75 mg, 60%).
1H NMR (400 MHz, DMSO-
d6) δ 10.40 (s, 1H), 9.30 (d,
J = 2.4 Hz, 1H), 9.00 (d,
J = 2.5 Hz, 1H), 8.64 (t,
J = 5.5 Hz, 1H), 8.60 (s, 1H), 8.56 (d,
J = 2.3 Hz, 1H), 8.13 (d,
J = 2.4 Hz, 1H), 7.74 (td,
J = 8.5, 6.2 Hz, 1H), 7.58 (ddd,
J = 11.3, 9.3, 2.5 Hz, 1H), 7.20 (td,
J = 8.5, 2.5 Hz, 1H), 3.63 (s, 3H), 3.58 (q,
J = 7.0 Hz, 2H), 1.66 (p,
J = 7.3 Hz, 2H), 1.41 (h,
J = 7.4 Hz, 2H), 0.94 (t,
J = 7.4 Hz, 3H).
13C NMR (100 MHz, DMSO-
d6) δ 166.85, 164.27 (d,
J = 11.9 Hz), 161.25, 161.12, 158.81 (d,
J = 14.4 Hz), 157.95, 153.82, 143.45, 135.59, 132.28 (d,
J = 10.5 Hz), 129.61 (d,
J = 24.0 Hz), 126.64, 125.70 (d,
J = 14.6 Hz), 120.31, 112.20 (d,
J = 22.3 Hz), 109.85, 106.22 (t,
J = 26.2 Hz), 53.89, 41.11, 31.07, 20.22, 14.24.
1H and
13C NMR spectra were shown in
Figure S15. HRMS (ESI)
m/
z: [M – H]
– calcd for C
23H
21F
2N
6O
3S, 499.1442; found, 499.1388.
N-(5-(4-((2-(dimethylamino)ethyl)amino)pyrido[2,3-
d]pyrimidin-6-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide (
17d): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (87 mg, 68%).
1H NMR (400 MHz, DMSO-
d6) δ 10.35–10.02 (m, 1H), 9.31 (d,
J = 2.4 Hz, 1H), 9.24 (s, 1H), 9.14 (d,
J = 2.6 Hz, 1H), 8.64 (s, 1H), 8.60 (d,
J = 2.4 Hz, 1H), 8.12 (d,
J = 2.4 Hz, 1H), 7.75 (td,
J = 8.4, 6.1 Hz, 1H), 7.65–7.54 (m, 1H), 7.20 (td,
J = 8.5, 2.5 Hz, 1H), 3.94 (q,
J = 5.1 Hz, 2H), 3.63 (s, 3H), 3.43 (t,
J = 5.7 Hz, 2H), 2.88 (s, 6H).
13C NMR (100 MHz, DMSO-d6) δ 166.80, 166.68, 164.28, 164.16, 161.21, 161.07, 158.65, 158.52, 158.26, 157.52, 155.53, 154.93, 142.98, 140.76, 137.86, 134.76, 133.72, 132.39, 132.27, 129.48, 125.79, 125.67, 124.28, 123.99, 121.78, 120.56, 115.14, 112.37, 112.14, 106.50, 106.24, 105.98, 66.59, 60.22, 57.56, 53.90, 53.82, 38.04, 24.95, 21.23, 14.55.
1H and
13C NMR spectra were shown in
Figure S16. HRMS (ESI)
m/
z: [M – H]
– calcd for C
23H
22F
2N
7O
3S, 514.1551; found, 514.1471.
N-(2-methoxy-5-(4-morpholinopyrido[2,3-
d]pyrimidin-6-yl)pyridin-3-yl)2,4-difluorobenzenesulfonamide (
17e): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (91 mg, 71%).
1H NMR (400 MHz, DMSO-
d6) δ 10.36 (s, 1H), 9.28 (d,
J = 2.5 Hz, 1H), 8.72 (s, 1H), 8.54–8.45 (m, 2H), 8.08 (d,
J = 2.4 Hz, 1H), 7.84–7.69 (m, 1H), 7.58 (ddd,
J = 11.4, 9.1, 2.5 Hz, 1H), 7.21 (td,
J = 8.6, 2.5 Hz, 1H), 3.92 (t,
J = 4.7 Hz, 4H), 3.77 (t,
J = 4.7 Hz, 4H), 3.67 (s, 3H).
13C NMR (100 MHz, DMSO-
d6) δ 166.83, 164.43, 164.19, 161.05, 159.64, 158.46, 157.00, 154.53, 143.66, 135.03, 133.51–130.58 (m), 129.04, 126.56, 125.70, 120.40, 112.30 (d,
J = 21.5 Hz), 110.21, 106.28 (t,
J = 26.1 Hz), 66.44, 53.94, 49.81.
1H and
13C NMR spectra were shown in
Figure S17. HRMS (ESI)
m/
z: [M – H]
– calcd for C
23H
19F
2N
6O
4S, 513.1235; found, 513.1203.
N-(2-methoxy-5-(4-(4-methylpiperidin-1-yl)pyrido[2,3-
d]pyrimidin-6-yl)pyridin-3-yl)-2,4-difluorobenzenesulfonamide (
17f): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (84 mg, 64%).
1H NMR (400 MHz, DMSO-
d6) δ 10.39–10.37 (m, 1H), 9.26 (d,
J = 2.4 Hz, 1H), 8.66 (s, 1H), 8.48 (d,
J = 2.4 Hz, 1H), 8.40 (d,
J = 2.5 Hz, 1H), 8.05 (d,
J = 2.4 Hz, 1H), 7.77 (td,
J = 8.6, 6.3 Hz, 1H), 7.57 (ddd,
J = 11.4, 9.2, 2.5 Hz, 1H), 7.21 (td,
J = 8.5, 2.5 Hz, 1H), 4.43 (d,
J = 13.1 Hz, 2H), 3.68 (s, 3H), 3.30 (d,
J = 14.4 Hz, 3H), 1.77 (t,
J = 7.8 Hz, 3H), 1.33 (td,
J = 13.6, 6.8 Hz, 2H), 0.97 (d,
J = 6.0 Hz, 3H).
13C NMR (100 MHz, DMSO-
d6) δ 170.80, 166.72 (d,
J = 11.2 Hz), 164.39, 164.25, 161.11 (d,
J = 13.4 Hz), 159.72, 158.55 (d,
J = 13.6 Hz), 158.31, 157.09, 154.22, 143.04, 134.32, 132.28, 132.18, 128.75, 126.60, 125.82, 120.87, 113.75–111.67 (m), 110.17, 107.45, 106.27 (t,
J = 26.1 Hz), 67.08, 60.22, 53.94, 49.82, 34.14, 30.87, 22.09, 14.55, 14.42.
1H and
13C NMR spectra were shown in
Figure S18. HRMS (ESI)
m/
z: [M – H]
– calcd for C
25H
23F
2N
6O
3S, 525.1599; found, 525.1539.
N-(2-methoxy-5-(4-((2-morpholinoethyl)amino)pyrido[2,3-
d]pyrimidin-6-yl)pyridin-3-yl) -2,4-difluorobenzenesulfonamide (
17g): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (96 mg, 69%).
1H NMR (400 MHz, DMSO-
d6) δ 10.40 (s, 1H), 9.30 (d,
J = 2.4 Hz, 1H), 8.99 (d,
J = 2.5 Hz, 1H), 8.71 (t,
J = 5.6 Hz, 1H), 8.62 (s, 1H), 8.52 (d,
J = 2.3 Hz, 1H), 7.76 (td,
J = 8.5, 6.2 Hz, 1H), 7.20 (td,
J = 8.5, 2.5 Hz, 1H), 3.73 (q,
J = 6.5 Hz, 2H), 3.65 (s, 3H), 3.59 (t,
J = 4.7 Hz, 4H), 2.64 (t,
J = 6.9 Hz, 2H), 2.56–2.44 (m, 4H).
13C NMR (100 MHz, DMSO-
d6) δ 170.80, 161.33, 158.77 (d,
J = 15.2 Hz), 157.94, 153.94, 142.75, 132.24 (d,
J = 10.8 Hz), 131.22, 129.70 (d,
J = 4.1 Hz), 126.56, 113.27–111.12 (m), 109.88, 106.16 (t,
J = 26.2 Hz), 66.59, 60.21, 57.29, 53.86, 38.58.
1H and
13C NMR spectra were shown in
Figure S19. HRMS (ESI)
m/
z: [M – H]
– calcd for C
25H
24F
2N
7O
4S, 556.1657; found, 556.1578.
N-(5-(4-((2-hydroxyethyl)amino)pyrido[2,3-
d]pyrimidin-6-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide (
17h): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (71 mg, 58%).
1H NMR (400 MHz, DMSO-
d6) δ 10.38 (s, 1H), 9.29 (d,
J = 2.4 Hz, 1H), 9.16 (d,
J = 2.5 Hz, 1H), 8.90 (d,
J = 5.4 Hz, 1H), 8.59 (s, 1H), 8.58 (d,
J = 3.3 Hz, 1H), 8.14 (d,
J = 2.3 Hz, 1H), 7.75 (td,
J = 8.5, 6.3 Hz, 1H), 7.63–7.49 (m, 1H), 7.19 (td,
J = 8.5, 2.5 Hz, 1H), 4.94 (d,
J = 5.5 Hz, 1H), 3.66 (d,
J = 2.6 Hz, 4H), 3.63 (s, 3H).
13C NMR (100 MHz, DMSO-
d6) δ 161.48, 158.80, 158.70, 157.97, 153.80, 132.28 (d,
J = 11.1 Hz), 129.99, 129.51, 126.55, 112.19 (d,
J = 22.1 Hz), 109.99, 106.18, 63.27, 59.52, 53.87, 44.32.
1H and
13C NMR spectra were shown in
Figure S20. HRMS (ESI)
m/
z: [M – H]
– calcd for C
21H
19F
2N
6O
4S, 487.1078; found, 487.1025.
N-(2-methoxy-5-(4-((3-methoxypropyl)amino)pyrido[2,3-
d]pyrimidin-6-yl)pyridin-3-yl)- 2,4-difluorobenzenesulfonamide (
17i): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (90 mg, 70%).
1H NMR (400 MHz, DMSO-
d6) δ 10.43 (s, 1H), 9.28 (d,
J = 2.4 Hz, 1H), 8.98 (d,
J = 2.5 Hz, 1H), 8.67 (t,
J = 5.5 Hz, 1H), 8.60 (s, 1H), 8.51 (d,
J = 2.3 Hz, 1H), 8.10 (d,
J = 2.4 Hz, 1H), 7.75 (td,
J = 8.6, 6.3 Hz, 1H), 7.55 (ddd,
J = 11.6, 9.3, 2.5 Hz, 1H), 7.19 (td,
J = 8.6, 2.5 Hz, 1H), 3.64 (s, 3H), 3.61 (d,
J = 6.9 Hz, 2H), 3.26 (s, 3H), 1.91 (p,
J = 6.5 Hz, 2H).
13C NMR (100 MHz, DMSO-
d6) δ 161.29, 158.84, 158.69, 157.95, 153.84, 132.24 (d,
J = 10.8 Hz), 129.69, 126.60, 113.46–111.45 (m), 109.89, 106.15 (t,
J = 26.1 Hz), 70.08, 58.40, 53.84, 38.68, 29.01.
1H and
13C NMR spectra were shown in
Figure S21. HRMS (ESI)
m/
z: [M – H]
– calcd for C
23H
21F
2N
6O
4S, 515.1391; found, 515.1345.
N-(2-methoxy-5-(4-((2-methoxyethyl)amino)pyrido[2,3-
d]pyrimidin-6-yl)pyridin-3-yl)-2,4-difluorobenzenesulfonamide (
17j): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (93 mg, 74%).
1H NMR (400 MHz, DMSO-
d6) δ 10.44–10.31 (m, 1H), 9.32 (d,
J = 2.4 Hz, 1H), 9.04 (d,
J = 2.5 Hz, 1H), 8.78 (t,
J = 5.6 Hz, 1H), 8.64–8.55 (m, 2H), 8.15 (d,
J = 2.4 Hz, 1H), 7.75 (td,
J = 8.5, 6.2 Hz, 1H), 7.58 (ddd,
J = 11.3, 9.3, 2.5 Hz, 1H), 7.20 (td,
J = 8.5, 2.5 Hz, 1H), 3.77 (q,
J = 5.5 Hz, 2H), 3.63 (s, 3H), 3.61 (t,
J = 5.9 Hz, 2H), 3.31 (s, 3H).
13C NMR (100 MHz, DMSO-
d6) δ 166.80 (d,
J = 11.6 Hz), 164.28 (d,
J = 11.8 Hz), 161.38, 161.18 (d,
J = 13.6 Hz), 158.75, 157.93, 153.92, 143.45, 135.60, 132.30 (d,
J = 10.8 Hz), 129.64 (d,
J = 19.0 Hz), 126.56, 125.67 (d,
J = 14.5 Hz), 120.28, 112.23 (d,
J = 20.9 Hz), 109.88, 106.22 (t,
J = 26.2 Hz), 70.46, 58.54, 53.90, 41.16.
1H and
13C NMR spectra were shown in
Figure S22. HRMS (ESI)
m/
z: [M + H]
+ calcd for C
22H
21F
2N
6O
4S, 503.1235; found, 503.1316.
N-(5-(4-((cyclohexylmethyl)amino)pyrido[2,3-
d]pyrimidin-6-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide (
17k): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (90 mg, 67%).
1H NMR (400 MHz, DMSO-
d6) δ 10.38 (s, 1H), 9.29 (d,
J = 2.4 Hz, 1H), 9.02 (d,
J = 2.5 Hz, 1H), 8.65 (t,
J = 5.7 Hz, 1H), 8.59 (s, 1H), 8.56 (d,
J = 2.4 Hz, 1H), 8.13 (d,
J = 2.3 Hz, 1H), 7.75 (td,
J = 8.5, 6.2 Hz, 1H), 7.57 (ddd,
J = 10.6, 9.2, 2.5 Hz, 1H), 7.19 (td,
J = 8.6, 2.5 Hz, 1H), 3.63 (s, 3H), 3.44 (t,
J = 6.1 Hz, 2H), 1.87–1.53 (m, 6H), 1.38–1.07 (m, 4H), 1.00 (t,
J = 11.9 Hz, 1H).
13C NMR (100 MHz, DMSO-
d6) δ 166.84, 164.26 (d,
J = 11.9 Hz), 161.45, 158.80 (d,
J = 10.9 Hz), 157.98, 153.82, 143.47, 135.58, 132.28 (d,
J = 11.1 Hz), 129.63 (d,
J = 25.7 Hz), 126.67, 125.71 (d,
J = 10.7 Hz), 120.32, 112.22 (d,
J = 22.5 Hz), 109.85, 106.21 (t,
J = 26.1 Hz), 53.88, 47.55, 37.33, 31.08, 26.51, 25.90.
1H and
13C NMR spectra were shown in
Figure S23. HRMS (ESI)
m/
z: [M – H]
– calcd for C
26H
25F
2N
6O
3S, 539.1755; found, 539.1725.
N-(5-(4-((cyclopropylmethyl)amino)pyrido[2,3-
d]pyrimidin-6-yl)-2-methoxypyridin-3-yl)-2,4-difluorobenzenesulfonamide (
17l): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (96 mg, 77%).
1H NMR (400 MHz, DMSO-
d6) δ 10.36 (s, 1H), 9.30 (d,
J = 2.4 Hz, 1H), 9.03 (d,
J = 2.5 Hz, 1H), 8.77 (t,
J = 5.5 Hz, 1H), 8.60 (s, 1H), 8.56 (d,
J = 2.4 Hz, 1H), 8.14 (d,
J = 2.4 Hz, 1H), 7.75 (td,
J = 8.5, 6.2 Hz, 1H), 7.57 (ddd,
J = 11.2, 9.3, 2.5 Hz, 1H), 7.20 (td,
J = 8.5, 2.5 Hz, 1H), 3.64 (s, 3H), 3.55–3.43 (m, 2H), 1.22 (ddd,
J = 14.0, 6.6, 2.8 Hz, 1H), 0.51 (dt,
J = 8.1, 2.9 Hz, 2H), 0.40–0.29 (m, 2H).
13C NMR (100 MHz, DMSO-
d6) δ 170.77, 166.79 (d,
J = 11.7 Hz), 164.27 (d,
J = 11.8 Hz), 161.27, 161.11, 158.78 (d,
J = 11.3 Hz), 158.00, 143.42, 135.47, 132.29 (d,
J = 10.8 Hz), 129.80, 129.54, 126.65, 125.72 (dd,
J = 14.6, 3.8 Hz), 120.36, 112.21 (dd,
J = 22.2, 3.7 Hz), 109.80, 106.21 (t,
J = 26.2 Hz), 67.48, 60.20, 53.88, 45.96, 25.58, 21.21, 14.54, 10.91, 4.06.
1H and
13C NMR spectra were shown in
Figure S24. HRMS (ESI)
m/
z: [M + H]
+ calcd for C
23H
21F
2N
6O
3S, 499.1286; found, 499.1369.
General Procedure C was used for the synthesis of 22a–l.
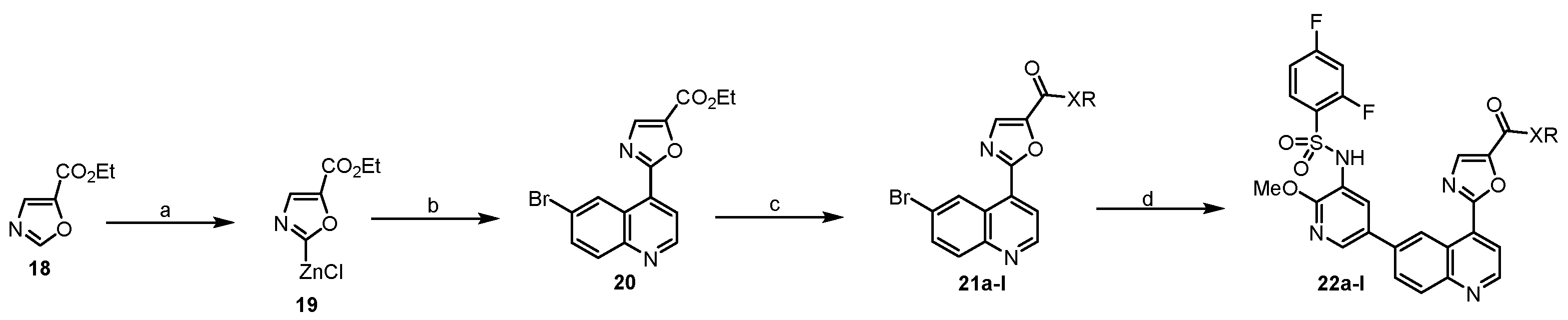
The synthesis of ethyl 2-(6-bromoquinolin-4-yl)oxazole-5-carboxylate (20). To a mixture of zinc chloride (1.0 M in THF, 6 mL,6 mmol) and compound 18 (242 μL, 2 mmol) in THF (10 mL) at −10 °C under argon, lithium hexamethyldisilazide (1.0 M in THF, 3 mL, 3 mmol) was added and the mixture was stirred at −10 °C for 1 h. Then, 6-bromo-4-iodoquinoline (333 mg, 1 mmol) and Pd(PPh3)4 (116 mg, 0.1 mmol) were added and the mixture was stirred at 60 °C for 15 h. It was cooled to room temperature and neutralized by saturated ammonium chloride (30 mL). Ethyl acetate (10 mL) was added and the mixture was stirred for 15 min. The separated organic layer was evaporated in vacuum and the residue was purified by flash column chromatography to obtain a white solid (20) (245mg, 71%). 1H NMR (400 MHz, DMSO-d6) δ 9.44 (d, J = 2.2 Hz, 1H), 9.10 (d, J = 4.6 Hz, 1H), 8.31 (s, 1H), 8.13 (d, J = 4.5 Hz, 1H), 8.05 (d, J = 8.9 Hz, 1H), 7.98 (dd, J = 9.0, 2.3 Hz, 1H), 4.40 (q, J = 7.1 Hz, 2H), 1.36 (t, J = 7.0 Hz, 3H).
The synthesis of 2-(6-bromoquinolin-4-yl)-N-isopropyloxazole-5-carboxamide (21c). In a sealed tube, a mixture of ester 20 (88 mg, 0.25 mmol) and isopropyl amine (1 mL) was stirred at 100 °C for 4 h. It was cooled to room temperature and H2O (5 mL) was added. The suspension was filtered and dried to give an ashy solid 21c (76.3 mg, 86%), which was used without purification.
21a–b, 21d–l were prepared by a similar procedure described for the synthesis of 21c.
The synthesis of 2-(6-(5-((2,4-difluorophenyl)sulfonamido)-6-methoxypyridin-3-yl)quinolin-4-yl)-
N-isopropyloxazole-5-carboxamide (
22c). Under argon protection, PdCl
2(dppf)•DCM (21 mg, 0.025 mmol) was added to a mixture of bromo compound
21c (76.3 mg, 0.21mmol), borate
5 (213 mg, 0.5 mmol), K
2CO
3 (52 mg, 0.375 mmol) in 1,4-dioxane (10 mL), and H
2O (2 mL). The mixture was refluxed for 4 h, then concentrated and purified by flash column chromatography (hexane: ethyl acetate = 1:1) to obtain the coupled
22c as a white solid (83.9 mg, 70%).
1H NMR (400 MHz, DMSO-
d6) δ 10.39 (s, 1H), 9.56 (d,
J = 2.0 Hz, 1H), 9.13 (d,
J = 4.6 Hz, 1H), 8.65 (d,
J = 7.9 Hz, 1H), 8.47 (d,
J = 2.3 Hz, 1H), 8.39 (d,
J = 4.6 Hz, 1H), 8.23 (d,
J = 8.8 Hz, 1H), 8.15 (dd,
J = 9.0, 2.2 Hz, 1H), 8.13 (s, 1H), 8.03 (d,
J = 2.3 Hz, 1H), 3.70 (s, 3H), 1.23 (d,
J = 6.6 Hz, 6H).
13C NMR (100 MHz, DMSO-
d6) δ 170.79, 166.83 (d,
J = 12.0 Hz), 164.30 (d,
J = 11.6 Hz), 161.13 (d,
J = 13.5 Hz), 159.62, 158.57 (d,
J = 13.5 Hz), 158.12, 155.80, 150.91, 148.42, 146.28, 143.02, 136.41, 134.18, 132.37 (d,
J = 10.7 Hz), 131.82, 131.18, 129.85, 129.55, 129.18, 125.53 (d,
J = 18.1 Hz), 124.02, 123.53, 121.89, 120.54, 113.19–111.28 (m), 106.32 (t,
J = 26.1 Hz), 60.21, 53.99, 41.32, 22.72, 21.22, 14.54.
1H and
13C NMR spectra were shown in
Figure S27. HRMS (ESI)
m/
z: [M – H]
– calcd for C
28H
22F
2N
5O
5S, 578.1388; found, 578.1355.
22a–b, 22d−l were prepared by a similar procedure described for the synthesis of 22c.
Ethyl-2-(6-(5-((2,4-difluorophenyl)sulfonamido)-6-methoxypyridin-3-yl)quinolin-4-yl)oxazole-5-carboxylate (
22a): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (99 mg, 68%).
1H NMR (400 MHz, DMSO-
d6) δ 10.37 (s, 1H), 9.41 (d,
J = 2.0 Hz, 1H), 9.10 (d,
J = 4.5 Hz, 1H), 8.46 (d,
J = 2.3 Hz, 1H), 8.36 (d,
J = 1.1 Hz, 1H), 8.23 (d,
J = 8.8 Hz, 1H), 8.20–8.13 (m, 2H), 8.03 (d,
J = 2.3 Hz, 1H), 7.80 (td,
J = 8.5, 6.2 Hz, 1H), 7.57 (ddd,
J = 11.2, 9.3, 2.5 Hz, 1H), 7.20 (td,
J = 8.5, 2.5 Hz, 1H), 4.42 (q,
J = 7.1 Hz, 2H), 3.70 (s, 3H), 1.36 (t,
J = 7.1 Hz, 3H).
13C NMR (100 MHz, DMSO-
d6) δ 166.83 (d,
J = 11.9 Hz), 164.30 (d,
J = 11.6 Hz), 161.47, 161.13 (d,
J = 13.9 Hz), 158.57 (d,
J = 13.6 Hz), 158.24, 157.51, 151.03, 148.42, 143.17, 142.93, 136.51, 135.97, 134.47, 132.36 (d,
J = 10.8 Hz), 131.26, 130.06–128.52 (m), 125.53 (d,
J = 14.3 Hz), 123.98, 123.24, 122.05, 120.43, 112.33 (d,
J = 19.3 Hz), 106.31 (t,
J = 26.2 Hz), 62.07, 53.98, 22.72, 14.58.
1H and
13C NMR spectra were shown in
Figure S25. HRMS (ESI)
m/
z: [M – H]
– calcd for C
27H
19F
2N
4O
6S, 565.1072; found, 565.1016.
2-(6-(5-((2,4-difluorophenyl)sulfonamido)-6-methoxypyridin-3-yl)quinolin-4-yl)-
N-methyloxazole-5-carboxamide (
22b): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (78 mg, 57%).
1H NMR (400 MHz, DMSO-
d6) δ 10.40 (s, 1H), 9.56 (d,
J = 2.1 Hz, 1H), 9.14 (d,
J = 4.6 Hz, 1H), 8.87 (q,
J = 4.5 Hz, 1H), 8.47 (d,
J = 2.4 Hz, 1H), 8.37 (d,
J = 4.5 Hz, 1H), 8.23 (d,
J = 8.8 Hz, 1H), 8.16 (dd,
J = 8.8, 2.1 Hz, 1H), 8.12 (s, 1H), 8.03 (d,
J = 2.4 Hz, 1H), 7.81 (td,
J = 8.6, 6.2 Hz, 1H), 7.63–7.53 (m, 1H), 7.20 (td,
J = 8.3, 2.3 Hz, 1H), 3.70 (s, 3H), 2.85 (d,
J = 4.6 Hz, 3H).
13C NMR (100 MHz, DMSO-
d6) δ 166.83 (d,
J = 11.8 Hz), 159.57, 158.16, 157.10, 150.94, 148.43, 146.22, 143.09, 136.44, 134.30, 132.36 (d,
J = 11.1 Hz), 131.63, 131.20, 129.83, 129.56, 129.23, 125.51 (d,
J = 14.0 Hz), 124.02, 123.50, 121.81, 120.50, 112.34 (d,
J = 23.7 Hz), 106.32 (t,
J = 26.0 Hz), 60.22, 54.00.
1H and
13C NMR spectra were shown in
Figure S26. HRMS (ESI)
m/
z: [M – H]
– calcd for C
26H
18F
2N
5O
5S, 550.1075; found, 550.1032.
N-cyclopropyl-2-(6-(5-((2,4-difluorophenyl)sulfonamido)-6-methoxypyridin-3-yl)quinolin-4-yl)oxazole-5-carboxamide (
22d): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (66 mg, 46%).
1H NMR (400 MHz, DMSO-
d6) δ 10.38 (s, 1H), 9.55 (d,
J = 2.0 Hz, 1H), 9.12 (d,
J = 4.6 Hz, 1H), 8.89 (d,
J = 4.0 Hz, 1H), 8.47 (d,
J = 2.3 Hz, 1H), 8.37 (d,
J = 4.5 Hz, 1H), 8.23 (d,
J = 8.8 Hz, 1H), 8.15 (dd,
J = 8.8, 2.1 Hz, 1H), 8.12 (s, 1H), 8.03 (d,
J = 2.4 Hz, 1H), 7.89–7.78 (m, 1H), 7.57 (ddd,
J = 11.4, 9.2, 2.5 Hz, 1H), 7.20 (td,
J = 8.5, 2.5 Hz, 1H), 3.71 (s, 3H), 3.08–2.83 (m, 1H), 0.77 (dt,
J = 6.9, 3.3 Hz, 2H), 0.69–0.61 (m, 2H).
13C NMR (100 MHz, DMSO-
d6) δ 159.68, 158.12, 157.83, 150.92, 148.42, 146.08, 143.01, 136.44, 134.15, 132.37 (d,
J = 10.8 Hz), 131.79, 131.19, 129.81, 129.54, 129.20, 125.52 (d,
J = 14.5 Hz), 124.01, 123.49, 121.89, 120.56, 112.35 (d,
J = 22.5 Hz), 106.31 (t,
J = 26.1 Hz), 60.22, 54.00, 22.98, 21.22, 14.55, 6.27.
1H and
13C NMR spectra were shown in
Figure S28. HRMS (ESI)
m/
z: [M + H]
+ calcd for C
28H
22F
2N
5O
5S, 578.1231; found, 578.1301.
2-(6-(5-((2,4-difluorophenyl)sulfonamido)-6-methoxypyridin-3-yl)quinolin-4-yl)-
N-(2-methoxyethyl)oxazole-5-carboxamide (
22e): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (106 mg, 71%).
1H NMR (400 MHz, DMSO-
d6) δ 10.38 (s, 1H), 9.56 (d,
J = 2.1 Hz, 1H), 9.13 (d,
J = 4.6 Hz, 1H), 8.99 (d,
J = 5.5 Hz, 1H), 8.47 (d,
J = 2.3 Hz, 1H), 8.39 (d,
J = 4.6 Hz, 1H), 8.23 (d,
J = 8.8 Hz, 1H), 8.19–8.11 (m, 2H), 8.03 (d,
J = 2.4 Hz, 1H), 7.81 (td,
J = 8.6, 6.2 Hz, 1H), 7.58 (ddd,
J = 10.5, 9.1, 2.5 Hz, 1H), 7.21 (td,
J = 8.6, 2.5 Hz, 1H), 3.71 (s, 3H), 3.50 (dt,
J = 6.2, 2.9 Hz, 4H), 3.29 (s, 3H).
13C NMR (100 MHz, DMSO-
d6) δ 170.79, 166.83 (d,
J = 11.9 Hz), 164.30 (d,
J = 11.6 Hz), 161.12 (d,
J = 13.4 Hz), 159.70, 158.56 (d,
J = 13.4 Hz), 158.13, 156.79, 150.91, 148.42, 146.06, 143.01, 136.43, 134.18, 132.37 (d,
J = 10.7 Hz), 131.96, 131.19, 129.82, 129.55, 129.20, 126.06–125.07 (m), 124.03, 123.50, 121.88, 120.56, 113.48–110.87 (m), 106.31 (t,
J = 26.0 Hz), 70.87, 67.48, 60.21, 58.43, 53.99, 22.98, 21.22, 14.55, 6.27.
1H and
13C NMR spectra were shown in
Figure S29. HRMS (ESI)
m/
z: [M + H]
+ calcd for C
28H
24F
2N
5O
6S, 596.1337; found, 596.1420.
2-(6-(5-((2,4-difluorophenyl)sulfonamido)-6-methoxypyridin-3-yl)quinolin-4-yl)-
N-(3-methoxypropyl)oxazole-5-carboxamide (
22f): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (90 mg, 59%).
1H NMR (400 MHz, DMSO-
d6) δ 10.38 (s, 1H), 9.55 (d,
J = 2.0 Hz, 1H), 9.13 (d,
J = 4.6 Hz, 1H), 8.90 (t,
J = 5.8 Hz, 1H), 8.47 (d,
J = 2.3 Hz, 1H), 8.37 (d,
J = 4.6 Hz, 1H), 8.23 (d,
J = 8.8 Hz, 1H), 8.16 (dd,
J = 8.8, 2.1 Hz, 1H), 8.13 (s, 1H), 8.03 (d,
J = 2.4 Hz, 1H), 7.81 (td,
J = 8.6, 6.2 Hz, 1H), 7.58 (ddd,
J = 10.5, 9.2, 2.5 Hz, 1H), 7.25–7.16 (m, 1H), 3.71 (s, 3H), 3.41 (t,
J = 6.2 Hz, 2H), 3.39–3.34 (m, 2H), 3.25 (s, 3H), 1.81 (p,
J = 6.6 Hz, 2H).
13C NMR (100 MHz, DMSO-
d6) δ 159.62, 158.14, 156.66, 150.93, 148.43, 146.20, 143.09, 136.43, 134.25, 132.42, 132.32, 131.78, 131.19, 129.86, 129.56, 129.21, 125.46, 124.03, 123.51, 121.85, 120.48, 112.46, 112.25, 106.58, 106.32, 106.06, 70.03, 60.21, 58.40, 54.00, 36.54, 29.67, 14.55.
1H and
13C NMR spectra were shown in
Figure S30. HRMS (ESI)
m/
z: [M – H]
– calcd for C
29H
24F
2N
5O
6S, 608.1494; found, 608.1464.
N-butyl-2-(6-(5-((2,4-difluorophenyl)sulfonamido)-6-methoxypyridin-3-yl)quinolin-4-yl)oxazole-5-carboxamide (
22g): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (95 mg, 64%).
1H NMR (400 MHz, DMSO-
d6) δ 10.40 (s, 1H), 9.55 (d,
J = 2.0 Hz, 1H), 9.13 (d,
J = 4.6 Hz, 1H), 8.87 (t,
J = 5.8 Hz, 1H), 8.47 (d,
J = 2.3 Hz, 1H), 8.37 (d,
J = 4.5 Hz, 1H), 8.23 (d,
J = 8.7 Hz, 1H), 8.15 (dd,
J = 8.8, 2.1 Hz, 1H), 8.12 (s, 1H), 8.03 (d,
J = 2.3 Hz, 1H), 7.81 (td,
J = 8.6, 6.2 Hz, 1H), 7.58 (ddd,
J = 11.2, 9.3, 2.5 Hz, 1H), 7.20 (td,
J = 8.5, 2.5 Hz, 1H), 3.70 (s, 3H), 3.30 (d,
J = 7.2 Hz, 2H), 1.64–1.50 (m, 2H), 1.36 (h,
J = 7.4 Hz, 2H), 0.92 (t,
J = 7.3 Hz, 3H).
13C NMR (100 MHz, DMSO-
d6) δ 170.80, 166.82 (d,
J = 11.7 Hz), 164.30 (d,
J = 11.6 Hz), 161.12 (d,
J = 13.4 Hz), 159.60, 158.56 (d,
J = 13.5 Hz), 158.13, 156.57, 150.92, 148.41, 146.23, 143.03, 136.42, 134.22, 132.37 (d,
J = 11.0 Hz), 131.73, 131.18, 129.84, 129.55, 129.20, 125.52 (dd,
J = 14.5, 3.7 Hz), 124.02, 123.51, 121.85, 120.54, 112.33 (dd,
J = 22.2, 3.5 Hz), 106.32 (t,
J = 26.2 Hz), 60.22, 53.99, 38.81, 31.67, 23.07, 21.22, 20.08, 14.54, 14.14.
1H and
13C NMR spectra were shown in
Figure S31. HRMS (ESI)
m/
z: [M – H]
– calcd for C
29H
24F
2N
5O
5S, 592.1544; found, 592.1503.
N-(cyclopropylmethyl)-2-(6-(5-((2,4-difluorophenyl)sulfonamido)-6-methoxypyridin-3-yl)quinolin-4-yl)oxazole-5-carboxamide (
22h): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (77 mg, 52%).
1H NMR (400 MHz, DMSO-
d6) δ 10.41–10.33 (m, 1H), 9.56 (s, 1H), 9.14 (d,
J = 4.6 Hz, 1H), 9.00 (t,
J = 5.7 Hz, 1H), 8.46 (s, 1H), 8.39 (d,
J = 4.5 Hz, 1H), 8.24 (d,
J = 8.7 Hz, 1H), 8.16 (d,
J = 10.0 Hz, 2H), 8.03 (s, 1H), 7.82 (q,
J = 7.9 Hz, 1H), 7.56 (t,
J = 10.1 Hz, 1H), 7.20 (t,
J = 8.0 Hz, 1H), 3.71 (s, 3H), 3.21 (t,
J = 6.4 Hz, 2H), 1.07 (d,
J = 8.1 Hz, 1H), 0.48 (d,
J = 7.7 Hz, 3H), 0.28 (d,
J = 5.0 Hz, 2H).
13C NMR (151 MHz, DMSO-
d6) δ 169.35, 166.35 (d,
J = 12.3 Hz), 164.66 (d,
J = 12.0 Hz), 160.69 (d,
J = 13.5 Hz), 159.66, 158.98 (d,
J = 13.5 Hz), 158.14, 156.62, 150.93, 148.42, 146.22, 142.78, 136.50, 133.98, 132.35 (d,
J = 10.9 Hz), 131.88, 131.19, 129.86, 129.54, 129.24, 125.66, 124.04, 123.51, 121.89, 120.82, 112.30 (d,
J = 22.1 Hz), 106.30 (t,
J = 26.2 Hz), 53.98, 43.57, 11.44, 11.30, 3.93, 3.66.
1H and
13C NMR spectra were shown in
Figure S32. HRMS (ESI)
m/
z: [M + H]
+ calcd for C
29H
24F
2N
5O
5S, 592.1388; found, 592.1471.
N-(cyclohexylmethyl)-2-(6-(5-((2,4-difluorophenyl)sulfonamido)-6-methoxypyridin-3-yl)quinolin-4-yl)oxazole-5-carboxamide (
22i): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (73 mg, 46%).
1H NMR (400 MHz, DMSO-
d6) δ 10.39 (s, 1H), 9.55 (d,
J = 2.1 Hz, 1H), 9.13 (d,
J = 4.5 Hz, 1H), 8.87 (t,
J = 6.0 Hz, 1H), 8.47 (d,
J = 2.3 Hz, 1H), 8.38 (d,
J = 4.6 Hz, 1H), 8.23 (d,
J = 8.8 Hz, 1H), 8.15 (dd,
J = 8.9, 2.1 Hz, 1H), 8.13 (s, 1H), 8.03 (d,
J = 2.4 Hz, 1H), 7.81 (td,
J = 8.5, 6.2 Hz, 1H), 7.58 (ddd,
J = 11.3, 9.3, 2.5 Hz, 1H), 7.20 (td,
J = 8.5, 2.5 Hz, 1H), 3.71 (s, 3H), 3.18 (s, 2H), 1.74 (d,
J = 14.6 Hz, 2H), 1.71–1.66 (m, 2H), 1.59 (ddd,
J = 18.4, 9.5, 5.8 Hz, 2H), 1.30–1.05 (m, 4H), 0.96–0.90 (m, 1H).
13C NMR (100 MHz, DMSO-
d6) δ 166.84 (d,
J = 11.8 Hz), 164.31 (d,
J = 11.8 Hz), 161.12 (d,
J = 13.7 Hz), 159.62, 158.56 (d,
J = 13.4 Hz), 158.12, 156.68, 150.93, 148.42, 146.25, 143.09, 136.41, 134.26, 132.38 (d,
J = 11.0 Hz), 131.75, 131.19, 129.87, 129.56, 129.21, 125.50 (d,
J = 10.6 Hz), 124.03, 123.52, 121.88, 120.46, 112.35 (d,
J = 21.0 Hz), 106.32 (t,
J = 26.0 Hz), 60.21, 53.99, 45.32, 38.01, 30.95, 26.48, 25.86, 21.22, 14.55.
1H and
13C NMR spectra were shown in
Figure S33. HRMS (ESI)
m/
z: [M – H]
– calcd for C
32H
28F
2N
5O
5S, 632.1857; found, 632.1823.
N-cyclohexyl-2-(6-(5-((2,4-difluorophenyl)sulfonamido)-6-methoxypyridin-3-yl)quinolin-4-yl)oxazole-5-carboxamide (
22j): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (76 mg, 49%).
1H NMR (400 MHz, DMSO-
d6) δ 10.40–10.36 (m, 1H), 9.55 (d,
J = 2.1 Hz, 1H), 9.13 (d,
J = 4.5 Hz, 1H), 8.63 (d,
J = 8.1 Hz, 1H), 8.48 (d,
J = 2.4 Hz, 1H), 8.40 (d,
J = 4.6 Hz, 1H), 8.23 (d,
J = 8.8 Hz, 2H), 8.18–8.14 (m, 1H), 8.14 (s, 1H), 8.03 (d,
J = 2.4 Hz, 1H), 7.81 (td,
J = 8.6, 6.3 Hz, 1H), 7.62–7.52 (m, 1H), 7.20 (td,
J = 8.5, 2.5 Hz, 1H), 3.84–3.78 (m, 1H), 3.71 (s, 3H), 1.92–1.73 (m, 4H), 1.68–1.40 (m, 2H), 1.38–1.26 (m, 4H).
13C NMR (100 MHz, DMSO-
d6) δ 161.19, 159.65, 158.56 (d,
J = 13.5 Hz), 158.12, 155.75, 150.92, 148.43, 146.30, 143.00, 136.42, 134.15, 132.37 (d,
J = 10.8 Hz), 131.86, 131.19, 129.88, 129.55, 129.20, 124.03, 123.54, 121.93, 120.59, 112.33 (d,
J = 24.6 Hz), 106.31 (t,
J = 26.2 Hz), 60.22, 53.98, 48.58, 32.86, 25.67, 25.38, 21.22, 14.55.
1H and
13C NMR spectra were shown in
Figure S34. HRMS (ESI)
m/
z: [M – H]
– calcd for C
31H
26F
2N
5O
5S, 618.1701; found, 618.1664.
2-(6-(5-((2,4-difluorophenyl)sulfonamido)-6-methoxypyridin-3-yl)quinolin-4-yl)-
N-(4-methoxyphenethyl)oxazole-5-carboxamide (
22k): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (92 mg, 55%).
1H NMR (400 MHz, DMSO-
d6) δ 10.39 (s, 1H), 9.56 (d,
J = 2.1 Hz, 1H), 9.14 (d,
J = 4.6 Hz, 1H), 8.99 (t,
J = 5.8 Hz, 1H), 8.47 (d,
J = 2.3 Hz, 1H), 8.37 (d,
J = 4.6 Hz, 1H), 8.24 (d,
J = 8.8 Hz, 1H), 8.17 (dd,
J = 8.8, 2.1 Hz, 1H), 8.13 (s, 1H), 8.03 (d,
J = 2.3 Hz, 1H), 7.81 (td,
J = 8.6, 6.3 Hz, 1H), 7.62–7.52 (m, 1H), 7.25–7.08 (m, 3H), 6.90–6.81 (m, 2H), 3.71 (d,
J = 4.7 Hz, 6H), 3.51 (dt,
J = 8.1, 6.2 Hz, 2H), 2.83 (t,
J = 7.5 Hz, 2H).
13C NMR (100 MHz, DMSO-
d6) δ 159.67, 158.25, 158.14, 156.60, 150.94, 148.44, 146.19, 136.49, 132.36 (d,
J = 11.1 Hz), 131.81, 131.49, 131.21, 130.09 129.86, 129.55, 129.25, 124.04, 123.50, 121.85, 114.32, 55.46, 53.99, 41.04, 34.69.
1H and
13C NMR spectra were shown in
Figure S35. HRMS (ESI)
m/
z: [M + H]
+ calcd for C
34H
28F
2N
5O
6S, 672.1650; found, 672.1731.
N-(4-chlorophenethyl)-2-(6-(5-((2,4-difluorophenyl)sulfonamido)-6-methoxypyridin-3-yl)quinolin-4-yl)oxazole-5-carboxamide (
22l): flash column chromatography (hexane: ethyl acetate = 1:1) as a white solid (103 mg, 61%).
1H NMR (400 MHz, DMSO-
d6) δ 10.39 (s, 1H), 9.55 (d,
J = 2.2 Hz, 1H), 9.14 (d,
J = 4.6 Hz, 1H), 9.00 (t,
J = 5.7 Hz, 1H), 8.47 (d,
J = 2.4 Hz, 1H), 8.35 (d,
J = 4.5 Hz, 1H), 8.24 (d,
J = 8.8 Hz, 1H), 8.16 (dd,
J = 8.8, 2.1 Hz, 1H), 8.13 (s, 1H), 8.03 (d,
J = 2.3 Hz, 1H), 7.81 (td,
J = 8.6, 6.2 Hz, 1H), 7.57 (ddd,
J = 11.3, 9.3, 2.5 Hz, 1H), 7.39–7.27 (m, 4H), 7.20 (td,
J = 8.3, 2.3 Hz, 1H), 3.70 (s, 3H), 3.59–3.50 (m, 2H), 2.89 (t,
J = 7.3 Hz, 2H).
13C NMR (100 MHz, DMSO-
d6) δ 159.69, 158.15, 156.65, 150.94, 148.43, 146.11, 138.71, 136.49, 132.36 (d,
J = 11.2 Hz), 131.88, 131.34, 131.20, 131.07, 129.85, 129.54, 129.25, 128.78, 124.03, 123.49, 121.85, 106.43 (d,
J = 26.2 Hz), 53.99, 34.78.
1H and
13C NMR spectra were shown in
Figure S36. HRMS (ESI)
m/
z: [M + H]
+ calcd for C
33H
25ClF
2N
5O
5S, 676.1155; found, 676.1230.


 and
and 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







