
Pharmacology and Emerging Therapies for Group 3 Pulmonary Hypertension Due to Chronic Lung Disease

Abstract
:1. Introduction
2. Classification
3. Pathogenesis
3.1. Pulmonary Vascular Remodeling
3.2. Hypoxia
3.3. Interstitial Lung Disease (ILD)
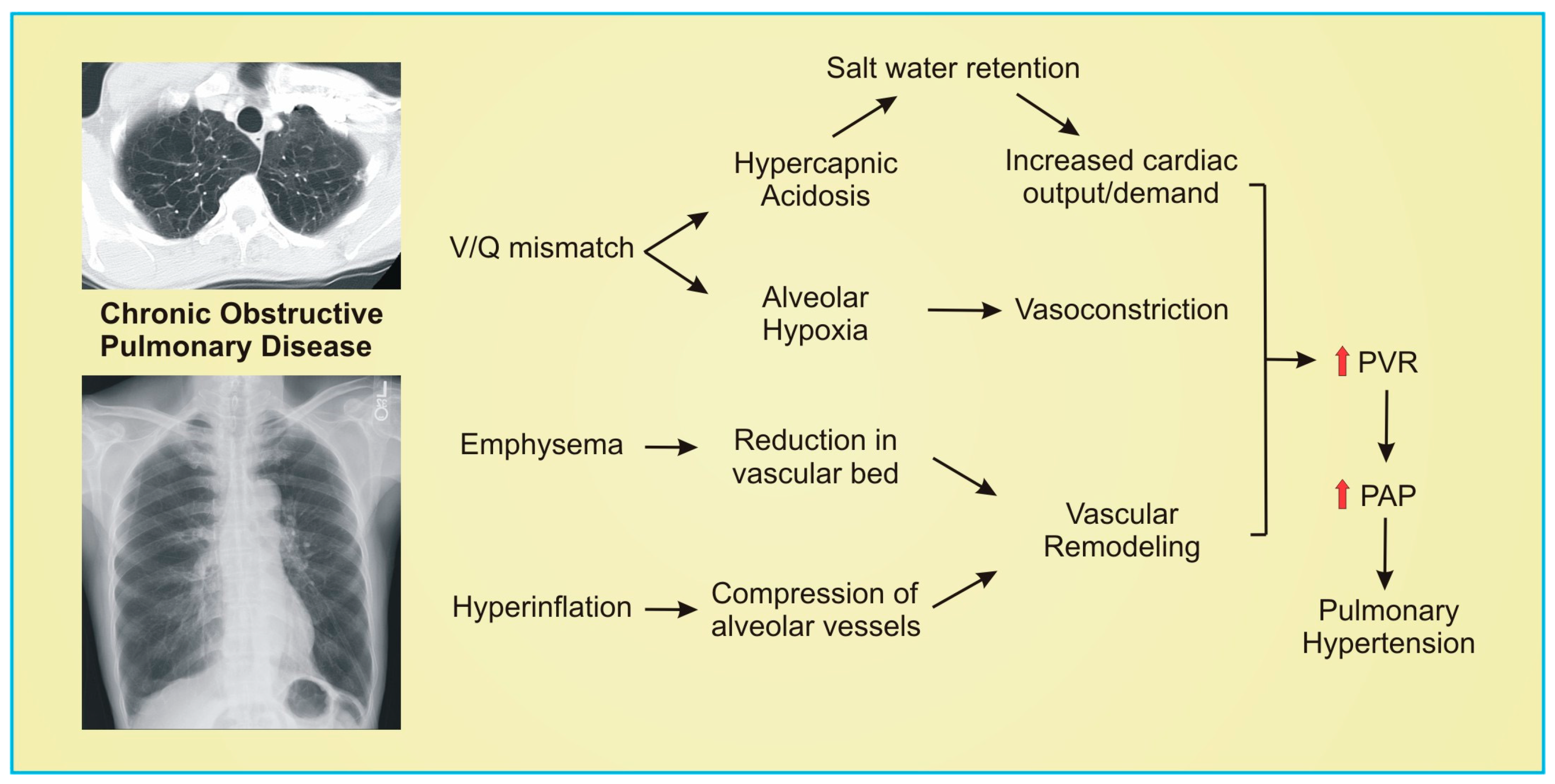
3.4. Chronic Obstructive Pulmonary Disease (COPD) and Emphysema
4. Group 3 Pulmonary Hypertension Therapies
4.1. Pulmonary-Arterial-Hypertension-Specific Therapies
4.1.1. Nitric Oxide (NO) Pathway
4.1.2. Endothelin (ET) Pathway

{kind=link}
{kind=link}
{kind=link}
| Trial Study | Therapy | Target | Outcome | Ref |
|---|---|---|---|---|
| Pulmonary Fibrosis-Associated Pulmonary Hypertension (PF-PH) | ||||
| STEP-IPF Zisman et al., 2010 | Sildenafil | NO | No improvement in 6MWD | [38] |
| Behr et al., 2021 | Sildenafil and Pirfenidone | NO | No improvement in 6MWD, respiratory hospitalization, or mortality | [39] |
| RISE-IIP Nathan et al., 2019 | Riociguat | NO | No improvement in 6MWD; increased adverse events and mortality | [44] |
| iNO-PF Nathan et al., 2020 | Pulsed inhaled NO | NO | Increased moderate/vigorous physical activity | [45] |
| ARTEMIS-IPF Raghu et al., 2013 | Ambrisentan | ET-1 | No improvement in lung function, respiratory hospitalization, or death; Increased harm | [57] |
| BPHIT Corte et al., 2014 | Bosentan | ET-1 | No decrease to PVR index of 20% or more | [58] |
| INCREASE Waxman et al., 2021 | Inhaled Treprostinil | Prostacyclin | Improvement in 6MWD | [8] |
| Chronic Obstructive Pulmonary Disease-Associated Pulmonary Hypertension (COPD-PH) | ||||
| Blanco et al., 2010 | Sildenafil | NO | Reduced mean PAP | [49] |
| Blanco et al., 2013 | Sildenafil and pulmonary rehabilitation | NO | No improvement in cycle endurance time | [50] |
| Goudie et al., 2014 | Tadalafil | NO | No improvement in 6MWD | [51] |
| SPHERIC-1 Vitulo et al., 2017 | Sildenafil | NO | Reduced PVR | [52] |
| Stolz et al., 2008 | Bosentan | ET-1 | No improvement in 6MWD | [59] |
| Valerio et al., 2009 | Bosentan | ET-1 | Reduced mean PAP and PVR, Increased 6MWD, and reduced BODE index | [60] |
4.1.3. Prostacyclin Pathway
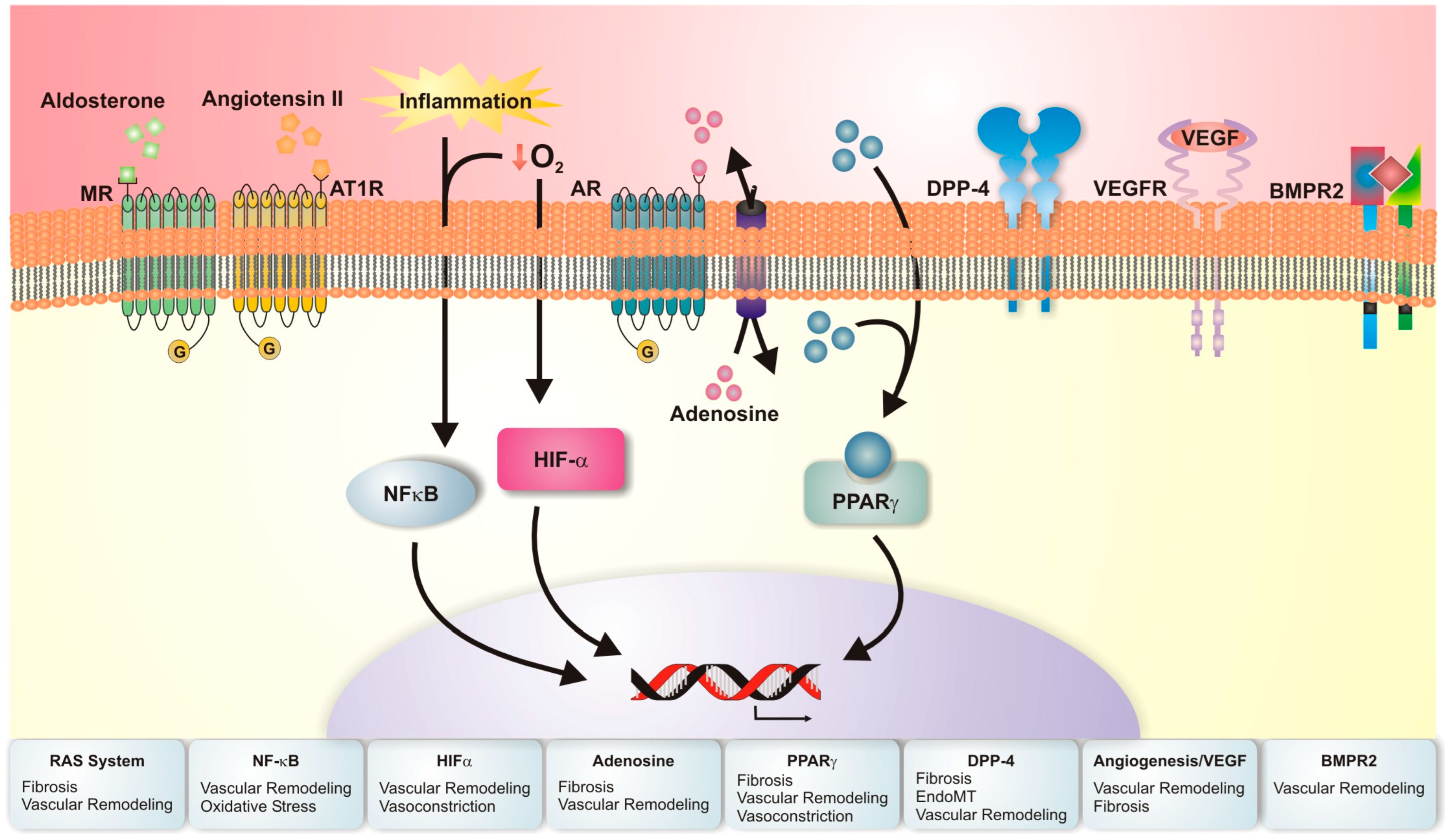
4.2. Emerging Molecular Targets for Pulmonary Hypertension Related to Chronic Lung Disease
4.2.1. Bone Morphogenic Protein Receptor Type II (BMPR2)
4.2.2. Angiogenesis and Vascular Endothelial Growth Factor (VEGF)
4.2.3. Nuclear Factor-Kappa B (NF-κB) Signaling and Oxidative Stress
4.2.4. Pulmonary Renin–Angiotensin System (RAS)
4.2.5. Peroxisome Proliferator-Activated Receptors (PPAR)
4.2.6. Endothelial to Mesenchymal Transition (EndoMT)
4.2.7. Hypoxia–Adrenergic Axis
4.2.8. Hypoxia-Inducible Factor
| Intervention | Target | Model Studied | Outcome | Ref |
|---|---|---|---|---|
| Pulmonary Fibrosis-Associated Pulmonary Hypertension (PF-PH) | ||||
| IL-6 -/- or soluble GP130 (IL-6 inhibitor) | BMPR2 | Bleomycin mice | Increased BMPR2 expression; abrogated development of PH; reduced development of PF | [67] |
| Recombinant BMP9 | BMPR2 | Bleomycin rat | Restored BMPR2 signaling; prevents bleomycin-induced PH and PF | [69] |
| Adenoviral delivery of VEGF | VEGF | Adenoviral delivery of TGFβ-1 in rats | Reduced PAP and pulmonary vascular remodeling; worsened PF | [77] |
| Ang-(1-7) or ACE2 overexpression | RAS | Bleomycin rat Monocrotaline rat | Prevented PH and PF | [95] |
| Recombinant ACE2 | RAS | Bleomycin mice | Attenuated pulmonary vascular remodeling | [96] |
| Compound 21 (AT2 receptor agonist) | RAS | Bleomycin rat | Reduced progression of PF, PH, and muscularization of pulmonary vessels | [98] |
| IVA337 (pan-PPAR agonist) | PPAR | Bleomycin mice Fra-2 transgenic mice | Prevented PF development; improves PH and vascular remodeling in Fra-2 transgenic mice | [112] |
| siRNA IL-11 | EndoMT | Bleomycin mice IL-11 treated mice | Attenuated PF, PH, and vascular remodeling; reduces evidence of EndoMT | [122] |
| Sitagliptin (DPP4 inhibitor) | EndoMT | MCT rat Bleomycin rat Chronic hypoxia rat | Attenuated PH, RV and pulmonary vascular remodeling, and EndoMT in MCT rats; prevented PH in bleomycin and chronic hypoxia rats | [125] |
| ADORA2B myeloid cell KO | Hypoxia-adrenergic axis | Bleomycin mice | Attenuated PF, improved lung function, and prevented PH | [129] |
| Endothelial HIF deficiency | HIF | Bleomycin mice | Prevented PH and RV and vascular remodeling | [132] |
| Chronic Obstructive Pulmonary Disease-Associated Pulmonary Hypertension (COPD-PH) | ||||
| Budesonide glycopyrronium formoterol fumarate therapy | NF-κB | Intratracheal elastase induced emphysema | Prevented PH development and COPD progression | [86] |
| ADORA2B blockade | Hypoxia-adrenergic axis | Adenosine deaminase deficient mice | Attenuated development of PH | [128] |
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADA | Adenosine deaminase |
| AR | Adenosine receptor |
| BMPR | Bone morphogentic protein receptor |
| COPD | Chronic obstructive pulmonary disease |
| DPP-4 | Dipeptidyl peptidase type 4 |
| EndoMT | Endothelial to mesenchymal transition |
| ET-1 | Endothelin-1 |
| ILD | Interstitial lung disease |
| IPF | Idiopathic pulmonary fibrosis |
| HIF | Hypoxia-inducible factor |
| LTOT | Long-term oxygen therapy |
| NF-κB | Nuclear factor kappa B |
| NO | Nitric oxide |
| PAH | Pulmonary arterial hypertension |
| PAP | Pulmonary arterial pressure |
| PCWP | Pulmonary capillary wedge pressure |
| PDE-5 | Phosphodiesterase type 5 |
| PF | Pulmonary fibrosis |
| PH | Pulmonary hypertension |
| PPAR | Peroxisome proliferator-activated receptor |
| PVR | Pulmonary vascular resistance |
| RAS | Renin–angiotensin system |
| RHC | Right heart catheterization |
| RV | Right ventricle |
| TGF-β | Transforming growth factor beta |
| VEGF | Vascular endothelial growth factor |
References
- Simonneau, G.; Montani, D.; Celermajer, D.S.; Denton, C.P.; Gatzoulis, M.A.; Krowka, M.; Williams, P.G.; Souza, R. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur. Respir. J. 2019, 53, 1801913. [Google Scholar] [CrossRef] [PubMed]
- Medrek, S.K.; Sharafkhaneh, A.; Spiegelman, A.M.; Kak, A.; Pandit, L.M. Admission for COPD Exacerbation Is Associated with the Clinical Diagnosis of Pulmonary Hypertension: Results from a Retrospective Longitudinal Study of a Veteran Population. COPD J. Chronic Obstr. Pulm. Dis. 2017, 14, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Hayes, D., Jr.; Black, S.M.; Tobias, J.D.; Kirkby, S.; Mansour, H.M.; Whitson, B.A. Influence of Pulmonary Hypertension on Patients with Idiopathic Pulmonary Fibrosis Awaiting Lung Transplantation. Ann. Thorac. Surg. 2016, 101, 246–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lettieri, C.J.; Nathan, S.D.; Barnett, S.D.; Ahmad, S.; Shorr, A.F. Prevalence and outcomes of pulmonary arterial hypertension in advanced idiopathic pulmonary fibrosis. Chest 2006, 129, 746–752. [Google Scholar] [CrossRef]
- Andersen, K.H.; Iversen, M.; Kjaergaard, J.; Mortensen, J.; Nielsen-Kudsk, J.E.; Bendstrup, E.; Videbaek, R.; Carlsen, J. Prevalence, predictors, and survival in pulmonary hypertension related to end-stage chronic obstructive pulmonary disease. J. Heart Lung Transplant. 2012, 31, 373–380. [Google Scholar] [CrossRef]
- Weitzenblum, E.; Hirth, C.; Ducolone, A.; Mirhom, R.; Rasaholinjanahary, J.; Ehrhart, M. Prognostic value of pulmonary artery pressure in chronic obstructive pulmonary disease. Thorax 1981, 36, 752–758. [Google Scholar] [CrossRef] [Green Version]
- Galiè, N.; Humbert, M.; Vachiery, J.L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Heart J. 2016, 37, 67–119. [Google Scholar]
- Waxman, A.; Restrepo-Jaramillo, R.; Thenappan, T.; Ravichandran, A.; Engel, P.; Bajwa, A.; Allen, R.; Feldman, J.; Argula, R.; Smith, P.; et al. Inhaled Treprostinil in Pulmonary Hypertension Due to Interstitial Lung Disease. N. Engl. J. Med. 2021, 384, 325–334. [Google Scholar] [CrossRef]
- Seeger, W.; Adir, Y.; Barberà, J.A.; Champion, H.; Coghlan, J.G.; Cottin, V.; De Marco, T.; Galiè, N.; Ghio, S.; Gibbs, S.; et al. Pulmonary hypertension in chronic lung diseases. J. Am. Coll. Cardiol. 2013, 62 (Suppl. S25), D109–D116. [Google Scholar] [CrossRef]
- Hamada, K.; Nagai, S.; Tanaka, S.; Handa, T.; Shigematsu, M.; Nagao, T.; Mishima, M.; Kitaichi, M.; Izumi, T. Significance of pulmonary arterial pressure and diffusion capacity of the lung as prognosticator in patients with idiopathic pulmonary fibrosis. Chest 2007, 131, 650–656. [Google Scholar] [CrossRef]
- Kimura, M.; Taniguchi, H.; Kondoh, Y.; Kimura, T.; Kataoka, K.; Nishiyama, O.; Aso, H.; Sakamoto, K.; Hasegawa, Y. Pulmonary hypertension as a prognostic indicator at the initial evaluation in idiopathic pulmonary fibrosis. Respiration 2013, 85, 456–463. [Google Scholar] [CrossRef] [PubMed]
- Behr, J.; Ryu, J.H. Pulmonary hypertension in interstitial lung disease. Eur. Respir. J. 2008, 31, 1357–1367. [Google Scholar] [CrossRef] [PubMed]
- Minai, O.A.; Santacruz, J.F.; Alster, J.M.; Budev, M.M.; McCarthy, K. Impact of pulmonary hemodynamics on 6-min walk test in idiopathic pulmonary fibrosis. Respir. Med. 2012, 106, 1613–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathan, S.D.; Shlobin, O.A.; Ahmad, S.; Koch, J.; Barnett, S.D.; Ad, N.; Burton, N.; Leslie, K. Serial development of pulmonary hypertension in patients with idiopathic pulmonary fibrosis. Respiration 2008, 76, 288–294. [Google Scholar] [CrossRef]
- Chaouat, A.; Bugnet, A.S.; Kadaoui, N.; Schott, R.; Enache, I.; Ducoloné, A.; Ehrhart, M.; Kessler, R. Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2005, 172, 189–194. [Google Scholar] [CrossRef] [Green Version]
- Bove, K.E.; Scott, R.C. The anatomy of chronic cor pulmonale secondary to intrinsic lung disease. Prog. Cardiovasc. Dis. 1966, 9, 227–238. [Google Scholar] [CrossRef]
- Tuder, R.M. Pulmonary vascular remodeling in pulmonary hypertension. Cell Tissue Res. 2017, 367, 643–649. [Google Scholar] [CrossRef] [Green Version]
- Hopkins, N.; McLoughlin, P. The structural basis of pulmonary hypertension in chronic lung disease: Remodelling, rarefaction or angiogenesis? J. Anat. 2002, 201, 335–348. [Google Scholar] [CrossRef]
- Colombat, M.; Mal, H.; Groussard, O.; Capron, F.; Thabut, G.; Jebrak, G.; Brugière, O.; Dauriat, G.; Castier, Y.; Lesèche, G.; et al. Pulmonary vascular lesions in end-stage idiopathic pulmonary fibrosis: Histopathologic study on lung explant specimens and correlations with pulmonary hemodynamics. Hum. Pathol. 2007, 38, 60–65. [Google Scholar] [CrossRef]
- Dorrington, K.L.; Clar, C.; Young, J.D.; Jonas, M.; Tansley, J.G.; Robbins, P.A. Time course of the human pulmonary vascular response to 8 hours of isocapnic hypoxia. Am. J. Physiol. 1997, 273 Pt 2, H1126–H1134. [Google Scholar] [CrossRef]
- Ebina, M.; Shimizukawa, M.; Shibata, N.; Kimura, Y.; Suzuki, T.; Endo, M.; Sasano, H.; Kondo, T.; Nukiwa, T. Heterogeneous increase in CD34-positive alveolar capillaries in idiopathic pulmonary fibrosis. Am. J. Respir Crit. Care Med. 2004, 169, 1203–1208. [Google Scholar] [CrossRef] [PubMed]
- Tachihara, A.; Jin, E.; Matsuoka, T.; Ghazizadeh, M.; Yoshino, S.; Takemura, T.; W, D.T.; Kawanami, O. Critical roles of capillary endothelial cells for alveolar remodeling in nonspecific and usual interstitial pneumonias. J. Nippon. Med. Sch. 2006, 73, 203–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farkas, L.; Gauldie, J.; Voelkel, N.F.; Kolb, M. Pulmonary hypertension and idiopathic pulmonary fibrosis: A tale of angiogenesis, apoptosis, and growth factors. Am. J. Respir. Cell Mol. Biol. 2011, 45, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gagermeier, J.; Dauber, J.; Yousem, S.; Gibson, K.; Kaminski, N. Abnormal vascular phenotypes in patients with idiopathic pulmonary fibrosis and secondary pulmonary hypertension. Chest 2005, 128 (Suppl. S6), 601s. [Google Scholar] [CrossRef]
- Mura, M.; Anraku, M.; Yun, Z.; McRae, K.; Liu, M.; Waddell, T.K.; Singer, L.G.; Granton, J.T.; Keshavjee, S.; de Perrot, M. Gene expression profiling in the lungs of patients with pulmonary hypertension associated with pulmonary fibrosis. Chest 2012, 141, 661–673. [Google Scholar] [CrossRef]
- Yu, P.N.; Lovejoy, F.W.; Jr Joos, H.A.; Nye, R.E.; Jr McCann, W.S. Studies of pulmonary hypertension. I. Pulmonary circulatory dynamics in patients with pulmonary emphysema at rest. J. Clin Investig. 1953, 32, 130–137. [Google Scholar] [CrossRef] [Green Version]
- Fishman, A.P.; Fritts, H.W., Jr.; Cournand, A. Effects of breathing carbon dioxide upon the pulmonary circulation. Circulation 1960, 22, 220–225. [Google Scholar] [CrossRef] [Green Version]
- Hemlin, M.; Ljungman, S.; Carlson, J.; Maljukanovic, S.; Mobini, R.; Bech-Hanssen, O.; Skoogh, B.E. The effects of hypoxia and hypercapnia on renal and heart function, haemodynamics and plasma hormone levels in stable COPD patients. Clin. Respir. J. 2007, 1, 80–90. [Google Scholar] [CrossRef]
- Anand, I.S.; Chandrashekhar, Y.; Ferrari, R.; Sarma, R.; Guleria, R.; Jindal, S.K.; Wahi, P.L.; Poole-Wilson, P.A.; Harris, P. Pathogenesis of congestive state in chronic obstructive pulmonary disease. Studies of body water and sodium, renal function, hemodynamics, and plasma hormones during edema and after recovery. Circulation 1992, 86, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Wrobel, J.P.; Thompson, B.R.; Williams, T.J. Mechanisms of pulmonary hypertension in chronic obstructive pulmonary disease: A pathophysiologic review. J. Heart Lung Transplant. 2012, 31, 557–564. [Google Scholar] [CrossRef]
- Hale, K.A.; Ewing, S.L.; Gosnell, B.A.; Niewoehner, D.E. Lung disease in long-term cigarette smokers with and without chronic air-flow obstruction. Am. Rev. Respir. Dis. 1984, 130, 716–721. [Google Scholar] [PubMed]
- Nathan, S.D.; Barbera, J.A.; Gaine, S.P.; Harari, S.; Martinez, F.J.; Olschewski, H.; Olsson, K.M.; Peacock, A.J.; Pepke-Zaba, J.; Provencher, S.; et al. Pulmonary hypertension in chronic lung disease and hypoxia. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weitzenblum, E.; Sautegeau, A.; Ehrhart, M.; Mammosser, M.; Pelletier, A. Long-term oxygen therapy can reverse the progression of pulmonary hypertension in patients with chronic obstructive pulmonary disease. Am. Rev. Respir. Dis. 1985, 131, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Klinger, J.R.; Kadowitz, P.J. The Nitric Oxide Pathway in Pulmonary Vascular Disease. Am. J. Cardiol. 2017, 120, S71–S79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giaid, A. Nitric oxide and endothelin-1 in pulmonary hypertension. Chest 1998, 114 (Suppl. S3), 208s–212s. [Google Scholar] [CrossRef]
- Barberà, J.A.; Peinado, V.I.; Santos, S.; Ramirez, J.; Roca, J.; Rodriguez-Roisin, R. Reduced expression of endothelial nitric oxide synthase in pulmonary arteries of smokers. Am. J. Respir. Crit. Care Med. 2001, 164, 709–713. [Google Scholar] [CrossRef]
- Ghofrani, H.A.; Wiedemann, R.; Rose, F.; Schermuly, R.T.; Olschewski, H.; Weissmann, N.; Gunther, A.; Walmrath, D.; Seeger, W.; Grimminger, F. Sildenafil for treatment of lung fibrosis and pulmonary hypertension: A randomised controlled trial. Lancet 2002, 360, 895–900. [Google Scholar] [CrossRef]
- Zisman, D.A.; Schwarz, M.; Anstrom, K.J.; Collard, H.R.; Flaherty, K.R.; Hunninghake, G.W. A controlled trial of sildenafil in advanced idiopathic pulmonary fibrosis. N. Engl. J. Med. 2010, 363, 620–628. [Google Scholar]
- Behr, J.; Nathan, S.D.; Wuyts, W.A.; Mogulkoc Bishop, N.; Bouros, D.E.; Antoniou, K.; Guiot, J.; Kramer, M.R.; Kirchgaessler, K.U.; Bengus, M.; et al. Efficacy and safety of sildenafil added to pirfenidone in patients with advanced idiopathic pulmonary fibrosis and risk of pulmonary hypertension: A double-blind, randomised, placebo-controlled, phase 2b trial. Lancet Respir. Med. 2021, 9, 85–95. [Google Scholar] [CrossRef]
- Ghofrani, H.A.; Galiè, N.; Grimminger, F.; Grünig, E.; Humbert, M.; Jing, Z.C.; Keogh, A.M.; Langleben, D.; Kilama, M.O.; Fritsch, A.; et al. Riociguat for the treatment of pulmonary arterial hypertension. N. Engl. J. Med. 2013, 369, 330–340. [Google Scholar] [CrossRef] [Green Version]
- Ghofrani, H.A.; D’Armini, A.M.; Grimminger, F.; Hoeper, M.M.; Jansa, P.; Kim, N.H.; Mayer, E.; Simonneau, G.; Wilkins, M.R.; Fritsch, A.; et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N. Engl. J. Med. 2013, 369, 319–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geschka, S.; Kretschmer, A.; Sharkovska, Y.; Evgenov, O.V.; Lawrenz, B.; Hucke, A.; Hocher, B.; Stasch, J.P. Soluble guanylate cyclase stimulation prevents fibrotic tissue remodeling and improves survival in salt-sensitive Dahl rats. PLoS ONE 2011, 6, e21853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyer, C.; Zenzmaier, C.; Palumbo-Zerr, K.; Mancuso, R.; Distler, A.; Dees, C.; Zerr, P.; Huang, J.; Maier, C.; Pachowsky, M.L.; et al. Stimulation of the soluble guanylate cyclase (sGC) inhibits fibrosis by blocking non-canonical TGFβ signalling. Ann. Rheum. Dis. 2015, 74, 1408–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathan, S.D.; Behr, J.; Collard, H.R.; Cottin, V.; Hoeper, M.M.; Martinez, F.J.; Corte, T.J.; Keogh, A.M.; Leuchte, H.; Mogulkoc, N.; et al. Riociguat for idiopathic interstitial pneumonia-associated pulmonary hypertension (RISE-IIP): A randomised, placebo-controlled phase 2b study. Lancet Respir. Med. 2019, 7, 780–790. [Google Scholar] [CrossRef] [PubMed]
- Nathan, S.D.; Flaherty, K.R.; Glassberg, M.K.; Raghu, G.; Swigris, J.; Alvarez, R.; Ettinger, N.; Loyd, J.; Fernandes, P.; Gillies, H.; et al. A Randomized, Double-Blind, Placebo-Controlled Study of Pulsed, Inhaled Nitric Oxide in Subjects at Risk of Pulmonary Hypertension Associated with Pulmonary Fibrosis. Chest 2020, 158, 637–645. [Google Scholar] [CrossRef]
- Khor, Y.H.; Adegunsoye, A. Inhaled Nitric Oxide for Fibrotic Interstitial Lung Disease: A Step Forward. Ann. Am. Thorac. Soc. 2022, 19, 536–538. [Google Scholar] [CrossRef]
- Rao, R.S.; Singh, S.; Sharma, B.B.; Agarwal, V.V.; Singh, V. Sildenafil improves six-minute walk distance in chronic obstructive pulmonary disease: A randomised, double-blind, placebo-controlled trial. Indian J. Chest Dis. Allied Sci. 2011, 53, 81–85. [Google Scholar]
- Lederer, D.J.; Bartels, M.N.; Schluger, N.W.; Brogan, F.; Jellen, P.; Thomashow, B.M.; Kawut, S.M. Sildenafil for chronic obstructive pulmonary disease: A randomized crossover trial. COPD J. Chronic Obstr. Pulm. Dis. 2012, 9, 268–275. [Google Scholar] [CrossRef] [Green Version]
- Blanco, I.; Gimeno, E.; Munoz, P.A.; Pizarro, S.; Gistau, C.; Rodriguez-Roisin, R.; Roca, J.; Barberà, J.A. Hemodynamic and gas exchange effects of sildenafil in patients with chronic obstructive pulmonary disease and pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2010, 181, 270–278. [Google Scholar] [CrossRef]
- Blanco, I.; Santos, S.; Gea, J.; Güell, R.; Torres, F.; Gimeno-Santos, E.; Rodriguez, D.A.; Vilaró, J.; Gómez, B.; Roca, J.; et al. Sildenafil to improve respiratory rehabilitation outcomes in COPD: A controlled trial. Eur. Respir. J. 2013, 42, 982–992. [Google Scholar] [CrossRef]
- Goudie, A.R.; Lipworth, B.J.; Hopkinson, P.J.; Wei, L.; Struthers, A.D. Tadalafil in patients with chronic obstructive pulmonary disease: A randomised, double-blind, parallel-group, placebo-controlled trial. Lancet Respir. Med. 2014, 2, 293–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitulo, P.; Stanziola, A.; Confalonieri, M.; Libertucci, D.; Oggionni, T.; Rottoli, P.; Paciocco, G.; Tuzzolino, F.; Martino, L.; Beretta, M.; et al. Sildenafil in severe pulmonary hypertension associated with chronic obstructive pulmonary disease: A randomized controlled multicenter clinical trial. J. Heart Lung. Transplant. 2017, 36, 166–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shioleno, A.M.; Ruopp, N.F. Group 3 Pulmonary Hypertension: A Review of Diagnostics and Clinical Trials. Clin. Chest Med. 2021, 42, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Galié, N.; Manes, A.; Branzi, A. The endothelin system in pulmonary arterial hypertension. Cardiovasc. Res. 2004, 61, 227–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giaid, A.; Yanagisawa, M.; Langleben, D.; Michel, R.P.; Levy, R.; Shennib, H.; Kimura, S.; Masaki, T.; Duguid, W.P.; Stewart, D.J. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N. Engl. J. Med. 1993, 328, 1732–1739. [Google Scholar] [CrossRef] [PubMed]
- Stolz, D.; Christ-Crain, M.; Morgenthaler, N.G.; Miedinger, D.; Leuppi, J.; Müller, C.; Bingisser, R.; Struck, J.; Müller, B.; Tamm, M. Plasma pro-adrenomedullin but not plasma pro-endothelin predicts survival in exacerbations of COPD. Chest 2008, 134, 263–272. [Google Scholar] [CrossRef]
- Raghu, G.; Behr, J.; Brown, K.K.; Egan, J.J.; Kawut, S.M.; Flaherty, K.R.; Martinez, F.J.; Nathan, S.D.; Wells, A.U.; Collard, H.R.; et al. Treatment of idiopathic pulmonary fibrosis with ambrisentan: A parallel, randomized trial. Ann. Intern. Med. 2013, 158, 641–649. [Google Scholar] [CrossRef]
- Corte, T.J.; Keir, G.J.; Dimopoulos, K.; Howard, L.; Corris, P.A.; Parfitt, L.; Foley, C.; Yanez-Lopez, M.; Babalis, D.; Marino, P.; et al. Bosentan in pulmonary hypertension associated with fibrotic idiopathic interstitial pneumonia. Am. J. Respir. Crit. Care Med. 2014, 190, 208–217. [Google Scholar] [CrossRef] [Green Version]
- Stolz, D.; Rasch, H.; Linka, A.; Di Valentino, M.; Meyer, A.; Brutsche, M.; Tamm, M. A randomised, controlled trial of bosentan in severe COPD. Eur. Respir. J. 2008, 32, 619–628. [Google Scholar] [CrossRef]
- Valerio, G.; Bracciale, P.; Grazia D’Agostino, A. Effect of bosentan upon pulmonary hypertension in chronic obstructive pulmonary disease. Ther. Adv. Respir. Dis. 2009, 3, 15–21. [Google Scholar] [CrossRef]
- Galiè, N.; Manes, A.; Branzi, A. Prostanoids for pulmonary arterial hypertension. Am. J. Respir. Med. 2003, 2, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Waxman, A.B.; Elia, D.; Adir, Y.; Humbert, M.; Harari, S. Recent advances in the management of pulmonary hypertension with interstitial lung disease. Eur. Respir. Rev. 2022, 31. [Google Scholar] [CrossRef] [PubMed]
- Nathan, S.D.; Cottin, V.; Behr, J.; Hoeper, M.M.; Martinez, F.J.; Corte, T.J.; Keogh, A.M.; Leuchte, H.; Mogulkoc, N.; Ulrich, S.; et al. Impact of lung morphology on clinical outcomes with riociguat in patients with pulmonary hypertension and idiopathic interstitial pneumonia: A post hoc subgroup analysis of the RISE-IIP study. J. Heart Lung. Transplant. 2021, 40, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Morrell, N.W.; Aldred, M.A.; Chung, W.K.; Elliott, C.G.; Nichols, W.C.; Soubrier, F.; Trembath, R.C.; Loyd, J.E. Genetics and genomics of pulmonary arterial hypertension. Eur. Respir. J. 2019, 53. [Google Scholar] [CrossRef] [Green Version]
- Tielemans, B.; Delcroix, M.; Belge, C.; Quarck, R. TGFβ and BMPRII signalling pathways in the pathogenesis of pulmonary arterial hypertension. Drug Discov. Today 2019, 24, 703–716. [Google Scholar] [CrossRef]
- Rajkumar, R.; Konishi, K.; Richards, T.J.; Ishizawar, D.C.; Wiechert, A.C.; Kaminski, N.; Ahmad, F. Genomewide RNA expression profiling in lung identifies distinct signatures in idiopathic pulmonary arterial hypertension and secondary pulmonary hypertension. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1235-48. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.Y.; S, D.C.; Luo, F.; Weng, T.; Le, T.T.; A, M.H.; Philip, K.; Molina, J.G.; Garcia-Morales, L.J.; Cao, Y.; et al. Macrophage bone morphogenic protein receptor 2 depletion in idiopathic pulmonary fibrosis and Group III pulmonary hypertension. Am. J. Physiol Lung Cell Mol. Physiol. 2016, 311, L238-54. [Google Scholar] [CrossRef] [Green Version]
- Bryant, A.J.; Robinson, L.J.; Moore, C.S.; Blackwell, T.R.; Gladson, S.; Penner, N.L.; Burman, A.; McClellan, L.J.; Polosukhin, V.V.; Tanjore, H.; et al. Expression of mutant bone morphogenetic protein receptor II worsens pulmonary hypertension secondary to pulmonary fibrosis. Pulm. Circ. 2015, 5, 681–690. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q.; Liu, C.; Liu, S.; Lu, W.; Li, Y.; Luo, X.; Ma, R.; Zhang, C.; Chen, H.; Chen, Y.; et al. Dysregulation of BMP9/BMPR2/SMAD signalling pathway contributes to pulmonary fibrosis and pulmonary hypertension induced by bleomycin in rats. Br. J. Pharmacol. 2021, 178, 203–216. [Google Scholar] [CrossRef]
- Yung, L.M.; Yang, P.; Joshi, S.; Augur, Z.M.; Kim, S.S.J.; Bocobo, G.A.; Dinter, T.; Troncone, L.; Chen, P.S.; McNeil, M.E.; et al. ACTRIIA-Fc rebalances activin/GDF versus BMP signaling in pulmonary hypertension. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Humbert, M.; McLaughlin, V.; Gibbs, J.S.R.; Gomberg-Maitland, M.; Hoeper, M.M.; Preston, I.R.; Souza, R.; Waxman, A.; Escribano Subias, P.; Feldman, J.; et al. Sotatercept for the Treatment of Pulmonary Arterial Hypertension. N. Engl. J. Med. 2021, 384, 1204–1215. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, G.P.; Brown, K.K.; Schiemann, W.P.; Serls, A.E.; Parr, J.E.; Geraci, M.W.; Schwarz, M.I.; Cool, C.D.; Worthen, G.S. Pigment epithelium-derived factor in idiopathic pulmonary fibrosis: A role in aberrant angiogenesis. Am. J. Respir. Crit Care Med. 2004, 170, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Ruffenach, G.; Hong, J.; Vaillancourt, M.; Medzikovic, L.; Eghbali, M. Pulmonary hypertension secondary to pulmonary fibrosis: Clinical data, histopathology and molecular insights. Respir. Res. 2020, 21, 303. [Google Scholar] [CrossRef] [PubMed]
- Tuder, R.M.; Flook, B.E.; Voelkel, N.F. Increased gene expression for VEGF and the VEGF receptors KDR/Flk and Flt in lungs exposed to acute or to chronic hypoxia. Modulation of gene expression by nitric oxide. J. Clin. Investig. 1995, 95, 1798–1807. [Google Scholar] [CrossRef] [Green Version]
- Partovian, C.; Adnot, S.; Eddahibi, S.; Teiger, E.; Levame, M.; Dreyfus, P.; Raffestin, B.; Frelin, C. Heart and lung VEGF mRNA expression in rats with monocrotaline- or hypoxia-induced pulmonary hypertension. Am. J. Physiol. 1998, 275, H1948-56. [Google Scholar] [CrossRef]
- Christou, H.; Yoshida, A.; Arthur, V.; Morita, T.; Kourembanas, S. Increased vascular endothelial growth factor production in the lungs of rats with hypoxia-induced pulmonary hypertension. Am. J. Respir. Cell Mol. Biol. 1998, 18, 768–776. [Google Scholar] [CrossRef] [Green Version]
- Farkas, L.; Farkas, D.; Ask, K.; Möller, A.; Gauldie, J.; Margetts, P.; Inman, M.; Kolb, M. VEGF ameliorates pulmonary hypertension through inhibition of endothelial apoptosis in experimental lung fibrosis in rats. J. Clin. Investig. 2009, 119, 1298–1311. [Google Scholar] [CrossRef] [Green Version]
- Barnes, P.J.; Karin, M. Nuclear factor-kappaB: A pivotal transcription factor in chronic inflammatory diseases. N. Engl. J. Med. 1997, 336, 1066–1071. [Google Scholar] [CrossRef]
- Hart, L.A.; Krishnan, V.L.; Adcock, I.M.; Barnes, P.J.; Chung, K.F. Activation and localization of transcription factor, nuclear factor-kappaB, in asthma. Am J. Respir Crit Care Med. 1998, 158 Pt 1, 1585–1592. [Google Scholar] [CrossRef] [Green Version]
- Di Stefano, A.; Caramori, G.; Oates, T.; Capelli, A.; Lusuardi, M.; Gnemmi, I.; Ioli, F.; Chung, K.F.; Donner, C.F.; Barnes, P.J.; et al. Increased expression of nuclear factor-kappaB in bronchial biopsies from smokers and patients with COPD. Eur. Respir. J. 2002, 20, 556–563. [Google Scholar] [CrossRef] [Green Version]
- Marok, R.; Winyard, P.G.; Coumbe, A.; Kus, M.L.; Gaffney, K.; Blades, S.; Mapp, P.I.; Morris, C.J.; Blake, D.R.; Kaltschmidt, C.; et al. Activation of the transcription factor nuclear factor-kappaB in human inflamed synovial tissue. Arthritis. Rheum. 1996, 39, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wei, C.; Kim, I.K.; Janssen-Heininger, Y.; Gupta, S. Inhibition of nuclear factor-κB in the lungs prevents monocrotaline-induced pulmonary hypertension in mice. Hypertension 2014, 63, 1260–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, L.C.; Caramori, G.; Perros, F.; Meng, C.; Gambaryan, N.; Dorfmuller, P.; Montani, D.; Casolari, P.; Zhu, J.; Dimopoulos, K.; et al. Nuclear factor κ-B is activated in the pulmonary vessels of patients with end-stage idiopathic pulmonary arterial hypertension. PLoS ONE 2013, 8, e75415. [Google Scholar] [CrossRef] [Green Version]
- De Martin, R.; Hoeth, M.; Hofer-Warbinek, R.; Schmid, J.A. The transcription factor NF-kappa B and the regulation of vascular cell function. Arterioscler. Thromb. Vasc. Biol. 2000, 20, E83–E88. [Google Scholar]
- Zuo, Z.T.; Ma, Y.; Sun, Y.; Bai, C.Q.; Zhou, H.Y.; Chen, B.H. Role of TLR4/NF-κB Signalling Pathway in Pulmonary Arterial Hypertension in Patients with Chronic Obstructive Pulmonary Disease. J. Coll Physicians. Surg. Pak. 2020, 30, 568–573. [Google Scholar] [PubMed]
- Suraya, R.; Nagano, T.; Ryanto, G.R.T.; Effendi, W.I.; Hazama, D.; Katsurada, N.; Yamamoto, M.; Tachihara, M.; Emoto, N.; Nishimura, Y.; et al. Budesonide/glycopyrronium/formoterol fumarate triple therapy prevents pulmonary hypertension in a COPD mouse model via NFκB inactivation. Respir. Res. 2022, 23, 173. [Google Scholar] [CrossRef]
- Eba, S.; Hoshikawa, Y.; Moriguchi, T.; Mitsuishi, Y.; Satoh, H.; Ishida, K.; Watanabe, T.; Shimizu, T.; Shimokawa, H.; Okada, Y.; et al. The nuclear factor erythroid 2-related factor 2 activator oltipraz attenuates chronic hypoxia-induced cardiopulmonary alterations in mice. Am. J. Respir. Cell Mol. Biol. 2013, 49, 324–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Condon, D.F.; Agarwal, S.; Chakraborty, A.; Auer, N.; Vazquez, R.; Patel, H.; Zamanian, R.T.; de Jesus Perez, V.A. Novel Mechanisms Targeted by Drug Trials in Pulmonary Arterial Hypertension. Chest 2022, 161, 1060–1072. [Google Scholar] [CrossRef]
- Hoshikawa, Y.; Ono, S.; Suzuki, S.; Tanita, T.; Chida, M.; Song, C.; Noda, M.; Tabata, T.; Voelkel, N.F.; Fujimura, S. Generation of oxidative stress contributes to the development of pulmonary hypertension induced by hypoxia. J. Appl. Physiol. 2001, 90, 1299–1306. [Google Scholar] [CrossRef] [Green Version]
- Mouradian, G.C.; Gaurav, R.; Pugliese, S.; El Kasmi, K.; Hartman, B.; Hernandez-Lagunas, L.; Stenmark, K.R.; Bowler, R.P.; Nozik-Grayck, E. Superoxide Dismutase 3 R213G Single-Nucleotide Polymorphism Blocks Murine Bleomycin-Induced Fibrosis and Promotes Resolution of Inflammation. Am. J. Respir. Cell Mol. Biol. 2017, 56, 362–371. [Google Scholar] [CrossRef] [Green Version]
- Van Rheen, Z.; Fattman, C.; Domarski, S.; Majka, S.; Klemm, D.; Stenmark, K.R.; Nozik-Grayck, E. Lung extracellular superoxide dismutase overexpression lessens bleomycin-induced pulmonary hypertension and vascular remodeling. Am. J. Respir. Cell Mol. Biol. 2011, 44, 500–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, R.P.; McAnulty, R.J.; Laurent, G.J. Angiotensin II is mitogenic for human lung fibroblasts via activation of the type 1 receptor. Am. J. Respir. Crit. Care Med. 2000, 161, 1999–2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhal, B.D.; Kim, J.K.; Li, X.; Molina-Molina, M. Angiotensin-TGF-beta 1 crosstalk in human idiopathic pulmonary fibrosis: Autocrine mechanisms in myofibroblasts and macrophages. Curr. Pharm. Des. 2007, 13, 1247–1256. [Google Scholar] [CrossRef] [PubMed]
- Orte, C.; Polak, J.M.; Haworth, S.G.; Yacoub, M.H.; Morrell, N.W. Expression of pulmonary vascular angiotensin-converting enzyme in primary and secondary plexiform pulmonary hypertension. J. Pathol. 2000, 192, 379–384. [Google Scholar] [CrossRef]
- Shenoy, V.; Ferreira, A.J.; Qi, Y.; Fraga-Silva, R.A.; Díez-Freire, C.; Dooies, A.; Jun, J.Y.; Sriramula, S.; Mariappan, N.; Pourang, D.; et al. The angiotensin-converting enzyme 2/angiogenesis-(1-7)/Mas axis confers cardiopulmonary protection against lung fibrosis and pulmonary hypertension. Am. J. Respir. Crit. Care Med. 2010, 182, 1065–1072. [Google Scholar] [CrossRef] [Green Version]
- Rathinasabapathy, A.; Bryant, A.J.; Suzuki, T.; Moore, C.; Shay, S.; Gladson, S.; West, J.D.; Carrier, E.J. rhACE2 Therapy Modifies Bleomycin-Induced Pulmonary Hypertension via Rescue of Vascular Remodeling. Front. Physiol. 2018, 9, 271. [Google Scholar] [CrossRef]
- Shenoy, V.; Kwon, K.C.; Rathinasabapathy, A.; Lin, S.; Jin, G.; Song, C.; Shil, P.; Nair, A.; Qi, Y.; Li, Q.; et al. Oral delivery of Angiotensin-converting enzyme 2 and Angiotensin-(1-7) bioencapsulated in plant cells attenuates pulmonary hypertension. Hypertension 2014, 64, 1248–1259. [Google Scholar] [CrossRef] [Green Version]
- Rathinasabapathy, A.; Horowitz, A.; Horton, K.; Kumar, A.; Gladson, S.; Unger, T.; Martinez, D.; Bedse, G.; West, J.; Raizada, M.K.; et al. The Selective Angiotensin II Type 2 Receptor Agonist, Compound 21, Attenuates the Progression of Lung Fibrosis and Pulmonary Hypertension in an Experimental Model of Bleomycin-Induced Lung Injury. Front. Physiol. 2018, 9, 180. [Google Scholar] [CrossRef] [Green Version]
- Hemnes, A.R.; Rathinasabapathy, A.; Austin, E.A.; Brittain, E.L.; Carrier, E.J.; Chen, X.; Fessel, J.P.; Fike, C.D.; Fong, P.; Fortune, N.; et al. A potential therapeutic role for angiotensin-converting enzyme 2 in human pulmonary arterial hypertension. Eur. Respir. J. 2018, 51. [Google Scholar] [CrossRef]
- Sommer, N.; Ghofrani, H.A.; Pak, O.; Bonnet, S.; Provencher, S.; Sitbon, O.; Rosenkranz, S.; Hoeper, M.M.; Kiely, D.G. Current and future treatments of pulmonary arterial hypertension. Br. J. Pharmacol. 2021, 178, 6–30. [Google Scholar] [CrossRef] [Green Version]
- Hart, C.M.; Roman, J.; Reddy, R.; Sime, P.J. PPARγ: A Novel Molecular Target in Lung Disease. J. Investig. Med. 2008, 56, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Ameshima, S.; Golpon, H.; Cool, C.D.; Chan, D.; Vandivier, R.W.; Gardai, S.J.; Wick, M.; Nemenoff, R.A.; Geraci, M.W.; Voelkel, N.F. Peroxisome proliferator-activated receptor gamma (PPARgamma) expression is decreased in pulmonary hypertension and affects endothelial cell growth. Circ. Res. 2003, 92, 1162–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonfield, T.L.; Farver, C.F.; Barna, B.P.; Malur, A.; Abraham, S.; Raychaudhuri, B.; Kavuru, M.S.; Thomassen, M.J. Peroxisome proliferator-activated receptor-gamma is deficient in alveolar macrophages from patients with alveolar proteinosis. Am. J. Respir. Cell Mol. Biol. 2003, 29, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Culver, D.A.; Barna, B.P.; Raychaudhuri, B.; Bonfield, T.L.; Abraham, S.; Malur, A.; Farver, C.F.; Kavuru, M.S.; Thomassen, M.J. Peroxisome proliferator-activated receptor gamma activity is deficient in alveolar macrophages in pulmonary sarcoidosis. Am. J. Respir. Cell Mol. Biol. 2004, 30, 1–5. [Google Scholar] [CrossRef]
- Kökény, G.; Calvier, L.; Legchenko, E.; Chouvarine, P.; Mózes, M.M.; Hansmann, G. PPARγ is a gatekeeper for extracellular matrix and vascular cell homeostasis: Beneficial role in pulmonary hypertension and renal/cardiac/pulmonary fibrosis. Curr. Opin. Nephrol. Hypertens. 2020, 29, 171–179. [Google Scholar] [CrossRef]
- Guignabert, C.; Alvira, C.M.; Alastalo, T.P.; Sawada, H.; Hansmann, G.; Zhao, M.; Wang, L.; El-Bizri, N.; Rabinovitch, M. Tie2-mediated loss of peroxisome proliferator-activated receptor-gamma in mice causes PDGF receptor-beta-dependent pulmonary arterial muscularization. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L1082-90. [Google Scholar] [CrossRef] [Green Version]
- Calvier, L.; Boucher, P.; Herz, J.; Hansmann, G. LRP1 Deficiency in Vascular SMC Leads to Pulmonary Arterial Hypertension That Is Reversed by PPARγ Activation. Circ. Res. 2019, 124, 1778–1785. [Google Scholar] [CrossRef]
- Samah, M.; El-Aidy Ael, R.; Tawfik, M.K.; Ewais, M.M. Evaluation of the antifibrotic effect of fenofibrate and rosiglitazone on bleomycin-induced pulmonary fibrosis in rats. Eur. J. Pharmacol. 2012, 689, 186–193. [Google Scholar] [CrossRef]
- Aoki, Y.; Maeno, T.; Aoyagi, K.; Ueno, M.; Aoki, F.; Aoki, N.; Nakagawa, J.; Sando, Y.; Shimizu, Y.; Suga, T.; et al. Pioglitazone, a peroxisome proliferator-activated receptor gamma ligand, suppresses bleomycin-induced acute lung injury and fibrosis. Respiration 2009, 77, 311–319. [Google Scholar] [CrossRef]
- Galuppo, M.; Di Paola, R.; Mazzon, E.; Esposito, E.; Paterniti, I.; Kapoor, A.; Thiemermann, C.; Cuzzocrea, S. GW0742, a high affinity PPAR-β/δ agonist reduces lung inflammation induced by bleomycin instillation in mice. Int. J. Immunopathol. Pharmacol. 2010, 23, 1033–1046. [Google Scholar] [CrossRef]
- Francque, S.M.; Bedossa, P.; Ratziu, V.; Anstee, Q.M.; Bugianesi, E.; Sanyal, A.J.; Loomba, R.; Harrison, S.A.; Balabanska, R.; Mateva, L.; et al. A Randomized, Controlled Trial of the Pan-PPAR Agonist Lanifibranor in NASH. N. Engl. J. Med. 2021, 385, 1547–1558. [Google Scholar] [CrossRef]
- Avouac, J.; Konstantinova, I.; Guignabert, C.; Pezet, S.; Sadoine, J.; Guilbert, T.; Cauvet, A.; Tu, L.; Luccarini, J.M.; Junien, J.L.; et al. Pan-PPAR agonist IVA337 is effective in experimental lung fibrosis and pulmonary hypertension. Ann. Rheum. Dis. 2017, 76, 1931–1940. [Google Scholar] [CrossRef] [PubMed]
- Derrett-Smith, E.; Clark, K.E.N.; Shiwen, X.; Abraham, D.J.; Hoyles, R.K.; Lacombe, O.; Broqua, P.; Junien, J.L.; Konstantinova, I.; Ong, V.H.; et al. The pan-PPAR agonist lanifibranor reduces development of lung fibrosis and attenuates cardiorespiratory manifestations in a transgenic mouse model of systemic sclerosis. Arthritis Res. Ther. 2021, 23, 234. [Google Scholar] [CrossRef]
- Hansmann, G.; de Jesus Perez, V.A.; Alastalo, T.P.; Alvira, C.M.; Guignabert, C.; Bekker, J.M.; Schellong, S.; Urashima, T.; Wang, L.; Morrell, N.W.; et al. An antiproliferative BMP-2/PPARgamma/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J. Clin. Investig. 2008, 118, 1846–1857. [Google Scholar] [CrossRef] [Green Version]
- Calvier, L.; Chouvarine, P.; Legchenko, E.; Hoffmann, N.; Geldner, J.; Borchert, P.; Jonigk, D.; Mozes, M.M.; Hansmann, G. PPARγ Links BMP2 and TGFβ1 Pathways in Vascular Smooth Muscle Cells, Regulating Cell Proliferation and Glucose Metabolism. Cell Metab. 2017, 25, 1118–1134e7. [Google Scholar] [CrossRef] [Green Version]
- Maquigussa, E.; Paterno, J.C.; de Oliveira Pokorny, G.H.; da Silva Perez, M.; Varela, V.A.; da Silva Novaes, A.; Schor, N.; Boim, M.A. Klotho and PPAR Gamma Activation Mediate the Renoprotective Effect of Losartan in the 5/6 Nephrectomy Model. Front. Physiol. 2018, 9, 1033. [Google Scholar] [CrossRef] [Green Version]
- Piera-Velazquez, S.; Jimenez, S.A. Endothelial to Mesenchymal Transition: Role in Physiology and in the Pathogenesis of Human Diseases. Physiol. Rev. 2019, 99, 1281–1324. [Google Scholar] [CrossRef]
- Tang, H.; Babicheva, A.; McDermott, K.M.; Gu, Y.; Ayon, R.J.; Song, S.; Wang, Z.; Gupta, A.; Zhou, T.; Sun, X.; et al. Endothelial HIF-2α contributes to severe pulmonary hypertension due to endothelial-to-mesenchymal transition. Am. J. Physiol Lung Cell Mol. Physiol. 2018, 314, L256–L275. [Google Scholar] [PubMed]
- Good, R.B.; Gilbane, A.J.; Trinder, S.L.; Denton, C.P.; Coghlan, G.; Abraham, D.J.; Holmes, A.M. Endothelial to Mesenchymal Transition Contributes to Endothelial Dysfunction in Pulmonary Arterial Hypertension. Am. J. Pathol. 2015, 185, 1850–1858. [Google Scholar] [CrossRef] [PubMed]
- Milara, J.; Ballester, B.; Morell, A.; Ortiz, J.L.; Escrivá, J.; Fernández, E.; Perez-Vizcaino, F.; Cogolludo, A.; Pastor, E.; Artigues, E.; et al. JAK2 mediates lung fibrosis, pulmonary vascular remodelling and hypertension in idiopathic pulmonary fibrosis: An experimental study. Thorax 2018, 73, 519–529. [Google Scholar] [CrossRef]
- Milara, J.; Escrivá, J.; Ortiz, J.L.; Juan, G.; Artigues, E.; Morcillo, E.; Cortijo, J. Vascular effects of sildenafil in patients with pulmonary fibrosis and pulmonary hypertension: An ex vivo/in vitro study. Eur. Respir. J. 2016, 47, 1737–1749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milara, J.; Roger, I.; Montero, P.; Artigues, E.; Escrivá, J.; Cortijo, J. IL-11 system participates in pulmonary artery remodeling and hypertension in pulmonary fibrosis. Respir. Res. 2022, 23, 313. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, N.; Phan, S.H.; Imaizumi, K.; Matsuo, M.; Nakashima, H.; Kawabe, T.; Shimokata, K.; Hasegawa, Y. Endothelial-mesenchymal transition in bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2010, 43, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Tada, Y.; Gladson, S.; Nishimura, R.; Shimomura, I.; Karasawa, S.; Tatsumi, K.; West, J. Vildagliptin ameliorates pulmonary fibrosis in lipopolysaccharide-induced lung injury by inhibiting endothelial-to-mesenchymal transition. Respir. Res. 2017, 18, 177. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Wang, J.; He, M.; Han, H.; Xie, W.; Wang, H.; Kong, H. Dipeptidyl peptidase IV (DPP-4) inhibition alleviates pulmonary arterial remodeling in experimental pulmonary hypertension. Lab. Investig. 2018, 98, 1333–1346. [Google Scholar] [CrossRef]
- Zhou, Y.; Schneider, D.J.; Blackburn, M.R. Adenosine signaling and the regulation of chronic lung disease. Pharmacol. Ther. 2009, 123, 105–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Murthy, J.N.; Zeng, D.; Belardinelli, L.; Blackburn, M.R. Alterations in adenosine metabolism and signaling in patients with chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. PLoS ONE 2010, 5, e9224. [Google Scholar] [CrossRef] [Green Version]
- Karmouty-Quintana, H.; Weng, T.; Garcia-Morales, L.J.; Chen, N.Y.; Pedroza, M.; Zhong, H.; Molina, J.G.; Bunge, R.; Bruckner, B.A.; Xia, Y.; et al. Adenosine A2B receptor and hyaluronan modulate pulmonary hypertension associated with chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2013, 49, 1038–1047. [Google Scholar] [CrossRef] [Green Version]
- Karmouty-Quintana, H.; Philip, K.; Acero, L.F.; Chen, N.Y.; Weng, T.; Molina, J.G.; Luo, F.; Davies, J.; Le, N.B.; Bunge, I.; et al. Deletion of ADORA2B from myeloid cells dampens lung fibrosis and pulmonary hypertension. Faseb. J. 2015, 29, 50–60. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Morales, L.J.; Chen, N.Y.; Weng, T.; Luo, F.; Davies, J.; Philip, K.; Volcik, K.A.; Melicoff, E.; Amione-Guerra, J.; Bunge, R.R.; et al. Altered Hypoxic-Adenosine Axis and Metabolism in Group III Pulmonary Hypertension. Am. J. Respir. Cell Mol. Biol. 2016, 54, 574–583. [Google Scholar] [CrossRef] [Green Version]
- Dzhalilova, D.; Makarova, O. Differences in Tolerance to Hypoxia: Physiological, Biochemical, and Molecular-Biological Characteristics. Biomedicines 2020, 8, 428. [Google Scholar] [CrossRef] [PubMed]
- Bryant, A.J.; Carrick, R.P.; McConaha, M.E.; Jones, B.R.; Shay, S.D.; Moore, C.S.; Blackwell, T.R.; Gladson, S.; Penner, N.L.; Burman, A.; et al. Endothelial HIF signaling regulates pulmonary fibrosis-associated pulmonary hypertension. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L249–L262. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzales, J.; Fraidenburg, D.R. Pharmacology and Emerging Therapies for Group 3 Pulmonary Hypertension Due to Chronic Lung Disease. Pharmaceuticals 2023, 16, 418. https://doi.org/10.3390/ph16030418
Gonzales J, Fraidenburg DR. Pharmacology and Emerging Therapies for Group 3 Pulmonary Hypertension Due to Chronic Lung Disease. Pharmaceuticals. 2023; 16(3):418. https://doi.org/10.3390/ph16030418
Chicago/Turabian StyleGonzales, Janae, and Dustin R. Fraidenburg. 2023. "Pharmacology and Emerging Therapies for Group 3 Pulmonary Hypertension Due to Chronic Lung Disease" Pharmaceuticals 16, no. 3: 418. https://doi.org/10.3390/ph16030418