
Human Mesenchymal Stem Cell (hMSC) Donor Potency Selection for the “First in Cystic Fibrosis” Phase I Clinical Trial (CEASE-CF)
 , ,
, ,
Abstract
:1. Introduction
2. Results
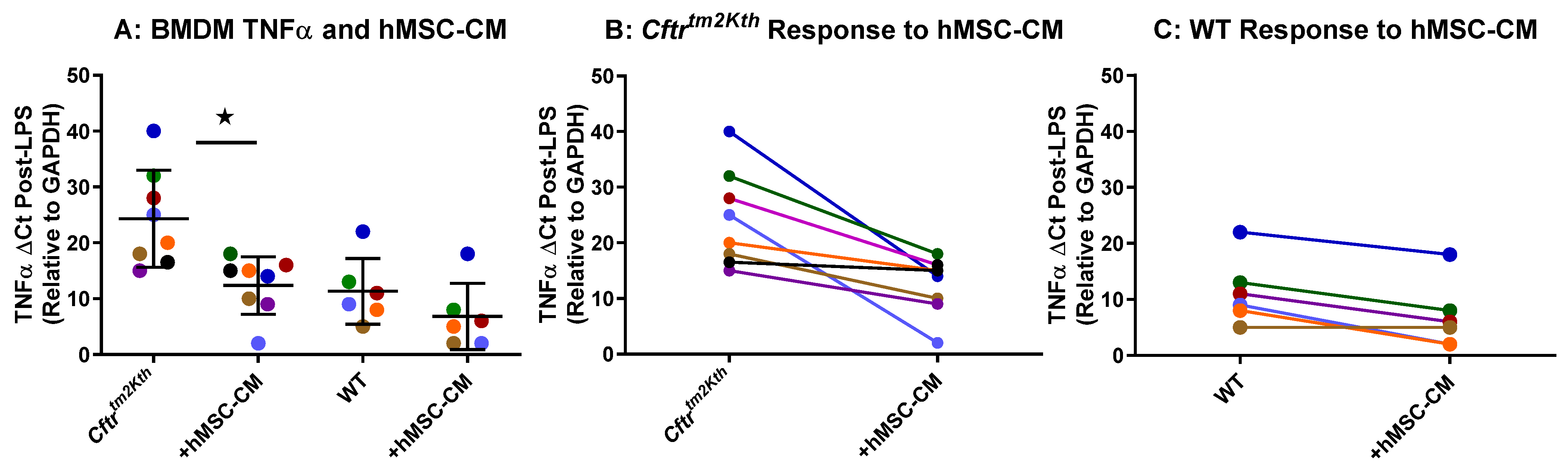
2.1. hMSC Anti-Inflammatory Activity on BMDM Target Cells
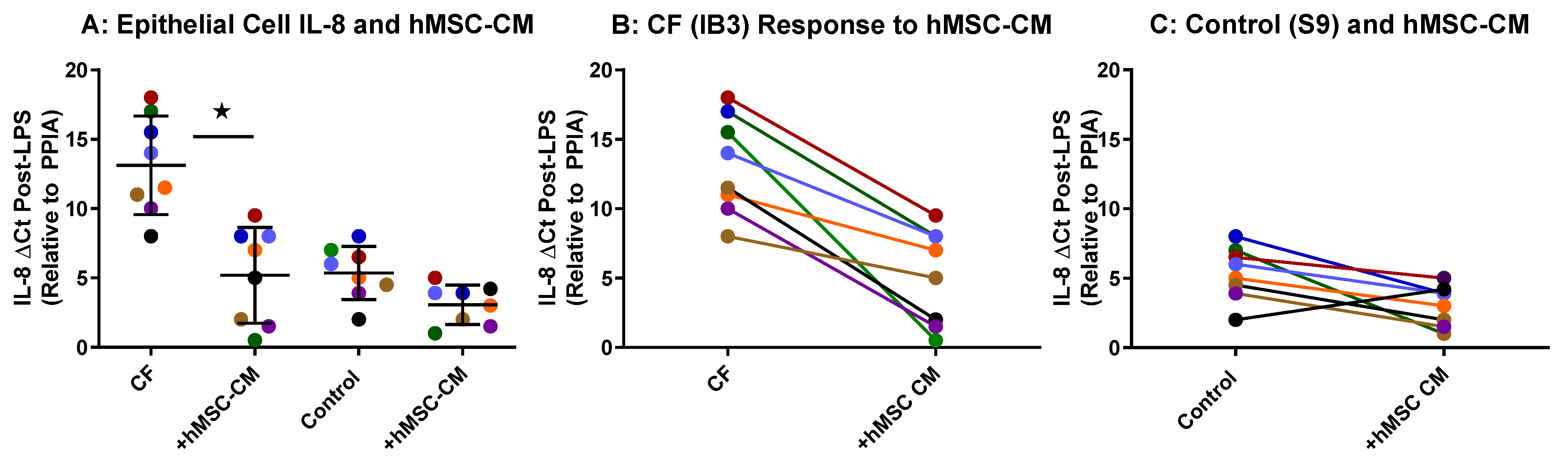
2.2. hMSC Anti-Inflammatory Activity on Airway Epithelial Cells
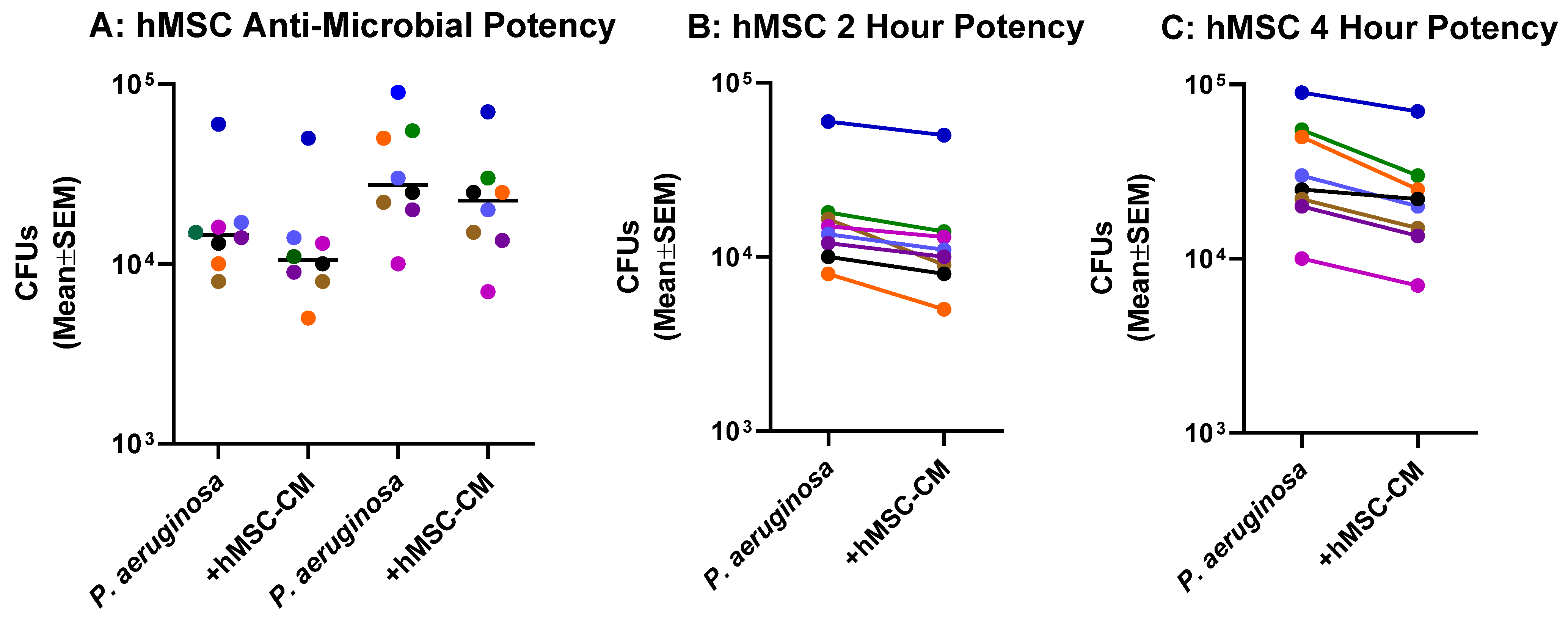
2.3. Anti-Microbial Potency of hMSCs against P. aeruginosa
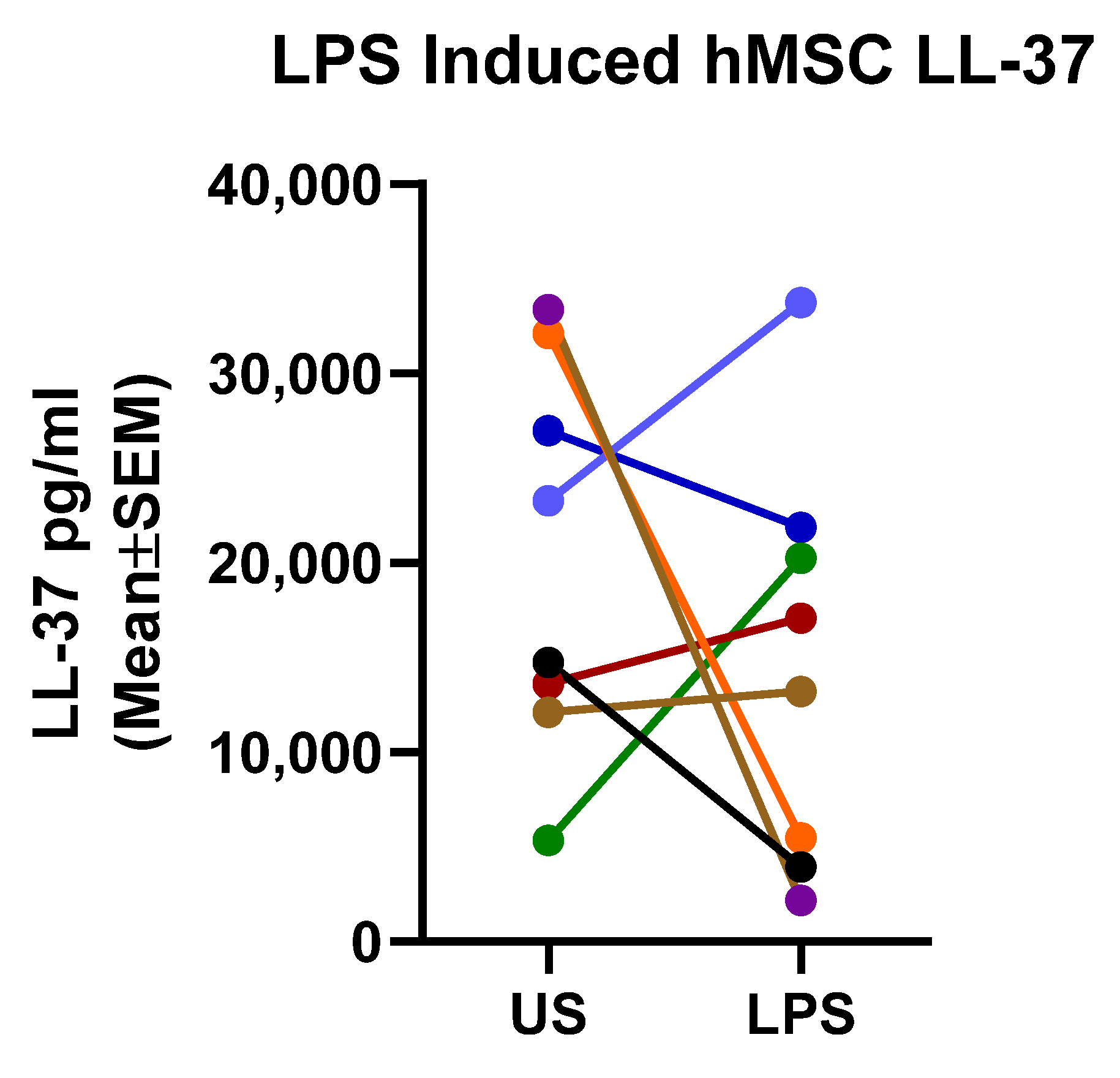
2.4. Mechanistic Fingerprinting of the Pre-GMP Donor Preparations
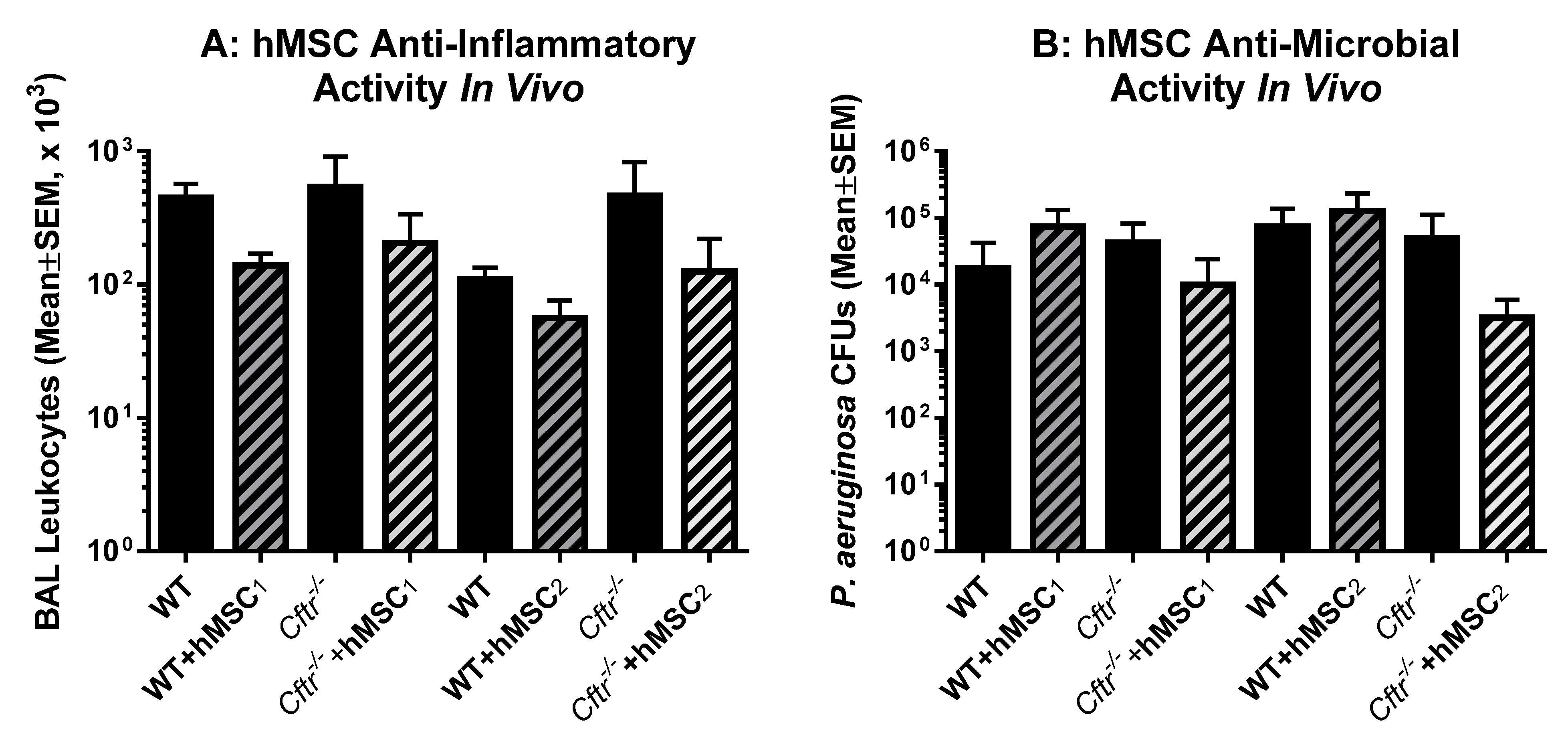
2.5. In Vivo Testing of Each of the Donor hMSCs
3. Discussion
4. Materials and Methods
4.1. Cell Sources
4.1.1. hMSCs
4.1.2. Human Transformed Airway Epithelial Cell Models
4.1.3. Bone Marrow-Derived Macrophages (BMDM)
4.2. In Vitro Assays
4.2.1. Anti-Microbial Potency Assays
4.2.2. Anti-Inflammatory Potency Assays
4.3. In Vivo Models
4.3.1. LL-37 Assay
4.3.2. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roesch, E.A.; Nichols, D.P.; Chmiel, J.F. Inflammation in Cystic Fibrosis: An Update. Pediatr Pulmonol. 2018, 53, S30–S50. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.C.; Mall, M.A.; Gutierrez, H.; Macek, M.; Madge, S.; Davies, J.C.; Burgel, P.R.; Tullis, E.; Castaños, C.; Castellani, C.; et al. The Future of Cystic Fibrosis Care: A Global Perspective. Lancet Respir. Med. 2019, 8, 65–124. [Google Scholar] [CrossRef] [PubMed]
- Cabrini, G. Innovative Therapies for Cystic Fibrosis: The Road from Treatment to Cure. Mol. Diagn. Ther. 2019, 23, 263–279. [Google Scholar] [CrossRef]
- Clancy, J.P.; Cotton, C.U.; Donaldson, S.H.; Solomon, G.M.; VanDevanter, D.R.; Boyle, M.P.; Gentzsch, M.; Nick, J.A.; Illek, B.; Wallenburg, J.C.; et al. CFTR modulator theratyping: Current status, gaps and future directions. J. Cyst. Fibros. 2019, 18, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Torphy, T.J.; Allen, J.; Cantin, A.M.; Konstan, M.W.; Accurso, F.J.; Joseloff, E.; Ratjen, F.A.; Chmiel, J.F. Considerations for the Conduct of Clinical Trials with Antiinflammatory Agents in Cystic Fibrosis. A Cystic Fibrosis Foundation Workshop Report 3. Ann. Am. Thorac. Soc. 2015, 12, 1398–1406. [Google Scholar] [CrossRef]
- Jain, R.; Beckett, V.V.; Konstan, M.W.; Accurso, F.J.; Burns, J.L.; Mayer-Hamblett, N.; Milla, C.; VanDevanter, D.R.; Chmiel, J.F. KB001-A.; a novel anti-inflammatory, found to be safe and well-tolerated in cystic fibrosis patients infected with Pseudomonas aeruginosa. J. Cyst. Fibros. 2018, 17, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Leyendecker, A.; Pinheiro, C.C.G.; Amano, M.T.; Bueno, D.F. The use of human mesenchymal stem cells as therapeutic agents for the in vivo treatment of immune-related diseases: A systematic review. Front. Immunol. 2018, 9, 2056. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.I.; Correa, D. The MSC: An Injury Drugstore. Cell Stem. Cell 2011, 9, 11–15. [Google Scholar] [CrossRef]
- van Heeckeren, A.M.; Sutton, M.T.; Fletcher, D.R.; Hodges, C.A.; Caplan, A.I.; Bonfield, T.L. Enhancing Cystic Fibrosis Immune Regulation. Front. Pharmacol. 2021, 12, 269. [Google Scholar] [CrossRef]
- Wang, M.; Yuan, Q.; Xie, L. Mesenchymal stem cell-based immunomodulation: Properties and clinical application. Stem. Cells Int. Hindawi Ltd. 2018, 2018, 3057624. [Google Scholar] [CrossRef]
- Wang, S.; Zhu, R.; Li, H.; Li, J.; Han, Q.; Zhao, R.C. Mesenchymal Stem Cells and Immune Disorders: From Basic Science to Clinical Transition. Front. Med. 2018, 13, 138–151. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.I. Cell-Based Therapies: The Nonresponder. Stem. Cells Transl. Med. 2018, 7, 762–766. [Google Scholar] [CrossRef]
- Bonfield, T.L.; Sutton, M.T.; Fletcher, D.R.; Folz, M.A.; Ragavapuram, V.; Somoza, R.A.; Caplan, A.I. Donor-defined mesenchymal stem cell antimicrobial potency against nontuberculous mycobacterium. Stem. Cells Transl. Med. 2021, 10, 1202–1216. [Google Scholar] [CrossRef] [PubMed]
- Abreu, S.C.; Rolandsson Enes, S.; Dearborn, J.; Goodwin, M.; Coffey, A.; Borg, Z.D.; Dos Santos, C.C.; Wargo, M.J.; Cruz, F.F.; Loi, R.; et al. Lung inflammatory environments differentially alter mesenchymal stromal cell behavior. Am. J. Physiol.–Lung Cell Mol. Physiol. 2019, 317, L823–L831. [Google Scholar] [CrossRef] [PubMed]
- Abreu, S.C.; Hampton, T.H.; Hoffman, E.; Dearborn, J.; Ashare, A.; Singh Sidhu, K.; Matthews, D.E.; McKenna, D.H.; Amiel, E.; Barua, J.; et al. Differential effects of the cystic fibrosis lung inflammatory environment on mesenchymal stromal cells. Am. J. Physiol.–Lung Cell Mol. Physiol. 2020, 319, L908–L925. [Google Scholar] [CrossRef] [PubMed]
- Bonfield, T.L.; Lennon, D.; Ghosh, S.K.; Di Marino, A.M.; Sadeghi, S.; Hijaz, A.; Wienberg, A.; Cpalan, A.I. Cell Based Therapy Aides in Infection and Inflammation Resolution in The Murine Model of Cystic Fibrosis Lung Disease. Stem. Cell Discov. 2013, 3, 138–159. [Google Scholar] [CrossRef]
- Paroni, M.; Moalli, F.; Nebuloni, M.; Pasqualini, F.; Bonfield, T.; Nonis, A.; Mantovani, A.; Garlanda, C.; Bragonzi, A. Response of cftr-deficient mice to long-term chronic pseudomonas aeruginosa infection and PTX3 therapy. J. Infect Dis. 2013, 208, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Matuska, B.; Comhair, S.; Farver, C.; Chmiel, J.; Midura, R.J.; Bonfield, T.; Lauer, M.E. Pathological Hyaluronan Matrices in Cystic Fibrosis Airways and Secretions. Am J Respir Cell Mol Biol 2016, 55, 576–585. [Google Scholar] [CrossRef]
- Darrah, R.; Bonfield, T.L.; LiPuma, P.; Hodges, C.A.; Jacono, F.; Drumm, M. Cystic Fibrosis Mice Develop Spontaneous Chronic Bordetella Airway Infections. J. Infect Pulm. Dis. 2017, 3, 128. [Google Scholar]
- Roesch , E.A.; Bonfield , T.L.; Lazarus , H.M.; Reese , J.; Hilliard, K.; Hilliard, J.; Khan, U.; Heltshe , S.; Gluvna , A.; Dasenbrook , E.; et al. A phase I study assessing the safety and tolerability of allogeneic mesenchymal stem cell infusion in adults with cystic fibrosis . J. Cyst. Fibros. 2020. [Google Scholar] [CrossRef]
- Sutton, M.T.; Fletcher, D.; Episalla, N.; Auster, L.; Kaur, S.; Gwin, M.C.; Folz, M.; Veasquez, D.; Roy, V.; van Heeckeren, R.; et al. Mesenchymal Stem Cell Soluble Mediators and Cystic Fibrosis. J. Stem. Cell Res. Ther. 2017, 7, 2157–7633. [Google Scholar] [CrossRef] [Green Version]
- Sutton, M.T.; Fletcher, D.; Ghosh, S.K.; Weinberg, A.; van Heeckeren, R.; Kaur, S.; Sadeghi, Z.; Hijaz, A.; Reese, J.; Lazarus, H.M.; et al. Antimicrobial Properties of Mesenchymal Stem Cells: Therapeutic Potential for Cystic Fibrosis Infection, and Treatment. Stem. Cells Int. 2016, 2016, 5303048. [Google Scholar] [CrossRef]
- Konstan, M.W.; Flume, P.A. Clinical care for cystic fibrosis: Preparing for the future now. Lancet Respir. Med. 2020, 8, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Tyndall, A.; Walker, U.A.; Cope, A.; Dazzi, F.; De Bari, C.; Fibbe, W.; Guiducci, S.; Jones, S.; Jorgensen, C.; Le Blanc, K.; et al. Immunomodulatory properties of mesenchymal stem cells: A review based on an interdisciplinary meeting held at the Kennedy Institute of Rheumatology Division, London, UK, 31 October 2005. Arthritis Res. Ther. 2007, 9, 301. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.I.; Dennis, J.E. Mesenchymal stem cells as trophic mediators. J. Cell Biochem. 2006, 98, 1076–1084. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.C.; Kang, I.; Yu, K.R. Therapeutic features and updated clinical trials of mesenchymal stem cell (Msc)-derived exosomes. J. Clin. Med. 2021, 10, 711. [Google Scholar] [CrossRef]
- Wu, X.; Jiang, J.; Gu, Z.; Zhang, J.; Chen, Y.; Liu, X. Mesenchymal stromal cell therapies: Immunomodulatory properties and clinical progress. Stem Cell Res. Ther. BioMed Cent. 2020, 11, 345. [Google Scholar] [CrossRef]
- Khoury, M.; Cuenca, J.; Cruz, F.F.; Figueroa, F.E.; Rocco, P.R.M.; Weiss, D.J. Current Status of Cell-Based Therapies for Respiratory Virus Infections: Applicability to COVID-19. Eur. Respir. J. 2020, 55, 2000858. [Google Scholar] [CrossRef] [PubMed]
- Duchesneau, P.; Waddell, T.K.; Karoubi, G. Cell-based therapeutic approaches for cystic fibrosis. Int. J. Mol. Sci. 2020, 21, 5219. [Google Scholar] [CrossRef]
- Marangi, M.; Pistritto, G. Innovative Therapeutic Strategies for Cystic Fibrosis: Moving Forward to CRISPR Technique. Front. Pharmacol. 2020, 9, 396. [Google Scholar] [CrossRef]
- Bernut, A.; Herrmann, J.-L.; Ordway, D.; Kremer, L. The Diverse Cellular and Animal Models to Decipher the Physiopathological Traits of Mycobacterium abscessus Infection. Front. Cell Infect Microbiol. 2017, 7, 100. [Google Scholar] [CrossRef] [Green Version]
- Mohammadipoor, A.; Antebi, B.; Batchinsky, A.I.; Cancio, L.C. Therapeutic potential of products derived from mesenchymal stem/stromal cells in pulmonary disease. Respir. Res. 2018, 19, 218. [Google Scholar] [CrossRef] [PubMed]
- Ryan, A.L.; Ikonomou, L.; Atarod, S.; Bölükbas, D.A.; Collins, J.; Freishtat, R.; Hawkins, F.; Gilpin, S.E.; Uhl, F.E.; Uriarte, J.J.; et al. Stem cells, cell therapies, and bioengineering in lung biology and diseases 2017. Am. J. Respir. Cell Mol. Biol. 2019, 61, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Krasnodembskaya, A.; Song, Y.; Fang, X.; Gupta, N.; Serikov, V.; Lee, J.W.; Matthay, M.A. Antibacterial effect of human mesenchymal stem cells is mediated in part from secretion of the antimicrobial peptide LL-37. Stem. Cells 2010, 28, 2229–2238. [Google Scholar] [CrossRef] [PubMed]
- Yasir, M.; Dutta, D.; Willcox, M.D.P. Comparative mode of action of the antimicrobial peptide melimine and its derivative Mel4 against Pseudomonas aeruginosa. Sci. Rep. 2019, 9, 7063. [Google Scholar] [CrossRef]
- Weiskopf, D.; Weinberger, B.; Grubeck-Loebenstein, B. The aging of the immune system. Transpl. Int. 2009, 22, 1041–1050. [Google Scholar] [CrossRef]
- Weinberg, A.; Sieg, S.; McCormick, T.S. The Yin and Yang of human beta-defensins in health and disease. Front. Immunol. 2012, 3, 294. [Google Scholar] [CrossRef]
- Alvayaga-Miranda, F.; Cuenca, J.; Khoury, M. Antimicrobial Activity of Mesenchymal Stem Cells: Current Status and New Perspectives of Antimicrobial Peptide-Based Therapies. Front Immunol. 2017, 8, 339. [Google Scholar]
- Yang, B.; Good, D.; Mosaiab, T.; Liu, W.; Ni, G.; Kaur, J.; Liu, X.; Jessop, C.; Yang, L.; Fadhil, R. Significance of LL-37 on Immunomodulation and Disease Outcome. BioMed Res. Int. 2020, 2020, 8349712. [Google Scholar] [CrossRef]
- Han, W.; Wei, Z.; Camesano, T.A. New antimicrobial peptide-antibiotic combinations strategies for Pseudomonas aeruginosa inactivation. Biointerphases 2022, 17, 041002. [Google Scholar] [CrossRef]
- Geitani, R.; Moubareck, A.A.; Tougui, L.; Sarkis, D.K. Cationic antimicrobial peptides: Alternatives and/or adjuvants to antibiotics active against methicillin-resistant Staphylococcus aureus and multidrug-resistant pseudomonas aeruginosa. BMC Microbiol. 2019, 19, 544. [Google Scholar] [CrossRef]
- Murphy, M.B.; Moncivais, K.; Caplan, A.I. Mesenchymal stem cells: Environmentally responsive therapeutics for regenerative medicine. Exp. Mol. Med. 2013, 45, e54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva-Carvalho, A.E.; Cardoso, M.H.; Alencar-Silva, T.; Bogea, G.M.R.; Carvalho, J.L.; Franco, O.L.; Saldanha-Araujo, F. Dissecting the relationship between antimicrobial peptides and mesenchymal stem cells. Pharmacol. Ther. 2022, 233, 108021. [Google Scholar] [CrossRef] [PubMed]
- Caplan, A.I. Mesenchymal Stem Cells: Time to Change the Name! Stem. Cells Transl. Med. 2017, 6, 1445–1451. [Google Scholar] [CrossRef]
- Saldaña, L.; Bensiamar, F.; Vallés, G.; Mancebo, F.J.; García-Rey, E.; Vilaboa, N. Immunoregulatory potential of mesenchymal stem cells following activation by macrophage-derived soluble factors. Stem. Cell Res. Ther. 2019, 10, 58. Available online: https://stemcellres.biomedcentral.com/articles/10.1186/s13287-019-1156-6 (accessed on 7 March 2019). [CrossRef] [PubMed]
- Lennon, D.P.; Caplan, A.I. Isolation of human marrow-derived mesenchymal stem cells. Exp. Hematol. 2006, 34, 1604–1605. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Lin, Y.; Lennon, D.P.; Correa, D.; Schluchter, M.; Caplan, A.I. Efficient lentiviral transduction of human mesenchymal stem cells that preserves proliferation and differentiation capabilities. Stem. Cells Transl. Med. 2012, 1, 886–897. [Google Scholar] [CrossRef] [PubMed]
- Bonfield, T.L.; Nolan, M.T.; Lennon, D.P.; Caplan, A.I. Defining human mesenchymal stem cell efficacy in vivo. J. Inflamm. 2010, 7, 51. [Google Scholar] [CrossRef]
- Czapla, J.; Matuszczak, S.; Kulik, K.; Wiśniewska, E.; Pilny, E.; Jarosz-Biej, M.; Smolarczyk, R.; Sirek, T.; Zembala, M.O.; Zembala, M.; et al. The effect of culture media on large-scale expansion and characteristic of adipose tissue-derived mesenchymal stromal cells. Stem. Cell Res. Ther. 2019, 10, 235. [Google Scholar] [CrossRef]
- Breitman, M.; Bonfield, T.L.; Caplan, A.I.; Lazarus, H.M.; Haghiac, M.; LaSalvia, S.; Reese-Koc, J.; Singer, N.G. Optimization of Human Mesenchymal Stem Cells for Rheumatoid Arthritis: Implications for Improved Therapeutic Outcomes. ACR Open RHeumatol. 2022, 4, 152–160. [Google Scholar] [CrossRef]
- Bonfield, T.L.; Hodges, C.A.; Cotton, C.U.; Drumm, M.L. Absence of cystic fibrosis transmembrane regulator (Cftr) from myeloid-derived cells slows resolution of inflammation and infection. J. Leukoc. Biol. 2012, 92, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Van Heeckeren, A.M.; Schluchter, M.D. Murine models of chronic Pseudomonas aeruginosa lung infection. Lab. Anim. 2002, 36, 291–312. [Google Scholar] [CrossRef]
- Van Heeckeren, A.; Walenga, R.; Konstan, M.W.; Bonfield, T.; Davis, P.B.; Ferkol, T. Excessive inflammatory response of cystic fibrosis mice to bronchopulmonary infection with Pseudomonas aeruginosa. J. Clin. Invest. 1997, 100, 2810–2815. [Google Scholar] [CrossRef] [PubMed]
- Chmiel, J.F.; Konstan, M.W.; Knesebeck, J.E.; Hilliard, J.B.; Bonfield, T.L.; Dawson, D.V.; Berger, M. IL-10 attenuates excessive inflammation in chronic pseudomonas infection in mice. Am. J. Respir. Crit. Care Med. 1999, 160, 2040–2047. [Google Scholar] [CrossRef] [PubMed]
- Soltys, J.; Bonfield, T.; Chmiel, J.; Berger, M. Functional IL-10 deficiency in the lung of cystic fibrosis (cftr(−/−)) and IL-10 knockout mice causes increased expression and function of B7 costimulatory molecules on alveolar macrophages. J. Immunol. 2002, 168, 1903–1910. [Google Scholar] [CrossRef] [PubMed]
- Peppers, B.P.; Vatsayan, A.; Dalal, J.; Bonfield, T.; Tcheurekdjian, H.; Hostoffer, R. A case series: Association of anaphylaxis with a significant decrease in platelet levels and possible secondary risk of thrombosis. Immun. Inflamm. Dis. 2018, 6, 377–381. [Google Scholar] [CrossRef]
- McNemar, Q. Note on the sampling error of the differences between correlated estimators of the odds ratio. Biometrika 2002, 65, 191–202. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Donor ID | Age | Sex |
|---|---|---|
| 786 | 35 | Male |
| 822 | 53 | Male |
| 829 | 33 | Male |
| 875 | 37 | Male |
| 882 | 28 | Female |
| TB001 | 36 | Female |
| TB002 | 34 | Male |
| TB003 | 27 | Female |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonfield, T.L.; Sutton, M.T.; Fletcher, D.R.; Reese-Koc, J.; Roesch, E.A.; Lazarus, H.M.; Chmiel, J.F.; Caplan, A.I. Human Mesenchymal Stem Cell (hMSC) Donor Potency Selection for the “First in Cystic Fibrosis” Phase I Clinical Trial (CEASE-CF). Pharmaceuticals 2023, 16, 220. https://doi.org/10.3390/ph16020220
Bonfield TL, Sutton MT, Fletcher DR, Reese-Koc J, Roesch EA, Lazarus HM, Chmiel JF, Caplan AI. Human Mesenchymal Stem Cell (hMSC) Donor Potency Selection for the “First in Cystic Fibrosis” Phase I Clinical Trial (CEASE-CF). Pharmaceuticals. 2023; 16(2):220. https://doi.org/10.3390/ph16020220
Chicago/Turabian StyleBonfield, Tracey L., Morgan T. Sutton, David R. Fletcher, Jane Reese-Koc, Erica A. Roesch, Hillard M. Lazarus, James F. Chmiel, and Arnold I. Caplan. 2023. "Human Mesenchymal Stem Cell (hMSC) Donor Potency Selection for the “First in Cystic Fibrosis” Phase I Clinical Trial (CEASE-CF)" Pharmaceuticals 16, no. 2: 220. https://doi.org/10.3390/ph16020220