

Bio-Oriented Synthesis and Molecular Docking Studies of 1,2,4-Triazole Based Derivatives as Potential Anti-Cancer Agents against HepG2 Cell Line
 ,
,  , , and
, , and
Abstract
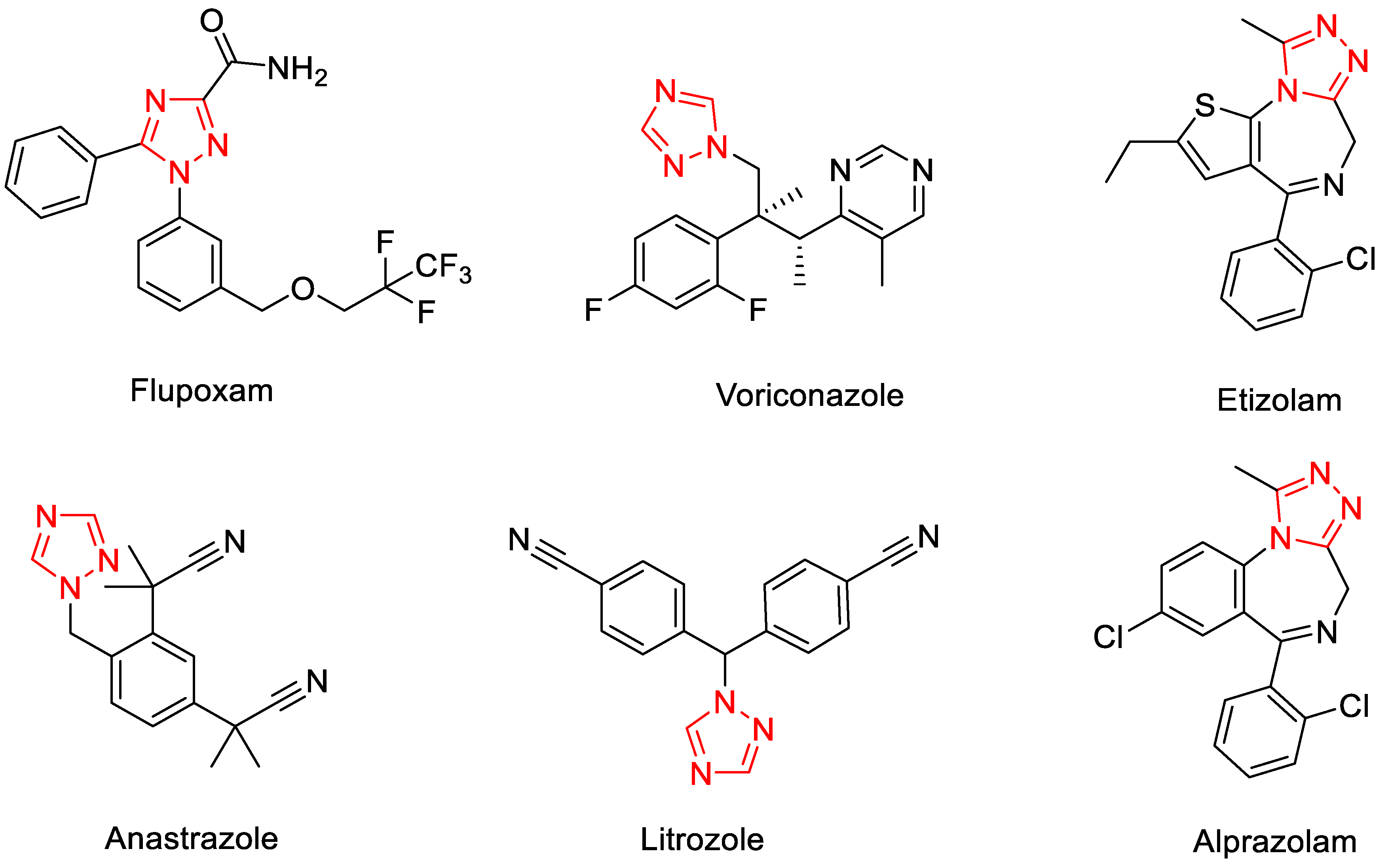
:1. Introduction
2. Results
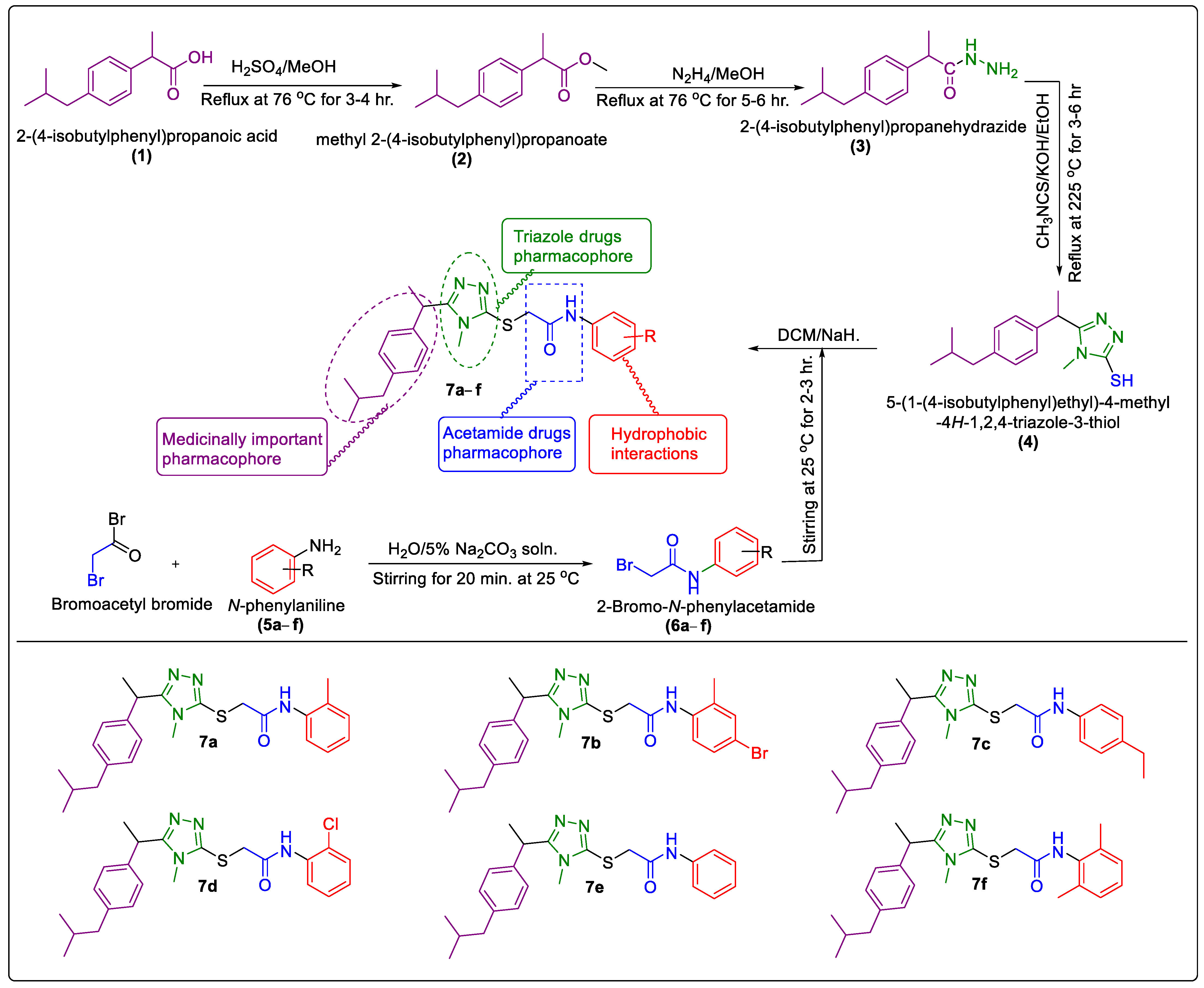
2.1. Chemistry
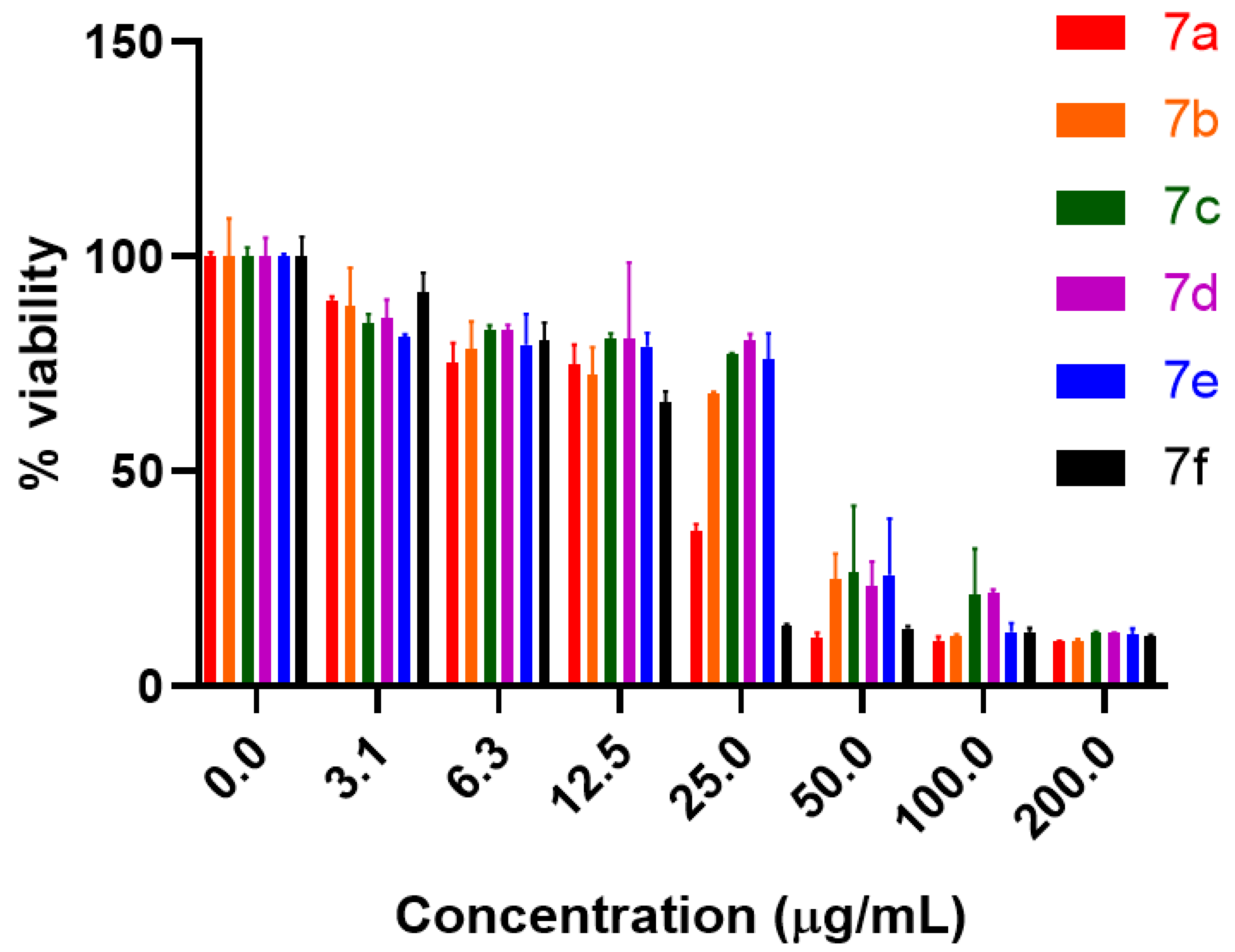
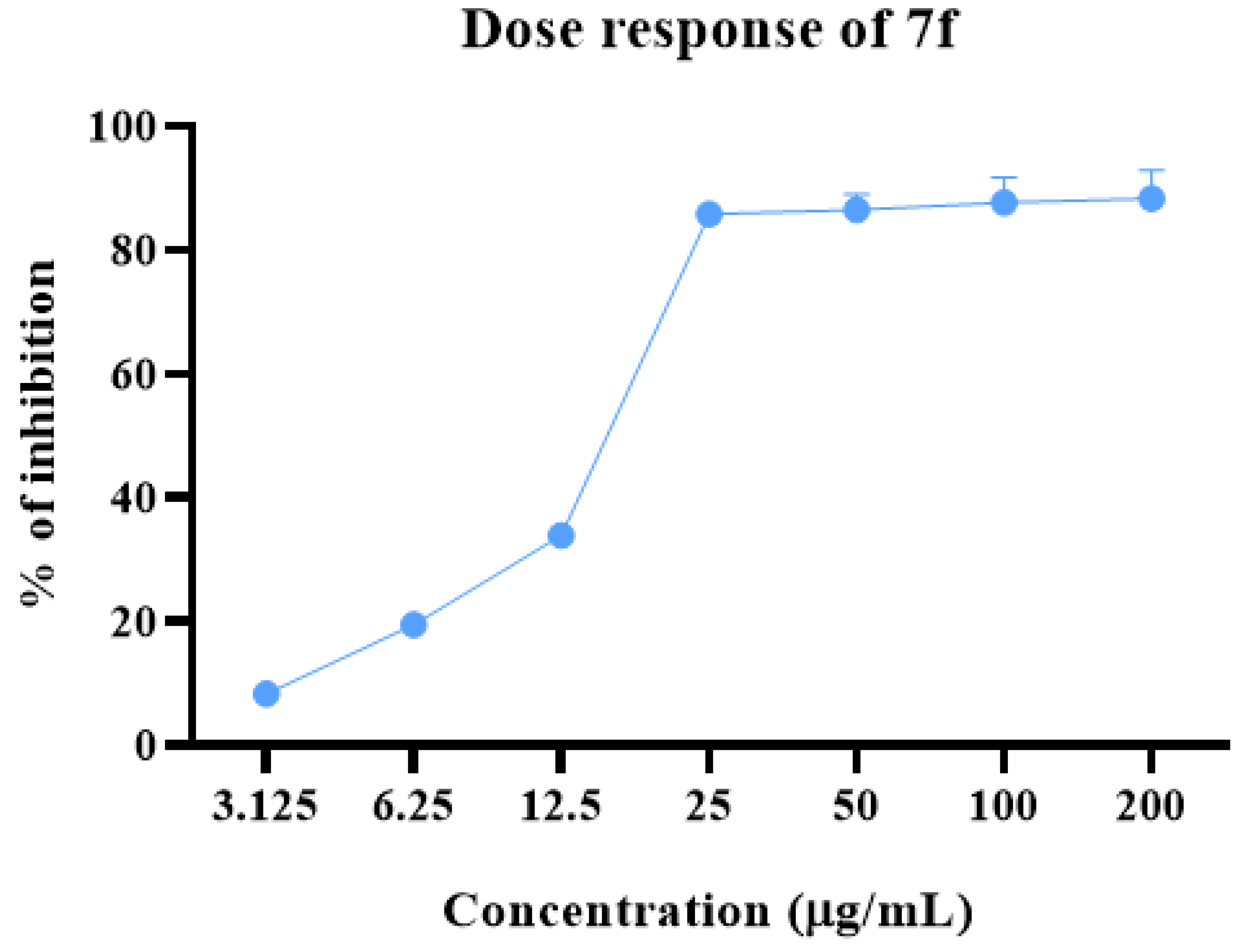
2.2. Anti-proliferative Potential
2.3. Hemolytic Activity Potential
2.4. Structure–Activity Relationship of 7a–f
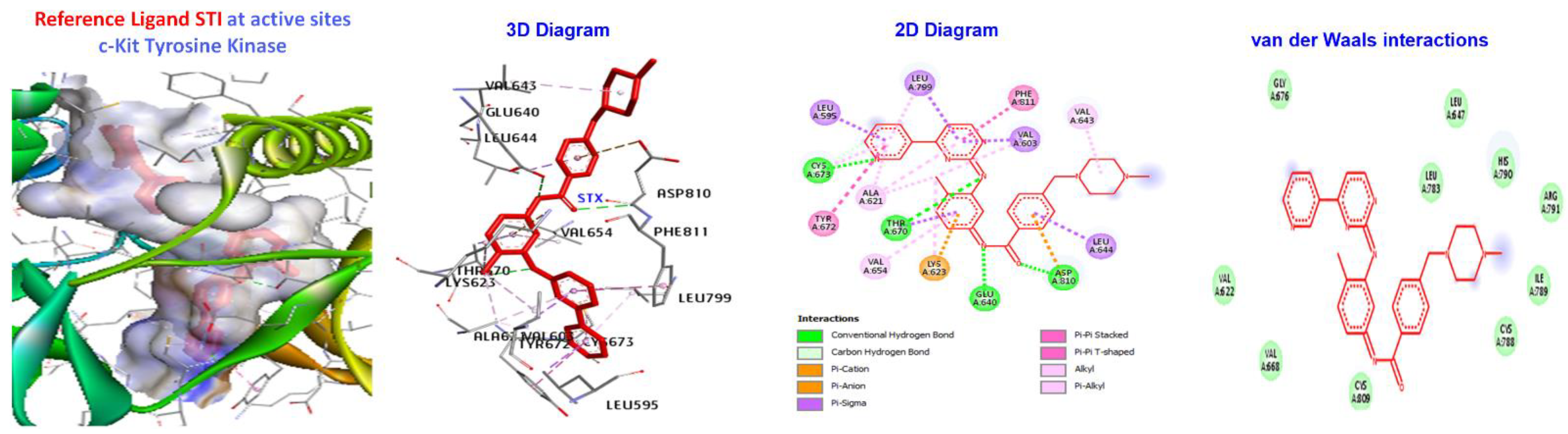
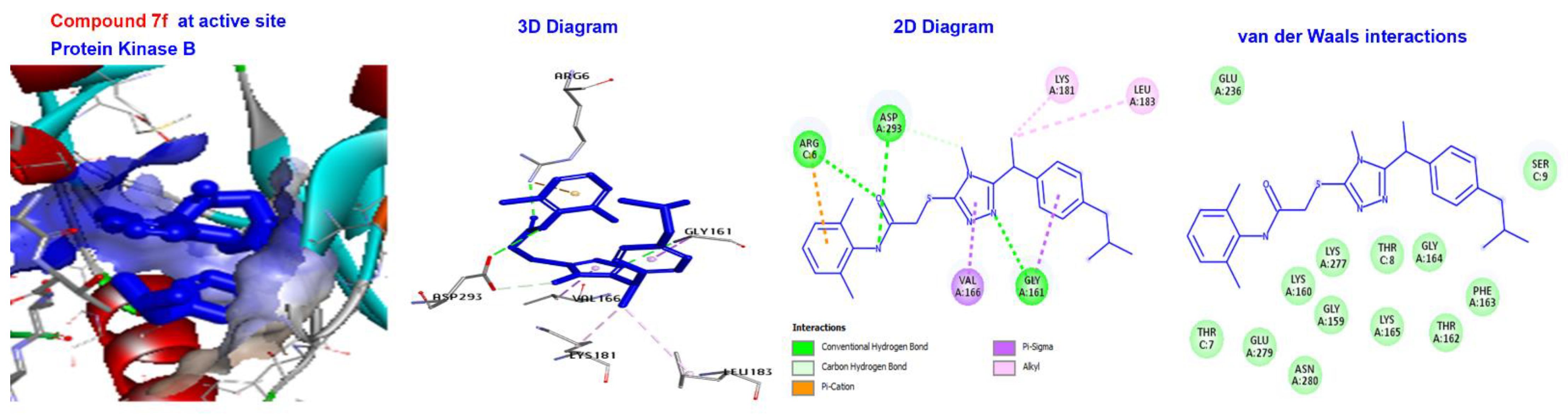
2.5. Molecular Docking
3. Materials and Methods
3.1. General
3.2. General Procedure for the Synthesis of Synthesized N-Arylated 5-Aryl-1,2,4-Triazole-Coupled Acetamide Scaffolds 7a–f
3.2.1. Synthesis of Methyl 2-(4-Isobutylphenyl)Propanoate (2)
3.2.2. Synthesis of 2-(4-Isobutylphenyl)Propanehydrazide (3)
3.2.3. Synthesis of 5-(1-(4- Isobutylphenyl)Ethyl)-1,2,4-Triazole -2-Thiol (4)
3.2.4. Synthesis of N–Aryl/Alkyl 2-Bromoroacetamides 6a–f
3.2.5. Synthesis of N-Arylated 5-(1-(4-Isobutylphenyl)Ethyl)-1,2,4-Triazole-2-yl- 2-Sulfanyl Coupled Acetamide Derivatives 7a–f
3.2.6. N-(2-Methylphenyl)-2-((5-(1-(4-isobutylphenyl)ethyl)-4-methyl-4H-1,2,4-triazol-3-yl)thio)Acetamide (7a)
3.2.7. N-(4-Bromo-2-Mthylphenyl)-2-((5-(1-(4-Isobutylphenyl)Ethyl)-4-Methyl-4H-1,2,4-Triazol-3-yl)Thio)Acetamide (7b)
3.2.8. N-(4-Ethylphenyl)-2-((5-(1-(4-Isobutylphenyl)Ethyl)-4-Methyl-4H-1,2,4-Triazol-3-yl)Thio)Acetamide (7c)
3.2.9. N-(2-Chlorophenyl)-2-((5-(1-(4-Isobutylphenyl)Ethyl)-4-Methyl-4H-1,2,4-Triazol-3-yl)Thio)Acetamide (7d)
3.2.10. N-(Phenyl)-2-((5-(1-(4-Isobutylphenyl)Ethyl)-4-Methyl-4H-1,2,4-Triazol-3-yl)Thio)Acetamide (7e)
3.2.11. N-(2,6-Dimethylphenyl)-2-((5-(1-(4-Isobutylphenyl)Ethyl)-4-Methyl-4H-1,2,4-Triazol-3-yl)Thio)Acetamide (7f)
3.3. Experimental Procedures for Biological Activities
3.3.1. Cell Culture and Treatment
3.3.2. Evaluation of Cell Viability
3.3.3. Hemolytic Activity Potential
3.4. Molecular Docking of Triazole-Coupled Acetamides
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Kamal, A.; Bharathi, E.V.; Reddy, J.S.; Ramaiah, M.J.; Dastagiri, D.; Reddy, M.K.; Viswanath, A.; Reddy, T.L.; Shaik, T.B.; Pushpavalli, S.; et al. Synthesis and biological evaluation of 3,5-diaryl isoxazoline/isoxazole linked 2,3-dihydroquinazolinone hybrids as anticancer agents. Eur. J. Med. Chem. 2011, 46, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, H.T.; Purohit, A.P.; Desai, S.C.; Jarwani, P.B. Retrospective analysis of histopathological spectrum of premalignant and malignant colorectal lesions. Cancer Res. Stat. Treat. 2021, 4, 472–478. [Google Scholar] [CrossRef]
- Jang, J.-W.; Song, Y.; Kim, K.M.; Kim, J.-S.; Choi, E.K.; Kim, J.; Seo, H. Hepatocellular carcinoma-targeted drug discovery through image-based phenotypic screening in co-cultures of HCC cells with hepatocytes. BMC Cancer 2016, 16, 810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rumgay, H.; Arnold, M.; Ferlay, J.; Lesi, O.; Cabasag, C.J.; Vignat, J.; Laversanne, M.; McGlynn, K.A.; Soerjomataram, I. Global burden of primary liver cancer in 2020 and predictions to 2040. J. Hepatol. 2022, 77, 1598–1606. [Google Scholar] [CrossRef]
- Zheng, Z.; Liu, Q.; Kim, W.; Tharmalingam, N.; Fuchs, B.B.; Mylonakis, E. Antimicrobial activity of 1,3,4-oxadiazole derivatives against planktonic cells and biofilm of Staphylococcus aureus. Futur. Med. Chem. 2018, 10, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-S.; Chou, C.-H.; Wu, Y.-H.; Yang, M.-H.; Chu, S.-H.; Chao, Y.-S.; Chen, C.-N. CC-01 (chidamide plus celecoxib) modifies the tumor immune microenvironment and reduces tumor progression combined with immune checkpoint inhibitor. Sci. Rep. 2022, 12, 1100. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, B.; Wang, Y.; Wang, X.; Gou, S. Discovery of phthalazino[1,2-b]-quinazolinone derivatives as multi-target HDAC inhibitors for the treatment of hepatocellular carcinoma via activating the p53 signal pathway. Eur. J. Med. Chem. 2021, 229, 114058. [Google Scholar] [CrossRef]
- Chen, C.; Li, X.; Zhao, H.; Liu, M.; Du, J.; Zhang, J.; Yang, X.; Hou, X.; Fang, H. Discovery of DNA-Targeting HDAC Inhibitors with Potent Antitumor Efficacy In Vivo That Trigger Antitumor Immunity. J. Med. Chem. 2022, 65, 3667–3683. [Google Scholar] [CrossRef]
- Khan, I.; Ibrar, A.; Abbas, N.; Saeed, A. Recent advances in the structural library of functionalized quinazoline and quinazolinone scaffolds: Synthetic approaches and multifarious applications. Eur. J. Med. Chem. 2014, 76, 193–244. [Google Scholar] [CrossRef]
- Khwaza, V.; Mlala, S.; Oyedeji, O.; Aderibigbe, B. Pentacyclic Triterpenoids with Nitrogen-Containing Heterocyclic Moiety, Privileged Hybrids in Anticancer Drug Discovery. Molecules 2021, 26, 2401. [Google Scholar] [CrossRef] [PubMed]
- Gomha, S.M.; Abdel-Aziz, H.M.; El-Reedy, A.A.M. Facile Synthesis of Pyrazolo[3,4-c]pyrazoles Bearing Coumarine Ring as Anticancer Agents. J. Heterocycl. Chem. 2018, 55, 1960–1965. [Google Scholar] [CrossRef]
- Kaproń, B.; Czarnomysy, R.; Wysokiński, M.; Andrys, R.; Musilek, K.; Angeli, A.; Supuran, C.T.; Plech, T. 1,2,4-Triazole-based anticonvulsant agents with additional ROS scavenging activity are effective in a model of pharmacoresistant epilepsy. J. Enzym. Inhib. Med. Chem. 2020, 35, 993–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.-S.; Tian, H.; Zhao, D.-S.; Hu, D.-K.; Liu, X.-Y.; Jin, H.-W.; Song, G.-P.; Cui, Z.-N. Synthesis and bioactivity of pyrazole and triazole derivatives as potential PDE4 inhibitors. Bioorganic Med. Chem. Lett. 2016, 26, 3632–3635. [Google Scholar] [CrossRef]
- de Andrade, M.A.R.; Lima, B.J.S.; de Jesus, A.A.; Teixeira, L.D.A.C.; de Souza, C.R.S.; Fontes, R.B.; Mendonça, N.P.V.; Gonçalves, H.S.; Cesar, A.S.; Aragão, M.T. Burnout Syndrome in COVID-19: An analysis of physicians from the public and private health system in the state of Sergipe. Res. Soc. Dev. 2022, 11, e3911729602. [Google Scholar] [CrossRef]
- Kumar, S.; Khokra, S.L.; Yadav, A. Triazole analogues as potential pharmacological agents: A brief review. Futur. J. Pharm. Sci. 2021, 7, 106. [Google Scholar] [CrossRef]
- Han, S.; Zhang, F.-F.; Xie, X.; Chen, J.-Z. Design, synthesis, biological evaluation, and comparative docking study of 1,2,4-triazolones as CB1 receptor selective antagonists. Eur. J. Med. Chem. 2014, 74, 73–84. [Google Scholar] [CrossRef]
- Zhang, W.; Yuan, J. Poly(1-Vinyl-1,2,4-triazolium) Poly(Ionic Liquid)s: Synthesis and the Unique Behavior in Loading Metal Ions. Macromol. Rapid Commun. 2016, 37, 1124–1129. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Wang, W.; Wang, S.; Bao, L. Asymmetric synthesis of novel triazole derivatives and their in vitro antiviral activity and mechanism of action. Eur. J. Med. Chem. 2017, 139, 718–725. [Google Scholar] [CrossRef]
- Hu, G.; Wang, C.; Xin, X.; Li, S.; Li, Z.; Zhao, Y.; Gong, P. Design, synthesis and biological evaluation of novel 2,4-diaminopyrimidine derivatives as potent antitumor agents. New J. Chem. 2019, 43, 10190–10202. [Google Scholar] [CrossRef]
- Ni, T.; Ding, Z.; Xie, F.; Hao, Y.; Bao, J.; Zhang, J.; Yu, S.; Jiang, Y.; Zhang, D. Design, Synthesis, and In Vitro and In Vivo Antifungal Activity of Novel Triazoles Containing Phenylethynyl Pyrazole Side Chains. Molecules 2022, 27, 3370. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.-F.; Liu, X.; Zhang, S.; Pan, B.; Liu, M.-L. Ciprofloxacin derivatives and their antibacterial activities. Eur. J. Med. Chem. 2018, 146, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xu, L.; Zhu, L.; Zhao, Y.; Hu, T.; Yin, B.; Liu, Y.; Hou, Y. Design, synthesis and biological evaluation of novel pteridinone derivatives possessing a hydrazone moiety as potent PLK1 inhibitors. Bioorganic Med. Chem. Lett. 2020, 30, 127329. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, T.; Hameed, S.; Al-Masoudi, N.P.; Khan, K.M. Synthesis and anti-HIV activity of new chiral 1,2,4-triazoles and 1,3,4-thiadiazoles. Heteroat. Chem. 2007, 18, 316–322. [Google Scholar] [CrossRef]
- Irfan, A.; Faiz, S.; Rasul, A.; Zafar, R.; Zahoor, A.F.; Kotwica-Mojzych, K.; Mojzych, M. Exploring the Synergistic Anticancer Potential of Benzofuran–Oxadiazoles and Triazoles: Improved Ultrasound- and Microwave-Assisted Synthesis, Molecular Docking, Hemolytic, Thrombolytic and Anticancer Evaluation of Furan-Based Molecules. Molecules 2022, 27, 1023. [Google Scholar] [CrossRef]
- Gul, S.; Rehman, A.U.; Abbasi, M.A.; Khan, K.M.; Nafeesa, K.; Siddiqa, A.; Akhtar, M.N.; Shahid, M.; Subhani, Z. Synthesis, antimicrobial evaluation and hemolytic activity of 2-[[5-alkyl/aralkyl substituted-1,3,4-oxadiazol-2-yl]thio]-N-[4-(4-morpholinyl)phenyl]acetamide derivatives. J. Saudi Chem. Soc. 2014, 21, S425–S433. [Google Scholar] [CrossRef]
- Khan, S.G.; Bokhari, T.H.; Anjum, F.; Akhter, N.; Rasool, S.; Shah, S.A.A.; Shahid, M.; Arshad, A. Synthesis, characterization, antibacterial, hemolytic and thrombolytic activity evaluation of 5-(3-chlorophenyl)-2-((N-(substituted)-2-acetamoyl) sulfanyl)-1, 3, 4-oxadiazole derivatives. Pak. J. Pharm. Sci. 2020, 33, 871–876. [Google Scholar]
- Mahmood, S.; Khan, S.G.; Rasul, A.; Christensen, J.B.; Abourehab, M.A.S. Ultrasound Assisted Synthesis and In Silico Modelling of 1,2,4-Triazole Coupled Acetamide Derivatives of 2-(4-Isobutyl phenyl)propanoic acid as Potential Anticancer Agents. Molecules 2022, 27, 7984. [Google Scholar] [CrossRef]
- Aziz-ur-Rehman; Khan, S.G.; Naqvi, S.A.R.; Ahmad, M.; Akhtar, N.; Bokhari, T.H.; Irfan, M.; Usman, A.; Batool, S.; Rasool, S. Synthesis, spectral analysis and biological evaluation of 2-{[(morpholin-4-yl) ethyl] thio}-5-phenyl/aryl-1, 3, 4-oxadiazole derivatives. Pak. J. Pharm. Sci. 2021, 34, 441–446. [Google Scholar] [CrossRef]
- Amewu, R.K.; Sakyi, P.O.; Osei-Safo, D.; Addae-Mensah, I. Synthetic and Naturally Occurring Heterocyclic Anticancer Compounds with Multiple Biological Targets. Molecules 2021, 26, 7134. [Google Scholar] [CrossRef]
- Zabiulla; Gulnaz, A.; Mohammed, Y.H.E.; Khanum, S.A. Design, synthesis and molecular docking of benzophenone conjugated with oxadiazole sulphur bridge pyrazole pharmacophores as anti-inflammatory and analgesic agents. Bioorganic Chem. 2019, 92, 103220. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Jamal, Q.; Iqbal, J.; Afreen, M.S.; Sandhu, M.Z.A.; Dar, E.; Farooq, U.; Mushtaq, M.F.; Arshad, N.; Iqbal, M.M. Synthesis of N-substituted acetamide derivatives of azinane-bearing 1,3,4-oxadiazole nucleus and screening for antibacterial activity. Trop. J. Pharm. Res. 2017, 16, 429. [Google Scholar] [CrossRef] [Green Version]
- Thongnest, S.; Chawengrum, P.; Keeratichamroen, S.; Lirdprapamongkol, K.; Eurtivong, C.; Boonsombat, J.; Kittakoop, P.; Svasti, J.; Ruchirawat, S. Vernodalidimer L, a sesquiterpene lactone dimer from Vernonia extensa and anti-tumor effects of vernodalin, vernolepin, and vernolide on HepG2 liver cancer cells. Bioorganic Chem. 2019, 92, 103197. [Google Scholar] [CrossRef] [PubMed]
- Shahzadi, I.; Parveen, B.; Ahmad, S.; Zahoor, A.F.; Rasul, A.; Zahid, F.M. In-vitro cytotoxic evaluation, hemolytic and thrombolytic potential of newly designed acefylline based hydrazones as potent anti-cancer agents against human lung cancer cell line (A549). Pak. J. Pharm. Sci. 2022, 35, 885–889. [Google Scholar] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. 2019, 47, W357–W364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mol, C.D.; Dougan, D.R.; Schneider, T.R.; Skene, R.J.; Kraus, M.L.; Scheibe, D.N.; Snell, G.P.; Zou, H.; Sang, B.-C.; Wilson, K.P. Structural Basis for the Autoinhibition and STI-571 Inhibition of c-Kit Tyrosine Kinase. J. Biol. Chem. 2004, 279, 31655–31663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McHardy, T.; Caldwell, J.J.; Cheung, K.-M.; Hunter, L.J.; Taylor, K.; Rowlands, M.; Ruddle, R.; Henley, A.; Brandon, A.D.H.; Valenti, M.; et al. Discovery of 4-Amino-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamides As Selective, Orally Active Inhibitors of Protein Kinase B (Akt). J. Med. Chem. 2010, 53, 2239–2249. [Google Scholar] [CrossRef] [PubMed]
- Elkins, J.M.; Santaguida, S.; Musacchio, A.; Knapp, S. Crystal Structure of Human Aurora B in Complex with INCENP and VX-680. J. Med. Chem. 2012, 55, 7841–7848. [Google Scholar] [CrossRef]
- Degorce, S.L.; Boyd, S.; Curwen, J.O.; Ducray, R.; Halsall, C.T.; Jones, C.D.; Lach, F.; Lenz, E.M.; Pass, M.; Pass, S.; et al. Discovery of a Potent, Selective, Orally Bioavailable, and Efficacious Novel 2-(Pyrazol-4-ylamino)-pyrimidine Inhibitor of the Insulin-like Growth Factor-1 Receptor (IGF-1R). J. Med. Chem. 2016, 59, 4859–4866. [Google Scholar] [CrossRef]
- Zhang, Q.; Sang, F.; Qian, J.; Lyu, S.; Wang, W.; Wang, Y.; Li, Q.; Du, L. Identification of novel potential PI3Kα inhibitors for cancer therapy. J. Biomol. Struct. Dyn. 2020, 39, 3721–3732. [Google Scholar] [CrossRef]
- Bai, L.; Zhou, H.; Xu, R.; Zhao, Y.; Chinnaswamy, K.; McEachern, D.; Chen, J.; Yang, C.-Y.; Liu, Z.; Wang, M.; et al. A Potent and Selective Small-Molecule Degrader of STAT3 Achieves Complete Tumor Regression In Vivo. Cancer Cell 2019, 36, 498–511.e17. [Google Scholar] [CrossRef] [PubMed]
- Sardar, A.; Abid, O.-U.; Daud, S.; Fakhar-E-Alam, M.; Siddique, M.H.; Ashraf, M.; Shahid, W.; Ejaz, S.A.; Atif, M.; Ahmad, S.; et al. Design, synthesis, in vitro and in silico studies of naproxen derivatives as dual lipoxygenase and α-glucosidase inhibitors. J. Saudi Chem. Soc. 2022, 26, 101468. [Google Scholar] [CrossRef]
- Alderawy MQ, A.; Alrubaie LA, R.; Sheri, F.H. Synthesis, Characterization of Ibuprofen N-Acyl-1, 3, 4-Oxadiazole Derivatives and Anti-cancer Activity against MCF-7 Cell Line. Syst. Rev. Pharm. 2020, 11, 681–689. [Google Scholar] [CrossRef]
- Shaikh, M.H.; Subhedar, D.D.; Nawale, L.; Sarkar, D.; Khan, F.A.K.; Sangshetti, J.N.; Shingate, B.B. 1,2,3-Triazole derivatives as antitubercular agents: Synthesis, biological evaluation and molecular docking study. MedChemComm 2015, 6, 1104–1116. [Google Scholar] [CrossRef]
- Abutaha, N.; Al-Mekhlafi, F.A.; Almutairi, B.O.; Wadaan, M.A. S-phase cell cycle arrest, and apoptotic potential of Echium arabicum phenolic fraction in hepatocellular carcinoma HepG2 cells. J. King Saud Univ.—Sci. 2021, 34, 101735. [Google Scholar] [CrossRef]
- Win, T.S.; Malik, A.A.; Prachayasittikul, V.S.; Wikberg, J.E.; Nantasenamat, C.; Shoombuatong, W. HemoPred: A web server for predicting the hemolytic activity of peptides. Futur. Med. Chem. 2017, 9, 275–291. [Google Scholar] [CrossRef]
- Le, P.T.; Cheng, H.; Ninkovic, S.; Plewe, M.; Huang, X.; Wang, H.; Bagrodia, S.; Sun, S.; Knighton, D.R.; Rogers, C.M.L.; et al. Design and synthesis of a novel pyrrolidinyl pyrido pyrimidinone derivative as a potent inhibitor of PI3Kα and mTOR. Bioorganic Med. Chem. Lett. 2012, 22, 5098–5103. [Google Scholar] [CrossRef]
- Thomsen, R.; Christensen, M.H. MolDock: A New Technique for High-Accuracy Molecular Docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Alkyl/Aryl | Cell Viability IC50 Value (µg/mL) | Hemolytic Activity (Mean% ± S.D) |
|---|---|---|---|
| 7a | 2-methyl phenyl | 20.667 | 2.46 ± 0.31 |
| 7b | 2-methyl-4-bromo phenyl | 33.565 | 2.43 ± 0.11 |
| 7c | 4-ethyl phenyl | 39.002 | 4.32 ± 0.24 |
| 7d | 2-chloro phenyl | 39.667 | 7.33 ± 0.42 |
| 7e | phenyl | 39.105 | 4.19 ± 0.02 |
| 7f | 2,6-dimethyl phenyl | 16.782 | 1.19 ± 0.02 |
| Sorafenib | 05.971 | ||
| PBS | 0.00 ± 0.0 | ||
| Triton-X-100 | 100 ± 0.0 |
| Concentration (µg/mL) | 7a | 7b | 7c | 7d | 7e | 7f |
|---|---|---|---|---|---|---|
| 200 | 89.40 ± 0.34 | 89.50 ± 0.06 | 87.51 ± 0.20 | 87.70 ± 0.18 | 88.00 ± 1.44 | 88.36 ± 0.31 |
| 100 | 88.17 ± 0.26 | 89.39 ± 0.91 | 78.82 ± 10.78 | 78.33 ± 0.78 | 87.34 ± 2.09 | 87.71 ± 1.34 |
| 50 | 75.18 ± 5.99 | 88.64 ± 1.11 | 73.66 ± 15.54 | 76.69 ± 5.63 | 74.24 ± 13.16 | 86.57 ± 0.56 |
| 25 | 63.98 ± 1.67 | 31.90 ± 0.30 | 22.82 ± 0.29 | 19.52 ± 1.44 | 24.01 ± 6.12 | 85.90 ± 0.30 |
| 12.5 | 27.70 ± 6.46 | 25.23 ± 4.67 | 19.14 ± 1.25 | 19.11 ± 17.71 | 20.99 ± 3.13 | 33.96 ± 2.47 |
| 6.25 | 21.44 ± 6.24 | 24.75 ± 4.62 | 17.35 ± 1.32 | 17.32 ± 1.40 | 20.52 ± 7.11 | 19.55 ± 4.07 |
| 3.125 | 11.57 + 8.85 | 10.33 + 0.96 | 15.40 ± 2.01 | 14.27 ± 4.27 | 18.65 ± 0.50 | 8.40 ± 4.54 |
| DMSO (-ve Control) | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| Targets | Protein Kinase B (Akt) (PKB) | c-Kit Tyrosine Kinase (c-Kit) | Human Aurora B Kinase (AURKB) | Phosphatidylinositol 3-Kinase Alpha (PI3Kalpha) | Signal Transducer and Activator of Transcription 3 (STAT3) |
|---|---|---|---|---|---|
| PDB ID | 2X39 | 1T46 | 4AF3 | 4FA6 | 6NJS |
| Center of docking | X:43 | X:28 | X:21 | X:44 | X:13 |
| Coordinates | Y:31 | Y:26 | Y:-22 | Y:14 | Y:56 |
| Z:111 | Z:39 | Z:-10 | Z:31 | Z:0.32 | |
| Reference Ligand | X39 | STI | VX6 | 0TA | KQV |
| Ligands | Mol. Dock Score (Kcal/mol) | Mol. Dock Score (Kcal/mol) | Mol. Dock Score (Kcal/mol) | Mol. Dock Score (Kcal/mol) | Mol. Dock Score (Kcal/mol) |
| 7a 7b | −166.843 −166.371 | −173.411 −167.882 | −145.234 −138.33 | −139.389 −136.267 | −125.105 −120.348 |
| 7c | −162.234 | −167.814 | −137.943 | −134.596 | −118.623 |
| 7d | −154.675 | −158.747 | −132.083 | −124.135 | −107.246 |
| 7e | −156.207 | −161.394 | −136.421 | −131.706 | −113.82 |
| 7f | −170.066 | −176.749 | −149.617 | −149.36 | −125.441 |
| Reference Molecules | −130.624 | −181.533 | −144.231 | −112.819 | −197.521 |
| Ligand | (ACE) (kcal/mol) | Category | Types | Interacting Residues |
|---|---|---|---|---|
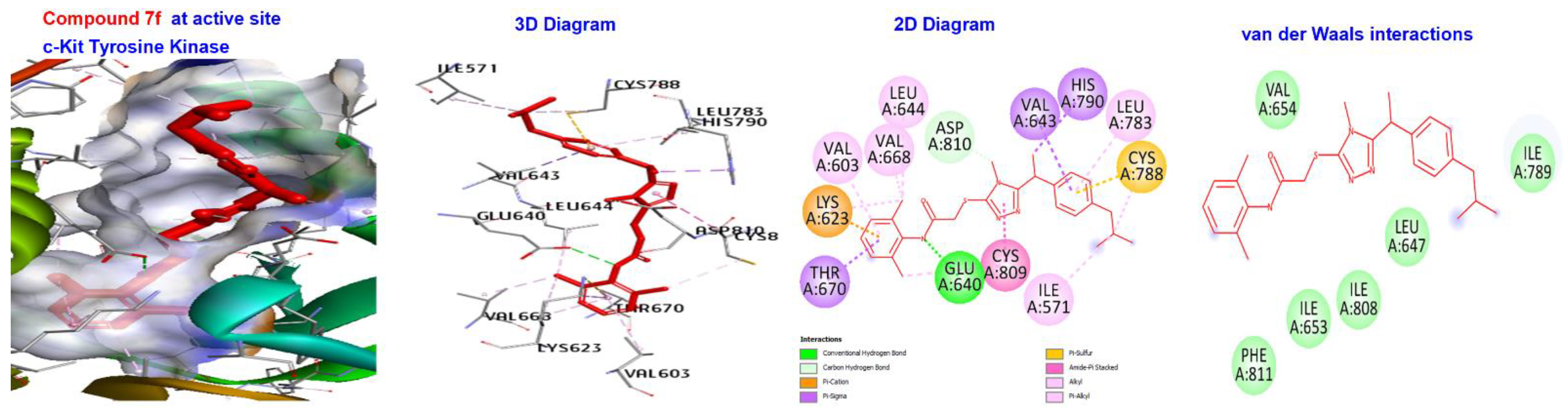
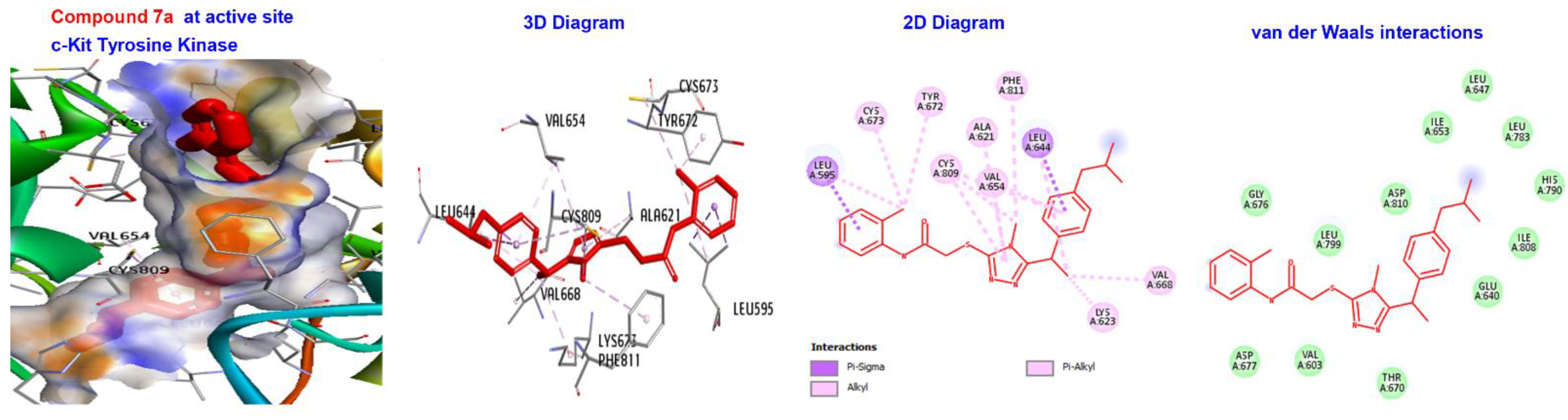
| 7a | −173.411 | H-bond | Alkyl | LEU595, LYS623, VAL654, LEU644, LEU595,LEU644, CYS673, CYS809, and VAL668. |
| Hydrophobic | Pi-alkyl | TYR672. | ||
| 7b | −167.882 | H-bond | Sulfur-X | CYS809. |
| Hydrophobic | C-alkyl Pi-alkyl | LEU595, VAL654, LEU644, CYS809, LEU595, ILE808, LEU644, AL654. TYR672, HIS790, and PHE811. | ||
| 7c | −167.814 | H-bond | Conventional | GLU640. |
| Hydrophobic | Pi-Alkyl | HIS790. | ||
| Hydrophobic | C-alkyl | VAL643, VAL603, LYS623, VAL668, LEU783, CYS788, LYS623, LEU644, VAL668, ALA621, and CYS788. | ||
| 7d | −158.747 | H-bond | Conventional | GLU640 and ASP810. |
| Hydrophobic | C-alkyl Pi-alkyl | ILE808. VAL603, VAL643, LEU783, CYS788, CYS809, LEU595, VAL603, VAL643, and LEU783. | ||
| 7e | −161.394 | H-bond | Conventional | CYS673, |
| Hydrophobic | C-alkyl Pi-alkyl | LEU595, VAL654, LEU644, CYS809, LYS623, LEU644, LEU644, TYR672, and VAL668. | ||
| 7f | −176.749 | H-bond | Conventional H-bond Pi-sigma | GLU640, ASP810, And HIS790. |
| Hydrophobic | Alkyl | VAL603, LYS623, VAL643, LEU783, CYS788, LYS623, LEU644, VAL668, CYS809, ILE571, and CYS788. | ||
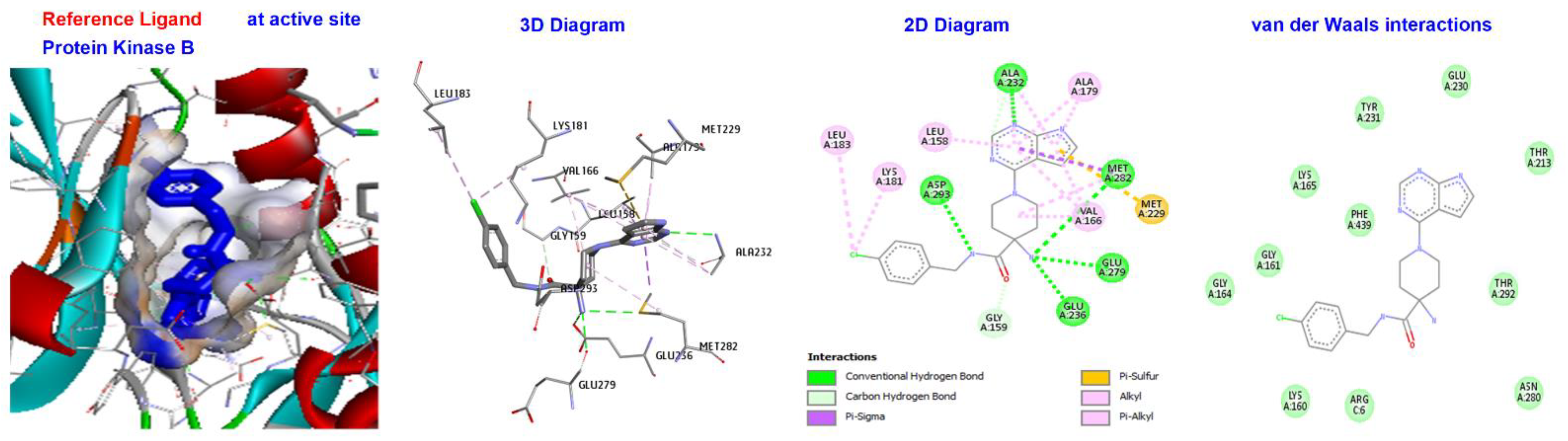
| Reference Ligand | H-bond | Conventional | ALA232, GLU236, MET282, ASP293, GLU279, | |
| STI | −181.533 | Other | C-H bond | MET229, GLY159, |
| Hydrophobic | C-alkyl | VAL166, LEU158, ALA179, LYS181, and LEU183. |
| Ligand | (ACE) (kcal/mol) | Category | Types | Interacting Residues |
|---|---|---|---|---|
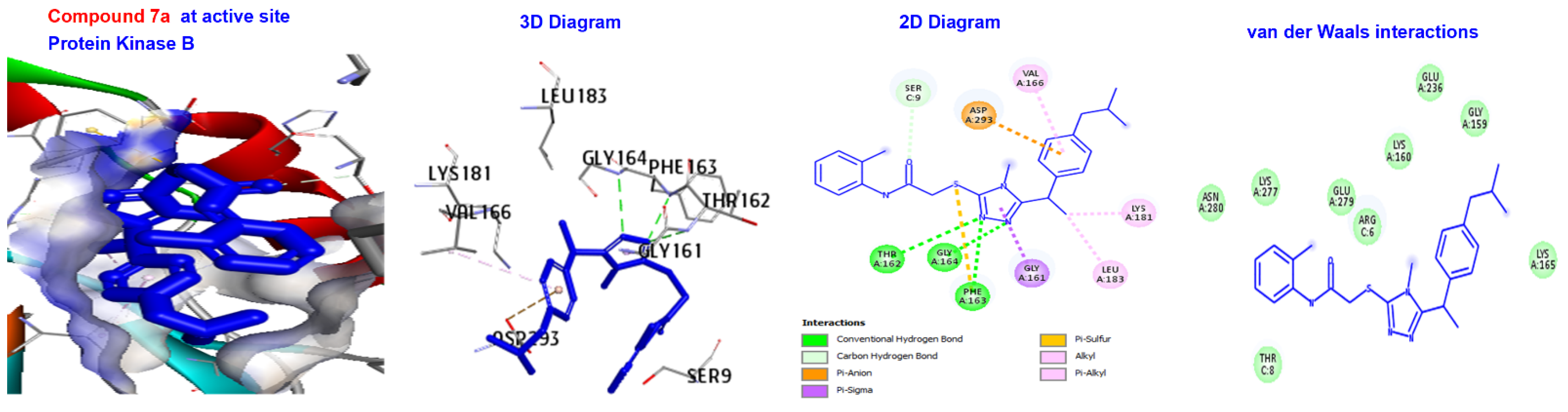
| 7a | −166.843 | H-bond | Conventional C-H bond | GLY164. SER9. |
| Other Hydrophobic | Pi-sulfur C-alkyl | PHE163. VAL166, LYS181, and EU183. | ||
| 7b | −166.371 | H-bond | Conventional | CYS809. |
| Hydrophobic | C-alkyl | LYS181, VAL166, and LEU296. | ||
| 7c | −162.234 | Hydrophobic | Alkyl | LYS181, VAL166, and LEU183. |
| 7d | −154.675 | H-bond | Conventional | GLY161, LEU158. |
| Hydrophobic | C-H bond C-alkyl Pi-alkyl | ASP293. VAL166, LYS181. PHE239, and PHE439. | ||
| 7e | −156.207 | H-bond | C-H bond | CYS673. |
| Hydrophobic | Alkyl | LEU296. | ||
| 7f | −170.066 | H-bond | Conventional C-H bond | GLY161, ASP293, and ARG6. ASP293. |
| Hydrophobic | C-alkyl | LYS181 and LEU183. | ||
| Reference Ligand | H-bond | Conventional | ALAA232, ASPA293, META282, GLUA279, GLUA236 | |
| X39 | −130.624 | Other | C-H bond | MET229, GLY159, |
| Hydrophobic | C-alkyl | VAL166, LEU A183, LEU A158, ALA179, LYS and A181. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akhter, N.; Batool, S.; Khan, S.G.; Rasool, N.; Anjum, F.; Rasul, A.; Adem, Ş.; Mahmood, S.; Rehman, A.u.; Nisa, M.u.; et al. Bio-Oriented Synthesis and Molecular Docking Studies of 1,2,4-Triazole Based Derivatives as Potential Anti-Cancer Agents against HepG2 Cell Line. Pharmaceuticals 2023, 16, 211. https://doi.org/10.3390/ph16020211
Akhter N, Batool S, Khan SG, Rasool N, Anjum F, Rasul A, Adem Ş, Mahmood S, Rehman Au, Nisa Mu, et al. Bio-Oriented Synthesis and Molecular Docking Studies of 1,2,4-Triazole Based Derivatives as Potential Anti-Cancer Agents against HepG2 Cell Line. Pharmaceuticals. 2023; 16(2):211. https://doi.org/10.3390/ph16020211
Chicago/Turabian StyleAkhter, Naheed, Sidra Batool, Samreen Gul Khan, Nasir Rasool, Fozia Anjum, Azhar Rasul, Şevki Adem, Sadaf Mahmood, Aziz ur Rehman, Mehr un Nisa, and et al. 2023. "Bio-Oriented Synthesis and Molecular Docking Studies of 1,2,4-Triazole Based Derivatives as Potential Anti-Cancer Agents against HepG2 Cell Line" Pharmaceuticals 16, no. 2: 211. https://doi.org/10.3390/ph16020211