
Long-Term Sleep Deprivation-Induced Myocardial Remodeling and Mitochondrial Dysfunction in Mice Were Attenuated by Lipoic Acid and N-Acetylcysteine

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
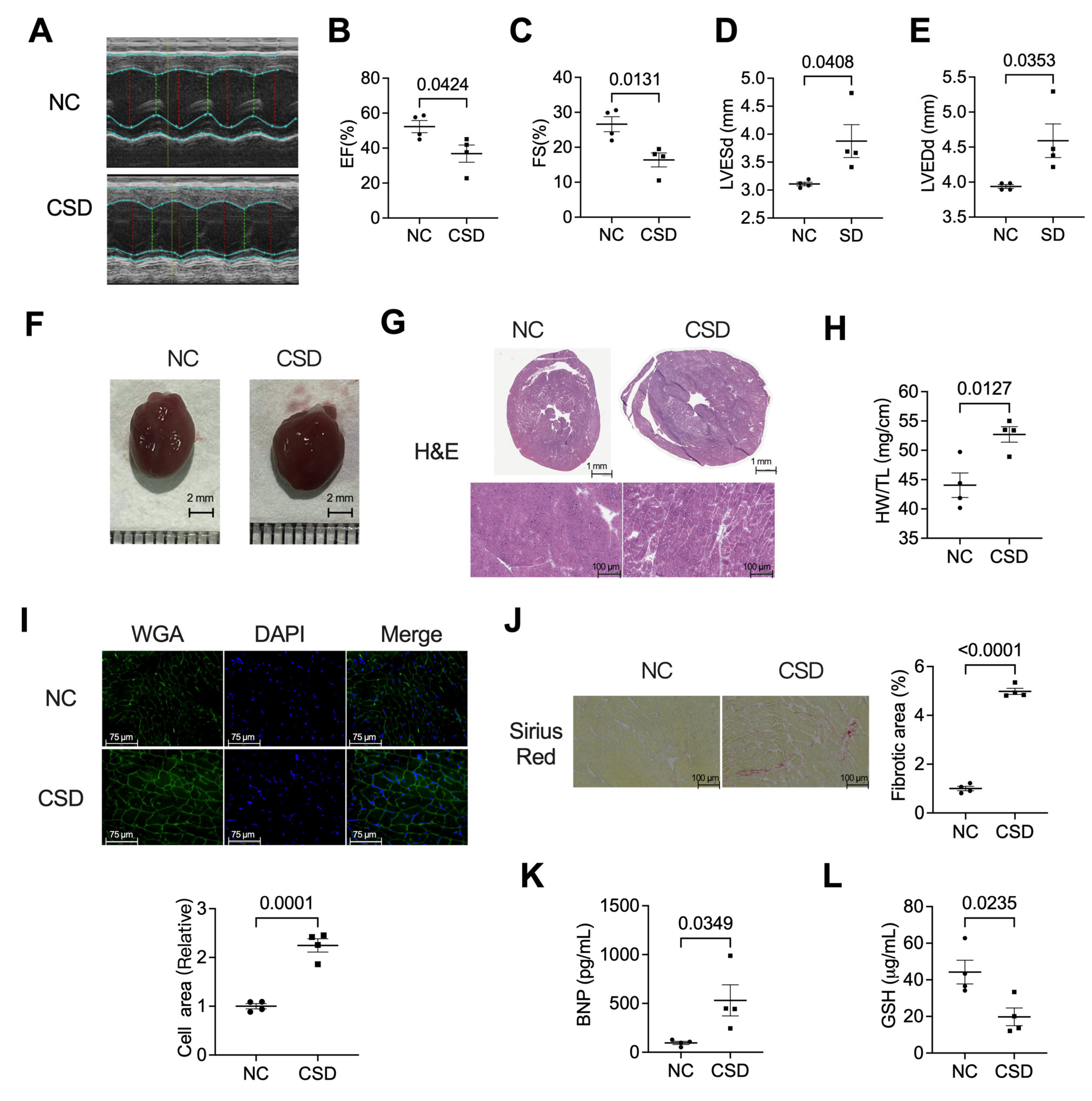
2.1. Chronic Sleep Deprivation Decreased Cardiac Function, Induced Hypertrophic Cardiomyopathy, and Myocardial Fibroblast
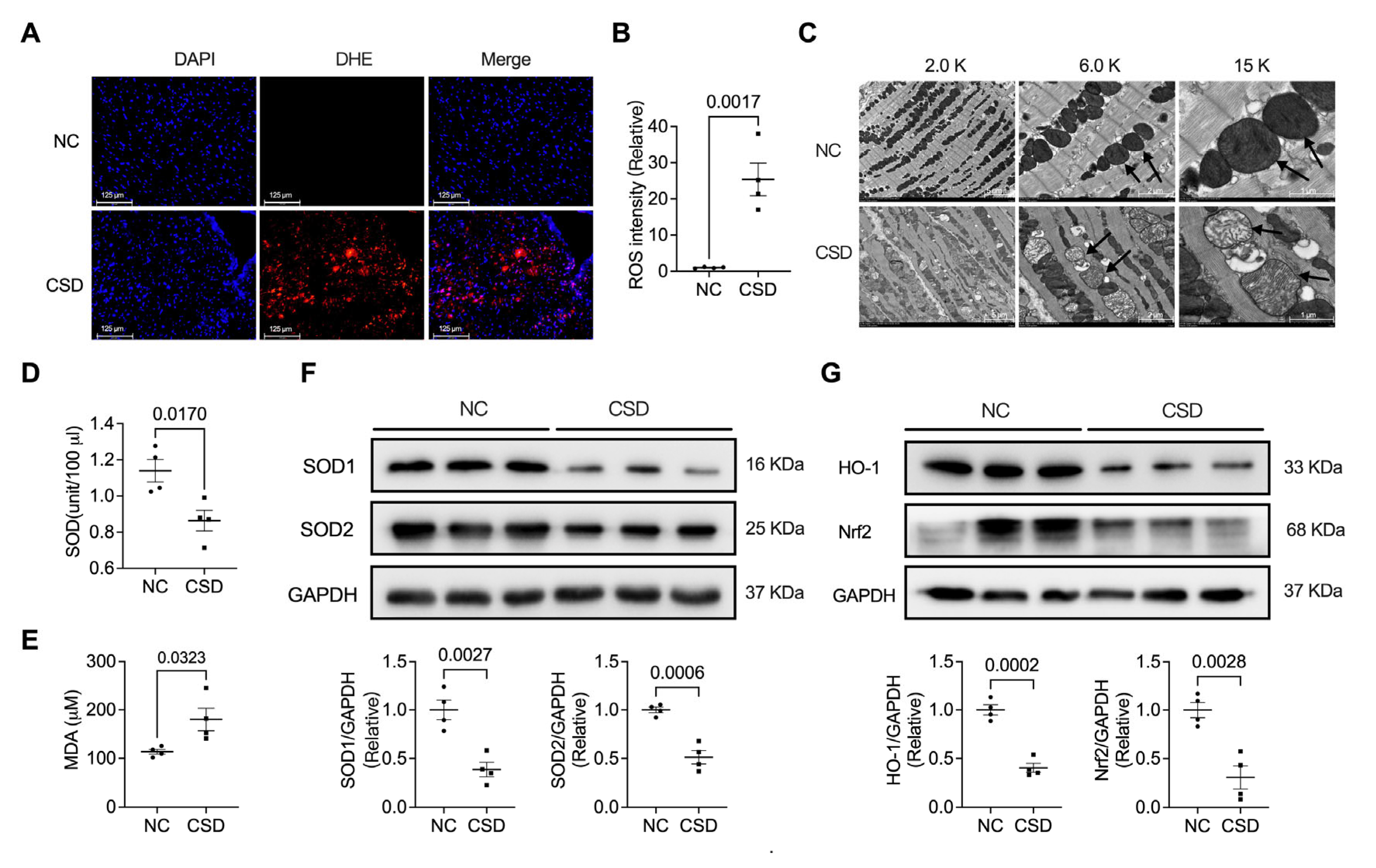
2.2. Chronic Sleep Deprivation Accumulated ROS and Induced Mitochondrial Dysfunction in the Heart
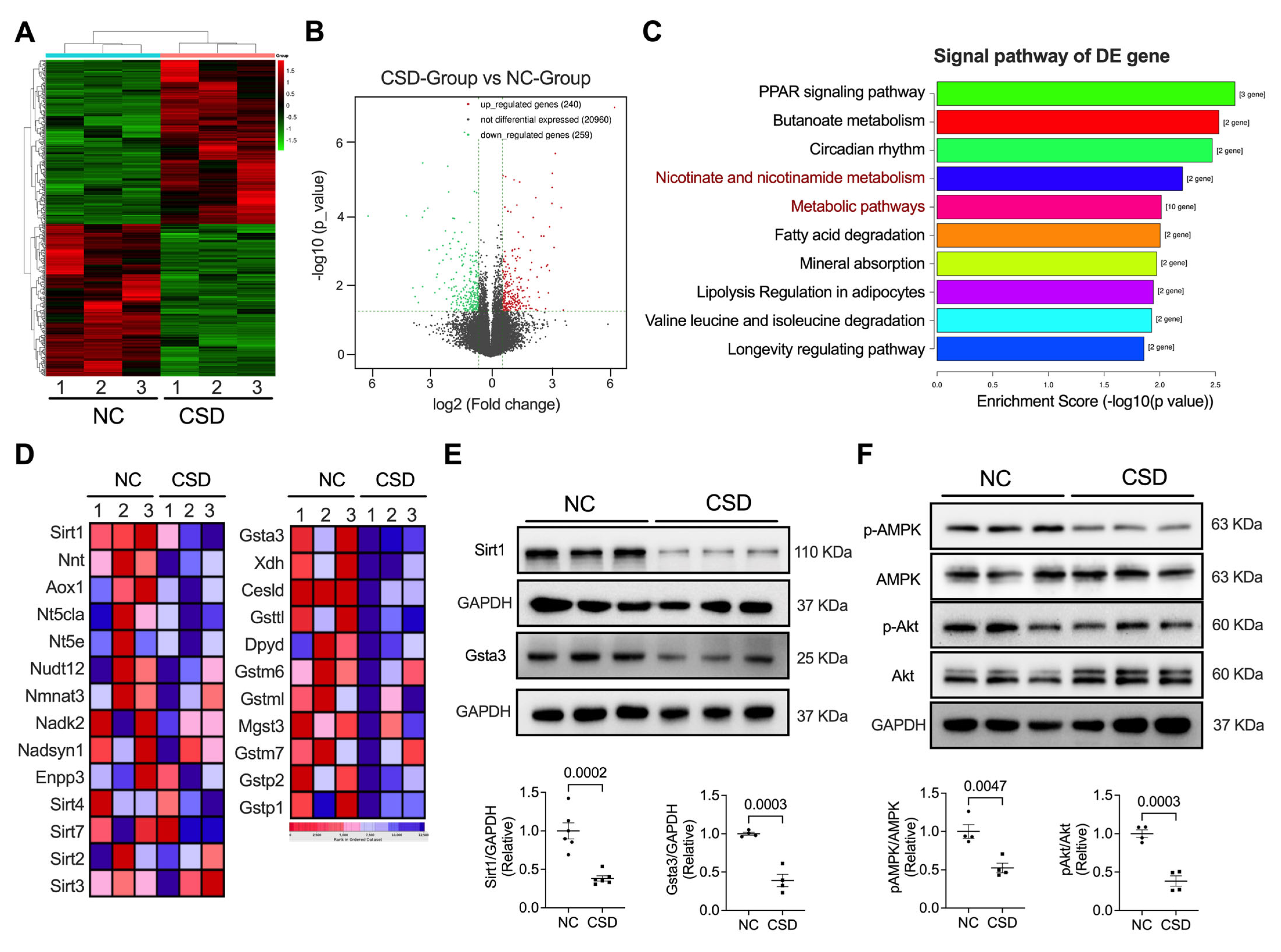
2.3. Sirt1 and Gsta3 Were Involved in CSD-Induced Cardiac Dysfunction
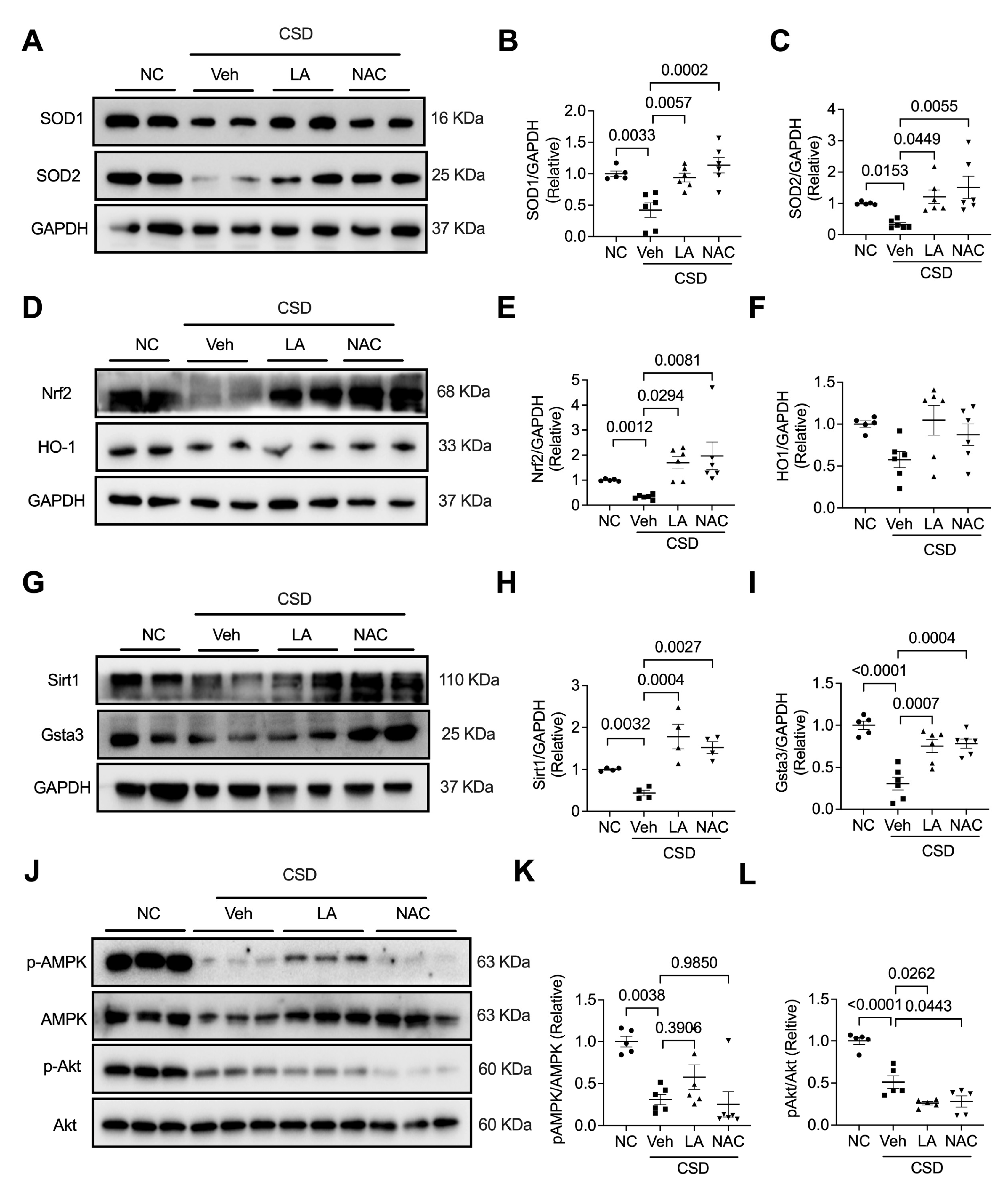
2.4. Administration of Antioxidants Ameliorated CSD-Induced Dysfunction of and Hypertrophic Cardiomyopathy
2.5. Antioxidants Protected against Mitochondria Dysfunction Induced by CSD
2.6. Antioxidants Reversed CSD-Induced Cardiac Dysfunction by Regulating the Nrf2/Sir1/Gsta3 Axis
3. Discussion
4. Materials and Methods
4.1. Induction of Chronic Sleep Deprivation
4.2. Antioxidant Feeding
4.3. Echocardiography Evaluation
4.4. Hematoxylin-Eosin (H&E) Staining
4.5. Picrosirius Red Staining
4.6. ROS Determination
4.7. Immunofluorescence Staining
4.8. Measurement of Serum Levels of BNP, GSH, SOD, and MDA
4.9. RNA-Sequencing
4.10. Western Blot Analysis
4.11. Transmission Electron Microscopy
4.12. Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kohansieh, M.; Makaryus, A.N. Sleep Deficiency and Deprivation Leading to Cardiovascular Disease. Int. J. Hypertens. 2015, 2015, 615681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medic, G.; Wille, M.; Hemels, M.E. Short- and long-term health consequences of sleep disruption. Nat. Sci. Sleep 2017, 9, 151–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangwisch, J.E.; Heymsfield, S.B.; Boden-Albala, B.; Buijs, R.M.; Kreier, F.; Pickering, T.G.; Rundle, A.G.; Zammit, G.K.; Malaspina, D. Short sleep duration as a risk factor for hypertension: Analyses of the first National Health and Nutrition Examination Survey. Hypertension 2006, 47, 833–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldabal, L.; Bahammam, A.S. Metabolic, endocrine, and immune consequences of sleep deprivation. Open Respir. Med. J. 2011, 5, 31–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullington, J.M.; Haack, M.; Toth, M.; Serrador, J.M.; Meier-Ewert, H.K. Cardiovascular, inflammatory, and metabolic consequences of sleep deprivation. Prog. Cardiovasc. Dis. 2009, 51, 294–302. [Google Scholar] [CrossRef] [Green Version]
- Knutson, K.L.; Spiegel, K.; Penev, P.; Van Cauter, E. The metabolic consequences of sleep deprivation. Sleep Med. Rev. 2007, 11, 163–178. [Google Scholar] [CrossRef] [Green Version]
- Ma, B.; Chen, J.; Mu, Y.; Xue, B.; Zhao, A.; Wang, D.; Chang, D.; Pan, Y.; Liu, J. Proteomic analysis of rat serum revealed the effects of chronic sleep deprivation on metabolic, cardiovascular and nervous system. PLoS ONE 2018, 13, e0199237. [Google Scholar] [CrossRef] [Green Version]
- Almeida, F.R.; Perry, J.C.; Futuro-Neto, H.A.; Almeida, V.R.; Sebastiao, R.M.; Andersen, M.L.; Tufik, S.; Campos, R.R.; Bergamaschi, C.T. Cardiovascular function alterations induced by acute paradoxical sleep deprivation in rats. Clin. Exp. Hypertens. 2014, 36, 567–571. [Google Scholar] [CrossRef]
- Vaara, J.; Kyrolainen, H.; Koivu, M.; Tulppo, M.; Finni, T. The effect of 60-h sleep deprivation on cardiovascular regulation and body temperature. Eur. J. Appl. Physiol. 2009, 105, 439–444. [Google Scholar] [CrossRef]
- Chen, W.R.; Liu, H.B.; Sha, Y.; Shi, Y.; Wang, H.; Yin, D.W.; Chen, Y.D.; Shi, X.M. Effects of Statin on Arrhythmia and Heart Rate Variability in Healthy Persons with 48-Hour Sleep Deprivation. J. Am. Heart Assoc. 2016, 5, e003833. [Google Scholar] [CrossRef]
- Cincin, A.; Sari, I.; Oguz, M.; Sert, S.; Bozbay, M.; Atas, H.; Ozben, B.; Tigen, K.; Basaran, Y. Effect of acute sleep deprivation on heart rate recovery in healthy young adults. Sleep Breath. 2015, 19, 631–636. [Google Scholar] [CrossRef]
- Periasamy, S.; Hsu, D.Z.; Fu, Y.H.; Liu, M.Y. Sleep deprivation-induced multi-organ injury: Role of oxidative stress and inflammation. Excli J. 2015, 14, 672–683. [Google Scholar] [CrossRef]
- Li, S.; Ning, K.; Zhou, J.; Guo, Y.; Zhang, H.; Zhu, Y.; Zhang, L.; Jia, C.; Chen, Y.; Sol Reinach, P.; et al. Sleep deprivation disrupts the lacrimal system and induces dry eye disease. Exp. Mol. Med. 2018, 50, e451. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb, D.J.; Redline, S.; Nieto, F.J.; Baldwin, C.M.; Newman, A.B.; Resnick, H.E.; Punjabi, N.M. Association of usual sleep duration with hypertension: The Sleep Heart Health Study. Sleep 2006, 29, 1009–1014. [Google Scholar] [CrossRef]
- Cappuccio, F.P.; Stranges, S.; Kandala, N.B.; Miller, M.A.; Taggart, F.M.; Kumari, M.; Ferrie, J.E.; Shipley, M.J.; Brunner, E.J.; Marmot, M.G. Gender-specific associations of short sleep duration with prevalent and incident hypertension: The Whitehall II Study. Hypertension 2007, 50, 693–700. [Google Scholar] [CrossRef]
- Sabanayagam, C.; Shankar, A. Sleep duration and cardiovascular disease: Results from the National Health Interview Survey. Sleep 2010, 33, 1037–1042. [Google Scholar] [CrossRef]
- Korostovtseva, L.; Bochkarev, M.; Sviryaev, Y. Sleep and Cardiovascular Risk. Sleep Med. Clin. 2021, 16, 485–497. [Google Scholar] [CrossRef]
- Parsa, H.; Imani, A.; Faghihi, M.; Riahi, E.; Badavi, M.; Shakoori, A.; Rastegar, T.; Aghajani, M.; Rajani, S.F. Acute sleep deprivation preconditions the heart against ischemia/reperfusion injury: The role of central GABA-A receptors. Iran. J. Basic Med. Sci. 2017, 20, 1232–1241. [Google Scholar] [CrossRef]
- Chen, H.; Zhao, Z.; Zheng, J.; Chen, X.; Zou, J.; Shi, Y.; Liu, Z. The effect of IGF-1 on symptoms of sleep deprivation in a rat model of inflammatory heart disease and metabolic syndrome. Biochem. Biophys. Res. Commun. 2014, 446, 843–849. [Google Scholar] [CrossRef]
- Chen, W.R.; Shi, X.M.; Yang, T.S.; Zhao, L.C.; Gao, L.G. Protective effect of metoprolol on arrhythmia and heart rate variability in healthy people with 24 h of sleep deprivation. J. Interv. Card. Electrophysiol. 2013, 36, 267–272. [Google Scholar] [CrossRef]
- Irwin, M.R.; Ziegler, M. Sleep deprivation potentiates activation of cardiovascular and catecholamine responses in abstinent alcoholics. Hypertension 2005, 45, 252–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faraci, F.M.; Didion, S.P. Vascular protection: Superoxide dismutase isoforms in the vessel wall. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1367–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Z.Q.; Zhou, H.Z.; Cecchini, G.; Gray, M.O.; Karliner, J.S. MnSOD in mouse heart: Acute responses to ischemic preconditioning and ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2986–H2994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Wu, H.; He, J.; Zhuang, J.; Liu, Z.; Yang, Y.; Huang, L.; Zhao, Z. Frontal cortical mitochondrial dysfunction and mitochondria-related beta-amyloid accumulation by chronic sleep restriction in mice. Neuroreport 2016, 27, 916–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, C.S.; Vaziri, N.D. The effects of iron dextran on the oxidative stress in cardiovascular tissues of rats with chronic renal failure. Kidney Int. 2004, 65, 1802–1809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siwik, D.A.; Colucci, W.S. Regulation of matrix metalloproteinases by cytokines and reactive oxygen/nitrogen species in the myocardium. Heart Fail. Rev. 2004, 9, 43–51. [Google Scholar] [CrossRef]
- Ma, Y.; Iyer, R.P.; Jung, M.; Czubryt, M.P.; Lindsey, M.L. Cardiac Fibroblast Activation Post-Myocardial Infarction: Current Knowledge Gaps. Trends Pharmacol. Sci. 2017, 38, 448–458. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y. New insights into epithelial-mesenchymal transition in kidney fibrosis. J. Am. Soc. Nephrol. 2010, 21, 212–222. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar] [CrossRef] [Green Version]
- Elorza, A.A.; Soffia, J.P. mtDNA Heteroplasmy at the Core of Aging-Associated Heart Failure. An Integrative View of OXPHOS and Mitochondrial Life Cycle in Cardiac Mitochondrial Physiology. Front. Cell Dev. Biol. 2021, 9, 625020. [Google Scholar] [CrossRef]
- Zhuang, L.; Jia, K.; Chen, C.; Li, Z.; Zhao, J.; Hu, J.; Zhang, H.; Fan, Q.; Huang, C.; Xie, H.; et al. DYRK1B-STAT3 Drives Cardiac Hypertrophy and Heart Failure by Impairing Mitochondrial Bioenergetics. Circulation 2022, 145, 829–846. [Google Scholar] [CrossRef]
- Zaki, S.M.; Abdalla, I.L.; Sadik, A.O.E.; Mohamed, E.A.; Kaooh, S. Protective Role of N-Acetylcysteine on Isoprenaline-Induced Myocardial Injury: Histological, Immunohistochemical and Morphometric Study. Cardiovasc. Toxicol. 2018, 18, 9–23. [Google Scholar] [CrossRef]
- Dekhuijzen, P.N. Antioxidant properties of N-acetylcysteine: Their relevance in relation to chronic obstructive pulmonary disease. Eur. Respir. J. 2004, 23, 629–636. [Google Scholar] [CrossRef] [Green Version]
- Fishbane, S. N-acetylcysteine in the prevention of contrast-induced nephropathy. Clin. J. Am. Soc. Nephrol. 2008, 3, 281–287. [Google Scholar] [CrossRef] [Green Version]
- Yin, J.; Ren, W.; Yang, G.; Duan, J.; Huang, X.; Fang, R.; Li, C.; Li, T.; Yin, Y.; Hou, Y.; et al. L-Cysteine metabolism and its nutritional implications. Mol. Nutr. Food Res. 2016, 60, 134–146. [Google Scholar] [CrossRef]
- Atkuri, K.R.; Mantovani, J.J.; Herzenberg, L.A.; Herzenberg, L.A. N-Acetylcysteine--a safe antidote for cysteine/glutathione deficiency. Curr. Opin. Pharmacol. 2007, 7, 355–359. [Google Scholar] [CrossRef] [Green Version]
- Rosic, G.; Selakovic, D.; Joksimovic, J.; Srejovic, I.; Zivkovic, V.; Tatalovic, N.; Orescanin-Dusic, Z.; Mitrovic, S.; Ilic, M.; Jakovljevic, V. The effects of N-acetylcysteine on cisplatin-induced changes of cardiodynamic parameters within coronary autoregulation range in isolated rat hearts. Toxicol. Lett. 2016, 242, 34–46. [Google Scholar] [CrossRef]
- Marinho, P.M.; Salomon, T.B.; Andrade, A.S.; Behling, C.S.; Putti, J.S.; Benfato, M.S.; Hackenhaar, F.S. The effect of n-3 long-chain polyunsaturated fatty acids and lipoic acid on the heart in the ovariectomized rat model of menopause. Free Radic. Res. 2019, 53, 669–679. [Google Scholar] [CrossRef]
- Gomes, M.B.; Negrato, C.A. Alpha-lipoic acid as a pleiotropic compound with potential therapeutic use in diabetes and other chronic diseases. Diabetol. Metab. Syndr. 2014, 6, 80. [Google Scholar] [CrossRef] [Green Version]
- Moini, H.; Packer, L.; Saris, N.E. Antioxidant and prooxidant activities of alpha-lipoic acid and dihydrolipoic acid. Toxicol. Appl. Pharmacol. 2002, 182, 84–90. [Google Scholar] [CrossRef]
- Tian, S.; Nakamura, J.; Hiller, S.; Simington, S.; Holley, D.W.; Mota, R.; Willis, M.S.; Bultman, S.J.; Luft, J.C.; DeSimone, J.M.; et al. New insights into immunomodulation via overexpressing lipoic acid synthase as a therapeutic potential to reduce atherosclerosis. Vascul. Pharmacol. 2020, 133–134, 106777. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Tian, L.; Shen, T.; Sun, H.; Liu, P. Alpha-Lipoic Acid Protects Human Aortic Endothelial Cells Against H2O2-Induced Injury and Inhibits Atherosclerosis in Ovariectomized Low Density Lipoprotein Receptor Knock-Out Mice. Cell. Physiol. Biochem. 2018, 47, 2261–2277. [Google Scholar] [CrossRef] [PubMed]
- Ying, Z.; Kherada, N.; Farrar, B.; Kampfrath, T.; Chung, Y.; Simonetti, O.; Deiuliis, J.; Desikan, R.; Khan, B.; Villamena, F.; et al. Lipoic acid effects on established atherosclerosis. Life Sci. 2010, 86, 95–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinelli, I.; Tomassoni, D.; Roy, P.; Di Cesare Mannelli, L.; Amenta, F.; Tayebati, S.K. Antioxidant Properties of Alpha-Lipoic (Thioctic) Acid Treatment on Renal and Heart Parenchyma in a Rat Model of Hypertension. Antioxidants 2021, 10, 1006. [Google Scholar] [CrossRef] [PubMed]
- Tayebati, S.K.; Tomassoni, D.; Di Cesare Mannelli, L.; Amenta, F. Effect of treatment with the antioxidant alpha-lipoic (thioctic) acid on heart and kidney microvasculature in spontaneously hypertensive rats. Clin. Exp. Hypertens. 2016, 38, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Midaoui, A.E.; Elimadi, A.; Wu, L.; Haddad, P.S.; de Champlain, J. Lipoic acid prevents hypertension, hyperglycemia, and the increase in heart mitochondrial superoxide production. Am. J. Hypertens. 2003, 16, 173–179. [Google Scholar] [CrossRef] [Green Version]
- Pop, C.; Stefan, M.G.; Muntean, D.M.; Stoicescu, L.; Gal, A.F.; Kiss, B.; Morgovan, C.; Loghin, F.; Rochette, L.; Lauzier, B.; et al. Protective Effects of a Discontinuous Treatment with Alpha-Lipoic Acid in Obesity-Related Heart Failure with Preserved Ejection Fraction, in Rats. Antioxidants 2020, 9, 1073. [Google Scholar] [CrossRef]
- Li, W.; Yin, L.; Sun, X.; Wu, J.; Dong, Z.; Hu, K.; Sun, A.; Ge, J. Alpha-lipoic acid protects against pressure overload-induced heart failure via ALDH2-dependent Nrf1-FUNDC1 signaling. Cell Death Dis. 2020, 11, 599. [Google Scholar] [CrossRef]
- Lee, J.; Cho, J.H. Letter: Effects of High-Dose alpha-Lipoic Acid on Heart Rate Variability of Type 2 Diabetes Mellitus Patients with Cardiac Autonomic Neuropathy in Korea (Diabetes Metab. J. 2017;41:275–283). Diabetes Metab. J. 2017, 41, 417–419. [Google Scholar] [CrossRef]
- Yi, X.; Maeda, N. alpha-Lipoic acid prevents the increase in atherosclerosis induced by diabetes in apolipoprotein E-deficient mice fed high-fat/low-cholesterol diet. Diabetes 2006, 55, 2238–2244. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, F.; Lin, J.; Zhang, H.; Guo, Y.; Mao, Y.; Liu, Z.; Li, G.; Wang, Y. Long-Term Sleep Deprivation-Induced Myocardial Remodeling and Mitochondrial Dysfunction in Mice Were Attenuated by Lipoic Acid and N-Acetylcysteine. Pharmaceuticals 2023, 16, 51. https://doi.org/10.3390/ph16010051
Song F, Lin J, Zhang H, Guo Y, Mao Y, Liu Z, Li G, Wang Y. Long-Term Sleep Deprivation-Induced Myocardial Remodeling and Mitochondrial Dysfunction in Mice Were Attenuated by Lipoic Acid and N-Acetylcysteine. Pharmaceuticals. 2023; 16(1):51. https://doi.org/10.3390/ph16010051
Chicago/Turabian StyleSong, Fei, Jiale Lin, Houjian Zhang, Yuli Guo, Yijie Mao, Zuguo Liu, Gang Li, and Yan Wang. 2023. "Long-Term Sleep Deprivation-Induced Myocardial Remodeling and Mitochondrial Dysfunction in Mice Were Attenuated by Lipoic Acid and N-Acetylcysteine" Pharmaceuticals 16, no. 1: 51. https://doi.org/10.3390/ph16010051