
Dehydroeburicoic Acid, a Dual Inhibitor against Oxidative Stress in Alcoholic Liver Disease
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
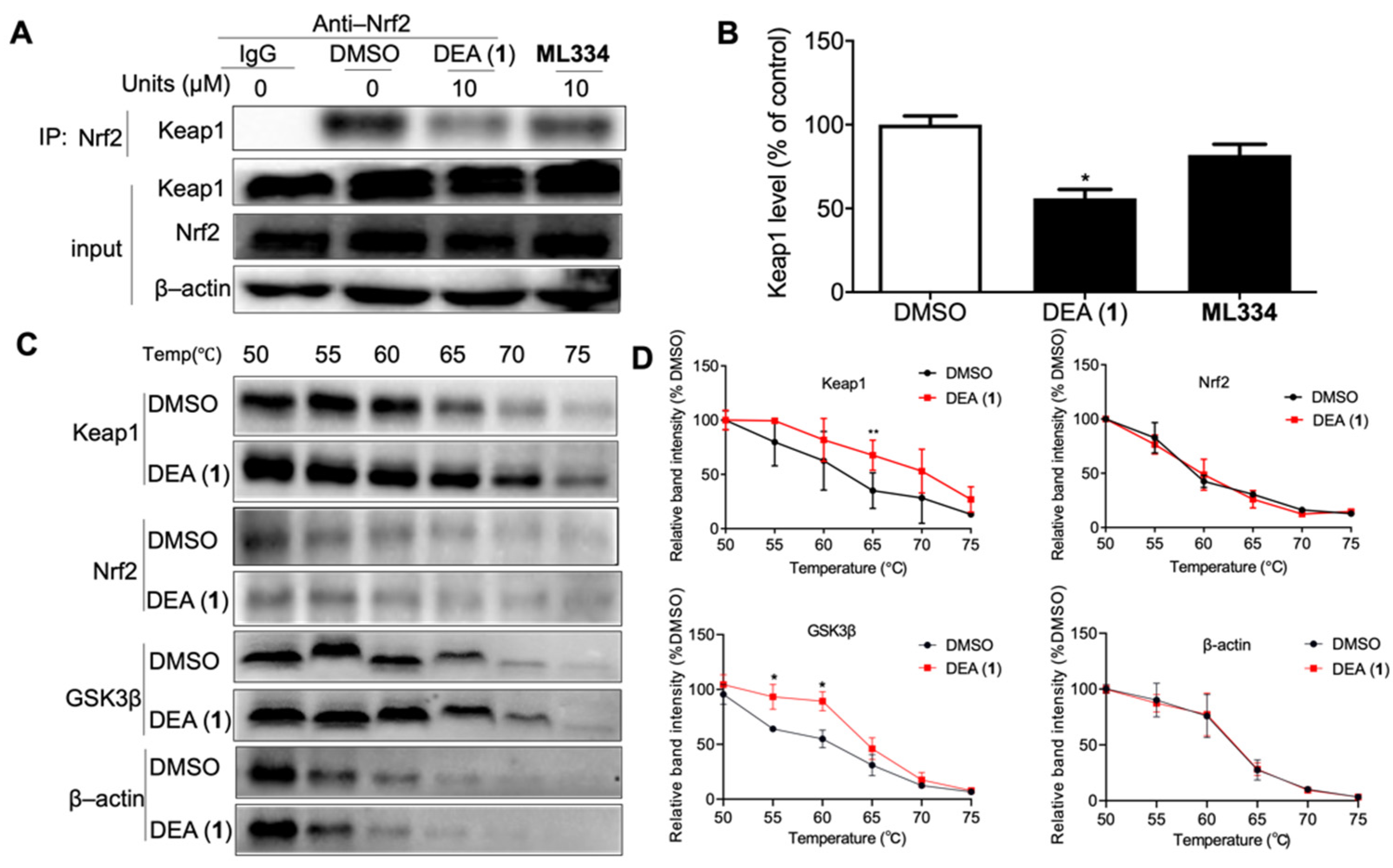
2.1. DEA (1) Inhibits the Keap1–Nrf2 PPI and GSK3β Activity
2.2. DEA (1) Exhibits Low Cytotoxicity In Cellulo
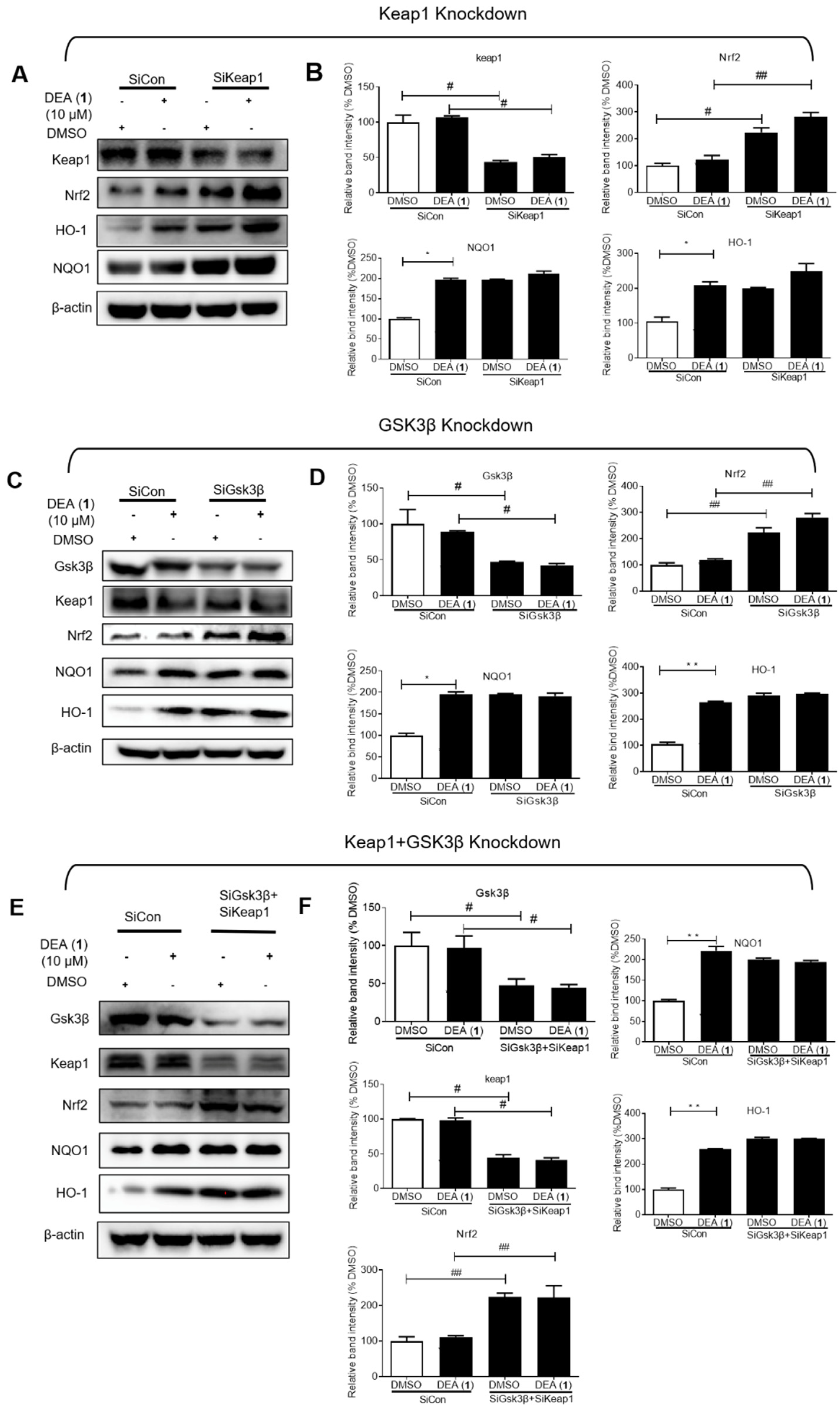
2.3. DEA (1) as a Dual Inhibitor of Keap1 and GSK3β
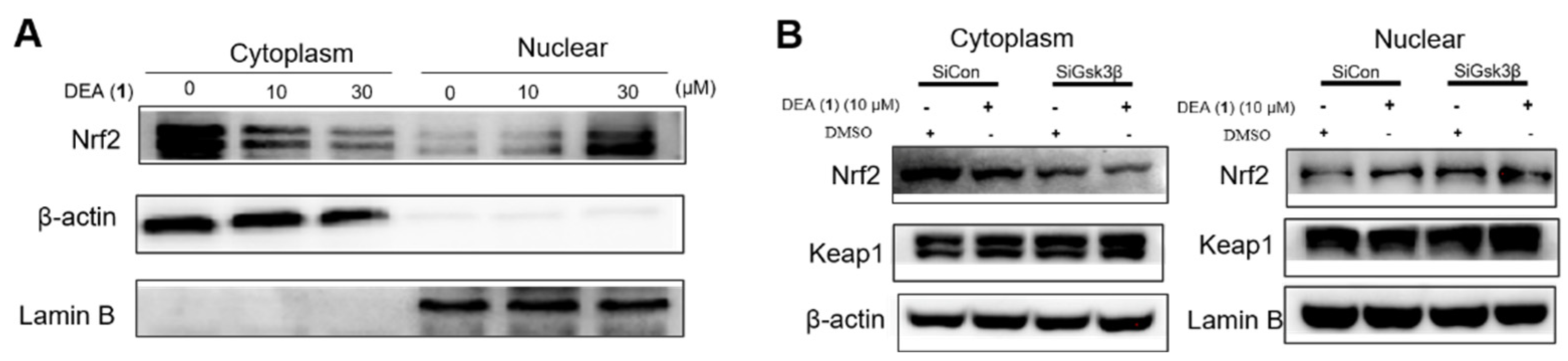
2.4. DEA (1) Targets Keap1 and GSK3β to Induce Nrf2 Accumulation in the Nucleus and the Expression of Downstream Antioxidant Proteins
2.5. DEA (1) Activates Nrf2 Downstream Antioxidant Genes in ALD Model Cells
2.6. DEA (1) Repairs the EtOH Induced Mitochondrial Dysfunction and Improves the Antioxidant Activity in LO2 Cells
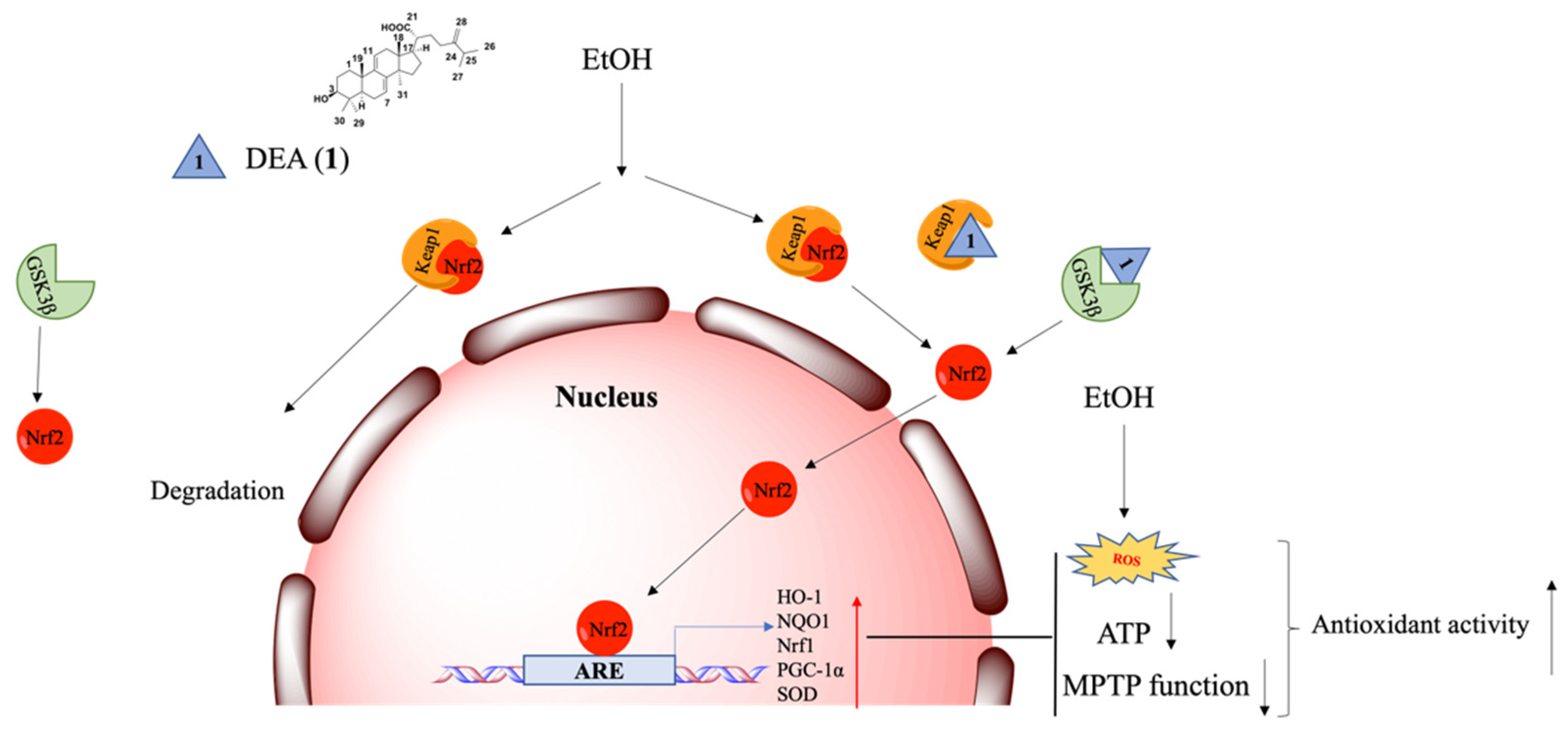
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture
4.2. Chemical
4.3. Fluorescence Polarization Assay
4.4. Western Blot (WB) and Co–Immunoprecipitation (Co–IP)
4.5. Real-Time Quantitative Polymerase Chain Reaction (RT–qPCR)
4.6. MTT Assay
4.7. Detection of ROS and Antioxidant Activity
4.8. Nuclear and Cytoplasmic Extraction
4.9. GSK3β Kinase Assay
4.10. Dual Luciferase Reporter Gene Assay
4.11. Cellular Thermal Shift Assay (CETSA)
4.12. Mitochondrial Permeability Transition Pore (MPTP) Assay
4.13. SiRNA Gene Knockdown
4.14. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Avila, M.A.; Dufour, J.F.; Gerbes, A.L.; Zoulim, F.; Bataller, R.; Burra, P.; Cortez-Pinto, H.; Gao, B.; Gilmore, I.; Mathurin, P.; et al. Recent advances in alcohol-related liver disease (ALD): Summary of a Gut round table meeting. Gut 2019, 69, 764–780. [Google Scholar] [CrossRef] [PubMed]
- Bakhautdin, B.; Das, D.; Mandal, P.; Roychowdhury, S.; Danner, J.; Bush, K.; Pollard, K.; Kaspar, J.W.; Li, W.; Salomon, R.G.; et al. Protective role of HO-1 and carbon monoxide in ethanol-induced hepatocyte cell death and liver injury in mice. J. Hepatol. 2014, 61, 1029–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higuera-de Tijera, F.; Servín-Caamaño, A.; Serralde-Zúñiga, A.E.; Cruz-Herrera, J.; Pérez-Torres, E.; Abdo-Francis, J.M.; Salas-Gordillo, F.; Pérez-Hernández, J.L. Metadoxine improves the three-and six-month survival rates in patients with severe alcoholic hepatitis. World J. Gastroenterol. 2015, 21, 4975–4985. [Google Scholar] [CrossRef] [PubMed]
- Triantafyllou, K.; Vlachogiannakos, J.; Ladas, S.D. Gastrointestinal and liver side effects of drugs in elderly patients. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Li, X.X.; Jiang, Z.H.; Zhou, B.; Chen, C. Hepatoprotective effect of gastrodin against alcohol-induced liver injury in mice. J. Physiol. Biochem. 2019, 75, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Nassir, F.; Ibdah, J.A. Role of mitochondria in alcoholic liver disease. World J. Gastroenterol. 2014, 20, 2136–2342. [Google Scholar] [CrossRef]
- Abdallah, M.A.; Singal, A.K. Mitochondrial dysfunction and alcohol-associated liver disease: A novel pathway and therapeutic target. Signal Transduct. Target. Ther. 2020, 5, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; Zhao, Y.; He, S.; Guo, T.S.; Song, Q.; Guo, N.; Yuan, Z.Y. Overexpression of FGF19 alleviates hypoxia/reoxygenation-induced injury of cardiomyocytes by regulating GSK-3β/Nrf2/ARE signaling. Biochem. Biophys. Res. Commun. 2018, 503, 2355–2362. [Google Scholar] [CrossRef]
- Han, D.; Ybanez, M.D.; Johnson, H.S.; McDonald, J.N.; Mesropyan, L.; Sancheti, H.; Martin, G.; Martin, A.; Lim, A.M.; Dara, L.; et al. Dynamic adaptation of liver mitochondria to chronic alcohol feeding in mice biogenesis, remodeling, and functional alterations. J. Biol. Chem. 2021, 287, 42165–42179. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Cui, W.; Meng, F.; Xia, Q.; Li, X.; Hou, M.; Jia, L.; Zhang, J. Glucopyranose from Pleurotus geesteranus prevent alcoholic liver diseases by regulating Nrf2/HO-1-TLR4/NF-κB signalling pathways and gut microbiota. Food Funct. 2022, 13, 2441–2455. [Google Scholar] [CrossRef]
- Jiang, Z.Y.; Lu, M.C.; You, Q.D. Discovery and development of Kelch-like ECH-associated protein 1. nuclear factor erythroid 2-related factor 2 (KEAP1: NRF2) protein–protein interaction inhibitors: Achievements, challenges, and future directions. J. Med. Chem. 2016, 59, 10837–10858. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zheng, Q.; Chen, Z. The Nrf2 Pathway in Liver Diseases. Front. Cell Dev. Biol. 2022, 10, 826204. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaličková, D.; Hrnčíř, T.; Canová, N.K.; Slanař, O. Targeting Keap1/Nrf2/ARE signaling pathway in multiple sclerosis. Eur. J. Pharmacol. 2020, 873, 172973–172988. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, A.; Reiner, Ž.; Ruscica, M.; Tedeschi-Reiner, E.; Radbakhsh, S.; Bagheri Ekta, M.; Sahebkar, A. Antioxidant Effects of Statins by Modulating Nrf2 and Nrf2/HO-1 Signaling in Different Diseases. J. Clin. Med. 2022, 11, 1313. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Xiong, H.; Pang, J.; Su, Z.W.; Lai, L.; Lin, H.Q.; Jian, B.Q.; He, W.H.; Yang, H.D.; Zheng, Y.Q. Nrf2 activation protects auditory hair cells from cisplatin-induced ototoxicity independent on mitochondrial ROS production. Toxicol. Lett. 2020, 331, 1–10. [Google Scholar] [CrossRef]
- Xu, D.; Xu, M.; Jeong, S.S.; Qian, Y.H.; Wu, H.L.; Xia, Q.; Kong, X.N. The role of Nrf2 in liver disease: Novel molecular mechanisms and therapeutic approaches. Front. Pharmacol. 2019, 8, 1428–1435. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Wang, Y.; Xie, S.; Lai, Y.; Mo, C.; Zeng, T.; Kuang, S.; Deng, G.; Zhou, C.; Chen, Y.; et al. Hepatic TGFβr1 Deficiency Attenuates Lipopolysaccharide/D-Galactosamine-Induced Acute Liver Failure Through Inhibiting GSK3β-Nrf2-Mediated Hepatocyte Apoptosis and Ferroptosis. Cell Mol. Gastroenterol. Hepatol. 2022, 13, 1649–1672. [Google Scholar] [CrossRef]
- Shu, G.; Qiu, Y.; Hao, J.; Fu, Q.; Deng, X. γ-Oryzanol alleviates acetaminophen-induced liver injury: Roles of modulating AMPK/GSK3β/Nrf2 and NF-κB signaling pathways. Food Funct. 2019, 10, 6858–6872. [Google Scholar] [CrossRef]
- Rojo, A.I.; Medina-Campos, O.N.; RadaP, P.; Zúñiga-Toalác, A.; López-Gazcón, A.; Espada, S.; Pedraza-Chaverri, J.; Cuadradoa, A. Signaling pathways activated by the phytochemical nordihydroguaiaretic acid contribute to a Keap1-independent regulation of Nrf2 stability: Role of glycogen synthase kinase-3. Free Radic. Biol. Med. 2012, 52, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zou, L.; Li, L.; Wu, T. The protective effect of glycyrrhetinic acid on carbon tetrachloride-induced chronic liver fibrosis in mice via upregulation of Nrf2. PLoS ONE 2013, 8, e53662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 system: A thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mundal, S.B.; Rakner, J.J.; Silva, G.B.; Gierman, L.M.; Austdal, M.; Basnet, P.; Elschot, M.; Bakke, S.S.; Ostrop, J.; Thomsen, L.C.V.; et al. Divergent Regulation of Decidual Oxidative-Stress Response by NRF2 and KEAP1 in Preeclampsia with and without Fetal Growth Restriction. Int. J. Mol. Sci. 2022, 10, 1966. [Google Scholar] [CrossRef] [PubMed]
- Liby, K.T.; Yore, M.M.; Sporn, M.B. Triterpenoids and rexinoids as multifunctional agents for the prevention and treatment of cancer. Nat. Rev. Cancer 2007, 7, 357–369. [Google Scholar] [CrossRef]
- Lu, M.; Zhou, H.S.; You, Q.D.; Jiang, Z. Design, synthesis, and initial evaluation of affinity-based small-molecule probes for fluorescent visualization and specific detection of Keap1. J. Med. Chem. 2016, 59, 7305–7310. [Google Scholar] [CrossRef]
- Kim, S.; Indu Viswanath, A.N.; Park, J.H.; Lee, H.E.; Park, A.Y.; Choi, J.W.; Kim, H.J.; Londhe, A.M.; Jang, B.K.; Lee, J.; et al. Nrf2 activator via interference of Nrf2-Keap1 interaction has antioxidant and anti-inflammatory properties in Parkinson’s disease animal model. Neuropharmacology 2020, 167, 107989. [Google Scholar] [CrossRef]
- Cuadrado, A.; Rojo, A.I.; Wells, G.; Hayes, J.D.; Cousin, S.P.; Rumsey, W.L.; Attucks, O.C.; Franklin, F.; Levonen, A.L.; Kensler, T.W.; et al. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019, 18, 295–317. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Z.Y.; Xu, L.L.; Lu, M.C.; Chen, Z.Y.; Yuan, Z.W.; Xu, X.L.; Guo, X.K.; Zhang, X.J.; Sun, H.P.; You, Q.D. Structure–activity and structure–property relationship and exploratory in vivo evaluation of the nanomolar Keap1–Nrf2 protein–protein interaction inhibitor. J. Med. Chem. 2015, 58, 6410–6421. [Google Scholar] [CrossRef]
- Li, G.D.; Liu, H.; Feng, R.B.; Kang, T.S.; Wang, W.H.; Ko, C.N.; Wong, C.Y.; Ye, M.; Ma, D.L.; Wan, J.B.; et al. A bioactive ligand-conjugated iridium (III) metal-based complex as a Keap1–Nrf2 protein-protein interaction inhibitor against acetaminophen-induced acute liver injury. Redox Biol. 2021, 48, 102129. [Google Scholar] [CrossRef]
- Sun, C.; Han, B.; Zhai, Y.; Zhao, H.; Li, X.; Qian, J.; Hao, X.; Liu, Q.; Shen, J.; Kai, G. Dihydrotanshinone I inhibits ovarian tumor growth by activating oxidative stress through Keap1-mediated Nrf2 ubiquitination degradation. Free Radic. Biol. Med. 2022, 20, 220–235. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.H.; Zhang, J.T.; Yang, G.J.; Liu, H.; Han, Q.B.; Ma, D.L. Emerging Screening Approaches in the Development of Nrf2–Keap1 Protein–Protein Interaction Inhibitors. Int. J. Mol. Sci. 2019, 20, 4445–4464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, R.B.; Wang, Y.; He, C.W.; Yang, Y.; Wan, J.B. Gallic acid, a natural polyphenol, protects against tert-butyl hydroperoxide-induced hepatotoxicity by activating ERK-Nrf2-Keap1-mediated antioxidative response. Food Chem. Toxicol. 2018, 119, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Noori, M.S.; Bhatt, P.M.; Courreges, M.C.; Ghazanfari, D.; Cuckler, C.; Orac, C.M.; McMills, M.C.; Schwartz, F.L.; Deosarkar, S.P.; Bergmeier, S.C.; et al. Identification of a novel selective and potent inhibitor of glycogen synthase kinase-3. Am. J. Physiol.-Cell Physiol. 2019, 317, C1289–C1303. [Google Scholar] [CrossRef]
- Chang, T.T.; Chou, W.N. Antrodia cinnamomea reconsidered and A. salmonea sp. nov. on Cunninghamia konishii in Taiwan. Bot. Bull. Acad. Sin. 2004, 45, 347–352. [Google Scholar]
- Li, H.X.; Wang, J.J.; Lu, C.L.; Gao, Y.J.; Gao, L.; Yang, Z.Q. Review of Bioactivity, Isolation, and Identification of Active Compounds from Antrodia cinnamomea. Bioengineering 2022, 9, 494. [Google Scholar] [CrossRef]
- Li, B.; Kuang, Y.; He, J.B.; Tang, R.; Xu, L.L.; Leung, C.H.; Ma, D.L.; Qiao, X.; Ye, M. Antcamphorols A-K, Cytotoxic and ROS Scavenging Triterpenoids from Antrodia camphorata. J. Nat. Prod. 2020, 83, 45–54. [Google Scholar] [CrossRef]
- Yang, X.; Wang, X.; Lin, J.; Lim, S.; Cao, Y.; Chen, S.; Xu, P.; Xu, C.; Zheng, H.; Fu, K.C.; et al. Structure and Anti-Inflammatory Activity Relationship of Ergostanes and Lanostanes in Antrodia cinnamomea. Foods 2022, 11, 1831. [Google Scholar] [CrossRef]
- Xu, L.; Peng, A.K.; Cao, Y.N.; Qiao, X.; Yue, S.S.; Ye, M.; Qi, R. Protective Effects of Antrodia cinnamomea and Its Constituent Compound Dehydroeburicoic Acid 32 against Alcoholic Fatty Liver Disease. Curr. Mol. Pharmacol. 2021, 14, 871–882. [Google Scholar] [CrossRef]
- Cao, Y.N.; Yue, S.S.; Wang, A.Y.; Xu, L.; Hu, Y.T.; Qiao, X.; Wu, T.Y.; Ye, M.; Wu, Y.C.; Qi, R. Antrodia cinnamomea and its compound dehydroeburicoic acid attenuate nonalcoholic fatty liver disease by upregulating ALDH2 activity. J. Ethnopharmacol. 2022, 28, 115146. [Google Scholar] [CrossRef]
- To, C.; Roy, A.; Chan, E.; Prado, M.A.M.; Guglielmo, G.M.D. Synthetic triterpenoids inhibit GSK3β activity and localization and affect focal adhesions and cell migration. Biochim. Biophys. Acta-Mol. Cell Res. 2017, 1864, 1274–1284. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, K.M.; Bhave, S.R.; Ferraro, D.J.; Jaboin, J.J.; Hallahan, D.E.; Thotala, D. GSK-3β: A Bifunctional Role in Cell Death Pathways. Int J Cell Biol. 2012, 2012, 930710–930722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venè, R.; Larghero, P.; Arena, G.; Sporn, M.B.; Albini, A.; Tosetti, F. Glycogen synthase kinase 3beta regulates cell death induced by synthetic triterpenoids. Cancer Res. 2008, 68, 6987–6996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamble, S.M.; Patel, H.M.; Goyal, S.N.; Noolvi, M.N.; Mahajan, U.B.; Ojha, S.; Patil, C.R. In silico Evidence for Binding of Pentacyclic Triterpenoids to Keap1-Nrf2 Protein-Protein Binding Site. Comb. Chem. High Throughput Screen. 2017, 20, 215–234. [Google Scholar] [CrossRef]
- Kou, R.W.; Xia, B.; Han, R.; Li, Z.Q.; Yang, J.R.; Yin, X.; Gao, Y.Q.; Gao, J.M. Neuroprotective effects of a new triterpenoid from edible mushroom on oxidative stress and apoptosis through the BDNF/TrkB/ERK/CREB and Nrf2 signaling pathway in vitro and in vivo. Food Funct. 2022, 13, 12121–12134. [Google Scholar] [CrossRef]
- Patyar, S.; Prakash, A.; Medhi, B. Dual inhibition: A novel promising pharmacological approach for different disease conditions. J. Pharm. Pharmacol. 2011, 63, 459–471. [Google Scholar] [CrossRef]
- Wang, L.; Lewis, T.; Zhang, Y.L.; Khodier, C.; Magesh, S.; Chen, L.; Inoyama, D.; Chen, Y.; Zhen, J.; Hu, L.Q. The identification and characterization of non-reactive inhibitor of Keap1-Nrf2 interaction through HTS using a fluorescence polarization assay. In Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2010. [Google Scholar]
- Kobayashi, M.; Yamamoto, M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid. Redox Signal. 2005, 7, 385–394. [Google Scholar] [CrossRef]
- Lu, M.; Wang, P.; Qiao, Y.J.; Ge, Y.; Flickinger, B.; Malhotrac, D.K.; Dworkin, L.D.; Liu, Z.S.; Gong, R.J. GSK3β-mediated Keap1-independent regulation of Nrf2 antioxidant response: A molecular rheostat of acute kidney injury to chronic kidney disease transition. Redox Biol. 2019, 26, 101275–101291. [Google Scholar] [CrossRef]
- Qiu, L.; Cai, C.; Zhao, X.; Fang, Y.; Tang, W.; Guo, C. Inhibition of aldose reductase ameliorates ethanol-induced steatosis in HepG2 cells. Mol. Med. Rep. 2017, 15, 2732–2736. [Google Scholar] [CrossRef] [Green Version]
- Lledías, F.; Hansberg, W. Oxidation of human catalase by singlet oxygen in myeloid leukemia cells. Photochem. Photobiol. 1999, 70, 887–892. [Google Scholar] [CrossRef]
- Ren, J.; Sha, W.; Shang, S.; Yuan, E. Hepatoprotective peptides purified from Corbicula fluminea and its effect against ethanol-induced LO2 cells injury. Int. J. Food Sci. Technol. 2021, 56, 352–361. [Google Scholar] [CrossRef]
- Kang, T.C. Nuclear Factor-Erythroid 2-Related Factor 2 (Nrf2) and Mitochondrial Dynamics/Mitophagy in Neurological Diseases. Antioxidants 2020, 9, 617–637. [Google Scholar] [CrossRef] [PubMed]
- Ping, Z.; Zhang, L.F.; Cui, Y.J.; Chang, Y.M.; Jiang, C.W.; Meng, Z.Z.; Xu, P.; Liu, H.Y.; Wang, D.Y.; Cao, X.B. The protective effects of salidroside from exhaustive exercise-induced heart injury by enhancing the PGC-1α–NRF1/NRF2 pathway and mitochondrial respiratory function in rats. Oxidative Med. Cell. Longev. 2015, 2015, 876825–876834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeligar, S.M.; Machida, K.; Kalra, V.K. Ethanol-induced HO-1 and NQO1 are differentially regulated by HIF-1alpha and Nrf2 to attenuate inflammatory cytokine expression. J. Biol. Chem. 2010, 285, 35359–35373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Assal, O.; Hong, F.; Kim, W.H.; Radaeva, S.; Gao, B. IL-6-deficient mice are susceptible to ethanol-induced hepatic steatosis: IL-6 protects against ethanol-induced oxidative stress and mitochondrial permeability transition in the liver. Cell. Mol. Immunol. 2004, 1, 205–211. [Google Scholar] [PubMed]
- Dudonne, S.; Vitrac, X.; Coutiere, P.; Woillez, M.; Mérillon, J.M. Comparative study of antioxidant properties and total phenolic content of 30 plant extracts of industrial interest using DPPH, ABTS, FRAP, SOD, and ORAC assays. J. Agric. Food Chem. 2009, 57, 1768–1774. [Google Scholar] [CrossRef]
- Wang, M.; Chen, W.Y.; Zhang, J.; Gobejishvili, L.; Barve, S.S.; McClain, C.J.; Joshi-Barve, S. Elevated Fructose and Uric Acid through Aldose Reductase Contribute to Experimental and Human Alcoholic Liver Disease. Hepatology. 2020, 72, 1617–1637. [Google Scholar] [CrossRef]
- Zhou, S.; Wang, P.; Qiao, Y.; Ge, Y.; Wang, Y.Z.; Quan, S.X.; Yao, R.; Zhuang, S.G.; Wang, L.J.; Du, Y.; et al. Genetic and pharmacologic targeting of glycogen synthase kinase 3β reinforces the Nrf2 antioxidant defense against podocytopathy. J. Am. Soc. Nephrol. 2016, 27, 2289–2308. [Google Scholar] [CrossRef] [Green Version]
- Bryan, H.K.; Olayanju, A.; Goldring, C.E.; Park, B.K. The Nrf2 cell defence pathway: Keap1-dependent and-independent mechanisms of regulation. Biochem. Pharmacol. 2013, 85, 705–717. [Google Scholar] [CrossRef] [Green Version]
- Soni, D.; Kumar, P. GSK-3β-mediated regulation of Nrf2/HO-1 signaling as a new therapeutic approach in the treatment of movement disorders. Pharmacol. Rep. 2022, 74, 557–569. [Google Scholar] [CrossRef]
- Yousef, M.H.; Salama, M.; El-Fawal, H.A.N.; Abdelnaser, A. Selective GSK3β Inhibition Mediates an Nrf2-Independent Anti-inflammatory Microglial Response. Mol. Neurobiol. 2022, 59, 5591–5611. [Google Scholar] [CrossRef] [PubMed]
- Li, G.D.; Boyle, J.W.; Ko, C.N.; Zeng, W.; Wong, V.K.W.; Wan, J.B.; Chan, P.W.H.; Ma, D.L.; Leung, C.H. Aurone derivatives as Vps34 inhibitors that modulate autophagy. Acta Pharm. Sin. B 2019, 9, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Chou, M.C.; Chang, R.; Hung, Y.H.; Chen, Y.C.; Chiu, C.H. Antrodia camphorata ameliorates high-fat-diet induced hepatic steatosis via improving lipid metabolism and antioxidative status. J. Funct. Foods 2013, 5, 1317–1325. [Google Scholar] [CrossRef]
- Tien, A.J.; Chien, C.Y.; Chen, Y.H.; Lin, L.C.; Chien, C.T. Fruiting Bodies of Antrodia cinnamomea and Its Active Triterpenoid, Antcin K, Ameliorates N-Nitrosodiethylamine-Induced Hepatic Inflammation, Fibrosis and Carcinogenesis in Rats. Am. J. Chin. Med. 2017, 45, 173–198. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, W.H.; Zhang, R.; Ge, Y.P.; Yang, S.D.; Liu, W.; Wu, Q.P.; Cheng, X.H. Study on Characteristics of Triterpenoids and Hepatoprotective Effects of Fruit Body of Stout Camphor Mushroom, Taiwanofungus camphoratus (Agaricomycetes), Cultivated with Apple-Wood. Int. J. Med. Mushrooms 2022, 24, 53–65. [Google Scholar] [CrossRef]
- Huang, G.J.; Deng, J.S.; Huang, S.S.; Lee, C.Y.; Hou, W.C.; Wang, S.Y.; Sung, P.J.; Kuo, Y.H. Hepatoprotective effects of eburicoic acid and dehydroeburicoic acid from Antrodia camphorata in a mouse model of acute hepatic injury. Food Chem. 2013, 141, 3020–3027. [Google Scholar] [CrossRef]
- Yi, Z.; Liu, X.; Liang, L.; Wang, G.; Xiong, Z.; Zhang, H.; Song, X.; Ai, L.; Xia, Y. Antrodin A from Antrodia camphorata modulates the gut microbiome and liver metabolome in mice exposed to acute alcohol intake. Food Funct. 2021, 12, 2925–2937. [Google Scholar] [CrossRef]
- Huang, C.H.; Chang, Y.Y.; Liu, C.W.; Kang, W.Y.; Lin, Y.L.; Chang, H.C.; Chen, Y.C. Fruiting body of Niuchangchih (Antrodia camphorata) protects livers against chronic alcohol consumption damage. J. Agric. Food Chem. 2010, 58, 3859–3866. [Google Scholar] [CrossRef]
- Qin, J.J.; Cheng, X.D.; Zhang, J.; Zhang, W.D. Dual roles and therapeutic potential of Keap1-Nrf2 pathway in pancreatic cancer: A systematic review. Cell Commun. Signal. 2019, 17, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.C.; Zhao, J.; Liu, Y.T.; Liu, T.; Tao, M.M.; You, Q.D.; Jiang, Z.Y. CPUY192018, a potent inhibitor of the Keap1-Nrf2 protein-protein interaction, alleviates renal inflammation in mice by restricting oxidative stress and NF-κB activation. Redox Biol. 2019, 26, 101266. [Google Scholar] [CrossRef]
- Sun, Y.; He, L.; Wang, T.; Hua, W.; Qin, H.; Wang, J.; Wang, L.; Gu, W.; Li, T.; Li, N.; et al. Activation of p62-Keap1-Nrf2 Pathway Protects 6-Hydroxydopamine-Induced Ferroptosis in Dopaminergic Cells. Mol. Neurobiol. 2020, 57, 4628–4641. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Huang, J.; Chen, Y.; Shang, H.; Zhang, W.; Yu, J.; He, L.; Xing, C.; Zhuang, C. Direct inhibition of Keap1-Nrf2 Protein-Protein interaction as a potential therapeutic strategy for Alzheimer’s disease. Bioorganic Chem. 2020, 103, 104172. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, R.M.C.; Pruccoli, L.; Bisi, A.; Gobbi, S.; Rampa, A.; Martinez, A.; Pérez, C.; Martinez-Gonzalez, L.; Paglione, M.; Di Schiavi, E.; et al. Novel Curcumin-Diethyl Fumarate Hybrid as a Dualistic GSK-3β Inhibitor/Nrf2 Inducer for the Treatment of Parkinson’s Disease. ACS Chem. Neurosci. 2020, 11, 2728–2740. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Lin, X.; Qiao, X.; Ji, S.; Liu, K.D.; Yeh, C.T.; Tzeng, Y.M.; Guo, D.; Ye, M. Antcamphins A-L, ergostanoids from Antrodia camphorate. J. Nat. Prod. 2014, 77, 118–124. [Google Scholar] [CrossRef]
- Yang, G.J.; Ko, C.N.; Zhong, H.J.; Leung, C.H.; Ma, D.L. Structure-Based Discovery of a Selective KDM5A Inhibitor that Exhibits Anti-Cancer Activity via Inducing Cell Cycle Arrest and Senescence in Breast Cancer Cell Lines. Cancers 2019, 11, 92–107. [Google Scholar] [CrossRef] [Green Version]
- Taylor, S.; Wakem, M.; Dijkman, G.; Alsarraj, M.; Nguyen, M. A practical approach to RT-qPCR-Publishing data that conform to the MIQE guidelines. Methods 2010, 50, S1–S5. [Google Scholar] [CrossRef]
- Dai, W.; Zhao, F.; Liu, J.; Liu, H. ASCT2 Is Involved in SARS-Mediated β-Casein Synthesis of Bovine Mammary Epithelial Cells with Methionine Supply. J. Agric. Food Chem. 2019, 68, 13038–13045. [Google Scholar] [CrossRef]
- Devling, T.W.; Lindsay, C.D.; McLellan, L.I.; McMahon, M.; Hayes, J.D. Utility of siRNA against Keap1 as a strategy to stimulate a cancer chemopreventive phenotype. Proc. Natl. Acad. Sci. USA 2005, 102, 7280–7285. [Google Scholar] [CrossRef] [Green Version]
- Ciotti, S.; Iuliano, L.; Cefalù, S.; Comelli, M.; Mavelli, I.; Di Giorgio, E.; Brancolini, C. GSK3β is a key regulator of the ROS-dependent necrotic death induced by the quinone DMNQ. Cell Death Dis. 2020, 11, 2. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, S.; Kuang, Y.; Li, G.; Wu, J.; Ko, C.-N.; Wang, W.; Ma, D.-L.; Ye, M.; Leung, C.-H. Dehydroeburicoic Acid, a Dual Inhibitor against Oxidative Stress in Alcoholic Liver Disease. Pharmaceuticals 2023, 16, 14. https://doi.org/10.3390/ph16010014
Cheng S, Kuang Y, Li G, Wu J, Ko C-N, Wang W, Ma D-L, Ye M, Leung C-H. Dehydroeburicoic Acid, a Dual Inhibitor against Oxidative Stress in Alcoholic Liver Disease. Pharmaceuticals. 2023; 16(1):14. https://doi.org/10.3390/ph16010014
Chicago/Turabian StyleCheng, Shasha, Yi Kuang, Guodong Li, Jia Wu, Chung-Nga Ko, Wanhe Wang, Dik-Lung Ma, Min Ye, and Chung-Hang Leung. 2023. "Dehydroeburicoic Acid, a Dual Inhibitor against Oxidative Stress in Alcoholic Liver Disease" Pharmaceuticals 16, no. 1: 14. https://doi.org/10.3390/ph16010014