
Concept of Hybrid Drugs and Recent Advancements in Anticancer Hybrids
 , , , , , ,
, , , , , ,  , , and
, , and
Abstract
:1. Introduction
2. The Concept of Hybrid Drugs in Anticancer Agent Development
3. Recent Advances in Anticancer Hybrids
3.1. Quinazoline Based Hybrids
Quinazoline-Based Hybrids That Are FDA Approved or under Clinical Trial
3.2. Indole-Based Hybrids
3.3. Indole-Based Hybrids That Are FDA Approved or under Clinical Trial
3.4. Carbazole Based Hybrids
Carbazole-Based Hybrids That Are FDA Approved or under Clinical Trial
3.5. Pyrimidine-Based Hybrids
FDA Approved Pyrimidine Based Hybrids
3.6. Quinoline-Based Hybrids
Quinoline-Based FDA Approved Drugs
3.7. Quinone Hybrids
Quinone Containing FDA Approved Drugs
3.8. Imidazole Based Hybrids
Imidazole Based Hybrids That Are FDA Approved or under clinical Trial
3.9. Selenium-Based Hybrids
Selenium-Based Hybrids That Are FDA Approved or under Clinical Trials
3.10. Platinum-Based Hybrids
Platinum Based Drugs That Are FDA Approved or under Clinical Trial
3.11. Hydroxamic Acid Hybrids
Hydroxamic Acid Based Hybrids That Are FDA Approved or under Clinical Trial
3.12. Ferrocene Hybrids
3.13. Curcumin-Based Hybrids
3.14. Triazole-Based Hybrids
3.15. Benzimidazole-Based Hybrids
Benzimidazole Based Hybrids That Are FDA Approved or under Clinical Trials
3.16. Isatin Containing Hybrids
Isatin Containing FDA-Approved Hybrids
3.17. Pyrrolo-Benzodiazepines Based Hybrids
FDA Approved Drugs Containing Pyrrolo-Benzodiazepines
3.18. Chalcone-Based Hybrids
3.19. Coumarin-Based Hybrids
3.20. Nitrogen Mustard Based Hybrids
3.21. Pyrazole-Based Hybrids
3.22. Pyridine-Based Hybrids
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| EGFR | Epidermal Growth Factor Receptor |
| EMA | European Medicines Agency |
| FDA | Food and Drug Administration |
| HDAC | Histone Deacetylases |
| MDR | Multidrug Resistance |
| PDGF | Platelet Derived Growth Factor |
| SAHA | Suberoylanilide Hydroxamic Acid |
| VEGFR-2 | Vascular Endothelial Growth Factor Receptor-2 |
| WHO | World Health Organization |
References
- Rana, A.; Alex, J.M.; Chauhan, M.; Joshi, G.; Kumar, R. A review on pharmacophoric designs of antiproliferative agents. Med. Chem. Res. 2015, 24, 903–920. [Google Scholar] [CrossRef]
- Kori, S. An overview: Several causes of breast cancer. Epidemol. Int. J. 2018, 2, 000107. [Google Scholar] [CrossRef]
- Hassanpour, S.H.; Dehghani, M. Review of cancer from perspective of molecular. J. Cancer Res. Pract. 2017, 4, 127–129. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Philip, C.C.; Mathew, A.; John, M.J. Cancer care: Challenges in the developing world. Cancer Res. Treat. 2018, 1, 58–62. [Google Scholar]
- Penny, L.K.; Wallace, H.M. The challenges for cancer chemoprevention. Chem. Soc. Rev. 2015, 44, 8836–8847. [Google Scholar] [CrossRef]
- Zugazagoitia, J.; Guedes, C.; Ponce, S.; Ferrer, I.; Molina-Pinelo, S.; Paz-Ares, L. Current challenges in cancer treatment. Clin. Ther. 2016, 38, 1551–1566. [Google Scholar] [CrossRef]
- Zhong, L.; Li, Y.; Xiong, L.; Wang, W.; Wu, M.; Yuan, T.; Yang, W.; Tian, C.; Miao, Z.; Wang, T. Small molecules in targeted cancer therapy: Advances, challenges, and future perspectives. Signal Transduct. Target. Ther. 2021, 6, 201. [Google Scholar] [CrossRef]
- Chakraborty, S.; Rahman, T. The difficulties in cancer treatment. Ecancermedicalscience 2012, 6, ed16. [Google Scholar]
- Shalini; Kumar, V. Have molecular hybrids delivered effective anticancer treatments and what should future drug discovery focus on? Expert Opin. Drug Discov. 2021, 16, 335–363. [Google Scholar] [CrossRef]
- Bass, A.K.; El-Zoghbi, M.S.; Nageeb, E.S.M.; Mohamed, M.F.; Badr, M.; Abuo-Rahma, G.E.D.A. Comprehensive review for anticancer hybridized multitargeting HDAC inhibitors. Eur. J. Med. Chem. 2021, 209, 112904. [Google Scholar] [CrossRef]
- Gediya, L.K.; Njar, V.C. Promise and challenges in drug discovery and development of hybrid anticancer drugs. Expert Opin. Drug Discov. 2009, 4, 1099–1111. [Google Scholar] [CrossRef]
- Moustafa, A.M.Y.; Bakare, S.B. Synthesis of some hybrid 7-hydroxy quinolinone derivatives as anti breast cancer drugs. Res. Chem. Intermed. 2019, 45, 3895–3912. [Google Scholar] [CrossRef]
- Nepali, K.; Sharma, S.; Sharma, M.; Bedi, P.; Dhar, K. Rational approaches, design strategies, structure activity relationship and mechanistic insights for anticancer hybrids. Eur. J. Med. Chem. 2014, 77, 422–487. [Google Scholar] [CrossRef]
- Decker, M. Hybrid molecules incorporating natural products: Applications in cancer therapy, neurodegenerative disorders and beyond. Curr. Med. Chem. 2011, 18, 1464–1475. [Google Scholar] [CrossRef]
- Kerru, N.; Singh, P.; Koorbanally, N.; Raj, R.; Kumar, V. Recent advances (2015–2016) in anticancer hybrids. Eur. J. Med. Chem. 2017, 142, 179–212. [Google Scholar] [CrossRef]
- Szumilak, M.; Wiktorowska-Owczarek, A.; Stanczak, A. Hybrid drugs—A strategy for overcoming anticancer drug resistance? Molecules 2021, 26, 2601. [Google Scholar] [CrossRef]
- Abbot, V.; Sharma, P.; Dhiman, S.; Noolvi, M.N.; Patel, H.M.; Bhardwaj, V. Small hybrid heteroaromatics: Resourceful biological tools in cancer research. RSC Adv. 2017, 7, 28313–28349. [Google Scholar] [CrossRef]
- Fortin, S.; Bérubé, G. Advances in the development of hybrid anticancer drugs. Expert Opin. Drug Discov. 2013, 8, 1029–1047. [Google Scholar] [CrossRef]
- Mishra, S.; Singh, P. Hybrid molecules: The privileged scaffolds for various pharmaceuticals. Eur. J. Med. Chem. 2016, 124, 500–536. [Google Scholar]
- Zheng, W.; Zhao, Y.; Luo, Q.; Zhang, Y.; Wu, K.; Wang, F. Multi-targeted anticancer agents. Curr. Top. Med. Chem. 2017, 17, 3084–3098. [Google Scholar] [CrossRef] [PubMed]
- Chamseddine, I.M.; Rejniak, K.A. Hybrid modeling frameworks of tumor development and treatment. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, 12, e1461. [Google Scholar] [CrossRef] [PubMed]
- Alagarsamy, V.; Chitra, K.; Saravanan, G.; Solomon, V.R.; Sulthana, M.; Narendhar, B. An overview of quinazolines: Pharmacological significance and recent developments. Eur. J. Med. Chem. 2018, 151, 628–685. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Hong, J. Recent advancements of 4-aminoquinazoline derivatives as kinase inhibitors and their applications in medicinal chemistry. Eur. J. Med. Chem. 2019, 170, 55–72. [Google Scholar] [CrossRef]
- Cheng, W.; Zhu, S.; Ma, X.; Qiu, N.; Peng, P.; Sheng, R.; Hu, Y. Design, synthesis and biological evaluation of 6-(nitroimidazole-1H-alkyloxyl)-4-anilinoquinazolines as efficient EGFR inhibitors exerting cytotoxic effects both under normoxia and hypoxia. Eur. J. Med. Chem. 2015, 89, 826–834. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, H.; Liu, R.; Liu, J.; Chen, L.; Li, X.; Zhao, L.; Wang, W.; Li, B. Quinazoline-1-deoxynojirimycin hybrids as high active dual inhibitors of EGFR and α-glucosidase. Bioorg. Med. Chem. Lett. 2017, 27, 4309–4313. [Google Scholar] [CrossRef]
- Zhang, H.Q.; Gong, F.H.; Ye, J.Q.; Zhang, C.; Yue, X.H.; Li, C.G.; Xu, Y.G.; Sun, L.P. Design and discovery of 4-anilinoquinazoline-urea derivatives as dual TK inhibitors of EGFR and VEGFR-2. Eur. J. Med. Chem. 2017, 125, 245–254. [Google Scholar] [CrossRef]
- Yadav, R.R.; Guru, S.K.; Joshi, P.; Mahajan, G.; Mintoo, M.J.; Kumar, V.; Bharate, S.S.; Mondhe, D.M.; Vishwakarma, R.A.; Bhushan, S. 6-Aryl substituted 4-(4-cyanomethyl) phenylamino quinazolines as a new class of isoform-selective PI3K-alpha inhibitors. Eur. J. Med. Chem. 2016, 122, 731–743. [Google Scholar] [CrossRef]
- Ding, H.W.; Deng, C.L.; Li, D.D.; Liu, D.D.; Chai, S.M.; Wang, W.; Zhang, Y.; Chen, K.; Li, X.; Wang, J. Design, synthesis and biological evaluation of novel 4-aminoquinazolines as dual target inhibitors of EGFR-PI3Kα. Eur. J. Med. Chem. 2018, 146, 460–470. [Google Scholar] [CrossRef]
- Fan, Y.H.; Ding, H.W.; Liu, D.D.; Song, H.R.; Xu, Y.N.; Wang, J. Novel 4-aminoquinazoline derivatives induce growth inhibition, cell cycle arrest and apoptosis via PI3Kα inhibition. Bioorg. Med. Chem. 2018, 26, 1675–1685. [Google Scholar] [CrossRef]
- Fröhlich, T.; Reiter, C.; Ibrahim, M.M.; Beutel, J.; Hutterer, C.; Zeitträger, I.; Bahsi, H.; Leidenberger, M.; Friedrich, O.; Kappes, B. Synthesis of novel hybrids of quinazoline and artemisinin with high activities against Plasmodium falciparum, human cytomegalovirus, and leukemia cells. ACS Omega 2017, 2, 2422–2431. [Google Scholar] [CrossRef]
- Yang, S.M.; Urban, D.J.; Yoshioka, M.; Strovel, J.W.; Fletcher, S.; Wang, A.Q.; Xu, X.; Shah, P.; Hu, X.; Hall, M.D. Discovery and lead identification of quinazoline-based BRD4 inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 3483–3488. [Google Scholar] [CrossRef]
- Lee, H.A.; Hyun, S.A.; Byun, B.; Chae, J.H.; Kim, K.S. Electrophysiological mechanisms of vandetanib-induced cardiotoxicity: Comparison of action potentials in rabbit Purkinje fibers and pluripotent stem cell-derived cardiomyocytes. PLoS ONE 2018, 13, e0195577. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.C.; Chang, J.; Huang, S.C.C.; Lin, H.C.; Ho, A.S.; Lim, K.H.; Chang, C.C.; Huang, L.; Chang, Y.C.; Chang, Y.F. YM155 as an inhibitor of cancer stemness simultaneously inhibits autophosphorylation of epidermal growth factor receptor and G9a-mediated stemness in lung cancer cells. PLoS ONE 2017, 12, e0182149. [Google Scholar] [CrossRef]
- Han, W.; Pan, H.; Chen, Y.; Sun, J.; Wang, Y.; Li, J.; Ge, W.; Feng, L.; Lin, X.; Wang, X. EGFR tyrosine kinase inhibitors activate autophagy as a cytoprotective response in human lung cancer cells. PLoS ONE 2011, 6, e18691. [Google Scholar] [CrossRef]
- Tabasum, S.; Singh, R.P. Fisetin suppresses migration, invasion and stem-cell-like phenotype of human non-small cell lung carcinoma cells via attenuation of epithelial to mesenchymal transition. Chem. Biol. Interact. 2019, 303, 14–21. [Google Scholar] [CrossRef]
- Eno, M.R.; El-Gendy, B.E.D.M.; Cameron, M.D. P450 3A-catalyzed O-dealkylation of lapatinib induces mitochondrial stress and activates Nrf2. Chem. Res. Toxicol. 2016, 29, 784–796. [Google Scholar] [CrossRef]
- Pham, H.T.T.; Maurer, B.; Prchal-Murphy, M.; Grausenburger, R.; Grundschober, E.; Javaheri, T.; Nivarthi, H.; Boersma, A.; Kolbe, T.; Elabd, M. STAT5B N642H is a driver mutation for T cell neoplasia. J. Clin. Investig. 2018, 128, 387–401. [Google Scholar] [CrossRef]
- Sun, H.; Mediwala, S.N.; Szafran, A.T.; Mancini, M.A.; Marcelli, M. CUDC-101, a novel inhibitor of full-length androgen receptor (flAR) and androgen receptor variant 7 (AR-V7) activity: Mechanism of action and in vivo efficacy. Horm. Cancer 2016, 7, 196–210. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, Y.; Ni, H.M.; Ding, W.X.; Zhong, H. Nrf2 but not autophagy inhibition is associated with the survival of wild-type epidermal growth factor receptor non-small cell lung cancer cells. Toxicol. Appl. Pharmacol. 2016, 310, 140–149. [Google Scholar] [CrossRef]
- Kumari, A.; Singh, R.K. Medicinal chemistry of indole derivatives: Current to future therapeutic prospectives. Bioorg. Chem. 2019, 89, 103021. [Google Scholar] [CrossRef] [PubMed]
- Dadashpour, S.; Emami, S. Indole in the target-based design of anticancer agents: A versatile scaffold with diverse mechanisms. Eur. J. Med. Chem. 2018, 150, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, P.; Chou, C.J.; Liu, C.; Wang, X.; Xu, W. Development of N-hydroxycinnamamide-based histone deacetylase inhibitors with an indole-containing cap group. ACS Med. Chem. Lett. 2013, 4, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Inks, E.S.; Li, X.; Hou, J.; Chou, C.J.; Zhang, J.; Jiang, Y.; Zhang, Y.; Xu, W. Discovery of the first N-hydroxycinnamamide-based histone deacetylase 1/3 dual inhibitors with potent oral antitumor activity. J. Med. Chem. 2014, 57, 3324–3341. [Google Scholar] [CrossRef]
- Mehndiratta, S.; Hsieh, Y.L.; Liu, Y.M.; Wang, A.W.; Lee, H.Y.; Liang, L.Y.; Kumar, S.; Teng, C.M.; Yang, C.R.; Liou, J.P. Indole-3-ethylsulfamoylphenylacrylamides: Potent histone deacetylase inhibitors with anti-inflammatory activity. Eur. J. Med. Chem. 2014, 85, 468–479. [Google Scholar] [CrossRef]
- Panathur, N.; Dalimba, U.; Koushik, P.V.; Alvala, M.; Yogeeswari, P.; Sriram, D.; Kumar, V. Identification and characterization of novel indole based small molecules as anticancer agents through SIRT1 inhibition. Eur. J. Med. Chem. 2013, 69, 125–138. [Google Scholar] [CrossRef]
- Lee, J.; More, K.N.; Yang, S.A.; Hong, V.S. 3, 5-bis (aminopyrimidinyl) indole derivatives: Synthesis and evaluation of pim kinase inhibitory activities. Bull. Korean Chem. Soc. 2014, 35, 2123–2129. [Google Scholar] [CrossRef]
- Mirzaei, H.; Shokrzadeh, M.; Modanloo, M.; Ziar, A.; Riazi, G.H.; Emami, S. New indole-based chalconoids as tubulin-targeting antiproliferative agents. Bioorg. Chem. 2017, 75, 86–98. [Google Scholar] [CrossRef]
- Zhou, Y.; Duan, K.; Zhu, L.; Liu, Z.; Zhang, C.; Yang, L.; Li, M.; Zhang, H.; Yang, X. Synthesis and cytotoxic activity of novel hexahydropyrrolo [2, 3-b] indole imidazolium salts. Bioorg. Med. Chem. Lett. 2016, 26, 460–465. [Google Scholar] [CrossRef]
- Kumar, D.; Kumar, N.M.; Tantak, M.P.; Ogura, M.; Kusaka, E.; Ito, T. Synthesis and identification of α-cyano bis (indolyl) chalcones as novel anticancer agents. Bioorg. Med. Chem. Lett. 2014, 24, 5170–5174. [Google Scholar] [CrossRef]
- Kumar, S.; Gu, L.; Palma, G.; Kaur, M.; Singh-Pillay, A.; Singh, P.; Kumar, V. Design, synthesis, anti-proliferative evaluation and docking studies of 1 H-1, 2, 3-triazole tethered ospemifene–isatin conjugates as selective estrogen receptor modulators. New J. Chem. 2018, 42, 3703–3713. [Google Scholar] [CrossRef]
- Sharma, B.; Singh, A.; Gu, L.; Saha, S.T.; Singh-Pillay, A.; Cele, N.; Singh, P.; Kaur, M.; Kumar, V. Diastereoselective approach to rationally design tetrahydro-β-carboline–isatin conjugates as potential SERMs against breast cancer. RSC Adv. 2019, 9, 9809–9819. [Google Scholar] [CrossRef]
- Lindsay, C.R.; MacPherson, I.R.; Cassidy, J. Current status of cediranib: The rapid development of a novel anti-angiogenic therapy. Future Oncol. 2009, 5, 421–432. [Google Scholar] [CrossRef]
- Song, F.; Hu, B.; Cheng, J.W.; Sun, Y.F.; Zhou, K.Q.; Wang, P.X.; Guo, W.; Zhou, J.; Fan, J.; Chen, Z. Anlotinib suppresses tumor progression via blocking the VEGFR2/PI3K/AKT cascade in intrahepatic cholangiocarcinoma. Cell Death Dis. 2020, 11, 573. [Google Scholar] [CrossRef]
- Heldin, C.H.; Vanlandewijck, M.; Moustakas, A. Regulation of EMT by TGFβ in cancer. FEBS Lett. 2012, 586, 1959–1970. [Google Scholar] [CrossRef]
- Yaseen, A.; Chen, S.; Hock, S.; Rosato, R.; Dent, P.; Dai, Y.; Grant, S. Resveratrol sensitizes acute myelogenous leukemia cells to histone deacetylase inhibitors through reactive oxygen species-mediated activation of the extrinsic apoptotic pathway. Mol. Pharmacol. 2012, 82, 1030–1041. [Google Scholar] [CrossRef]
- Kato, Y.; Salumbides, B.C.; Wang, X.F.; Qian, D.Z.; Williams, S.; Wei, Y.; Sanni, T.B.; Atadja, P.; Pili, R. Antitumor effect of the histone deacetylase inhibitor LAQ824 in combination with 13-cis-retinoic acid in human malignant melanoma. Mol. Cancer Ther. 2007, 6, 70–81. [Google Scholar] [CrossRef]
- Głuszyńska, A. Biological potential of carbazole derivatives. Eur. J. Med. Chem. 2015, 94, 405–426. [Google Scholar] [CrossRef]
- Issa, S.; Prandina, A.; Bedel, N.; Rongved, P.; Yous, S.; Le Borgne, M.; Bouaziz, Z. Carbazole scaffolds in cancer therapy: A review from 2012 to 2018. J. Enzyme Inhib. Med. Chem. 2019, 34, 1321–1346. [Google Scholar] [CrossRef]
- Liu, L.X.; Wang, X.Q.; Zhou, B.; Yang, L.J.; Li, Y.; Zhang, H.B.; Yang, X.D. Synthesis and antitumor activity of novel N-substituted carbazole imidazolium salt derivatives. Sci. Rep. 2015, 5, 13101. [Google Scholar] [CrossRef]
- Mongre, R.K.; Mishra, C.B.; Prakash, A.; Jung, S.; Lee, B.S.; Kumari, S.; Hong, J.T.; Lee, M.S. Novel carbazole-piperazine hybrid small molecule induces apoptosis by targeting BCL-2 and inhibits tumor progression in lung adenocarcinoma in vitro and xenograft mice model. Cancers 2019, 11, 1245. [Google Scholar] [CrossRef] [PubMed]
- Rawjewski, R.A. Formulations of Indole-3-Carbinol Derived Antitumor Agents with Increased Oral Bioavailability. U.S. Patent 13/263,838, 19 July 2012. [Google Scholar]
- Tucker, J.; Sviridov, S.; Brodsky, L.; Burkhart, C.; Purmal, A.; Gurova, K.; Gudkov, A. Carbazole Compounds and Therapeutic Uses of the Compounds. U.S. Patent 8,765,738, 4 June 2015. [Google Scholar]
- Narayanan, R.; Miller, D.D.; Ponnusamy, T.; Hwang, D.J.; Pagadala, J.; Duke, C.B.; Coss, C.C.; Dalton, J.T.; He, Y. Selective Androgen Receptor Degrader (SARD) Ligands and Methods of Use Thereof. U.S. Patent 9,815,776 B2, 14 November 2017. [Google Scholar]
- Ruiz-Ceja, K.A.; Chirino, Y.I. Current FDA-approved treatments for non-small cell lung cancer and potential biomarkers for its detection. Biomed. Pharmacother. 2017, 90, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.M.; Manley, P.W.; Larson, R.A.; Capdeville, R. Midostaurin: Its odyssey from discovery to approval for treating acute myeloid leukemia and advanced systemic mastocytosis. Blood Adv. 2018, 2, 444–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.C.; Chao, D.K.; Chao, K.C.; Chen, Y.J. Oral small-molecule tyrosine kinase inhibitor midostaurin (PKC412) inhibits growth and induces megakaryocytic differentiation in human leukemia cells. Toxicol. Vitr. 2009, 23, 979–985. [Google Scholar] [CrossRef]
- Zhang, J.; Ren, L.; Yang, X.; White, M.; Greenhaw, J.; Harris, T.; Wu, Q.; Bryant, M.; Papoian, T.; Mattes, W. Cytotoxicity of 34 FDA approved small-molecule kinase inhibitors in primary rat and human hepatocytes. Toxicol. Lett. 2018, 291, 138–148. [Google Scholar] [CrossRef]
- Hawkins, W.; Mitchell, C.; McKinstry, R.; Gilfor, D.; Starkey, J.; Dai, Y.; Dawson, K.; Ramakrishnan, V.; Roberts, J.D.; Yacoub, A. Transient exposure of mammary tumors to PD184352 and UCN-01 causes tumor cell death in vivo and prolonged suppression of tumor re-growth. Cancer Biol. Ther. 2005, 4, 1275–1284. [Google Scholar] [CrossRef]
- Kumar, S.; Narasimhan, B. Therapeutic potential of heterocyclic pyrimidine scaffolds. Chem. Cent. J. 2018, 12, 38. [Google Scholar] [CrossRef]
- Kaur, R.; Kaur, P.; Sharma, S.; Singh, G.; Mehndiratta, S.; MS Bedi, P.; Nepali, K. Anticancer pyrimidines in diverse scaffolds: A review of patent literature. Recent Pat. Anticancer. Drug Discov. 2015, 10, 23–71. [Google Scholar] [CrossRef]
- Combs, A.P.; Li, Y.L.; Mei, S.; Yue, E.W. Substituted Diamino-Pyrimidine and Diamino-Pyridine Derivatives as PI3K Inhibitors. U.S. Patent 9,108,984, 18 August 2015. [Google Scholar]
- Boloor, A.; Cheung, M.; Harris, P.A.; Hinkle, K.; Laudeman, C.P.; Stafford, J.A.; Veal, J.M. Chemical Compounds. Patent GB201201758D0, 1 February 2012. [Google Scholar]
- Hogberg, M.; Dahlstedt, E.; Smitt, O.; Johansson, T. Novel Pyrimidine Derivatives. Patent WO2012127032, 27 September 2012. [Google Scholar]
- Liu, J.; Mao, L.; Wang, X.; Xu, X.; Zhao, L. Pyrazolopyrimidine Derivatives and Uses as Anticancer Agents. Patent WO2012/097196 A, 19 July 2012. [Google Scholar]
- Tanaka, M.; Zhang, C.; Shokat, K.M.; Burlingame, A.L.; Hansen, K.; Bateman, R.L.; DiMagno, S.G. Pyrazolo Pyrimidine Derivatives and Methods of Use Thereof. Patent US201313862348A, 12 March 2013. [Google Scholar]
- Liang, C. mTOR Selective Kinase Inhibitors. U.S. Patent US20130072481, 21 March 2013. [Google Scholar]
- Dorsch, D.; Hoelzemann, G.; Schiemann, K.; Wegener, A. Triazolo [4, 5-d] Pyrimidine Derivatives. Patent WO2013110309A1, 1 August 2013. [Google Scholar]
- El-Sayed, N.S.; El-Bendary, E.R.; El-Ashry, S.M.; El-Kerdawy, M.M. Synthesis and antitumor activity of new sulfonamide derivatives of thiadiazolo [3, 2-a] pyrimidines. Eur. J. Med. Chem. 2011, 46, 3714–3720. [Google Scholar] [CrossRef]
- Shao, H.; Shi, S.; Foley, D.W.; Lam, F.; Abbas, A.Y.; Liu, X.; Huang, S.; Jiang, X.; Baharin, N.; Fischer, P.M. Synthesis, structure–activity relationship and biological evaluation of 2, 4, 5-trisubstituted pyrimidine CDK inhibitors as potential anti-tumour agents. Eur. J. Med. Chem. 2013, 70, 447–455. [Google Scholar] [CrossRef]
- Fares, M.; Abou-Seri, S.M.; Abdel-Aziz, H.A.; Abbas, S.E.S.; Youssef, M.M.; Eladwy, R.A. Synthesis and antitumor activity of pyrido [2, 3-d] pyrimidine and pyrido [2, 3-d] [1, 2, 4] triazolo [4, 3-a] pyrimidine derivatives that induce apoptosis through G1 cell-cycle arrest. Eur. J. Med. Chem. 2014, 83, 155–166. [Google Scholar] [CrossRef]
- Kurumurthy, C.; Rao, P.S.; Rao, P.S.; Narsaiah, B.; Velatooru, L.; Pamanji, R.; Rao, J.V. Synthesis of novel alkyltriazole tagged pyrido [2, 3-d] pyrimidine derivatives and their anticancer activity. Eur. J. Med. Chem. 2011, 46, 3462–3468. [Google Scholar] [CrossRef]
- Nagender, P.; Reddy, G.M.; Kumar, R.N.; Poornachandra, Y.; Kumar, C.G.; Narsaiah, B. Synthesis, cytotoxicity, antimicrobial and anti-biofilm activities of novel pyrazolo [3, 4-b] pyridine and pyrimidine functionalized 1, 2, 3-triazole derivatives. Bioorg. Med. Chem. Lett. 2014, 24, 2905–2908. [Google Scholar] [CrossRef]
- Huang, Y.Y.; Wang, L.Y.; Chang, C.H.; Kuo, Y.H.; Kaneko, K.; Takayama, H.; Kimura, M.; Juang, S.H.; Wong, F.F. One-pot synthesis and antiproliferative evaluation of pyrazolo [3, 4-d] pyrimidine derivatives. Tetrahedron 2012, 68, 9658–9664. [Google Scholar] [CrossRef]
- Han, M.; Shen, J.; Wang, L.; Wang, Y.; Zhai, X.; Li, Y.; Liu, M.; Li, Z.; Zuo, D.; Wu, Y. 5-chloro-N4-(2-(isopropylsulfonyl) phenyl)-N2-(2-methoxy-4-(4-((4-methylpiperazin-1-yl) methyl)-1H-1, 2, 3-triazol-1-yl) phenyl) pyrimidine-2, 4-diamine (WY-135), a novel ALK inhibitor, induces cell cycle arrest and apoptosis through inhibiting ALK and its downstream pathways in Karpas299 and H2228 cells. Chem. Biol. Interact. 2018, 284, 24–31. [Google Scholar]
- The, I.; Ruijtenberg, S.; Bouchet, B.P.; Cristobal, A.; Prinsen, M.B.; van Mourik, T.; Koreth, J.; Xu, H.; Heck, A.J.; Akhmanova, A. Rb and FZR1/Cdh1 determine CDK4/6-cyclin D requirement in C. elegans and human cancer cells. Nat. Commun. 2015, 6, 5906. [Google Scholar] [CrossRef]
- Ceribelli, M.; Kelly, P.N.; Shaffer, A.L.; Wright, G.W.; Xiao, W.; Yang, Y.; Mathews Griner, L.A.; Guha, R.; Shinn, P.; Keller, J.M. Blockade of oncogenic IκB kinase activity in diffuse large B-cell lymphoma by bromodomain and extraterminal domain protein inhibitors. Proc. Natl. Acad. Sci. USA 2014, 111, 11365–11370. [Google Scholar] [CrossRef]
- Matsui, T.; Miyamoto, K.; Yamanaka, K.; Okai, Y.; Kaushik, E.P.; Harada, K.; Wagoner, M.; Shinozawa, T. Cell-based two-dimensional morphological assessment system to predict cancer drug-induced cardiotoxicity using human induced pluripotent stem cell-derived cardiomyocytes. Toxicol. Appl. Pharmacol. 2019, 383, 114761. [Google Scholar] [CrossRef]
- Chu, X.M.; Wang, C.; Liu, W.; Liang, L.L.; Gong, K.K.; Zhao, C.Y.; Sun, K.L. Quinoline and quinolone dimers and their biological activities: An overview. Eur. J. Med. Chem. 2019, 161, 101–117. [Google Scholar] [CrossRef]
- Zhao, D.; Hamilton, J.P.; Pham, G.M.; Crisovan, E.; Wiegert-Rininger, K.; Vaillancourt, B.; DellaPenna, D.; Buell, C.R. De novo genome assembly of Camptotheca acuminata, a natural source of the anticancer compound camptothecin. GigaScience 2017, 6, gix065. [Google Scholar] [CrossRef]
- Afzal, O.; Kumar, S.; Haider, M.R.; Ali, M.R.; Kumar, R.; Jaggi, M.; Bawa, S. A review on anticancer potential of bioactive heterocycle quinoline. Eur. J. Med. Chem. 2015, 97, 871–910. [Google Scholar] [CrossRef] [PubMed]
- Sidoryk, K.; Świtalska, M.; Jaromin, A.; Cmoch, P.; Bujak, I.; Kaczmarska, M.; Wietrzyk, J.; Dominguez, E.G.; Żarnowski, R.; Andes, D.R. The synthesis of indolo [2, 3-b] quinoline derivatives with a guanidine group: Highly selective cytotoxic agents. Eur. J. Med. Chem. 2015, 105, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Gedawy, E.M.; Kassab, A.E.; El-Malah, A.A. Synthesis and anticancer activity of novel tetrahydroquinoline and tetrahydropyrimidoquinoline derivatives. Med. Chem. Res. 2015, 24, 3387–3397. [Google Scholar] [CrossRef]
- González-Sánchez, I.; Solano, J.D.; Loza-Mejía, M.A.; Olvera-Vázquez, S.; Rodríguez-Sotres, R.; Morán, J.; Lira-Rocha, A.; Cerbón, M.A. Antineoplastic activity of the thiazolo [5, 4-b] quinoline derivative D3CLP in K-562 cells is mediated through effector caspases activation. Eur. J. Med. Chem. 2011, 46, 2102–2108. [Google Scholar] [CrossRef]
- Luniewski, W.; Wietrzyk, J.; Godlewska, J.; Switalska, M.; Piskozub, M.; Peczynska-Czoch, W.; Kaczmarek, L. New derivatives of 11-methyl-6-[2-(dimethylamino) ethyl]-6H-indolo [2, 3-b] quinoline as cytotoxic DNA topoisomerase II inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 6103–6107. [Google Scholar] [CrossRef]
- Jin, X.Y.; Chen, H.; Li, D.D.; Li, A.L.; Wang, W.Y.; Gu, W. Design, synthesis, and anticancer evaluation of novel quinoline derivatives of ursolic acid with hydrazide, oxadiazole, and thiadiazole moieties as potent MEK inhibitors. J. Enzyme Inhib. Med. Chem. 2019, 34, 955–972. [Google Scholar] [CrossRef]
- Solomon, V.R.; Pundir, S.; Lee, H. Examination of novel 4-aminoquinoline derivatives designed and synthesized by a hybrid pharmacophore approach to enhance their anticancer activities. Sci. Rep. 2019, 9, 6315. [Google Scholar] [CrossRef]
- Kumar, K.; Schniper, S.; González-Sarrías, A.; Holder, A.A.; Sanders, N.; Sullivan, D.; Jarrett, W.L.; Davis, K.; Bai, F.; Seeram, N.P. Highly potent anti-proliferative effects of a gallium (III) complex with 7-chloroquinoline thiosemicarbazone as a ligand: Synthesis, cytotoxic and antimalarial evaluation. Eur. J. Med. Chem. 2014, 86, 81–86. [Google Scholar] [CrossRef]
- Capozzi, M.; De Divitiis, C.; Ottaiano, A.; von Arx, C.; Scala, S.; Tatangelo, F.; Delrio, P.; Tafuto, S. Lenvatinib, a molecule with versatile application: From preclinical evidence to future development in anticancer treatment. Cancer Manag. Res. 2019, 11, 3847–3860. [Google Scholar] [CrossRef]
- Dong, H.; Wade, M.G. Application of a nonradioactive assay for high throughput screening for inhibition of thyroid hormone uptake via the transmembrane transporter MCT8. Toxicol. Vitr. 2017, 40, 234–242. [Google Scholar] [CrossRef]
- Pereyra, C.E.; Dantas, R.F.; Ferreira, S.B.; Gomes, L.P.; Silva, F.P., Jr. The diverse mechanisms and anticancer potential of naphthoquinones. Cancer Cell Int. 2019, 19, 207. [Google Scholar] [CrossRef]
- Powis, G. Free radical formation by antitumor quinones. Free Radic. Biol. Med. 1989, 6, 63–101. [Google Scholar] [CrossRef]
- Marković, V.; Debeljak, N.; Stanojković, T.; Kolundžija, B.; Sladić, D.; Vujčić, M.; Janović, B.; Tanić, N.; Perović, M.; Tešić, V. Anthraquinone–chalcone hybrids: Synthesis, preliminary antiproliferative evaluation and DNA-interaction studies. Eur. J. Med. Chem. 2015, 89, 401–410. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, M.; Song, S.; Xu, Y.; Miao, Z.; Zhang, A. Design, synthesis, and anticancer activities of new compounds bearing the quinone–pyran–lactone tricyclic pharmacophore. RSC Adv. 2015, 5, 27502–27508. [Google Scholar] [CrossRef]
- Ali, I.; Lone, M.N.; Aboul-Enein, H.Y. Imidazoles as potential anticancer agents. Med. Chem. Comm. 2017, 8, 1742–1773. [Google Scholar] [CrossRef]
- Wan, W.C.; Chen, W.; Liu, L.X.; Li, Y.; Yang, L.J.; Deng, X.Y.; Zhang, H.B.; Yang, X.D. Synthesis and cytotoxic activity of novel hybrid compounds between 2-alkylbenzofuran and imidazole. Med. Chem. Res. 2014, 23, 1599–1611. [Google Scholar] [CrossRef]
- Bhatnagar, A.; Sharma, P.; Kumar, N. A review on “Imidazoles”: Their chemistry and pharmacological potentials. Int. J. Pharm. Tech. Res. 2011, 3, 268–282. [Google Scholar]
- Narasimhan, B.; Sharma, D.; Kumar, P. Biological importance of imidazole nucleus in the new millennium. Med. Chem. Res. 2011, 20, 1119–1140. [Google Scholar] [CrossRef]
- Yang, X.D.; Wan, W.C.; Deng, X.Y.; Li, Y.; Yang, L.J.; Li, L.; Zhang, H.B. Design, synthesis and cytotoxic activities of novel hybrid compounds between 2-phenylbenzofuran and imidazole. Bioorg. Med. Chem. Lett. 2012, 22, 2726–2729. [Google Scholar] [CrossRef]
- Chen, W.; Deng, X.Y.; Li, Y.; Yang, L.J.; Wan, W.C.; Wang, X.Q.; Zhang, H.B.; Yang, X.D. Synthesis and cytotoxic activities of novel hybrid 2-phenyl-3-alkylbenzofuran and imidazole/triazole compounds. Bioorg. Med. Chem. Lett. 2013, 23, 4297–4302. [Google Scholar] [CrossRef]
- Al-Blewi, F.; Shaikh, S.A.; Naqvi, A.; Aljohani, F.; Aouad, M.R.; Ihmaid, S.; Rezki, N. Design and synthesis of novel imidazole derivatives possessing triazole pharmacophore with potent anticancer activity, and in silico ADMET with GSK-3β molecular docking investigations. Int. J. Mol. Sci. 2021, 22, 1162. [Google Scholar] [CrossRef]
- Hu, Y.; Li, N.; Zhang, J.; Wang, Y.; Chen, L.; Sun, J. Artemisinin-indole and artemisinin-imidazole hybrids: Synthesis, cytotoxic evaluation and reversal effects on multidrug resistance in MCF-7/ADR cells. Bioorg. Med. Chem. Lett. 2019, 29, 1138–1142. [Google Scholar] [CrossRef]
- Song, W.J.; Yang, X.D.; Zeng, X.H.; Xu, X.L.; Zhang, G.L.; Zhang, H.B. Synthesis and cytotoxic activities of novel hybrid compounds of imidazole scaffold-based 2-substituted benzofurans. RSC Adv. 2012, 2, 4612–4615. [Google Scholar] [CrossRef]
- Di Martino, S.; Rainone, A.; Troise, A.; Di Paolo, M.; Pugliese, S.; Zappavigna, S.; Grimaldi, A.; Valente, D. Overview of FDA-approved anti cancer drugs used for targeted therapy. WCRJ 2015, 2, e553WCRJ. [Google Scholar]
- Sun, J.; Wei, Q.; Zhou, Y.; Wang, J.; Liu, Q.; Xu, H. A systematic analysis of FDA-approved anticancer drugs. BMC Syst. Biol. 2017, 11, 87. [Google Scholar] [CrossRef]
- Khojasteh Poor, F.; Keivan, M.; Ramazii, M.; Ghaedrahmati, F.; Anbiyaiee, A.; Panahandeh, S.; Khoshnam, S.E.; Farzaneh, M. Mini review: The FDA-approved prescription drugs that target the MAPK signaling pathway in women with breast cancer. Breast Dis. 2021, 40, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.W.; Mo, H.Y.; Lau, A.T.; Xu, Y.M. Selenium species: Current status and potentials in cancer prevention and therapy. Int. J. Mol. Sci. 2018, 20, 75. [Google Scholar] [CrossRef]
- He, X.; Zhong, M.; Li, S.; Li, X.; Li, Y.; Li, Z.; Gao, Y.; Ding, F.; Wen, D.; Lei, Y. Synthesis and biological evaluation of organoselenium (NSAIDs-SeCN and SeCF3) derivatives as potential anticancer agents. Eur. J. Med. Chem. 2020, 208, 112864. [Google Scholar] [CrossRef]
- Gandin, V.; Khalkar, P.; Braude, J.; Fernandes, A.P. Organic selenium compounds as potential chemotherapeutic agents for improved cancer treatment. Free Radic. Biol. Med. 2018, 127, 80–97. [Google Scholar] [CrossRef]
- Jardim, G.A.; da Cruz, E.H.; Valença, W.O.; Lima, D.J.; Cavalcanti, B.C.; Pessoa, C.; Rafique, J.; Braga, A.L.; Jacob, C.; da Silva Júnior, E.N. Synthesis of selenium-quinone hybrid compounds with potential antitumor activity via Rh-catalyzed CH bond activation and click reactions. Molecules 2017, 23, 83. [Google Scholar] [CrossRef]
- An, B.; Zhang, S.; Hu, J.; Pan, T.; Huang, L.; Tang, J.C.O.; Li, X.; Chan, A.S. The design, synthesis and evaluation of selenium-containing 4-anilinoquinazoline hybrids as anticancer agents and a study of their mechanism. Org. Biomol. Chem. 2018, 16, 4701–4714. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Liang, Y.; Cheng, J.; Ding, K.; Wang, Y. Bifunctional chiral selenium-containing 1, 4-diarylazetidin-2-ones with potent antitumor activities by disrupting tubulin polymerization and inducing reactive oxygen species production. Eur. J. Med. Chem. 2021, 221, 113531. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Sheng, X.; Bian, M.; Yang, Z.; Lu, Y.; Liu, W. Synthesis and in vitro anticancer activities of selenium N-heterocyclic carbene compounds. Chem. Biol. Drug Des. 2021, 98, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Rottenberg, S.; Disler, C.; Perego, P. The rediscovery of platinum-based cancer therapy. Nat. Rev. Cancer 2021, 21, 37–50. [Google Scholar] [CrossRef]
- Ndagi, U.; Mhlongo, N.; Soliman, M.E. Metal complexes in cancer therapy–an update from drug design perspective. Drug Des. Dev. Ther. 2017, 11, 599–616. [Google Scholar] [CrossRef]
- Ciarimboli, G. Anticancer Platinum Drugs Update. Biomolecules 2021, 11, 1637. [Google Scholar] [CrossRef]
- Zajda, J.; Wróblewska, A.; Ruzik, L.; Matczuk, M. Methodology for characterization of platinum-based drug’s targeted delivery nanosystems. J. Control. Release 2021, 335, 178–190. [Google Scholar] [CrossRef]
- Johnstone, T.C.; Park, G.Y.; Lippard, S.J. Understanding and improving platinum anticancer drugs–phenanthriplatin. Anticancer Res. 2014, 34, 471–476. [Google Scholar]
- Graham, L.A.; Suryadi, J.; West, T.K.; Kucera, G.L.; Bierbach, U. Synthesis, aqueous reactivity, and biological evaluation of carboxylic acid ester-functionalized platinum–acridine hybrid anticancer agents. J. Med. Chem. 2012, 55, 7817–7827. [Google Scholar] [CrossRef]
- Zhao, J.; Gou, S.; Sun, Y.; Fang, L.; Wang, Z. Antitumor platinum (II) complexes containing platinum-based moieties of present platinum drugs and furoxan groups as nitric oxide donors: Synthesis, DNA interaction, and cytotoxicity. Inorg. Chem. 2012, 51, 10317–10324. [Google Scholar] [CrossRef]
- Liu, Z.; Li, Z.; Du, T.; Chen, Y.; Wang, Q.; Li, G.; Liu, M.; Zhang, N.; Li, D.; Han, J. Design, synthesis and biological evaluation of dihydro-2-quinolone platinum (IV) hybrids as antitumor agents displaying mitochondria injury and DNA damage mechanism. Dalton Trans. 2021, 50, 362–375. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, Q.; Li, Z.; Liu, Z.; Zhao, Y.; Zhang, J.; Liu, M.; Wang, Z.; Li, D.; Han, J. Naproxen platinum (iv) hybrids inhibiting cycloxygenases and matrix metalloproteinases and causing DNA damage: Synthesis and biological evaluation as antitumor agents in vitro and in vivo. Dalton Trans. 2020, 49, 5192–5204. [Google Scholar] [CrossRef]
- Cincinelli, R.; Musso, L.; Dallavalle, S.; Artali, R.; Tinelli, S.; Colangelo, D.; Zunino, F.; De Cesare, M.; Beretta, G.L.; Zaffaroni, N. Design, modeling, synthesis and biological activity evaluation of camptothecin-linked platinum anticancer agents. Eur. J. Med. Chem. 2013, 63, 387–400. [Google Scholar] [CrossRef]
- Zhou, J.; Kang, Y.; Chen, L.; Wang, H.; Liu, J.; Zeng, S.; Yu, L. The drug-resistance mechanisms of five platinum-based antitumor agents. Front. Pharmacol. 2020, 11, 343. [Google Scholar] [CrossRef]
- Dilruba, S.; Kalayda, G.V. Platinum-based drugs: Past, present and future. Cancer Chemother. Pharmacol. 2016, 77, 1103–1124. [Google Scholar] [CrossRef]
- Ha, V.T.; Kien, V.T.; Binh, L.H.; Tien, V.D.; My, N.T.T.; Nam, N.H.; Baltas, M.; Hahn, H.; Han, B.W.; Thao, D.T.; et al. Design, synthesis and biological evaluation of novel hydroxamic acids bearing artemisinin skeleton. Bioorg. Chem. 2016, 66, 63–71. [Google Scholar] [CrossRef]
- Ling, Y.; Guo, J.; Yang, Q.; Zhu, P.; Miao, J.; Gao, W.; Peng, Y.; Yang, J.; Xu, K.; Xiong, B.; et al. Development of novel β-carboline-based hydroxamate derivatives as HDAC inhibitors with antiproliferative and antimetastatic activities in human cancer cells. Eur. J. Med. Chem. 2018, 144, 398–409. [Google Scholar] [CrossRef]
- Mohamed, M.F.A.; Shaykoon, M.S.A.; Abdelrahman, M.H.; Elsadek, B.E.M.; Aboraia, A.S.; Abuo-Rahma, G. Design, synthesis, docking studies and biological evaluation of novel chalcone derivatives as potential histone deacetylase inhibitors. Bioorg. Chem. 2017, 72, 32–41. [Google Scholar] [CrossRef]
- Ding, C.; Chen, S.; Zhang, C.; Hu, G.; Zhang, W.; Li, L.; Chen, Y.Z.; Tan, C.; Jiang, Y. Synthesis and investigation of novel 6-(1,2,3-triazol-4-yl)-4-aminoquinazolin derivatives possessing hydroxamic acid moiety for cancer therapy. Bioorg. Med. Chem. 2017, 25, 27–37. [Google Scholar] [CrossRef]
- Dung, D.T.M.; Dung, P.T.P.; Oanh, D.T.K.; Vu, T.K.; Hahn, H.; Han, B.W.; Pyo, M.; Kim, Y.G.; Han, S.B.; Nam, N.H. Exploration of novel 5′(7′)-substituted-2′-oxospiro[1,3]dioxolane-2,3′-indoline-based N-hydroxypropenamides as histone deacetylase inhibitors and antitumor agents. Arab. J. Chem. 2017, 10, 465–472. [Google Scholar] [CrossRef]
- Bubna, A.K. Vorinostat-An Overview. Indian J. Dermatol. 2015, 60, 419. [Google Scholar] [CrossRef]
- Moore, D. Panobinostat (Farydak): A Novel Option for the Treatment of Relapsed Or Relapsed and Refractory Multiple Myeloma. Pharm. Ther. 2016, 41, 296–300. [Google Scholar]
- Tak, W.Y.; Ryoo, B.Y.; Lim, H.Y.; Kim, D.Y.; Okusaka, T.; Ikeda, M.; Hidaka, H.; Yeon, J.E.; Mizukoshi, E.; Morimoto, M.; et al. Phase I/II study of first-line combination therapy with sorafenib plus resminostat, an oral HDAC inhibitor, versus sorafenib monotherapy for advanced hepatocellular carcinoma in east Asian patients. Investig. New Drugs 2018, 36, 1072–1084. [Google Scholar] [CrossRef]
- Alam, M.A. Methods for Hydroxamic Acid Synthesis. Curr. Org. Chem. 2019, 23, 978–993. [Google Scholar] [CrossRef]
- Quirante, J.; Dubar, F.; González, A.; Lopez, C.; Cascante, M.; Cortés, R.; Forfar, I.; Pradines, B.; Biot, C. Ferrocene–indole hybrids for cancer and malaria therapy. J. Organomet. Chem. 2011, 696, 1011–1017. [Google Scholar] [CrossRef]
- Huang, X.F.; Wang, L.Z.; Tang, L.; Lu, Y.X.; Wang, F.; Song, G.Q.; Ruan, B.F. Synthesis, characterization and antitumor activity of novel ferrocene derivatives containing pyrazolyl-moiety. J. Organomet. Chem. 2014, 749, 157–162. [Google Scholar] [CrossRef]
- Smit, F.J.; Bezuidenhout, J.J.; Bezuidenhout, C.C.; N’Da, D.D. Synthesis and in vitro biological activities of ferrocenyl–chalcone amides. Med. Chem. Res. 2016, 25, 568–584. [Google Scholar] [CrossRef]
- Wei, J.N.; Jia, Z.D.; Zhou, Y.Q.; Chen, P.H.; Li, B.; Zhang, N.; Hao, X.Q.; Xu, Y.; Zhang, B. Synthesis, characterization and antitumor activity of novel ferrocene-coumarin conjugates. J. Organomet. Chem. 2019, 902, 120968. [Google Scholar] [CrossRef]
- Panaka, S.; Trivedi, R.; Jaipal, K.; Giribabu, L.; Sujitha, P.; Kumar, C.G.; Sridhar, B. Ferrocenyl chalcogeno (sugar) triazole conjugates: Synthesis, characterization and anticancer properties. J. Organomet. Chem. 2016, 813, 125–130. [Google Scholar] [CrossRef]
- Raghavan, S.; Manogaran, P.; Gadepalli Narasimha, K.K.; Kalpattu Kuppusami, B.; Mariyappan, P.; Gopalakrishnan, A.; Venkatraman, G. Synthesis and anticancer activity of novel curcumin–quinolone hybrids. Bioorg. Med. Chem. Lett. 2015, 25, 3601–3605. [Google Scholar] [CrossRef]
- Banuppriya, G.; Sribalan, R.; Padmini, V. Synthesis and characterization of curcumin-sulfonamide hybrids: Biological evaluation and molecular docking studies. J. Mol. Struct. 2018, 1155, 90–100. [Google Scholar] [CrossRef]
- Puneeth, H.R.; Ananda, H.; Kumar, K.S.S.; Rangappa, K.S.; Sharada, A.C. Synthesis and antiproliferative studies of curcumin pyrazole derivatives. Med. Chem. Res. 2016, 25, 1842–1851. [Google Scholar] [CrossRef]
- Qiu, P.; Xu, L.; Gao, L.; Zhang, M.; Wang, S.; Tong, S.; Sun, Y.; Zhang, L.; Jiang, T. Exploring pyrimidine-substituted curcumin analogues: Design, synthesis and effects on EGFR signaling. Bioorg. Med. Chem. 2013, 21, 5012–5020. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Gupta, M.K.; Saxena, A.K.; Bedi, P.M.S. Triazole linked mono carbonyl curcumin-isatin bifunctional hybrids as novel anti tubulin agents: Design, synthesis, biological evaluation and molecular modeling studies. Bioorg. Med. Chem. 2015, 23, 7165–7180. [Google Scholar] [CrossRef]
- Ma, L.Y.; Wang, B.; Pang, L.P.; Zhang, M.; Wang, S.Q.; Zheng, Y.C.; Shao, K.P.; Xue, D.Q.; Liu, H.M. Design and synthesis of novel 1,2,3-triazole–pyrimidine–urea hybrids as potential anticancer agents. Bioorg. Med. Chem. Lett. 2015, 25, 1124–1128. [Google Scholar] [CrossRef]
- Chandrashekhar, M.; Nayak, V.L.; Ramakrishna, S.; Mallavadhani, U.V. Novel triazole hybrids of myrrhanone C, a natural polypodane triterpene: Synthesis, cytotoxic activity and cell based studies. Eur. J. Med. Chem. 2016, 114, 293–307. [Google Scholar] [CrossRef]
- Najafi, Z.; Mahdavi, M.; Safavi, M.; Saeedi, M.; Alinezhad, H.; Pordeli, M.; Kabudanian Ardestani, S.; Shafiee, A.; Foroumadi, A.; Akbarzadeh, T. Synthesis and In vitro Cytotoxic Activity of Novel Triazole-Isoxazole Derivatives. J. Heterocycl. Chem. 2015, 52, 1743–1747. [Google Scholar] [CrossRef]
- Duan, Y.C.; Ma, Y.C.; Zhang, E.; Shi, X.J.; Wang, M.M.; Ye, X.W.; Liu, H.M. Design and synthesis of novel 1,2,3-triazole-dithiocarbamate hybrids as potential anticancer agents. Eur. J. Med. Chem. 2013, 62, 11–19. [Google Scholar] [CrossRef]
- Kumbhare, R.M.; Dadmal, T.L.; Ramaiah, M.J.; Kishore, K.S.V.; Pushpa Valli, S.N.C.V.L.; Tiwari, S.K.; Appalanaidu, K.; Rao, Y.K.; Bhadra, M.P. Synthesis and anticancer evaluation of novel triazole linked N-(pyrimidin-2-yl)benzo[d]thiazol-2-amine derivatives as inhibitors of cell survival proteins and inducers of apoptosis in MCF-7 breast cancer cells. Bioorg. Med. Chem. Lett. 2015, 25, 654–658. [Google Scholar] [CrossRef]
- Sivaramakarthikeyan, R.; Iniyaval, S.; Saravanan, V.; Lim, W.M.; Mai, C.W.; Ramalingan, C. Molecular Hybrids Integrated with Benzimidazole and Pyrazole Structural Motifs: Design, Synthesis, Biological Evaluation, and Molecular Docking Studies. ACS Omega 2020, 5, 10089–10098. [Google Scholar] [CrossRef]
- Shao, K.P.; Zhang, X.Y.; Chen, P.J.; Xue, D.Q.; He, P.; Ma, L.Y.; Zheng, J.X.; Zhang, Q.R.; Liu, H.M. Synthesis and biological evaluation of novel pyrimidine–benzimidazol hybrids as potential anticancer agents. Bioorg. Med. Chem. Lett. 2014, 24, 3877–3881. [Google Scholar] [CrossRef]
- Sharma, P.; Srinivasa Reddy, T.; Thummuri, D.; Senwar, K.R.; Praveen Kumar, N.; Naidu, V.G.M.; Bhargava, S.K.; Shankaraiah, N. Synthesis and biological evaluation of new benzimidazole-thiazolidinedione hybrids as potential cytotoxic and apoptosis inducing agents. Eur. J. Med. Chem. 2016, 124, 608–621. [Google Scholar] [CrossRef]
- Shi, L.; Wu, T.T.; Wang, Z.; Xue, J.Y.; Xu, Y.G. Discovery of quinazolin-4-amines bearing benzimidazole fragments as dual inhibitors of c-Met and VEGFR-2. Bioorg. Med. Chem. 2014, 22, 4735–4744. [Google Scholar] [CrossRef]
- Sireesha, R.; Sreenivasulu, R.; Chandrasekhar, C.; Jadav, S.S.; Pavani, Y.; Rao, M.V.B.; Subbarao, M. Design, synthesis, anticancer evaluation and binding mode studies of benzimidazole/benzoxazole linked β-carboline derivatives. J. Mol. Struct. 2021, 1226, 129351. [Google Scholar] [CrossRef]
- Kim, E.S. Abemaciclib: First Global Approval. Drugs 2017, 77, 2063–2070. [Google Scholar] [CrossRef]
- Ghisoni, E.; Giannone, G.; Tuninetti, V.; Genta, S.; Scotto, G.; Aglietta, M.; Sangiolo, D.; Mittica, G.; Valabrega, G. Veliparib: A new therapeutic option in ovarian cancer? Future Oncol. 2019, 15, 1975–1987. [Google Scholar] [CrossRef]
- Choi, Y.J.; Kim, H.S.; Park, S.H.; Kim, B.S.; Kim, K.H.; Lee, H.J.; Song, H.S.; Shin, D.Y.; Lee, H.Y.; Kim, H.G.; et al. Phase II Study of Dovitinib in Patients with Castration-Resistant Prostate Cancer (KCSG-GU11-05). Cancer Res. Treat. 2018, 50, 1252–1259. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Xiao, Z.; Zhang, L.L. Clinical efficacy and safety of nazartinib for epidermal growth factor receptor mutated non-small cell lung cancer: Study protocol for a prospective, multicenter, open-label. Medicine 2021, 100, e25992. [Google Scholar] [CrossRef]
- Karthikeyan, C.; Solomon, V.R.; Lee, H.; Trivedi, P.J.B.; Nutrition, P. Design, synthesis and biological evaluation of some isatin-linked chalcones as novel anti-breast cancer agents: A molecular hybridization approach. Biomed. Prev. Nutr. 2013, 3, 325–330. [Google Scholar] [CrossRef]
- Eldehna, W.M.; El Hassab, M.A.; Abo-Ashour, M.F.; Al-Warhi, T.; Elaasser, M.M.; Safwat, N.A.; Suliman, H.; Ahmed, M.F.; Al-Rashood, S.T.; Abdel-Aziz, H.A.J.B.C. Development of isatin-thiazolo [3, 2-a] benzimidazole hybrids as novel CDK2 inhibitors with potent in vitro apoptotic anti-proliferative activity: Synthesis, biological and molecular dynamics investigations. Bioorg. Chem. 2021, 110, 104748. [Google Scholar] [CrossRef]
- Abdel-Aziz, H.A.; Eldehna, W.M.; Keeton, A.B.; Piazza, G.A.; Kadi, A.A.; Attwa, M.W.; Abdelhameed, A.S.; Attia, M.I.J.D.D. Development; Therapy, Isatin-benzoazine molecular hybrids as potential antiproliferative agents: Synthesis and in vitro pharmacological profiling. Drug Des. Dev. Ther. 2017, 11, 2333–2346. [Google Scholar] [CrossRef] [PubMed]
- Meleddu, R.; Petrikaite, V.; Distinto, S.; Arridu, A.; Angius, R.; Serusi, L.; Škarnulytė, L.; Endriulaitytė, U.; Paškevičiūtė, M.; Cottiglia, F.J.A.M.C.L. Investigating the anticancer activity of isatin/dihydropyrazole hybrids. ACS Med. Chem. Lett. 2018, 10, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Eldehna, W.M.; Altoukhy, A.; Mahrous, H.; Abdel-Aziz, H.A. Design, synthesis and QSAR study of certain isatin-pyridine hybrids as potential anti-proliferative agents. Eur. J. Med. Chem. 2015, 90, 684–694. [Google Scholar] [CrossRef] [PubMed]
- Singh, H.; Singh, J.V.; Gupta, M.K.; Saxena, A.K.; Sharma, S.; Nepali, K.; Bedi, P.M.S.J.B.; Letters, M.C. Triazole tethered isatin-coumarin based molecular hybrids as novel antitubulin agents: Design, synthesis, biological investigation and docking studies. Bioorg. Med. Chem. Lett. 2017, 27, 3974–3979. [Google Scholar] [CrossRef]
- Al-Wabli, R.I.; Almomen, A.A.; Almutairi, M.S.; Keeton, A.B.; Piazza, G.A.; Attia, M.I. New Isatin–Indole Conjugates: Synthesis, Characterization, and a Plausible Mechanism of Their in vitro Antiproliferative Activity. Drug Des. Dev. Ther. 2020, 14, 483–495. [Google Scholar] [CrossRef]
- Panga, S.; Podila, N.K.; Asres, K.; Ciddi, V. Synthesis and anticancer activity of new isatin-benzoic acid conjugates. Ethiop. Pharm. J. 2016, 31, 75–92. [Google Scholar] [CrossRef]
- Cheke, R.S.; Patil, V.M.; Firke, S.D.; Ambhore, J.P.; Ansari, I.A.; Patel, H.M.; Shinde, S.D.; Pasupuleti, V.R.; Hassan, M.I.; Adnan, M.J.P. Therapeutic Outcomes of Isatin and Its Derivatives against Multiple Diseases: Recent Developments in Drug Discovery. Pharmaceuticals 2022, 15, 272. [Google Scholar] [CrossRef]
- Ferguson, L.; Bhakta, S.; Fox, K.R.; Wells, G.; Brucoli, F.J.M. Synthesis and Biological Evaluation of a Novel C8-Pyrrolobenzodiazepine (PBD) Adenosine Conjugate. A Study on the Role of the PBD Ring in the Biological Activity of PBD-Conjugates. Molecules 2020, 25, 1243. [Google Scholar] [CrossRef] [Green Version]
- Bose, D.S.; Idrees, M.; Todewale, I.K.; Jakka, N.; Rao, J.V. Hybrids of privileged structures benzothiazoles and pyrrolo [2, 1-c][1, 4] benzodiazepin-5-one, and diversity-oriented synthesis of benzothiazoles. Eur. J. Med. Chem. 2012, 50, 27–38. [Google Scholar] [CrossRef]
- Kamal, A.; Ramakrishna, G.; Nayak, V.L.; Raju, P.; Rao, A.S.; Viswanath, A.; Vishnuvardhan, M.; Ramakrishna, S.; Srinivas, G. Design and synthesis of benzo [c, d] indolone-pyrrolobenzodiazepine conjugates as potential anticancer agents. Bioorg. Med. Chem. 2012, 20, 789–800. [Google Scholar] [CrossRef]
- Li, Y.; Quan, J.; Song, H.; Li, D.; Ma, E.; Wang, Y.; Ma, C. Novel pyrrolo [2, 1-c][1, 4] benzodiazepine-3, 11-dione (PBD) derivatives as selective HDAC6 inhibitors to suppress tumor metastasis and invasion in vitro and in vivo. Bioorg. Chem. 2021, 114, 105081. [Google Scholar] [CrossRef]
- Chen, C.Y.; Lee, P.H.; Lin, Y.Y.; Yu, W.T.; Hu, W.P.; Hsu, C.C.; Lin, Y.T.; Chang, L.S.; Hsiao, C.T.; Wang, J.-J.; et al. Synthesis, DNA-binding abilities and anticancer activities of triazole-pyrrolo [2, 1-c][1, 4] benzodiazepines hybrid scaffolds. Bioorg. Med. Chem. Lett. 2013, 23, 6854–6859. [Google Scholar] [CrossRef]
- Al Zahrani, N.A.; El-Shishtawy, R.M.; Elaasser, M.M.; Asiri, A.M. Synthesis of novel chalcone-based phenothiazine derivatives as antioxidant and anticancer agents. Molecules 2020, 25, 4566. [Google Scholar] [CrossRef]
- Ouyang, Y.; Li, J.; Chen, X.; Fu, X.; Sun, S.; Wu, Q. Chalcone Derivatives: Role in Anticancer Therapy. Biomolecules 2021, 11, 894. [Google Scholar] [CrossRef]
- Sivapriya, S.; Sivakumar, K.; Manikandan, H. Anticancer effects of chalcone-benzoxadiazole hybrids on KB human cancer cells. Chem. Data Collect. 2021, 35, 100762. [Google Scholar] [CrossRef]
- Alswah, M.; Bayoumi, A.H.; Elgamal, K.; Elmorsy, A.; Ihmaid, S.; Ahmed, H.E.A. Design, synthesis and cytotoxic evaluation of novel chalcone derivatives bearing triazolo [4, 3-a]-quinoxaline moieties as potent anticancer agents with dual EGFR kinase and tubulin polymerization inhibitory effects. Molecules 2017, 23, 48. [Google Scholar] [CrossRef]
- Yepes, A.F.; Arias, J.D.; Cardona-G, W.; Herrera-R, A.; Moreno, G. New class of hybrids based on chalcone and melatonin: A promising therapeutic option for the treatment of colorectal cancer. Med. Chem. Res. 2021, 30, 2240–2255. [Google Scholar] [CrossRef]
- Ma, X.; Wang, D.; Wei, G.; Zhou, Q.; Gan, X. Synthesis and anticancer activity of chalcone–quinoxalin conjugates. Synth. Commun. 2021, 51, 1363–1372. [Google Scholar] [CrossRef]
- Paul, K.; Bindal, S.; Luxami, V. Synthesis of new conjugated coumarin–benzimidazole hybrids and their anticancer activity. Bioorg. Med. Chem. Lett. 2013, 23, 3667–3672. [Google Scholar] [CrossRef]
- An, R.; Hou, Z.; Li, J.T.; Yu, H.N.; Mou, Y.H.; Guo, C. Design, synthesis and biological evaluation of novel 4-substituted coumarin derivatives as antitumor agents. Molecules 2018, 23, 2281. [Google Scholar] [CrossRef]
- Elshemy, H.A.; Zaki, M.A. Design and synthesis of new coumarin hybrids and insight into their mode of antiproliferative action. Bioorg. Med. Chem. 2017, 25, 1066–1075. [Google Scholar] [CrossRef] [PubMed]
- Sanduja, M.; Gupta, J.; Singh, H.; Pagare, P.P.; Rana, A. Uracil-coumarin based hybrid molecules as potent anticancer and antibacterial agents. J. Saudi Chem. Soc. 2020, 24, 251–266. [Google Scholar] [CrossRef]
- Zhang, Z.; Bai, Z.W.; Ling, Y.; He, L.Q.; Huang, P.; Gu, H.X.; Hu, R.F. Design, synthesis and biological evaluation of novel furoxan-based coumarin derivatives as antitumor agents. Med. Chem. Res. 2018, 27, 1198–1205. [Google Scholar] [CrossRef]
- Xu, S.; Pei, L.; Wang, C.; Zhang, Y.K.; Li, D.; Yao, H.; Wu, X.; Chen, Z.S.; Sun, Y.; Xu, J. Novel hybrids of natural oridonin-bearing nitrogen mustards as potential anticancer drug candidates. ACS Med. Chem. Lett. 2014, 5, 797–802. [Google Scholar] [CrossRef]
- Łączkowski, K.Z.; Świtalska, M.; Baranowska-Łączkowska, A.; Plech, T.; Paneth, A.; Misiura, K.; Wietrzyk, J.; Czaplińska, B.; Mrozek-Wilczkiewicz, A.; Malarz, K. Thiazole-based nitrogen mustards: Design, synthesis, spectroscopic studies, DFT calculation, molecular docking, and antiproliferative activity against selected human cancer cell lines. J. Mol. Struct. 2016, 1119, 139–150. [Google Scholar] [CrossRef]
- Kolesinska, B.; Barszcz, K.; Kaminski, Z.J.; Drozdowska, D.; Wietrzyk, J.; Switalska, M. Synthesis and cytotoxicity studies of bifunctional hybrids of nitrogen mustards with potential enzymes inhibitors based on melamine framework. J. Enzyme Inhib. Med. Chem. 2012, 27, 619–627. [Google Scholar] [CrossRef]
- Acharya, P.C.; Bansal, R. Synthesis of androstene oxime-nitrogen mustard bioconjugates as potent antineoplastic agents. Steroids 2017, 123, 73–83. [Google Scholar] [CrossRef]
- Hassan, A.S.; Moustafa, G.O.; Awad, H.M.; Nossier, E.S.; Mady, M.F. Design, synthesis, anticancer evaluation, enzymatic assays, and a molecular modeling study of novel pyrazole–indole hybrids. ACS Omega 2021, 6, 12361–12374. [Google Scholar] [CrossRef]
- Abd El-Karim, S.S.; Anwar, M.M.; Mohamed, N.A.; Nasr, T.; Elseginy, S.A. Design, synthesis, biological evaluation and molecular docking studies of novel benzofuran–pyrazole derivatives as anticancer agents. Bioorg. Chem. 2015, 63, 1–12. [Google Scholar] [CrossRef]
- Abdalha, A.A.; Hekal, M.H. An efficient synthesis and evaluation of some novel quinazolinone-pyrazole hybrids as potential antiproliferative agents. Synth. Commun. 2021, 51, 2498–2509. [Google Scholar] [CrossRef]
- Akhtar, W.; Marella, A.; Alam, M.M.; Khan, M.F.; Akhtar, M.; Anwer, T.; Khan, F.; Naematullah, M.; Azam, F.; Rizvi, M.A. Design and synthesis of pyrazole–pyrazoline hybrids as cancer-associated selective COX-2 inhibitors. Archiv. Der. Pharmazie. 2021, 354, 2000116. [Google Scholar] [CrossRef]
- Verma, G.; Chashoo, G.; Ali, A.; Khan, M.F.; Akhtar, W.; Ali, I.; Akhtar, M.; Alam, M.M.; Shaquiquzzaman, M. Synthesis of pyrazole acrylic acid based oxadiazole and amide derivatives as antimalarial and anticancer agents. Bioorg. Chem. 2018, 77, 106–124. [Google Scholar] [CrossRef]
- Sangani, C.B.; Makawana, J.A.; Duan, Y.T.; Yin, Y.; Teraiya, S.B.; Thumar, N.J.; Zhu, H.L. Design, synthesis and molecular modeling of biquinoline–pyridine hybrids as a new class of potential EGFR and HER-2 kinase inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 4472–4476. [Google Scholar] [CrossRef]
- Hamza, E.K.; Hamdy, N.A.; Zarie, E.S.; Fakhr, I.M.; Elwahy, A.H.; Awad, H.M. Synthesis and in vitro evaluation of novel tetralin-pyrazolo [3, 4-b] pyridine hybrids as potential anticancer agents. J. Heterocycl. Chem. 2020, 57, 182–196. [Google Scholar] [CrossRef]

































































































































| Compound No. | R1 | R2 | R3 | R4 | n | EGFR (IC50 nM) | HT-29 (IC50 µM) | |
|---|---|---|---|---|---|---|---|---|
| Normoxia | Hypoxia | |||||||
| 1b | Cl | F | NO2 | H | 5 | 0.32 | 12.89 | 9.81 |
| 1c | Br | H | NO2 | H | 2 | 0.66 | 4.48 | 4.01 |
| 1d | ethynyl | H | NO2 | H | 3 | 0.56 | 10.08 | 5.96 |
| 1e | ethynyl | H | NO2 | H | 5 | 0.50 | 2.93 | 3.46 |
| Gefitinib | 0.45 | 3.63 | 5.21 | |||||
| Compound No. | R1 | R2 | R3 | EGFR (IC50 nM) | α-Glucosidase (IC50 µM) |
|---|---|---|---|---|---|
| 2b | 3-Cl, 4-(3-fluorobenzyloxy) |  |  | 4.53 | 0.14 |
| 2c | 3-ethynyl |  |  | 4.87 | 0.09 |
| 2d | 3-ethynyl |  |  | ND * | 6.25 |
| 2e | 3-Cl, 4-F |  |  | 10.71 | 4.34 |
| Gefitinib | 3.32 | ≥100 | |||
| Compound No. | R1 | R2 | X | EGFR (IC50 nM) | VEGFR-2 (IC50 nM) |
|---|---|---|---|---|---|
| 3b |  | m-Cl, p-F | Cl | 14 | 14 |
| 3c |  | m-CH3, p-CH3 | Cl | 78 | 51 |
| 3d |  | o-CH3 | Cl | 15 | 178 |
| 3e |  | H | Cl | 14 | 261 |
| vandetanib | 11 | 15 | |||
| Compound No. | Ar. | MCF-7 (GI50 μM) | PI3K (IC50 μM) | ||
|---|---|---|---|---|---|
| A | β | γ | |||
| 4b |  | 15 | 5.3 | 16 | 2.3 |
| 4c |  | 12 | 22.2 | 1.9 | 22.2 |
| 4d |  | 12 | 133 | 56 | 5.9 |
| 4e |  | 32 | 14.2 | 0.5 | 14.2 |
| Compounds No. | R1 | R2 | X | A549 (µM) | BT549 (µM) | HCT-116 (µM) | MCF-7 (µM) | SK-HEP-1 (µM) | SNU638 (µM) |
|---|---|---|---|---|---|---|---|---|---|
| 5b | 3-COOCH |  | CH | 1.10 | 1.08 | 0.40 | 10.1 | 2.40 | 1.12 |
| 5c | 3-COOCH3-4-Cl |  | CH | 2.13 | 2.36 | 3.13 | 1.43 | 7.06 | 2.20 |
| 5d | 4-OCH3 |  | CH | 3.58 | 1.88 | 3.49 | 1.79 | 1.61 | 3.97 |
| 5e | 2,4-diF |  | CH | 4.71 | 2.48 | 4.01 | 1.61 | 2.49 | 2.05 |
| Gefitinib | 8.27 | 6.56 | 5.98 | 26.7 | 10.1 | 7.56 | |||
| Dactolisib | 0.62 | 0.74 | 0.84 | 1.33 | 1.82 | 1.24 | |||
| Compound No. | R1 | R2 | HCT-116 (µM) | SK-HEP1 (µM) | MDA-MB-231 (µM) | SNU638 (µM) | A549 (µM) | MCF-7 (µM) |
|---|---|---|---|---|---|---|---|---|
| 6b |  |  | 1.44 | 4.72 | 0.71 | 0.62 | 0.94 | 1.02 |
| 6c |  |  | 0.49 | 0.86 | 0.88 | 1.26 | 3.52 | 4.73 |
| 6d |  |  | 0.59 | 0.44 | 0.42 | 0.61 | 1.56 | 10.8 |
| 6e |  |  | 1.74 | 1.14 | 2.58 | 0.98 | 3.14 | 4.59 |
| BEZ235 | 0.84 | 1.82 | 0.18 | 1.24 | 0.62 | 1.33 |
| Compound No. | R1 | BRD4 Kd (nM) | MV4-11, IC50 (µM) |
|---|---|---|---|
| 8b |  | 480 | 4.88 |
| 8c |  | 250 | 2.54 |
| 8d |  | 60 | 5.07 |
| 8e |  | 28 | 1.83 |
| BET760 | - | 37 | 0.80 |
| Company Name | Compound Name | Drug Target | Type of Cancer | Status | References |
|---|---|---|---|---|---|
| AstraZeneca | Vandetanib | Kinase inhibitor | Medullary thyroid cancer | Approved | [33] |
| Boehringer Ingelheim | Afatinib | Tyrosine kinase | Non-small cell lung Carcinoma | Approved | [34] |
| Pfizer | Dacomitinib | EGFR inhibitor | Non-small cell lung carcinoma | Approved | [10] |
| AstraZeneca and Teva | Gefitinib | EGFR inhibitor | Breast and Lung cancer | Approved | [35] |
| Roche Pharmaceuticals | Erlotinib | EGFR inhibitor | pancreatic cancer and non-small cell lung cancer | Approved | [36] |
| GlaxoSmith Kline (GSK) | Lapatinib | Dual tyrosine kinase inhibitor | solid tumors and Breast cancer | Approved | [37] |
| AstraZeneca | Sapitinib (AZD 8931) | Erb8 receptor tyrosine kinase | Breast cancer and metastatic cancer | Clinical trials | [10] |
| Array Biopharma | Tucatinib (ARRY 380) | Kinase inhibitor | Breast cancer | Approved | [10] |
| Selleck chemicals | Barasertib (AZD 1152) | Aurora Kinase | Tumor lymphoma, solid tumors and myeloid leukemia | Clinical trials | [38] |
| Spectrum Pharmaceuticals | Poziotinib | Tyrosine kinase | Breast cancer | Clinical trials | [10] |
| AstraZeneca | AZD 3759 | EGFR antagonist | Non-small cell lung Cancer | Clinical trials | [10] |
| Curis Inc. | CUDC-101 | By inhibiting Histone deacetylase, EGFR and HER2 | Advanced /Liver/Neck/Gastric/Head/non-small cell lung cancer and Breast | Clinical trials | [39] |
| Beta-Phama | Icotinib | EGFR-TK1 inhibitor | Non-small cell lung cancer | Approved | [40] |
| Compound No. | R | U937 (µM) | PC-3 (µM) | A549 (µM) | ES-2 (µM) | MDA-MB-231 (µM) | HCT116 (µM) |
|---|---|---|---|---|---|---|---|
| 9b |  | 3.1 | 10.5 | 11.8 | 29.2 | 7.2 | 6.0 |
| 9c |  | 2.2 | 10.4 | 4.2 | 25.1 | 4.5 | 3.8 |
| 9d |  | 2.2 | 5.8 | 1.6 | 4.4 | 6.8 | 5.9 |
| 9e |  | 2.7 | 5.4 | 7.0 | 8.9 | 7.2 | 2.4 |
| SAHA | - | 2.3 | 9.9 | 3.8 | 12.7 | 5.6 | 6.0 |
| Compound No. | R | U937 (µM) | K562 (µM) | HEL (µM) | KG1 (µM) | HL60 (µM) | MDA-MB-231 (µM) | PC-3 (µM) | MCF-7 (µM) | HCT116 (µM) | A549 (µM) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 10b |  | 0.33 | 0.79 | 0.20 | 0.39 | 2.11 | 0.24 | 0.33 | 3.47 | 0.37 | 3.39 |
| 10c |  | 0.32 | 0.68 | 0.27 | 0.72 | 1.59 | 0.41 | 0.53 | 2.95 | 0.57 | 3.91 |
| 10d |  | 0.18 | 1.01 | 0.19 | 0.24 | 1.04 | 0.27 | 0.51 | 2.7 | 0.37 | 2.96 |
| 10e |  | 0.34 | 0.89 | 0.16 | 0.47 | 1.68 | 0.15 | 0.29 | 2.32 | 0.22 | 3.27 |
| SAHA | 1.45 | 3.24 | 0.49 | 1.59 | 4.26 | 1.72 | 3.57 | 3.78 | 2.81 | 3.9 |
| Compound No. | R1 | R2 | R3 | HeLa Nuclear HDAC (nM) |
|---|---|---|---|---|
| 11b | H | H | 4′-(N-3-hydroxyacrylamide) | 2.8 |
| 11c | CH3 | H | 4′-(N-3-hydroxyacrylamide) | 3.3 |
| 11d | CH2CH3 | H | 4′-(N-3-hydroxyacrylamide) | 3.4 |
| 11e | H | CH3 | 4′-(N-3-hydroxyacrylamide) | 47.4 |
| LBH589.HCl | 7.5 |
| Compound No. | R1 | R2 | K562 (Upto %) | MDA-MB 231 (µM) | LNCaP (µM) |
|---|---|---|---|---|---|
| 12b | H |  | 88 | 2 | 32 |
| 12c | H |  | 87 | 28 | 38 |
| 12d | F |  | 64 | 58 | 24 |
| 12e | F |  | 67 | 68 | 35 |
| Compound No. | R | PIM (µM) | ||
|---|---|---|---|---|
| PIM1 | PIM2 | PIM3 | ||
| 13b | Me2N(CH2)2O | 0.30 | 1.40 | 0.50 |
| 13c | Et2N(CH2)2O | 0.14 | 0.84 | 0.27 |
| 13d | Et2N(CH2)3O | 0.11 | 0.38 | 0.081 |
| 13e | Et2N(CH2)3NH | 0.067 | 3.16 | 0.61 |
| Compound No. | R1 | R2 | R3 | A549 (µg/mL) | MCF7 (µg/mL) | SKOV3 (µg/mL) | NIH3T3 (µg/mL) |
|---|---|---|---|---|---|---|---|
| 14b | C2H5 | Br | Me | 4.9 | 50.1 | 68.8 | 52.0 |
| 14c | H | Br | n-Bu | 8.0 | 54.0 | 74.1 | 141.3 |
| 14d | Me | OMe | Me | 5.1 | 36.2 | 53.0 | _ |
| 14e | H | Br | Me | 27.9 | 28.0 | 52.0 | _ |
| Etoposide | 7.8 | 9.9 | 8.5 | 118.0 |
| Compound No. | R1 | R2 | R3 | HL-60 (µM) | SMMC-7721 (µM) | A-549 (µM) | MCF-7 (µM) | SW480 (µM) |
|---|---|---|---|---|---|---|---|---|
| 15b | Bn | Benzimidazole | 2-Bromobenzyl | 1.21 | 4.69 | 6.76 | 2.23 | 6.35 |
| 15c | Bn | Benzimidazole | 4-Methylbenzyl | 1.20 | 4.98 | 6.23 | 2.72 | 6.57 |
| 15d | Bn | 5,6-Dimethyl-benzimidazole | 2-Naphthylmethyl | 1.21 | 2.27 | 4.80 | 1.68 | 1.76 |
| 15e | Me | 5,6-Dimethyl-benzimidazole | 2-Naphthylmethyl | 1.35 | 4.03 | 6.18 | 1.84 | 4.5 |
| DPP | - | - | - | 1.16 | 8.08 | 7.10 | 10.45 | 8.88 |
| Compound No. | R1 | R2 | R3 | R4 | R5 | R6 | A549 (µM) | PC3 (µM) | PaCa2 (µM) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | |||||||
| 16b | H | H | H | OCH3 | H | H | 9.6 | 5.8 | 33.3 | 12.5 | 27.5 | >50 |
| 16c | H | OCH3 | H | H | H | H | 6.4 | 7.5 | >50 | 12.0 | 13.5 | >50 |
| 16d | H | OCH3 | OCH3 | H | H | H | 3.7 | 5.5 | 31.1 | 37.1 | >50 | >50 |
| 16e | 4.9 | 3.0 | 17.2 | 8.1 | 24.0 | >50 | ||||||
| Mitomycin C | 0.45 | |||||||||||
| Compound No. | R | MCF-7 (µM) | MDA-MB-231 (µM) |
|---|---|---|---|
| 17b |  | ≥100 | 71.40 |
| 17c |  | 16.50 | ≥100 |
| 17d |  | 10.99 | 71.40 |
| 17e |  | ≥100 | ≥100 |
| Ospemifene | 55 | 50 | |
| Tamoxifen | 3.5 | ≥100 | |
| Plumbagin | 75 | 4.4 |
| Compound No. | R | MCF-7 (µM) | MDA-MB-231 (µM) |
|---|---|---|---|
| 18b |  | 50 | >100 |
| 18c |  | >100 | >100 |
| Plumbagin | 3.5 | 4.4 | |
| Peganumine A | 38.5 | Not observed | |
| Tamoxifen | 50 | 75 |
| Company Name | Compound Name | Drug Target | Type of Cancer | Status | Reference |
|---|---|---|---|---|---|
| AstraZeneca | Cediranib | VEGFR tyrosine kinases | Glioblastoma | Approved | [53] |
| Chia-tai Tianqing Pharmaceutical Co. | AL 3818 (Anlotinib) | Tyrosine kinase | Synovial sarcoma, Advanced alveolar soft part sarcoma | Clinical trials | [54] |
| Janssen pharmaceuticals | Quisinostat | HDAC inhibitor | Multiple myeloma | Approved | [55] |
| Novartis | Panobinostat (LBH-589) | Non-selective HDAC inhibitor | Multiple myeloma | Approved | [56] |
| Selleck | Dacinostat (LAQ824) | Histone deacetylase inhibitor | Breast and Prostate cancer | Approved | [57] |
| Compound No. | n | R | R’ | HL-60 (µM) | SMMC-7721 (µM) | A549 (µM) | MCF-7 (µM) | SW480 (µM) |
|---|---|---|---|---|---|---|---|---|
| 19b | 2 | Benzimidazole | - | 3.11 | 3.21 | 12.36 | 5.06 | 18.25 |
| 19c | 2 | imidazole | 4-methylbenzyl | 0.84 | 5.74 | 3.92 | 2.24 | 9.56 |
| 19d | 2 | benzimidazole | 2-bromobenzyl | 0.71 | 3.66 | 3.58 | 2.14 | 3.08 |
| 19e | 3 | benzimidazole | 4-methylbenzyl | 0.57 | 2.55 | 2.65 | 2.82 | 3.19 |
| DDP | 1.32 | 6.24 | 11.83 | 15.17 | 12.95 |
| Company Name | Compound Name | Drug Target | Type of Cancer | Status | References |
|---|---|---|---|---|---|
| Novartis Pharmaceutical Corporation | Midostaurin | Kinase inhibitor | Advanced systemic mastocytosis, myelodysplastic syndrome | Approved | [67] |
| Chugai Pharmaceuticals Co. | Alectinib | Tyrosine kinase | Non-small cell lung cancer | Approved | [68] |
| Schwarz Pharma | CEP-2563 | Tyrosine kinase | Solid tumors | Clinical trials | [10] |
| Cayman Chemicals | UCN-01 | Tyrosine kinase | Pancreatic, malignant melanoma, ovarian Cancer and small cell lung | Clinical trials | [69] |
| Helsinn Healthcare | Becatecarin | Topoisomerase-I | Leukemia and gastric cancer | Clinical Trials | [10] |
| Pfizer Pharmaceutical Co. | Edotecarin | Topoisomerase-I | Oesophageal cancer and solid tumors | Clinical trials | [10] |
| Compound No. | R | R1 | DNA Displacement Assay (µg/mL) | DNA Binding Affinity |
|---|---|---|---|---|
| 29b | 4-H | 4-Br | 74 | High |
| 29c | 4-H | 4-NO2 | 81 | weak |
| 29d | 4-H | 4-OCH3 | 81 | Moderate |
| 29e | 4-H | 3,4-diOMe | 62 | High |
| Ethidium bromide | - | - | 1.4 | - |
| Compound No. | R1 | R2 | R3 | CDK9T1 (µM) | CDK1B (µM) | CDK2A (µM) | CDK7H (µM) | HCT-116 (µM) | MCF-7 (µM) |
|---|---|---|---|---|---|---|---|---|---|
| 30b | NH2 | F | m-SO2NH2 | 3 | 7 | 3 | 252 | 0.05 | 0.41 |
| 30c | NHMe | CN | m-SO2Me | 5 | 19 | 43 | 110 | 0.20 | 0.43 |
| 30d | NHMe | F | m-SO2NH(CH2)2OCH3 | 3 | 10 | 6 | 30 | 0.30 | 0.72 |
| 30e | NHMe | CN | p-CO-N-(1-methylpiperidin-4-yl) | 8 | 43 | 32 | 304 | 0.18 | 0.5 |
| Compound No. | Ar | R | A-549 (IC50 (µM) | PC-3 |
|---|---|---|---|---|
| 31b | 4-MeC6H4 | COCH3 | 19.33 | 16.92 |
| 31c | C6H5 | COOC2H5 | 26.64 | 33.56 |
| 31d | 4-SO2NH2C6H4 | COOC2H5 | 16.42 | 7.15 |
| 31d | 4-ClC6H4 | COCH3 | 30.56 | 22.90 |
| 5-FU | 4.21 | 12.00 |
| Compound No. | R1 | R2 | U937 (µg/mL) | THP-1 (µg/mL) | Colo205 (µg/mL) |
|---|---|---|---|---|---|
| 32b | H | CH3-(CH2)8-CH2- | 8.16 | 16.91 | 19.25 |
| 32c | CH3-(CH2)4-CH2- | 6F13-CH2-CH2- | 7.56 | - | 132.42 |
| 32c | (CH3)2CH- | 8F17-CH2-CH2- | 8.35 | 142.23 | - |
| 32d | C2H5 | CH3-(CH2)8-CH2- | 17.83 | 82.65 | - |
| Etoposide | 17.94 | 2.16 | 7.24 |
| Compound No. | R | A549 (µM) | MCF7 (µM) | DU145 (µM) | HeLa (µM) |
|---|---|---|---|---|---|
| 33b | CH3CH2OC(O)CH2- | 16.3 | 12.4 | 18.2 | 9.8 |
| 33c | CH3(CH2)6CH2- | 5.7 | 24.7 | 6.3 | 22.7 |
| 33d | CF3(CF2)7CH2CH2- | 33.7 | - | 37.7 | - |
| 33e |  | 4.1 | - | 4.7 | - |
| 5-FU | - | 1.3 | 1.4 | 1.5 | 1.3 |
| Compound No. | R1 | R2 | NCI-H226 (µM) | NPC-TW01 (µM) | Jurkat |
|---|---|---|---|---|---|
| 34b | Ph | p-Me-Ph | 35 | 49 | 48 |
| 34c | 2-Quinolinyl | p-Cl-Ph | 39 | 35 | 69 |
| 34d | 2-Quinolinyl | p-OMe-Ph | 37 | 36 | >100 |
| 34e | Ph | p-Cl-Ph | 18 | 23 | 36 |
| N0-(4-formyl-1,3-diphenyl-1H-pyrazol-5-yl)-N,N-dimethyl-methanimidamide | 9.3 | 31.4 | 23.5 |
| Company Name | Compound Name | Drug Target | Type of Cancer | Status | Reference |
|---|---|---|---|---|---|
| Novartis | Ceritinib | Abnormal ALK-gene | Non-Small cell lung Cancer | Approved | [85] |
| Pfizer Pharmaceutical company | Palbociclib | CDK4/6 inhibitor | Breast cancer | Approved | [86] |
| AbbVie Pharmaceuticals | Ibrutinib | Tyrosine kinase | Mantle cell lymphoma | Approved | [87,88] |
| Compounds No. | BALB/3T3 (µM) | A549 (µM) | MCF-7 (µM) | LoVo (µM) | KB (µM) |
|---|---|---|---|---|---|
| 35 | 30.04 | 3.24 | 0.81 | 9.38 | 0.87 |
| Doxorubicin | 1.08 | 0.33 | 0.44 | 0.11 | 0.84 |
| DiMIQ | 5.77 | 2.19 | 1.54 | 0.20 | 1.14 |
| Compound No. | HCT116 (µM) | MCF7 (µM) |
|---|---|---|
| 36 | 16.33 | 27.26 |
| Imatinib | 34.40 | - |
| Tamoxifien | -- | 34.30 |
| Compound No. | R1 | R2 | KB (µM) | A-549 (µM) | MCF-7 (µM) | Hs294T (µM) | BALB/3T3 (µM) |
|---|---|---|---|---|---|---|---|
| 38b |  | H | 0.15 | 0.81 | 0.79 | 0.64 | 0.67 |
| 38c |  | H | 0.36 | 0.29 | 0.99 | 0.72 | 0.60 |
| 38d | H |  | 0.64 | 0.17 | 0.47 | 0.35 | 0.34 |
| 38e | H |  | 0.08 | 0.19 | 0.66 | 0.76 | 0.57 |
| DIMIQ | 1.14 | 2.19 | 1.50 | 9.70 | 5.70 |
| Compound No. | R1 | R2 | MDA-MB-231(µM) | HeLa (µM) | SMMC-7721(µM) | QSG-7701(µM) |
|---|---|---|---|---|---|---|
| 39b | H | CH3 | 1.84 | 1.18 | 17.48 | 40.59 |
| 39c | OCH3 | CH3 | 1.42 | 0.83 | 17.65 | 45.20 |
| 39d | F | CH3 | 1.16 | 0.99 | 19.41 | >50 |
| 39e | H | n-C4H9 | >50 | >50 | >50 | Not tested |
| Etoposide | - | - | 5.26 | 2.98 | 3.48 | 28.75 |
| Compound No. | R1 | MB231 (GI50 µM) | MB468 (GI50 µM) | MCF-7 (GI50 µM) | 184B5 (GI50 µM) | MCF10A (GI50 µM) |
|---|---|---|---|---|---|---|
| 40b | 2,4-Dinitrophenyl | 24.3 | 19.2 | 10.8 | 37.8 | 35.4 |
| 40c | 3-Nitrophenyl | 32.2 | 18.6 | 9.4 | 17.7 | 15.4 |
| 40d | 2,4-Dichlorophenyl | 20.3 | 18.6 | 16.7 | 20.4 | 15.6 |
| 40e | Biphenyl | 27.2 | 20.5 | 14.8 | 19.1 | 15.5 |
| Chloroquine | - | 22.5 | 28.6 | 38.4 | 76.1 | 81.26 |
| Cisplatin | - | 23.7 | 31.0 | 25.8 | 25.5 | 51.51 |
| Company Name | Compound Name | Drug Target | Type of Cancer | Status | Reference |
|---|---|---|---|---|---|
| Eisai Co. | Lenvatinib | Kinase inhibitor | Thyroid cancer | Approved | [99] |
| Exelixis Inc. | Cabozantinib | Tyrosine-kinase | Thyroid cancer and renal carcinoma | Approved | [68] |
| Wyeth and Pfizer | Bosutinib | BCR and Src tyrosine kinase | myelogenous leukemia | Approved | [100] |
| Compound No. | R | HeLa (µM) | LS174 (µM) | A549 (µM) | MRC-5 (µM) |
|---|---|---|---|---|---|
| 42b | H | 2.41 | 4.56 | 26.20 | 33.57 |
| 42c | 2-CH3 | 2.36 | 3.13 | 29.05 | 41.87 |
| 42d | 3-CH3 | 2.45 | 11.79 | 33.70 | 52.00 |
| 42e | 4-CH3 | 2.64 | 22.63 | 24.15 | 38.49 |
| cisplatin | - | 2.10 | 5.54 | 11.92 | 14.21 |
| Compound No. | R | KB (µg/mL) | KB/VCR (µg/mL) | A549 (µg/mL) | HL60 (µg/mL) |
|---|---|---|---|---|---|
| 43b |  | 4.31 | 2.21 | 6.58 | 4.45 |
| 43c |  | >8.00 | >8.00 | >8.00 | >8.00 |
| 43d |  | >8.60 | 8.52 | 4.49 | 4.46 |
| Vincristine | - | 0.46 | 0.26 | 12.09 | - |
| Adriamycin I | - | - | - | - | 0.02 |
| Compound No. | R | X | SMMC-7721 (µM) | SW480 (µM) | MCF-7 (µM) | A549 (µM) | HL-60 (µM) |
|---|---|---|---|---|---|---|---|
| 44a | 2-Bromobenzyl | Br | 4.38 | 12.71 | 14.29 | 9.77 | 1.97 |
| 44b | Phenacyl | Br | 3.71 | 10.34 | 11.90 | 12.94 | 2.61 |
| 44c | 4-Bromophenacyl | Br | 3.39 | 2.85 | 2.84 | 8.46 | 3.15 |
| 44d | Naphthyl acyl | Br | 1.65 | 3.38 | 5.87 | 10.93 | 2.49 |
| 44e | 2′-Phenyl-phenacyl | Br | 3.31 | 6.93 | 6.90 | 6.79 | 2.70 |
| DPP | - | - | 8.86 | 15.92 | 16.65 | 11.68 | 1.81 |
| Compound No. | R1 | R2 | SMMC-7721 (µM) | SW480 (µM) | MCF-7 (µM) | A549 (µM) | HL-60 (µM) |
|---|---|---|---|---|---|---|---|
| 45a | Benzimidazole | 2-Bromobenzyl | 2.10 | 5.56 | 4.78 | 3.34 | 0.64 |
| 45b | 2-Ethylimidazole | 4-Hydroxyphenacyl | 11.81 | 5.69 | 3.17 | 12.90 | 0.58 |
| 45c | 2-Ethylimidazole | 4-Bromophenacyl | 6.07 | 3.58 | 2.89 | 12.76 | 0.72 |
| 45d | 2-Ethylimidazole | Naphthylacyl | 2.30 | 3.14 | 3.03 | 5.35 | 0.61 |
| 45e | 2-Ethylimidazole | 2-Bromobenzyl | 0.52 | 0.47 | 0.51 | 0.55 | 0.08 |
| DPP | 1.69 | 12.49 | 14.09 | 20.82 | 18.85 |
| Compound No. | R | Caco2 (µM) | HCT116 (µM) | HeLa (µM) | MCF-7 (µM) |
|---|---|---|---|---|---|
| 46a |  | 6.31 | 12.04 | 7.91 | 3.80 |
| 46b |  | 8.45 | 18.32 | 9.45 | 4.45 |
| 46c |  | 4.67 | 16.78 | 6.87 | 0.38 |
| 46d |  | 5.22 | 18.70 | 8.42 | 3.87 |
| 46e |  | 10.87 | 30.98 | 20.34 | 15.56 |
| Doxorubicin | - | 5.17 | 5.64 | 1.25 | 0.65 |
| Compound No. | R1 | R2 | R3 | n | MCF-7 (µM) | A549 (µM) | HEPG-2 (µM) | MDA-MB-231 (µM) | LO2 (µM) |
|---|---|---|---|---|---|---|---|---|---|
| 47a | H | CN | CN | 1 | 10.75 | 12.36 | 25.59 | >100 | 49.05 |
| 47b | H | NO2 | H | 2 | 12.86 | 20.95 | 39.30 | >100 | 46.37 |
| 47c | H | H | Br | 2 | 12.40 | 26.02 | 41.59 | 80.92 | >100 |
| 47d | H | H | NO2 | 2 | 12.69 | 19.97 | 33.33 | 80.92 | 34.32 |
| 47e | H | NO2 | H | 3 | 9.78 | 16.53 | 21.25 | 85.20 | 45.74 |
| Andriamycin | - | - | - | 0.67 | 1.13 | 0.85 | 2.94 | 0.79 |
| Compounds No. | R1 | R2 | R3 (Imidazole) | SKOV-3 (µM) | HL-60 (µM) | MCF-7 (µM) |
|---|---|---|---|---|---|---|
| 48a | H | H | Benzimidazole | 9.5 | 8.4 | 11.8 |
| 48b | H | Allyl | Benzimidazole | 7.9 | >40 | >40 |
| 48c | OMe | Allyl | Imidazole | 36.2 | >40 | >40 |
| 48d | OMe | Allyl | Benzimidazole | 9.3 | >40 | >40 |
| 48e | H | H | Imidazole | >40 | >40 | >40 |
| DDP | - | - | - | 8.9 | 5.5 | 13.0 |
| Brand/Company Name | Compound Name | Drug Target | Type of Cancer | Status | References |
|---|---|---|---|---|---|
| AdisInsight-springer/Ligand pharmaceuticals | Acadesine | AMP-activated protein kinase | Acute lymphoblastic leukemia | Phase-III | [105] |
| Tasigna/ Novartis | Nilotinib | BCR-ABL | Leukemia | FDA approved | [114] |
| Hikma Pharmaceuticals | Dacarbazine | DNA synthesis | Melanoma; Lymphoma | FDA approved | [115] |
| Janssen Pharmaceutical | Tipifarnib | Farnesyltransferase inhibitors (FTIs) | Breast cancer | phase II trials | [116] |
| Treanda Astellas Pharma | Bendamustine hydrochloride | DNA synthesis | Leukemia; Lymphoma | FDA approved | [115] |
| Oforta/ Sanofi pharma | Fludarabine phosphate | DNA synthesis | Leukemia | FDA approved Discontinued | [115] |
| Nova Laboratories, Ltd. | Azathioprine prodrug of mercaptopurine | Thiopurine S-methyltransferase (TPMT) | Childhood acute lymphoblastic leukemia | FDA approved | [115] |
| Iclusig/ Otsuka Pharmaceutical | Ponatinib | BCR-ABL | Leukemia | FDA approved | [115] |
| Temodar/ Ranbaxy (UK) | Temozolomide | DNA synthesis | Brain cancer | FDA approved | [115] |
| Puri-nethol/ German Remedies Limited | Mercaptopurine | HPRT1 | Leukemia | FDA approved | [115] |
| GlaxoSmithKline (GSK) | SB-431542 | Activin receptor-like kinase (ALK) receptors | Childhood acute lymphoblastic leukemia | No clinical trials | [105] |
| Compound No. | Structure | h | SW480 (µM) | HeLa (µM) | A549 (µM) | MCF-7 (µM) |
|---|---|---|---|---|---|---|
| 49a |  | 24 | 13.4 | 24.3 | 9.4 | 8.2 |
| 48 | 11.4 | 15.1 | 11.3 | 6.4 | ||
| 72 | 10.1 | 19.4 | 7.4 | 10.4 | ||
| 49b |  | 24 | 8.2 | 19.6 | 13.1 | 8.6 |
| 48 | 7.4 | 17.5 | 18.4 | 9.3 | ||
| 72 | 6.5 | 28.7 | 22.6 | 9.5 | ||
| 49c |  | 24 | 4.9 | 11.5 | 9.4 | 3.4 |
| 48 | 3.3 | 17.4 | 15.2 | 4.3 | ||
| 72 | 4.2 | 19.7 | 18.5 | 2.8 | ||
| Fluorouracil (5FU) | - | 24 | 15.3 | 20.6 | 25.3 | 8.5 |
| 48 | 12.4 | 15.5 | 22.5 | 10.4 | ||
| 72 | 13.1 | 12.7 | 17.3 | 12.6 |
| Compound No. | R | HL-60 (µM) | HCT-116 (µM) | SF295 (µM) | NCIH-460 (µM) | PC3 (µM) | L929 (µM) |
|---|---|---|---|---|---|---|---|
| 50a |  | 0.81 | 78.0 | 2.60 | 2.06 | 2.03 | 0.52 |
| 50b |  | 0.59 | 0.37 | 1.48 | 1.32 | 1.06 | 0.36 |
| 50c |  | 1.0 | 2.03 | 3.12 | 3.26 | 2.70 | 0.61 |
| 50d |  | 0.53 | 78.0 | 2.13 | 2.75 | 2.47 | 3.16 |
| 50e |  | 0.71 | 0.97 | 3.43 | 2.64 | 1.64 | 2.12 |
| DOXO | - | 0.02 | 0.21 | 0.41 | 0.15 | 0.76 | 1.72 |
| Compound No. | R1 | R2 | R3 | R4 | RKO (nM) | HGPG2 (nM) | MCF7 (nM) | HELA (nM) | HCT116 (nM) | MGC803 (nM) |
|---|---|---|---|---|---|---|---|---|---|---|
| 51a | H | H | -CN | Cl | 3.39 | 5.24 | 9.98 | 2.09 | 4.97 | 3.54 |
| 51b | CH3 | H | -CN | Cl | 6.01 | 6.79 | 18.9 | 8.73 | 22.7 | 16.5 |
| 51c | Cl | H | -CN | Cl | 3.42 | 6.78 | 10.6 | 6.78 | 9.17 | 9.67 |
| 51d | F | H | -Se-CH3 | Cl | 7.87 | 13.2 | 22.5 | 9.28 | 12.3 | 14.8 |
| 51e | H | H | -Se-CH3 | -OCH3 | 7.85 | 8.92 | 22.7 | 9.15 | 11.4 | 4.78 |
| EP128495 | - | - | - | - | 4.22 | 6.47 | 5.61 | 5.11 | 8.27 | 6.23 |
| Comound No. | R | HeLa (nM) | HUH-7 (nM) | SKOV3 (nM) | A2780 (nM) |
|---|---|---|---|---|---|
| 52a | OH | 3.3 | 2.3 | 1.6 | 2.0 |
| 52b | F | 2.0 | 6.7 | 8.5 | 7.9 |
| 52c | Cl | 6.1 | 3.7 | 1.0 | 5.9 |
| 52d | I | 9.2 | 17.1 | 9.4 | 8.2 |
| Paclitaxol | - | 2.1 | 3.2 | 2.2 | 3.2 |
| Compound No. | R1 | R2 | A 2780 (nM) | IOSE 80(nM) |
|---|---|---|---|---|
| 53a | OMe | Et | 9.7 | 51.3 |
| 53b | OMe |  | 6.4 | 11.0 |
| 53c | F |  | 19.1 | 34.8 |
| 53d | F |  | 13.0 | 29.4 |
| 53e | Br | Et | 21.1 | 43.7 |
| Ebselen | - | - | 25.4 | 55.4 |
| Compound Name | Mode of Action | Type of Cancer | Dose (Conc.) * | References |
|---|---|---|---|---|
| Methylseleninic Acid (MSA) | Apoptosis mediated by caspases, ER stress, UPR, mitochondrial dysfunction/signaling and PARP cleavage Anoikis, whereby cell detachment is a prerequisite for caspase activation and PARP cleavage | Breast, Colon Lung, Lymphoma Pancreatic | Medium to Low In pancreatic Very low-Low | [119] |
| Ebselen and corresponding Analogues | Not determined. Compounds have antioxidant activity | Breast, Liver, Promyelocytic Leukemia, Prostate | Medium-high | [119] |
| Benzoselenazole- stilbene hybrids | Apoptosis mediated by thioredoxin reductase inhibition and oxidative stress | Breast, Cervical, Liver, Lung | Very low-low | [119] |
| Compound No. | R | m | OVCAR-3 (µM) | MCF-7 (µM) | MDA-MB231 (µM) | PANC1 (µM) | NCI-H460 (µM) |
|---|---|---|---|---|---|---|---|
| 54a | CH3 | 2 | 1.1 | 2.5 | 15 | 0.09 | 0.008 |
| 54b | CH3 | 3 | 33 | 11 | 37 | 4.4 | 0.052 |
| 54c | (CH2)(CO)OCH3 | 2 | - | - | - | - | 0.036 |
| 54d | (CH2)2(CO)OCH3 | 2 | 1.9 | 3.6 | 9.9 | 0.086 | 0.011 |
| 54e | (CH2)2(CO)OCH3 | 3 | 150 | 19 | 36 | 2.2 | 0.065 |
| Cisplatin | - | - | 3.3 | 12 | 60 | 6.6 | 1.2 |
| Compounds No. | HCT-116 (µM) | SGC (µM) |
|---|---|---|
| 55 | 64.06 | 217.93 |
| 56 | 57.21 | 94.23 |
| 57 | 39.43 | 34.64 |
| 58 | 111.91 | 248.07 |
| 59 | 142.15 | 59.10 |
| 60 | 87.06 | 70.83 |
| carboplatin | 273.05 | 58.11 |
| oxaliplatin | 57.04 | 17.35 |
| Compound No. | n | CT26 (μM) | SKOV-3 (μM) | HeLa (μM) | A549 (μM) | A549R (μM) |
|---|---|---|---|---|---|---|
| 61a | 01 | 5.0 | 4.0 | 3.2 | 9.8 | 23.4 |
| 61b | 03 | 11.7 | 2.7 | 2.6 | 8.6 | 8.3 |
| 61c | 04 | 9.2 | 3.5 | 2.5 | 6.1 | 8.9 |
| Cisplatin | - | 5.3 | 2.4 | 2.4 | 13.5 | 22.6 |
| Oxaliplatin | - | 3.9 | 5.3 | 7.4 | 26.8 | 22.2 |
| Compound No. | A549 (μM) | A549R (μM) | SKOV-3 (μM) | CT-26 (μM) | LO-2 (μM) |
|---|---|---|---|---|---|
| 62 | 2.2 | 19.7 | 14.4 | 0.2 | 1.9 |
| 63 | 5.2 | 4.8 | 8.5 | 2.9 | 4.8 |
| 64 | 47.2 | 62.2 | 26.0 | 26.1 | 39.9 |
| 65 | 10.2 | 12.0 | 11.3 | 8.2 | 15.3 |
| 66 | 27.3 | 83.0 | 48.9 | 48.9 | 26.3 |
| Cisplatin | 4.8 | 15.1 | 2.5 | 0.3 | 3.0 |
| Oxaliplatin | 8.4 | 7.3 | 9.4 | 2.3 | 3.6 |
| Carboplatin | 79.6 | 60.6 | 38.1 | 46.2 | 70.7 |
| Compound No. | H460 (μM) | U20S (μM) | A431 (μM) | IGROV-1 (μM) | A2780 (μM) |
|---|---|---|---|---|---|
| 67 | 1.37 | 0.48 | 0.075 | 1.03 | 0.036 |
| 68 | 0.4 | 1.09 | 0.185 | 1.24 | 0.22 |
| 69 | 0.44 | 1.36 | 0.28 | 2.24 | 0.08 |
| Topotecan (TPT) | 1.37 | 0.48 | 0.075 | 1.03 | 0.036 |
| cDDP | 22 | 20.5 | 19.57 | 14.8 | 4.35 |
| Compound Name | Chemical Structure | Type of Cancer | Status | References |
|---|---|---|---|---|
| Cisplatin |  | solid neoplasms, including ovarian, testicular, bladder, colorectal, lung and head and neck cancers | FDA-approved | [134] |
| Carboplatin |  | Retinoblastoma Lung cancer | FDA-approved | [134] |
| Oxaliplatin |  | Medulloblastoma Non-small cell lung cancer | FDA-approved | [134] |
| Nedaplatin |  | solid neoplasms, including ovarian, testicular | FDA-approved | [134] |
| Lobaplatin |  | metastatic breast cancer, chronic myelogenous leukemia and SCLC | Regional approval CHINA | [135] |
| Heptaplatin |  | gastric cancer | Regional approval (Republic of Korea) | [135] |
| Compound No. | n | HDAC Inhibiting | Cytotoxicity (µM) | ||
|---|---|---|---|---|---|
| HepG2 | MCF-2 | HC-60 | |||
| 70a | (-CH2)6 | 2.50 | 2.50 | 2.62 | 1.28 |
| 70b | (-CH2)2 | 14.17 | 14.17 | 10.98 | 6.41 |
| 70c | (-CH2)3 | >125 | 84.80 | 71.64 | 21.84 |
| SAHA | - | 0.35 | 0.31 | 1.90 | 0.18 |
| Compounds No. | R | In Vitro Antiproliferative Activity (IC50 µM) | ||||
|---|---|---|---|---|---|---|
| HCT116 | SUMM-7721 | HepG2 | Mcf-7 | Huh-7 | ||
| 71a | 3,4,5 (MeO)3-Ph | 0.82 | 1.06 | 0.65 | 2.25 | 1.52 |
| 71b | 3-MeO-Ph | 0.89 | 1.22 | 1.02 | 2.18 | 1.52 |
| 71c | 4-Me-Ph | 0.78 | 0.84 | 0.53 | 1.56 | 0.96 |
| 71d | 4-NO2-Ph | 1.41 | 2.61 | 1.51 | 4.03 | 2.78 |
| SAHA | - | 5.53 | 5.61 | 6.26 | 4.48 | 4.95 |
| Compound No. | R | R1 | Antiproliferative Activity (IC50 µM) | ||
|---|---|---|---|---|---|
| HeptG2 | MCF-7 | HCF-116 | |||
| 72a | 4-OH3 | OH | 0.620.04 | 2.050.48 | 2.920.52 |
| 72b | H | OH | 3.550.77 | 2.210.44 | 2.180.33 |
| 72c | 4-F | OH | 8.721.65 | 3.200.82 | 6.741.59 |
| 72d | 4-OCH3 |  | 7.172.01 | 12.992.99 | 3.020.81 |
| SAHA | - | - | 3.330.74 | 2.180.35 | 1.230.08 |
| Compound No. | R | n | Antiproliferative Activity (IC50 µM) | |
|---|---|---|---|---|
| A549 | BT-474 | |||
| 73a |  | 1 | 0.51 | 3.63 |
| 73b |  | 2 | 0.63 | 3.88 |
| 73c |  | 2 | 3.68 | 2.24 |
| 73d |  | 1 | 8.46 | 14.65 |
| Lapatinib | - | - | 1.74 | 0.10 |
| SAHA | - | - | 2.57 | 2.67 |
| Compound No. | R | Cytotoxicity Cell Line (IC50 µM) | ||
|---|---|---|---|---|
| SE620 | AsPc-1 | PC3 | ||
| 74a | 5-Br | 3.05 | 6.83 | 7.30 |
| 74b | 5-F | 3.30 | 7.28 | 11.50 |
| 74c | 5-Cl | 3.42 | 22.35 | 17.44 |
| 74d | 5-CH3 | 3.44 | 7.47 | 12.25 |
| SAHA | - | 1.44 | 7.04 | 5.30 |
| Company Name | Compound Name | Drug Structure | Drug Target | Type of Cancer | Status | References |
|---|---|---|---|---|---|---|
| Merck and Co., Inc | Vorinostat (SAHA) |  | histone deacetylase inhibitor | cutaneous T cell lymphoma | Approved | [141] |
| Novartis | Panobinostat |  | non-selective histone deacetylase inhibitor | multiple myeloma | Approved | [142] |
| 4SC AG | Resminostat |  | histone deacetylase inhibitor | hepatocellular carcinoma | Approved | [143] |
| Helsinn and MEI Pharma | Pracinostat |  | histone deacetylase inhibitor | Acute Myeloid Leukemia | Clinical Trial | [144] |
| Italfarmaco Group’s | Givinostat |  | histone deacetylase inhibitor | chronic lymphocytic leukemia | Clinical Trial | [144] |
| Xynomic Pharmaceuticals, Inc. | Abexinostat |  | histone deacetylase inhibitor | Hodgkin’s lymphoma | Clinical Trial | [144] |
| Compounds No. | R1 | R2 | X | A549 (µM) |
|---|---|---|---|---|
| 75a | OCH3 | H | H | 5 |
| 75b | H | Cl | H | 33 |
| 75c | Cl | H | N | 10 |
| 75d | NO2 | H | H | 14 |
| 5-Fluorouracil | <5 |
| Compound No. | R | Antitumor Activity (µM) | ||
|---|---|---|---|---|
| A549 | HepG2 | MDA-MB-45 | ||
| 76a |  | 4.47 | 26.70 | 4.50 |
| 76b |  | 5.31 | 10.89 | 5.39 |
| 76c |  | 4.44 | 20.82 | 4.89 |
| 76d |  | 7.93 | 8.43 | 6.98 |
| 5-FU | - | 16.8 | 17.6 | 2.80 |
| Cisplatin | - | 0.87 | 0.74 | 1.14 |
| Compound No. | R | Anticancer Activity IC50 (µM) | ||
|---|---|---|---|---|
| TK-10 | UACC-62 | MCF-7 | ||
| 77a |  | 2.4 | 3.0 | 2.5 |
| 77b |  | 2.3 | 6.0 | 2.8 |
| 77c |  | 2.5 | 3.0 | 2.8 |
| 77d |  | 2.5 | 3.5 | 3.0 |
| PTD | - | 6.4 | 15.0 | 5.8 |
| Compound No. | Name | Anticancer Activity IC50 (µM) | |||||
|---|---|---|---|---|---|---|---|
| BIU-87 | SGC-7901 | EC-9706 | ECa-109 | MCF-7 | Jurkat | ||
| 78a | 4-Methyl-7-hydroxy-8-nitrocoumarin-ferrocene butyrate conjugate | 1.09 | 1.061 | 25.89 | 36.38 | 12.10 | 53.01 |
| 78b | 4-Methyl-7-hydroxycoumarin-ferrocene butyrate conjugate | 4.48 | 16.61 | 41.26 | 27.97 | 15.34 | 54.90 |
| 78c | 7-Hydroxycoumarin-ferrocene butyrate conjugate | 5.24 | 7.46 | 82.83 | 42.47 | 28.10 | 55.77 |
| 78d | 7-Hydroxy-8-nitrocoumarin-ferrocene butyrate conjugate | 15.27 | 19.78 | N | 40.00 | 23.91 | 38.81 |
| Adriamycin | - | 6.09 | 5.44 | 8.56 | 6.52 | 7.90 | 4.50 |
| Compound No. | E | R | Anticancer Activity IC50 (µM) | ||||
|---|---|---|---|---|---|---|---|
| A549 | MDA-MB-231 | MCF-7 | HeLa | HEK-293T | |||
| 79a | Se (Selenium) |  | 2.9 | 3.35 | 5.85 | 11.6 | - |
| 79b | Se (Selenium) |  | 3.71 | >200 | >200 | 18.3 | -- |
| 79c | S (Sulphur) |  | 11.6 | >200 | >200 | 14.8 | - |
| 79d | S (Sulphur) |  | >200 | 9.7 | >200 | >200 | - |
| Doxorubicin | 0.39 | 0.47 | 0.98 | 0.89 | - | ||
| Compound No. | R | R1 | Anticancer Activity IC50 (µM) | |||
|---|---|---|---|---|---|---|
| A549 | MCF-9 | SKOV3 | H460 | |||
| 80a | -CH3 |  | 23.9 | 36.2 | 12.8 | 21.75 |
| 80b | -H |  | 21.3 | 151.56 | 19 | 25.4 |
| 80c | -OCH3 |  | 20.6 | >100 | 14.04 | 38.14 |
| 80d | -F |  | 40.45 | 25.0 | >100 | 44.3 |
| Compound No. | R1 | R2 | R3 | R4 | R5 | Anticancer Activity IC50 (µM) | |
|---|---|---|---|---|---|---|---|
| A549 | AGS | ||||||
| 81a | H | OMe | OH | H | H | 1.29 | 10.16 |
| 81b | H | H | Me | H | H | 6.25 | 11.94 |
| 81c | H | H | Cl | H | H | 12.56 | 22.31 |
| Curcumin | - | - | - | - | - | 25.33 | 20.76 |
| Compound No. | R | Anticancer Activity IC50 (µM) | |||
|---|---|---|---|---|---|
| HeLa | MCF-7 | K562 | HEK293K | ||
| 82a | o-Cl | 45.56 | 34.99 | 25.55 | >1000 |
| 82b | H | 42.56 | 52.88 | 36.99 | NT |
| 82c | o, p-diNO2 | 56.45 | 43.60 | 42.35 | NT |
| Paclitaxel | - | 0.0061 | 0.0053 | 0.0049 | NT |
| Compound. No. | R1 | R2 | R3 | R4 | Anticancer Activity IC50 (µM) | |
|---|---|---|---|---|---|---|
| HT29 | HCT116 | |||||
| 83a | OCH3 | OCH3 | H | CH2CH2OH | 7.10.4 | 6.21.2 |
| 83b | OCH3 | OCH3 | H | CH2(CH2)2OH | 12.81.9 | 9.96.9 |
| 83c | OCH3 | OCH3 | H | CH2CH3 | 38.525.4 | 46.423.6 |
| Compound No. | X1 | X2 | X3 | X4 | X5 | R | Anticancer Activity IC50 (µM) | |||
|---|---|---|---|---|---|---|---|---|---|---|
| THP-1 | CoLo-205 | HCT-116 | PC-3 | |||||||
| 84a | H | OCH3 | OCH3 | OCH3 | H | H | 2.87 | 4.15 | 1.12 | 5.67 |
| 84b | H | OCH3 | OCH3 | H | H | H | 4.31 | 5.78 | 2.92 | 6.44 |
| 84c | H | H | OCH3 | H | H | H | 4.96 | 6.72 | 3.45 | 8.95 |
| Compound No. | R1 | R2 | Anticancer Activity IC50 (µM) | |||
|---|---|---|---|---|---|---|
| EC-109 | MCF-7 | MGC-803 | B16-F10 | |||
| 85a | m-CF3 | p-Br | 2.96 | 3.11 | 3.60 | 4.55 |
| 85b | m-CF3 | 7-CH3 | 32.29 | 6.12 | 7.58 | 9.58 |
| 85c | m-CF3 | m,p,m-Tri-OCH3 | >64 | 8.14 | 8.21 | 24.43 |
| 85d | m-CF3 | p-CH(CH3)2 | 24.22 | 8.62 | >64 | 1.70 |
| 5-FU | - | - | 11.61 | 9.12 | 8.43 | 1.43 |
| Compound No. | R | Anticancer Activity IC50 (µM) | ||||
|---|---|---|---|---|---|---|
| A549 | Hela | MCF-7 | DU-I45 | HepG2 | ||
| 86a |  | 06.16 | 07.76 | 09.59 | 08.83 | 09.52 |
| 86b |  | 08.12 | 12.83 | 14.66 | 11.14 | 14.08 |
| 86c |  | 11.22 | 22.96 | 16.59 | 16.37 | 34.87 |
| Doxorubicin | - | 2.81 | 2.57 | 1.13 | 1.41 | 3.01 |
| Compound No. | Ar1 | Ar2 | Cytotoxic Activity IC50 (µm) | |
|---|---|---|---|---|
| MCF-7 | T47D | |||
| 87a | Ph |  | >100 | 27.7 ± 0.1 |
| 87b | Ph |  | 85.7 ± 4.5 | >100 |
| Etoposide | - | - | 7.5 ± 0.32 | 7.9 ± 0.45 |
| Compound No. | R | Anticancer Activity IC50 (µM) | |||
|---|---|---|---|---|---|
| MGC-803 | MCF-7 | PC-3 | EC-109 | ||
| 88a | o-F | 0.73 | 5.67 | 11.61 | 2.44 |
| 88b | o-Cl | 0.49 | 6.09 | 12.45 | 11.93 |
| 5-FU | - | 7.01 | 7.54 | 27.07 | 3.3 |
| Compound No. | R | Anticancer Activity IC50 (µM) | |||
|---|---|---|---|---|---|
| MCF-7 | A549 | A375 | MCF-10A | ||
| 89a | 5-(CF3)-1,2,4-thiadiazole | 2.12 | 5.48 | 4.7 | 29.33 |
| 89b | 4,5-CF3C6H4 | 2.52 | 4.97 | 4.67 | 2.5 |
| 89c | 2,4-FC6H3 | 3.0 | 4.45 | 6.02 | 2.2 |
| Doxorubicin | - | 0.12 | 3.13 | 7.2 | 24.0 |
| Paclitaxel | - | 2.58 | 4.9 | 8.0 | 38.0 |
| Compound No. | R1 | R2 | R3 | Anticancer Activity IC50 (µM) | ||
|---|---|---|---|---|---|---|
| SW1990 | AsPC1 | MRCS | ||||
| 90a | F | H | H | 30.9 | 32.8 | 80.0 |
| 90b | o-Me | H | H | 57.6 | 62.4 | >100 |
| Gemicitabine | - | - | - | 35.09 | 39.27 | 54.17 |
| Compound No. | R1 | R2 | Anticancer Activity IC50 (µM) | |||
|---|---|---|---|---|---|---|
| MCF-7 | MGC-803 | EC-9706 | SMMC-7721 | |||
| 91a | H | 4-CH3-C6H5-NH- | 1.43 | 1.33 | 3.33 | 20.50 |
| 91b | H | 4-CH3O-C6H5-NH- | 2.90 | 2.03 | 5.83 | 10.55 |
| 91c | H | 4-F-C6H5-NH- | 4.24 | 2.30 | 8.55 | 22.58 |
| 5-FU | - | - | 7.12 | 3.45 | 8.07 | 15.08 |
| Compound No. | NR1R2 | R3 | Anticancer Activity IC50 (µM) | ||||
|---|---|---|---|---|---|---|---|
| PC-3 | DU-145 | MDA MB-231 | A549 | MCF10A | |||
| 92a | R1 = H, R2 = 4-phenylthiazol | Methyl | 39.87 | 31.41 | 29.18 | 11.46 | >100 |
| 92b | 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline | Ethyl | 33.54 | >50 | >50 | 29.40 | ---- |
| 92c | 6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline | Methyl | 41.10 | 37.32 | 36.03 | 13.35 | >100 |
| 5-FU | - | - | 45.32 | 40.58 | 35.98 | 30.47 | - |
| Compound No. | R | Proliferative Inhibition IC50 (µM) | |
|---|---|---|---|
| Hep-G2 | MCF-7 | ||
| 93a | 4-F | 8.7 | 12.6 |
| 93b | 4-Cl | 15.8 | 3.6 |
| 93c | 4-Br | 24.7 | 8.3 |
| Galvatinib | - | 65.5 | 49.6 |
| Compound No. | R | Cytotoxicity of Compound IC50 (µM) | |||
|---|---|---|---|---|---|
| MCF-7 | A549 | Colo-205 | A2780 | ||
| 94a | 5,6-dicyano | 0.220 | 1.550 | 1.680 | 1.160 |
| 94b | 5,6-dimethoxy | 0.0920 | 0.720 | 0.340 | 1.230 |
| 94c | 5,6-dimethyl | 0.810 | 1.900 | 0.410 | 1.800 |
| Etoposide | - | 2.110 | 3.080 | 0.130 | 1.310 |
| Company Name | Compound Name | Drug Structure | Drug Target | Type of Cancer | Status | Reference |
|---|---|---|---|---|---|---|
| Eli Lilly and Company | Abemaciclib |  | cyclin dependent kinase-4 (CDK4) and CDK6 inhibitor | negative metastatic breast cancer | Approved | [165] |
| AbbVie | Veliparib |  | PARP inhibitor | ovarian cancer | Clinical Trial | [166] |
| Allarity Therapeutics | Dovitinib |  | pan tyrosine kinase inhibitor | prostate cancer | Clinical Trial | [167] |
| Novartis | Nazartinib |  | EGFR kinase inhibitor | non-small cell lung cancer | Clinical Trial | [168] |
| Compound No. | R | ZR-75(µM) | HT-29 (µM) | A-549 (µM) | Average IC50(µM) |
|---|---|---|---|---|---|
| 95a | H | 13.25 | 6.69 | 7.19 | 9.04 |
| 95b | F | 5.9 | 5.31 | 5.39 | 5.53 |
| 95c | Cl | 7.77 | 6.23 | 7.02 | 7.01 |
| Sunitinib | 8.31 | 10.14 | 5.87 | 8.11 |
| Compound No. | Ar | R | EC50 Values (µM) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| IGR39 | A549 | U87 | Fibroblasts | MCF-7 | BT474 | H1299 | BxPC-3 | SKOV-3 | |||
| 96a |  | 5-Cl | 0.33 | 0.34 | 0.38 | - | 0.07 | 0.09 | 0.0.1 | 0.06 | 0.06 |
| 96b |  | 5-OCH3 | 0.50 | 0.73 | 0.67 | 0.27 | 0.27 | 0.24 | 0.15 | 0.10 | - |
| 96c |  | 5-CH3 | 0.14 | 0.18 | 0.23 | 0.15 | 0.31 | - | - | 0.10 | - |
| 96d |  | 5-Cl | 2.97 | - | 5.76 | - | - | - | - | - | - |
| Sunitinib | - | - | - | - | 0.30 | 0.96 | 0.90 | 1.54 | 2.5 | 1.36 | |
| Compound No. | X | Ar | IC50 (µM) | ||
|---|---|---|---|---|---|
| A549 | HepG2 | MCF-7 | |||
| 97a | H |  | >200 | >200 | >200 |
| 97b | Cl |  | >200 | >200 | >200 |
| 97c | Br |  | >200 | >200 | >200 |
| 98 | - | - | 19.3 | 2.5 | 11.6 |
| 99a | H | - | 16.8 | >200 | 14.7 |
| 99b | F | - | 19.7 | 11.5 | 10.4 |
| 99c | Cl | - | 10.8 | 8.7 | 6.3 |
| Doxorubicin | - | - | 7.6 | 6.9 | 6.1 |
| Compound No. | R | n | IC50 (µM) | ||
|---|---|---|---|---|---|
| THP-1 | COLO-205 | HCT-116 | |||
| 100a | H | 1 | 0.73 | 3.45 | 3.04 |
| 100b | F | 1 | 1.99 | 6.67 | 5.41 |
| 100c | Cl | 1 | 5.47 | 8.87 | 5.77 |
| 100d | Br | 1 | 6.43 | 10.53 | 8.09 |
| Compound No. | X | R | IC50 (µM) | Average IC50 (µM) | ||
|---|---|---|---|---|---|---|
| ZR-75 | HT-29 | A-549 | ||||
| 101a | H | H | 22.73 | 19.74 | 12.98 | 18.48 |
| 101b | Br | H | 21.92 | 21.93 | 16.01 | 19.95 |
| 101c | OCH3 | CH3 | 1.48 | 5.73 | 1.93 | 3.05 |
| 101d | Cl |  | 0.74 | 2.02 | 0.76 | 1.17 |
| Sunitinib | - | - | 8.31 | 10.14 | 5.87 | 8.11 |
| Compound No. | R1 | R2 | R3 | R4 | R5 | R6 | IC50 (µM) | |
|---|---|---|---|---|---|---|---|---|
| HeLa | MCF-7 | |||||||
| 102a | I | H | Cl | H | OC2H | H | 9.24 | 6.57 |
| 102b | Br | Br | Cl | H | OC2H | H | 8.34 | 5.88 |
| 102c | Br | H | H | OCH3 | H |  | 16.68 | 11.32 |
| 102d | Br | Br | H | OCH3 | H |  | 10.44 | 9.46 |
| Vinblastine | - | - | - | - | - | 7.14 | 4.02 | |
| Compound No. | R | R1 | IC50 Values | |
|---|---|---|---|---|
| MCF | MDA-MB-231 | |||
| 103a | H | - | 2.02 | 6.50 |
| 103b | F | - | 16.83 | 42.64 |
| 103c | Br | - | 9.39 | 8.62 |
| 104a | H | CH3 | 3.01 | 2.60 |
| 104b | H |  | 9.80 | 18.06 |
| 104c | H |  | 1.27 | 13.73 |
| Staurosporine | - | - | 3.81 | 4.29 |
| Compound No. | R | IC50 Values (µM) | ||||
|---|---|---|---|---|---|---|
| THP-1 | U-937 | HL-60 | Jurkat | A-549 | ||
| 105a | F | 0.49 | 3.44 | 4.13 | 6.58 | nd |
| 105b | OMe | 2.14 | 6.03 | 6.39 | 11.27 | nd |
| 105c | H | 3.09 | 5.41 | 7.27 | 9.43 | nd |
| Etoposide | 2.16 | 17.94 | 1.83 | 5.35 | 17.94 | |
| Camptothecin | 0.07 | 1.98 | 0.60 | 0.026 | 0.008 | |
| Compound No. | X | n | IC50 (µM) | |||
|---|---|---|---|---|---|---|
| A431 | A549 | Colo-205 | PC-3 | |||
| 106a | CO | 1 | 3.04 | 13.85 | 8.65 | 7.67 |
| 106b | CO | 2 | 1.34 | 9.80 | 3.31 | 3.23 |
| 106c | CO | 3 | 3.71 | 21.94 | 9.76 | 15.45 |
| 106d | SO2 | 4 | 1.72 | 1.05 | 1.21 | 1.52 |
| DC-81 | - | - | 1.65 | 1.11 | 0.86 | 1.19 |
| Dox | - | - | 0.03 | 1.02 | 1.69 | 2.51 |
| Compound No. | R | m/n | IC50 Value (nM) |
|---|---|---|---|
| HDAC6 Enzyme | |||
| 107a | H | 0 | 38.80 |
| 107b | H | 1 | 147 |
| 107c | H | 3 | >300 |
| 108a | Cl | 5 | 23.77 |
| 108b | Cl | 6 | 27.80 |
| 108c | Cl | 7 | 137.77 |
| Tubacin | - | - | 5.36 |
| Tubastatin A | - | - | 22.36 |
| Compound No. | R | IC50 Value |
|---|---|---|
| A375 | ||
| 109a | H | 2.2 |
| 109b | 2-OCH3 | 4.2 |
| 109c | 3-OCH3 | 3.6 |
| 110a | H | 4.5 |
| 110b | 2-OCH3 | 5.2 |
| 110c | 3-OCH3 | 5.3 |
| Compound No. | Ar | IC50 (µM) | |
|---|---|---|---|
| MCF-7 | HepG-2 | ||
| 111a |  | 12 | 7.6 |
| 111b |  | 13.8 | 7.14 |
| 111c |  | 15.4 | 11.6 |
| 111d |  | 19.2 | 14.7 |
| Cisplatin | - | 5.71 | 3.67 |
| Doxorubicin | - | 0.35 | 0.36 |
| Compound No. | R | IC50 (µM) |
|---|---|---|
| KB | ||
| 112a | 4-OCH3 | 15 |
| 112b | 4-F | 15 |
| 112c | 4-Br | 15 |
| 112d | 4-Cl | 31 |
| Compound No. | R1 | R2 | R3 | R4 | R5 | IC50 (µM) | ||
|---|---|---|---|---|---|---|---|---|
| MCF-7 | HCT-116 | HepG-2 | ||||||
| 113a | H | H | OCH3 | H | H | 8.23 | 9.57 | 34.28 |
| 113b | H | H | H | H | H | 58.92 | 57.49 | 105.21 |
| 113c | OCH3 | H | H | H | H | 24.04 | 25.60 | 30.72 |
| 113d | OH | H | H | H | H | 22.17 | 19.76 | 17.32 |
| Doxorubicin | - | - | - | - | - | 0.27 | 1.55 | 0.22 |
| Compound No. | R | 24 h | 48 h | ||
|---|---|---|---|---|---|
| IC50 (µM) SW480 Cells | IC50 (µM) CHO-K1 Cells | IC50 (µM) SW480 Cells | IC50 (µM) CHOK1 Cells | ||
| 114a |  | 1.26 | 6.40 | 0.24 | 0.16 |
| 114b |  | 29.49 | 20.48 | 4.14 | 4.95 |
| 114c |  | 30.46 | 10.12 | 2.43 | 1.13 |
| 114d |  | 49.82 | >100 | 1.77 | 10.61 |
| 5-fluorouracil | - | - | 543.5 | 174.3 | 173.2 |
| Compound No. | Ar | IC50 Value (mM) | ||
|---|---|---|---|---|
| BPH-1 | PC12 | MCF-7 | ||
| 115a |  | 48.5 | 42.1 | 72.4 |
| 115b |  | 54.8 | 48.5 | 54.8 |
| 115c |  | 64.2 | 73.1 | 80.9 |
| 115d |  | 76.9 | 81.1 | 77.3 |
| 115e |  | 10.4 | 16.7 | 9.1 |
| Doxorubicin | 14.1 | 22.5 | 9.2 | |
| Compound No. | Substitution | % Growth Inhibition | Maximal Inhibition | |
|---|---|---|---|---|
| 116a | NR1R2 = Ethylenediamine |  | 20.5 | Colon Cancer |
| 33.98 | Breast Cancer | |||
| 116b | NR1R2 = Ethanolamine |  | 80.51 | Leukemia |
| 116c | NR1R2 = Morpholine |  | 62.12 | Breast Cancer |
| 116d | NR1R2 = Methylpiperazine |  | 56.39 | Leukemia |
| 116e | NR1R2 = 2-Aminoethylmorpholine |  | 23.26 | Small cell lung cancer |
| 22.91 | CNS Cancer | |||
| 5-FU | 47.9 | Leukemia | ||
| Compound No. | Substitution | IC50(mM) | IC50 normoxia/IC50 hypoxia | |
|---|---|---|---|---|
| Hypoxia | Normoxic | |||
| 117a |  | 23.47 | 24.41 | 1.04 |
| 117b |  | 75.21 | 73.77 | 0.98 |
| 117c |  | 6.72 | 6.78 | 1.01 |
| 117d |  | 0.03 | 1.34 | 46.31 |
| 117e |  | 73.82 | 91.61 | 1.24 |
| Dox | 0.60 | 1.07 | 1.79 | |
| Compound No. | Substitution | IC50 (µm) | ||||
|---|---|---|---|---|---|---|
| R1 | R2 | R3 | HEPG2 | Leukemia K562 | WI-38 | |
| 118a | H | OCH3 | H | 0.65 | 1.09 | 292.7 |
| 118b | OCH3 | OCH3 | H | 0.82 | O.93 | 33.09 |
| 118c | OCH3 | OCH3 | OCH3 | 2.02 | 1.36 | 9.55 |
| 119a | H | OCH3 | H | 0.77 | 3.96 | 48.4 |
| 119b | OCH3 | OCH3 | H | 0.93 | 8.71 | 3.3 |
| 119c | OCH3 | OCH3 | OCH3 | 1.57 | 0.49 | 11.1 |
| Cisplatin | 2.56 | 2.02 | NA | |||
| Compound No. | Substitution (R) | n | MCF-7(GI50-µM) |
|---|---|---|---|
| 120a | H | 1 | 10.99 |
| 120b | F | 1 | 1.55 |
| 120c | Cl | 1 | 2.57 |
| 120d | Br | 1 | 3.34 |
| 120e | I | 1 | 3.96 |
| 5-FU | 7.28 |
| Compound No. | Substitution® | IC50 (µM) |
|---|---|---|
| 121a |  | 32.32 |
| 121b |  | 4.85 |
| 121c |  | 5.16 |
| 121d |  | 5.95 |
| 121e |  | 4.72 |
| Dox | 10.21 |
| Compound No. | Substitution (R1) | IC50 (µM) against Bel-7502 |
|---|---|---|
| 122a |  | 8.35 |
| 122b |  | 0.50 |
| 122c |  | 1.43 |
| 122d |  | 7.91 |
| Chlorambucil | 49.31 |
| Compound No. | Substitution (Ar) | IC50 (µg/mL) | Log P |
|---|---|---|---|
| 123a |  | 3.63 | 6.15 |
| 123b |  | 3.95 | 5.74 |
| 123c |  | 3.29 | 6.80 |
| 123d |  | 4.26 | 6.35 |
| 123e |  | 2.17 | 6.53 |
| Cisplatin | 0.31 | - |
| Compound No. | Substitution (R) | IC50 (µg/mL) | ||
|---|---|---|---|---|
| Breast Cancer (T47D) | Prostate Cancer (LNCaP) | Colorectal Cancer (SW707) | ||
| 124a | CH3 | 27.86 | 15.78 | 32.98 |
| 124b | C6H5CH2 | 1.40 | 0.99 | 3.45 |
| 124c | CF3CH2 | 96.84 | 18.27 | 96.84 |
| 124d | C6H5 | 2.60 | 1.47 | 2.93 |
| Compound No. | Substitution (n) | GI50 (µM) | |
|---|---|---|---|
| K-562 | HL-60 | ||
| 125a | 0 | 3.90 | 7.01 |
| 125b | 1 | 13.6 | 36.0 |
| 126a | 0 | 12.4 | 32.2 |
| 126b | 1 | 40.1 | 61.2 |
| Compound No. | Substitution | IC50(µM) | ||||
|---|---|---|---|---|---|---|
| Ar | Ar1 | HCT-116 | MCF-7 | HepG2 | A549 | |
| 127a | C6H5 | C6H5 | 17.4 | 10.6 | 6.1 | 23.7 |
| 127b | 4-CH3-C6H4 | C6H5 | 19.6 | 14.5 | 7.9 | 14.1 |
| 127c | H | 4-CH3-O-C6H4 | 31.9 | 22.2 | 35.8 | 43.4 |
| 127d | C6H5 | 4-CH3-O-C6H4 | 25.3 | 17.4 | 27.2 | 58.7 |
| 127e | 4-CH3-C6H4 | 4-CH3-O-C6H4 | 37.4 | 16.2 | 25.8 | 40.8 |
| Dox | - | - | 40.0 | 64.8 | 24.7 | 58.1 |
| Compound No. | Substitution (R) | Cell Growth Promotion Percentage (At 10−5 M Concentration) | ||
|---|---|---|---|---|
| CCRM-CEM | A549/ATCC | HCT-116 | ||
| 128a | 4-Cl-C6H4- | - | - | - |
| 128b | Cyclohexyl | - | - | - |
| 128c | 2-pyroryl | 90.65 | 62.56 | 27.86 |
| 128d | 2-furyl | 78.58 | 93.57 | 74.8 |
| Compound No. | IC50 (µM) | ||
|---|---|---|---|
| HePG2 | HCT-116 | MCF-7 | |
| 129 | 65.14 | 45.95 | 79.88 |
| 130 | 4.99 | 6.83 | 4.63 |
| 131 | 16.18 | 11.78 | 18.72 |
| 132 | 6.85 | 3.46 | 9.07 |
| 133 | 73.11 | 88.60 | 69.16 |
| Compound No. | Substitution | IC50 (µM) | |||||
|---|---|---|---|---|---|---|---|
| R | R1 | MCF-7 | A549 | SiHa | COLO205 | HepG2 | |
| 134a | 4-Cl | 4-OCH3 | 31.38 | 15.30 | 4.54 | 12.95 | >50 |
| 134b | H | 4-OCH3 | 27.37 | 4.94 | 4.58 | 4.86 | 2.09 |
| 134c | H | 4-F | 29.31 | 10.27 | 5.67 | 6.69 | 8.56 |
| 134d | H | 4-Cl | 30.05 | 11.50 | 6.40 | 7.16 | >50 |
| 134e | H | H | >50 | 26.25 | >50 | 31.27 | >50 |
| 5-FU | 2.08 | 4.35 | 5.78 | 4.00 | 19.01 | ||
| Compound No. | Substitution | IC50 (µM) | ||||
|---|---|---|---|---|---|---|
| R | X | HCT-116 | SW-620 | MCF-7 | HT-29 | |
| 135a | 4-CH3 |  | 1.8 | 3.6 | 2.0 | 4.4 |
| 135b | 4-CH3 |  | 10 | >10 | 10.9 | 8.5 |
| 135c | H |  | 1.9 | 5.0 | 6.4 | 4.6 |
| Paclitaxel | 0.004 | 0.006 | 0.006 | 0.005 | ||
| Compound No. | Substitution | IC50 (µM) | |
|---|---|---|---|
| R1 | R2 | ||
| 136a | H | CN | 4.8 |
| 136b | OCH3 | CN | 5.4 |
| 136c | H | COOMe | 1.1 |
| 136d | CH3 | COOEt | 0.09 |
| 136e | OCH3 | COOEt | 0.2 |
| Erlotinib | 0.03 | ||
| Compounds No. | Substitution (X) | IC50 (µM) | ||
|---|---|---|---|---|
| HepG2 | A549 | MCF-7 | ||
| 138a | H | NA | 16.8 | 14.7 |
| 138b | F | 11.5 | 19.7 | 10.4 |
| 138c | Cl | 8.7 | 10.8 | 6.3 |
| 138d | Br | 59.1 | 85 | 14.9 |
| Dox | 6.9 | 7.6 | 6.1 | |




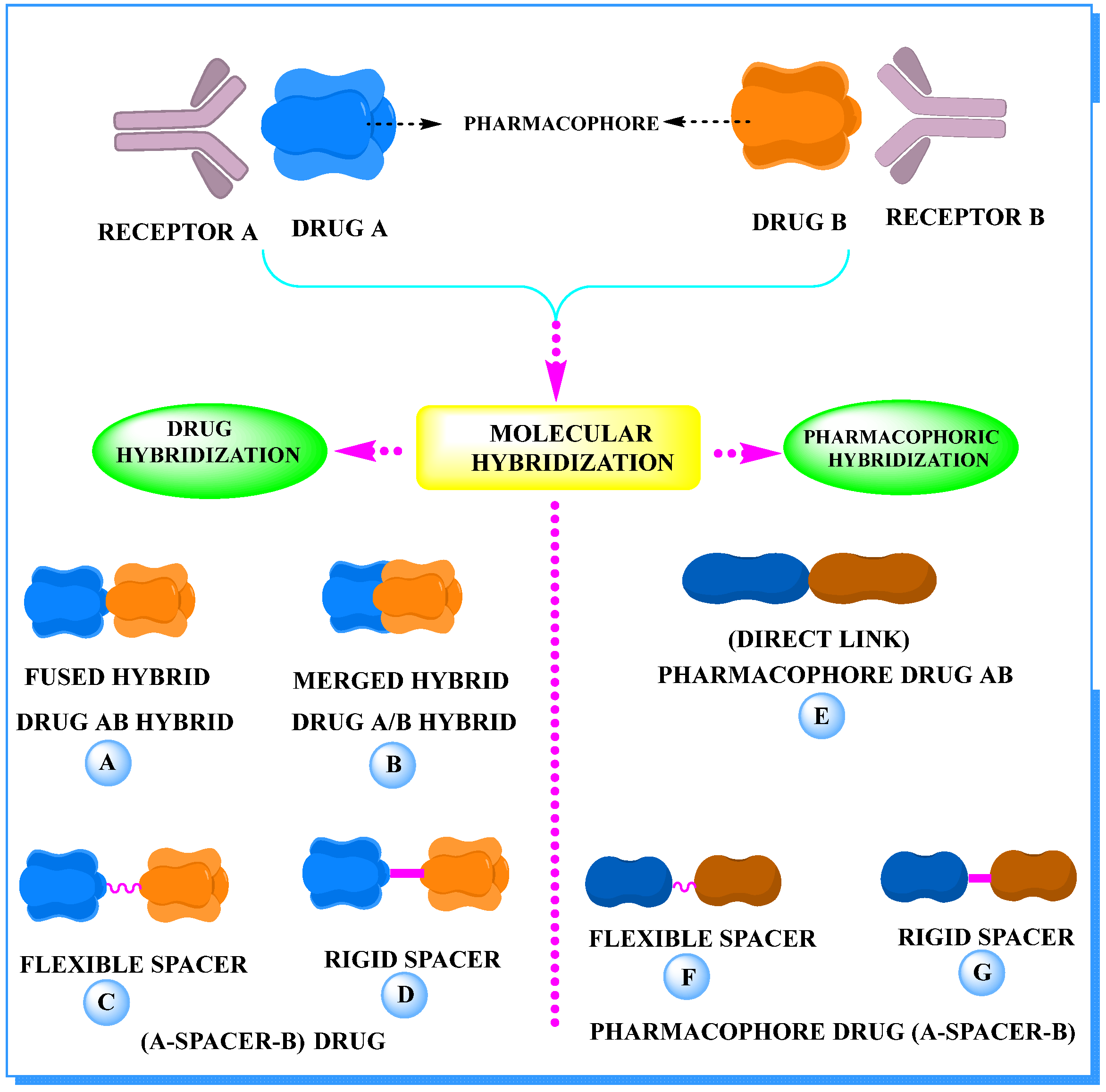{kind=link}
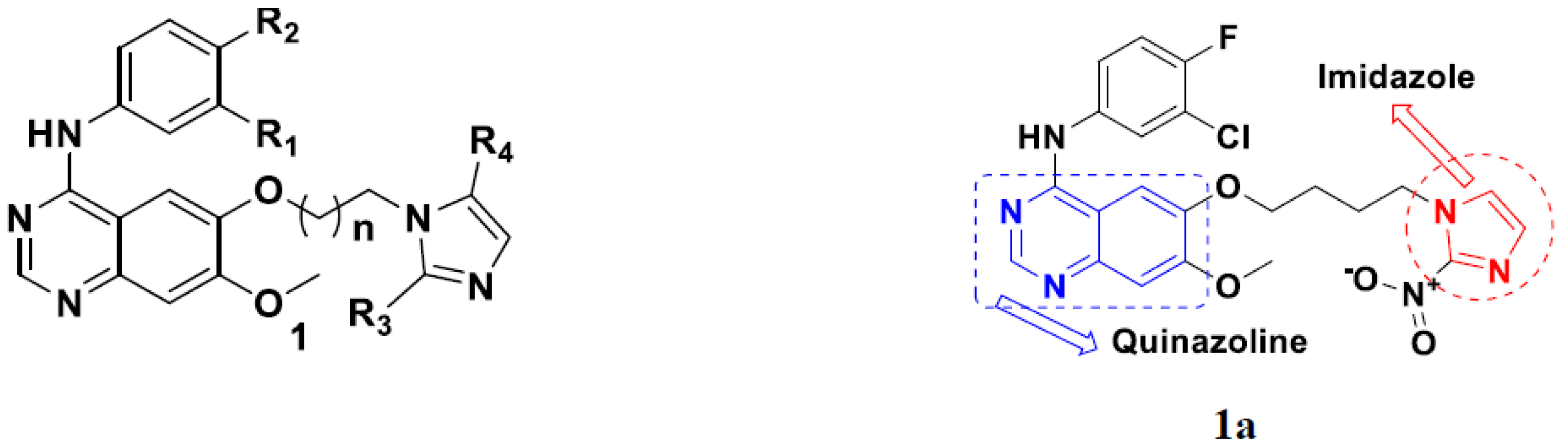{kind=link}
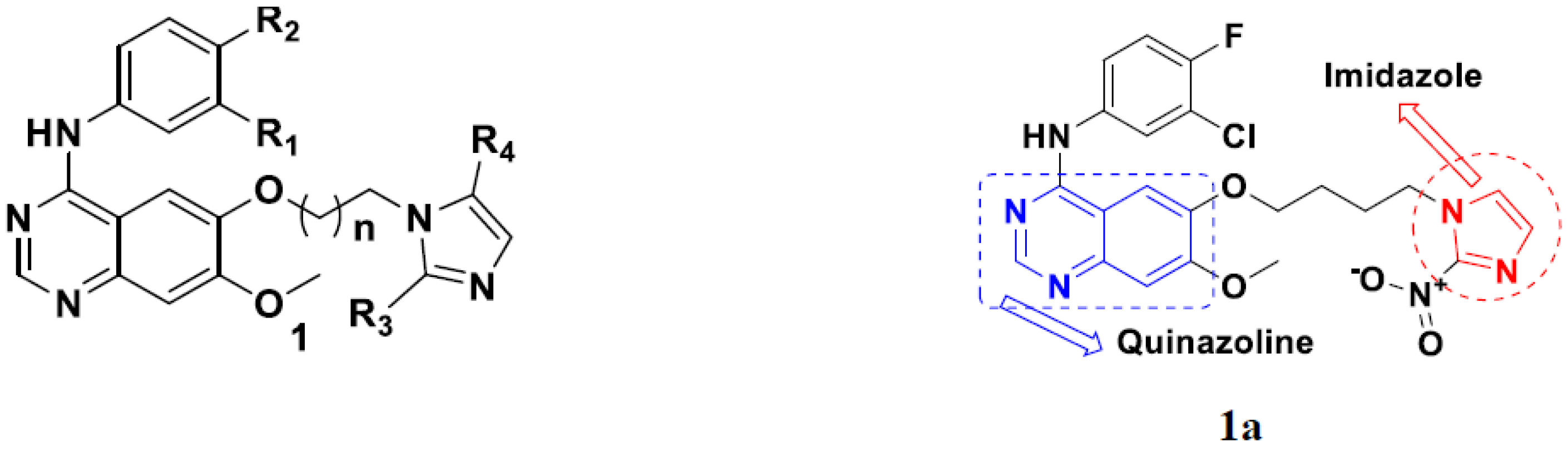{kind=link}
{kind=link}
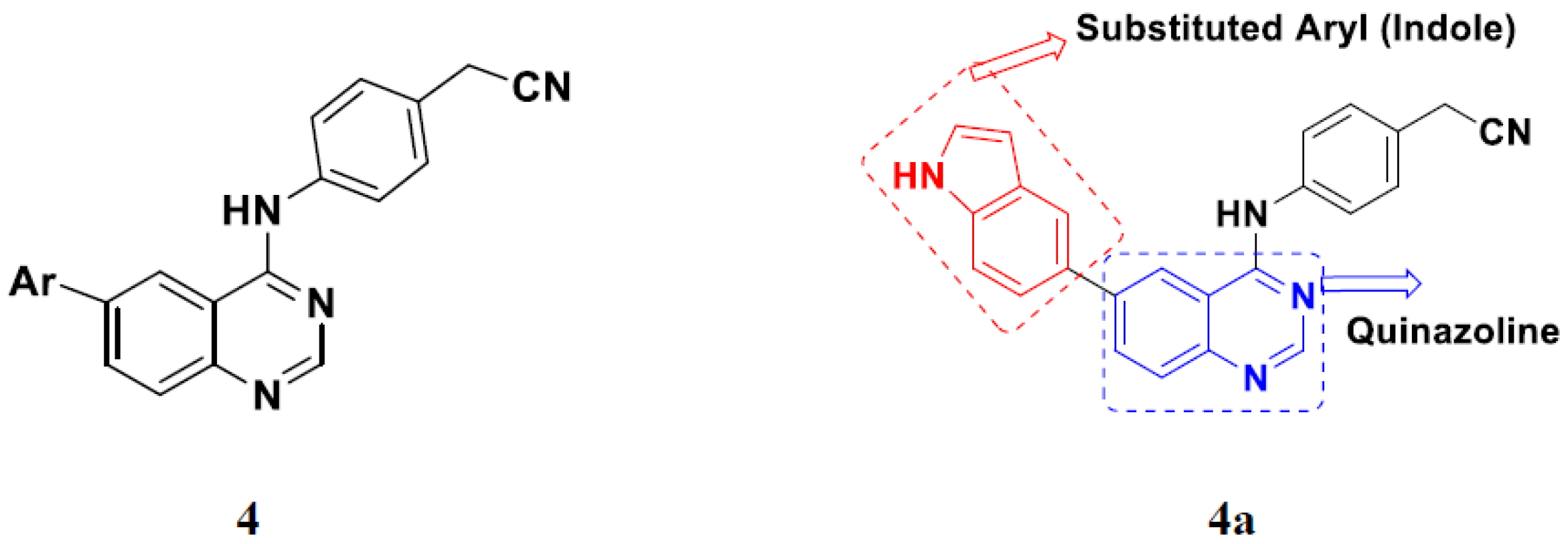{kind=link}
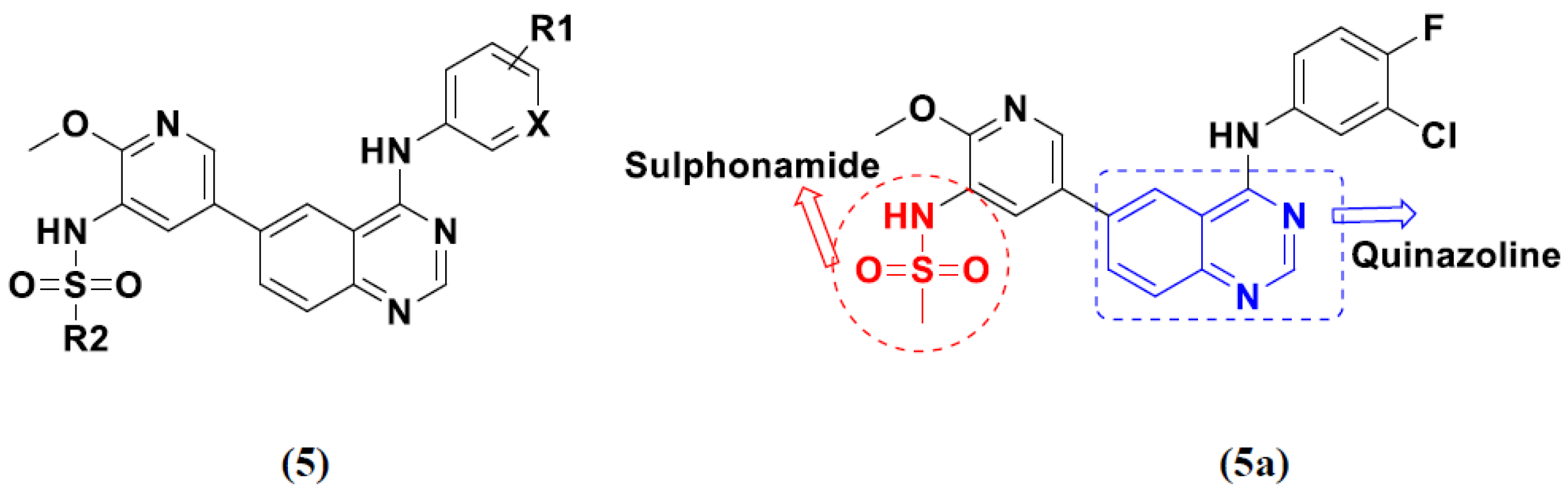{kind=link}
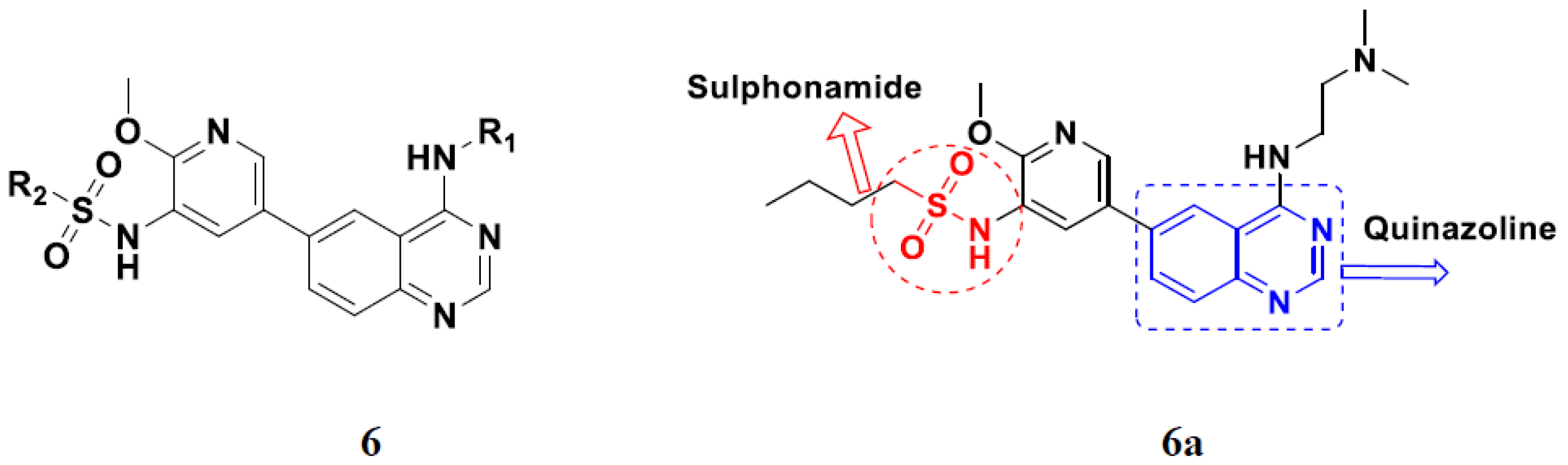{kind=link}
{kind=link}
{kind=link}
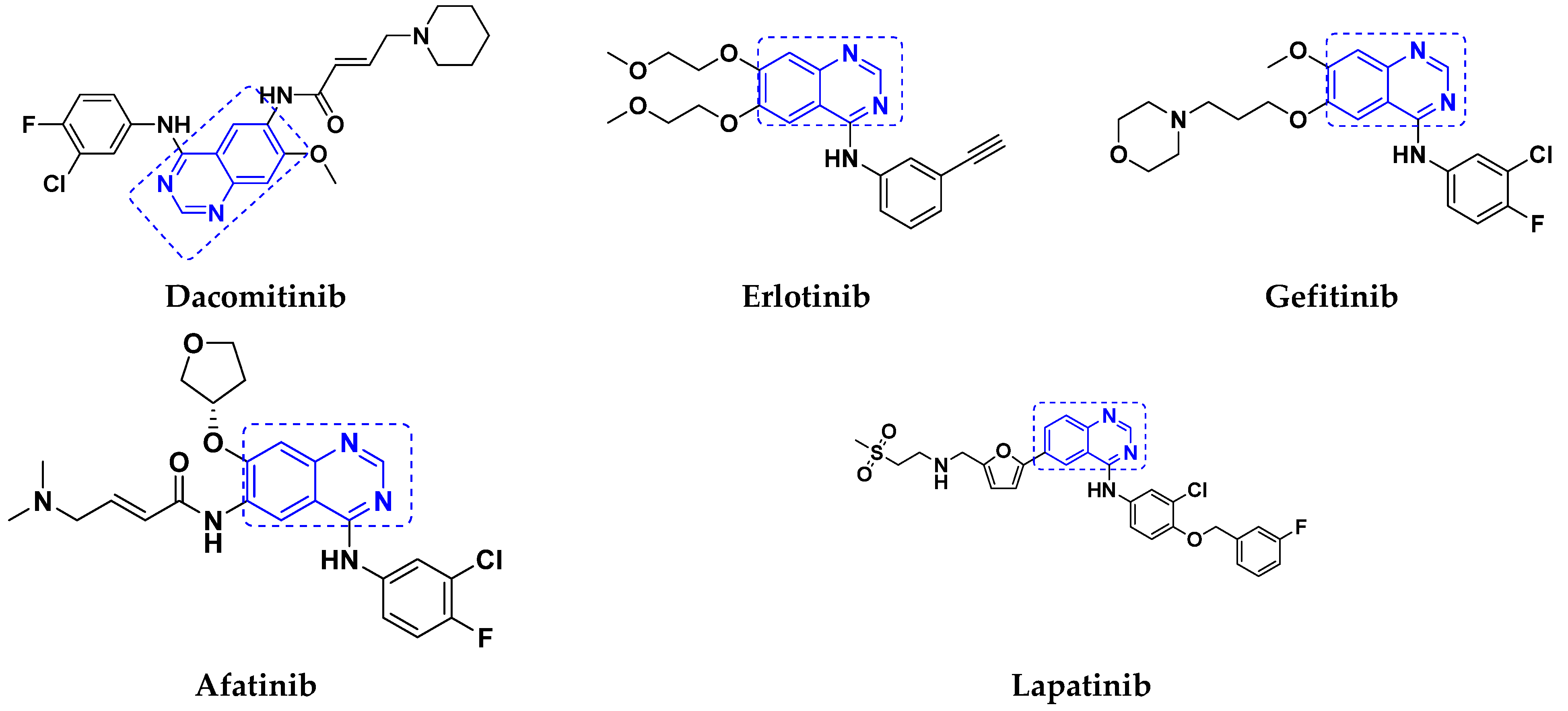{kind=link}
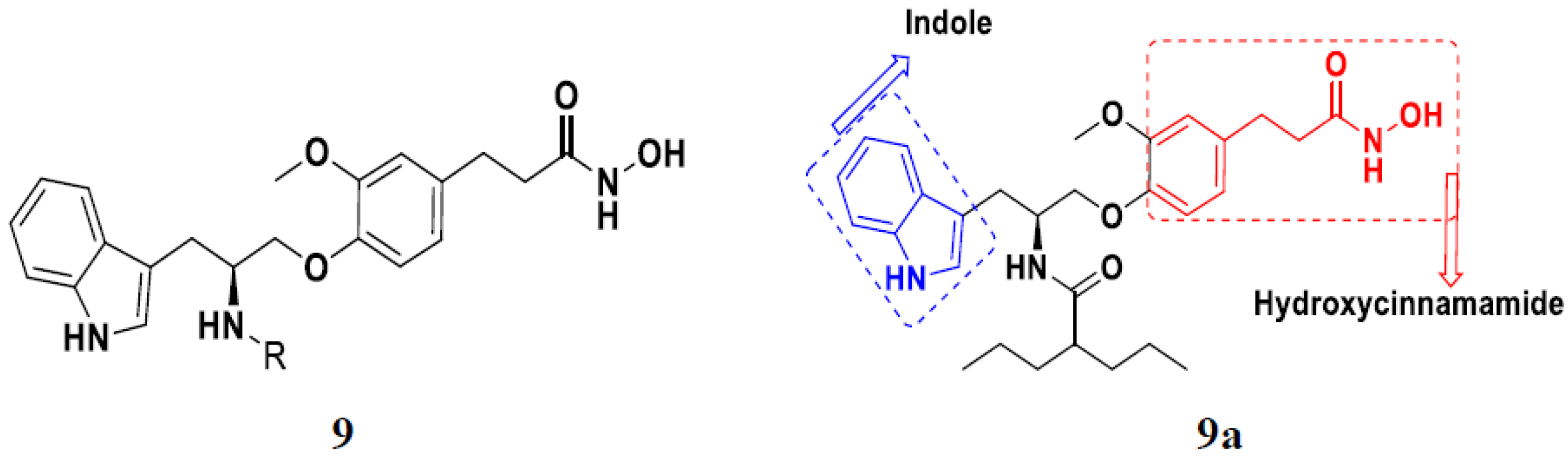{kind=link}
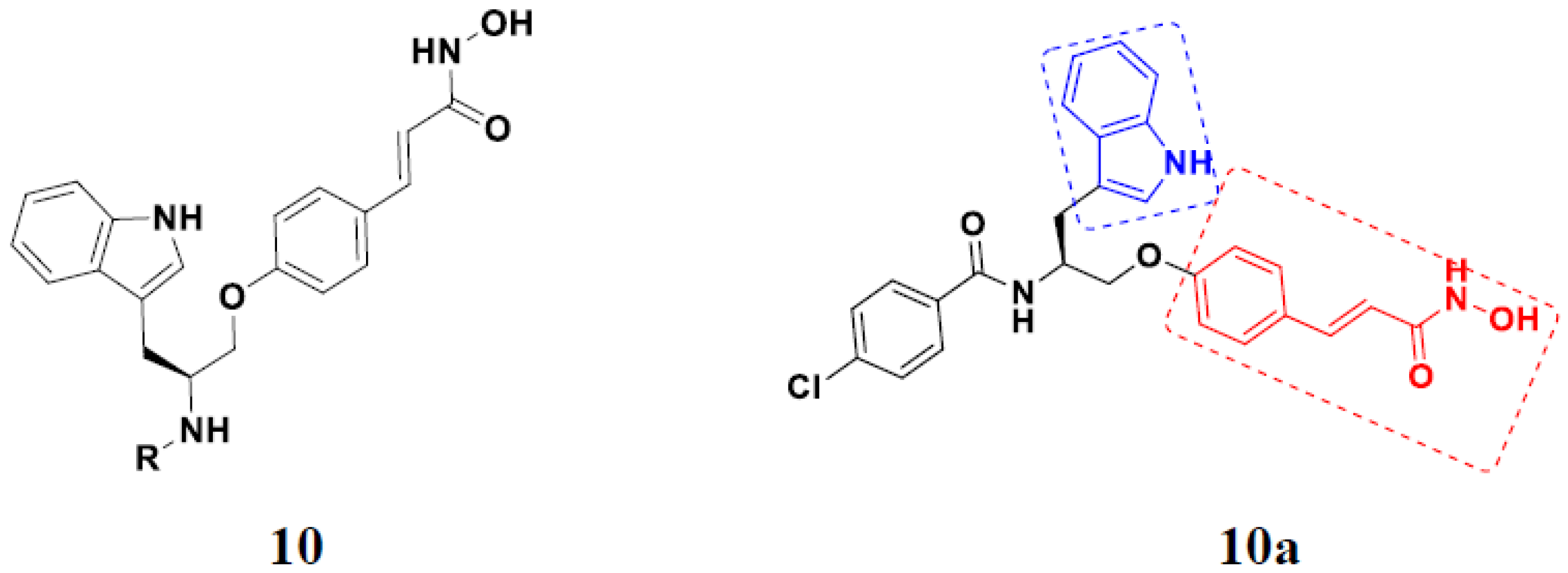{kind=link}
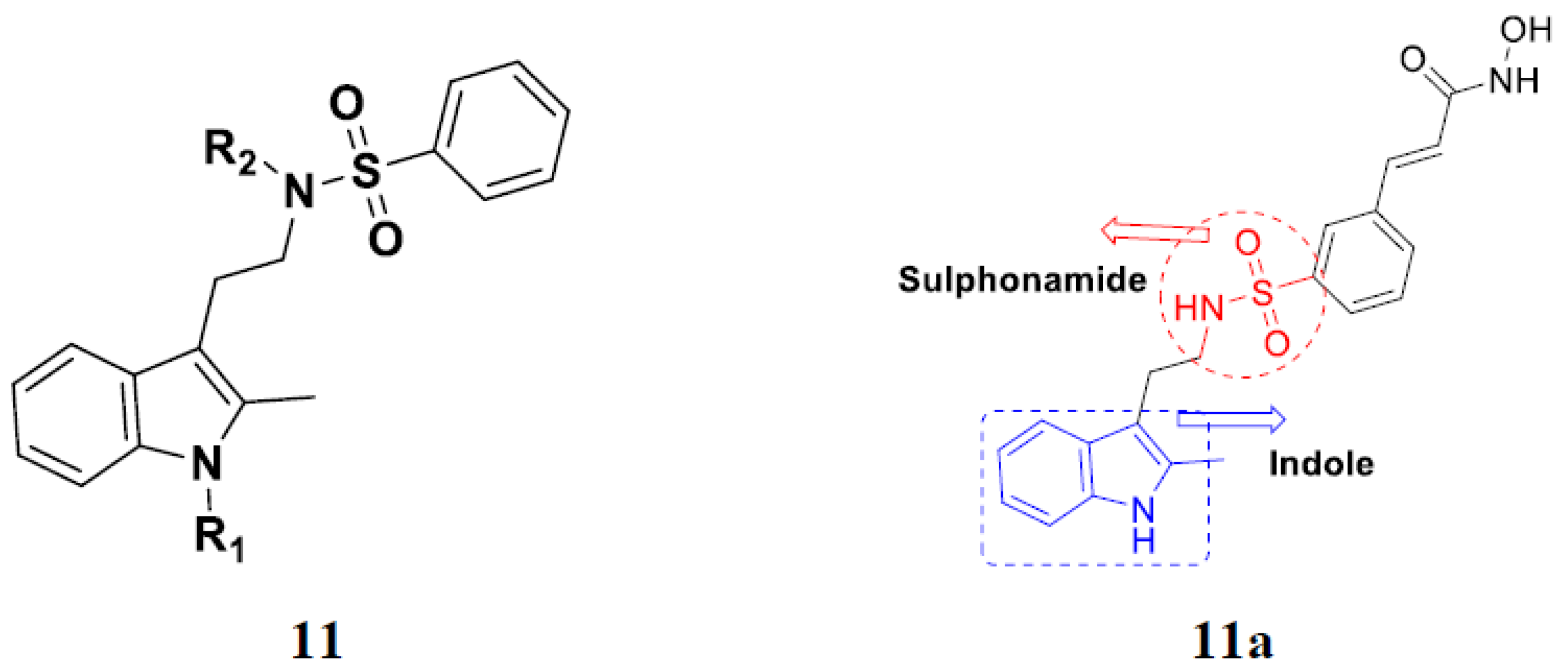{kind=link}
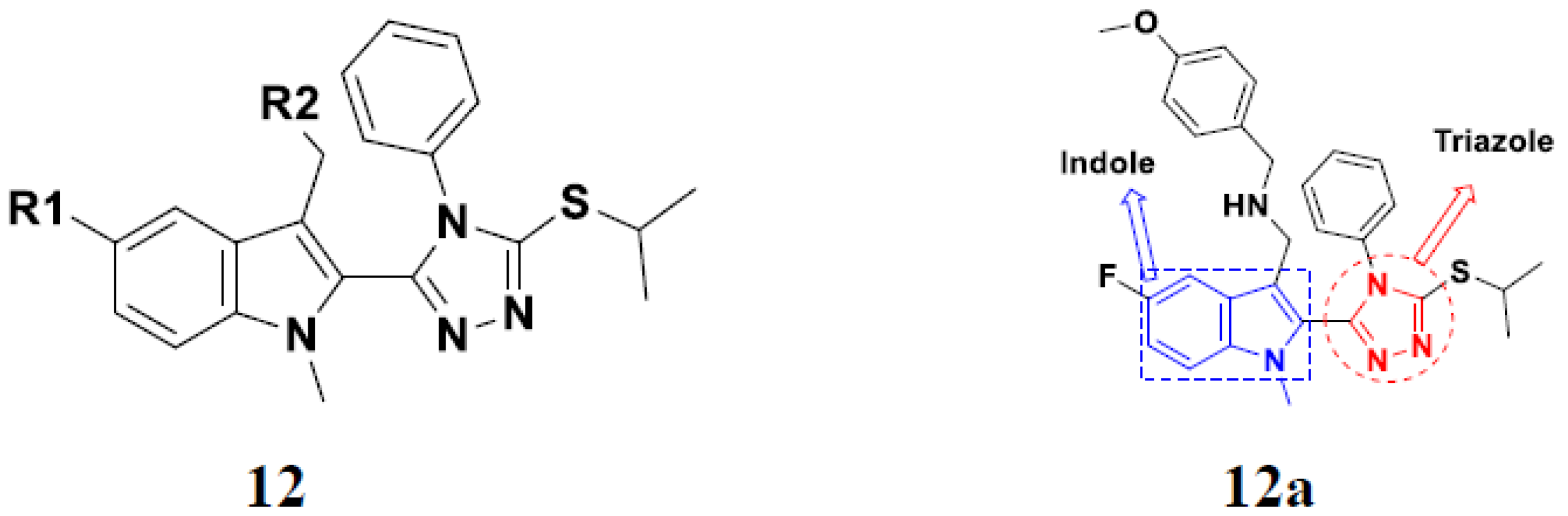{kind=link}
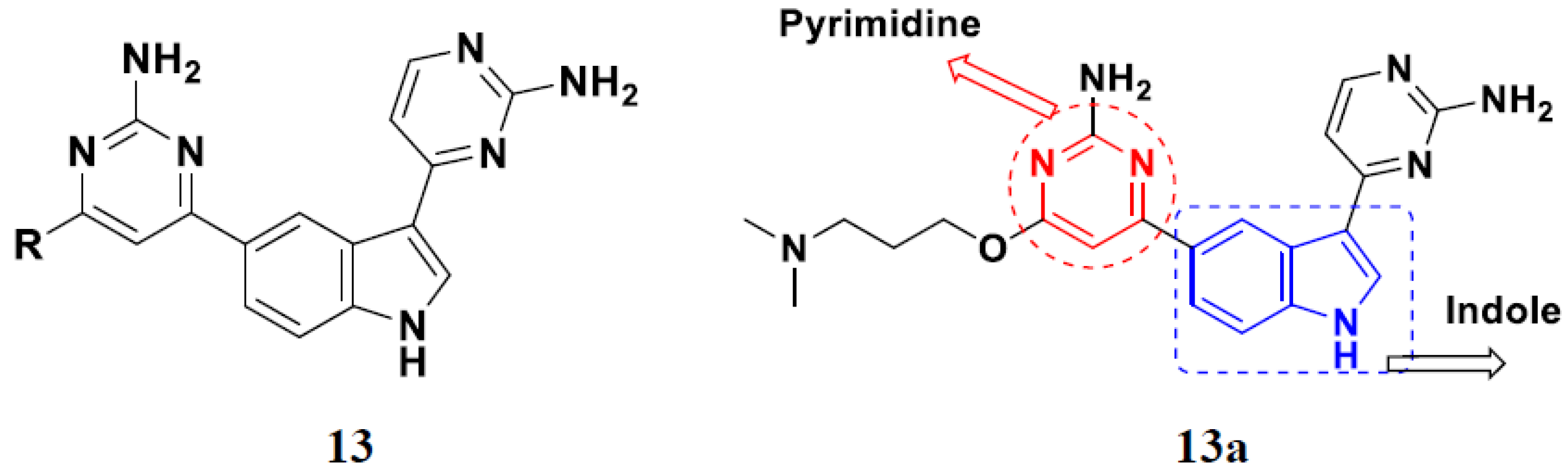{kind=link}
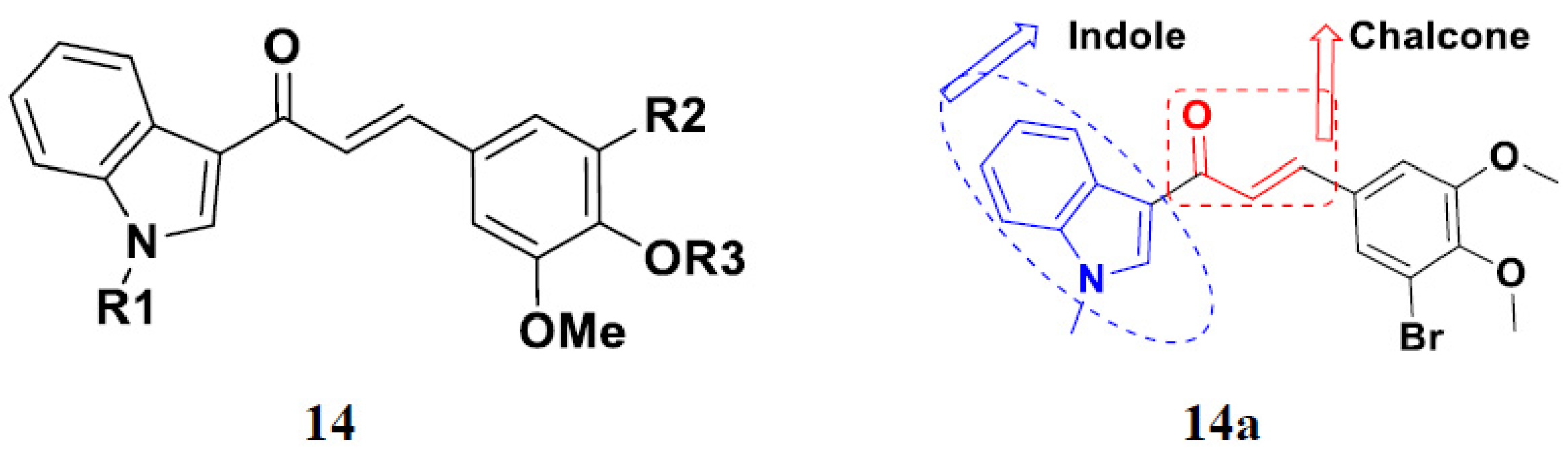{kind=link}
{kind=link}
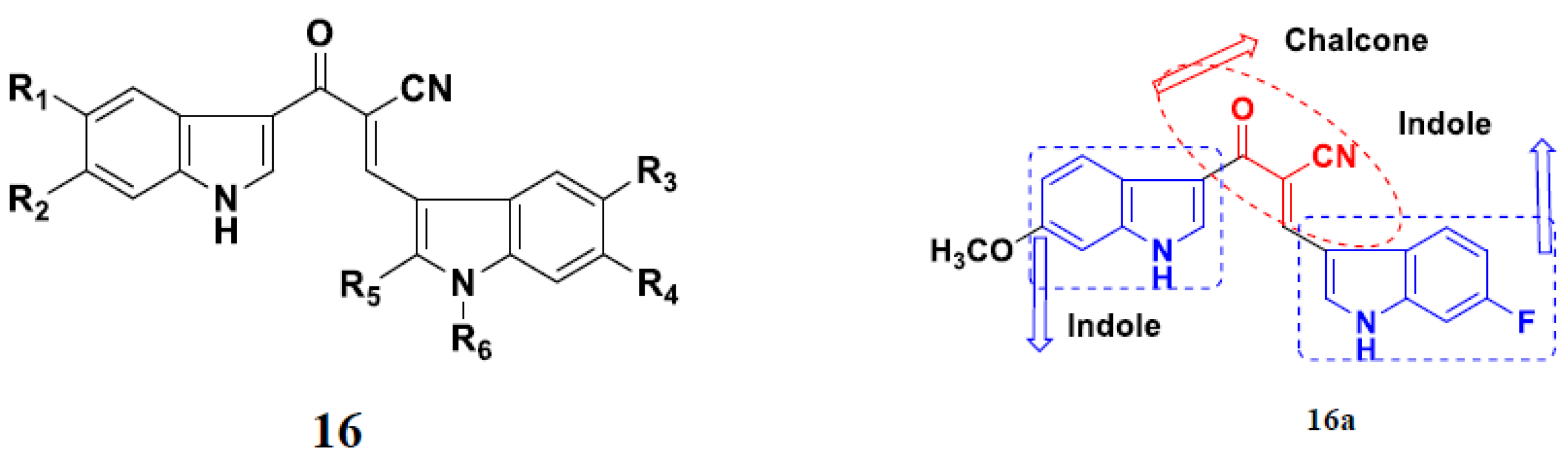{kind=link}
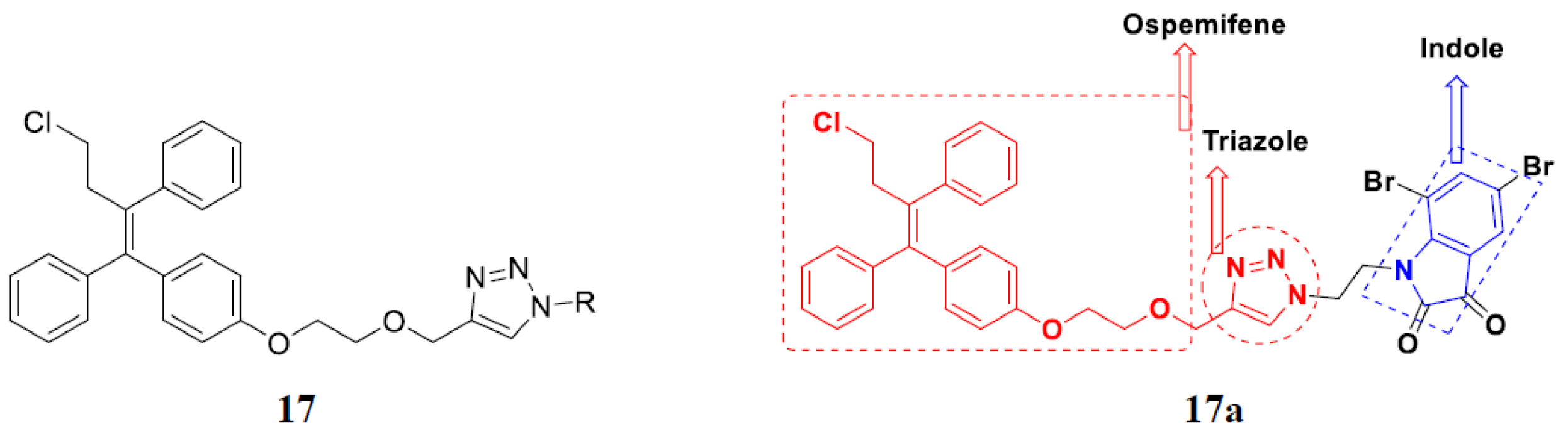{kind=link}
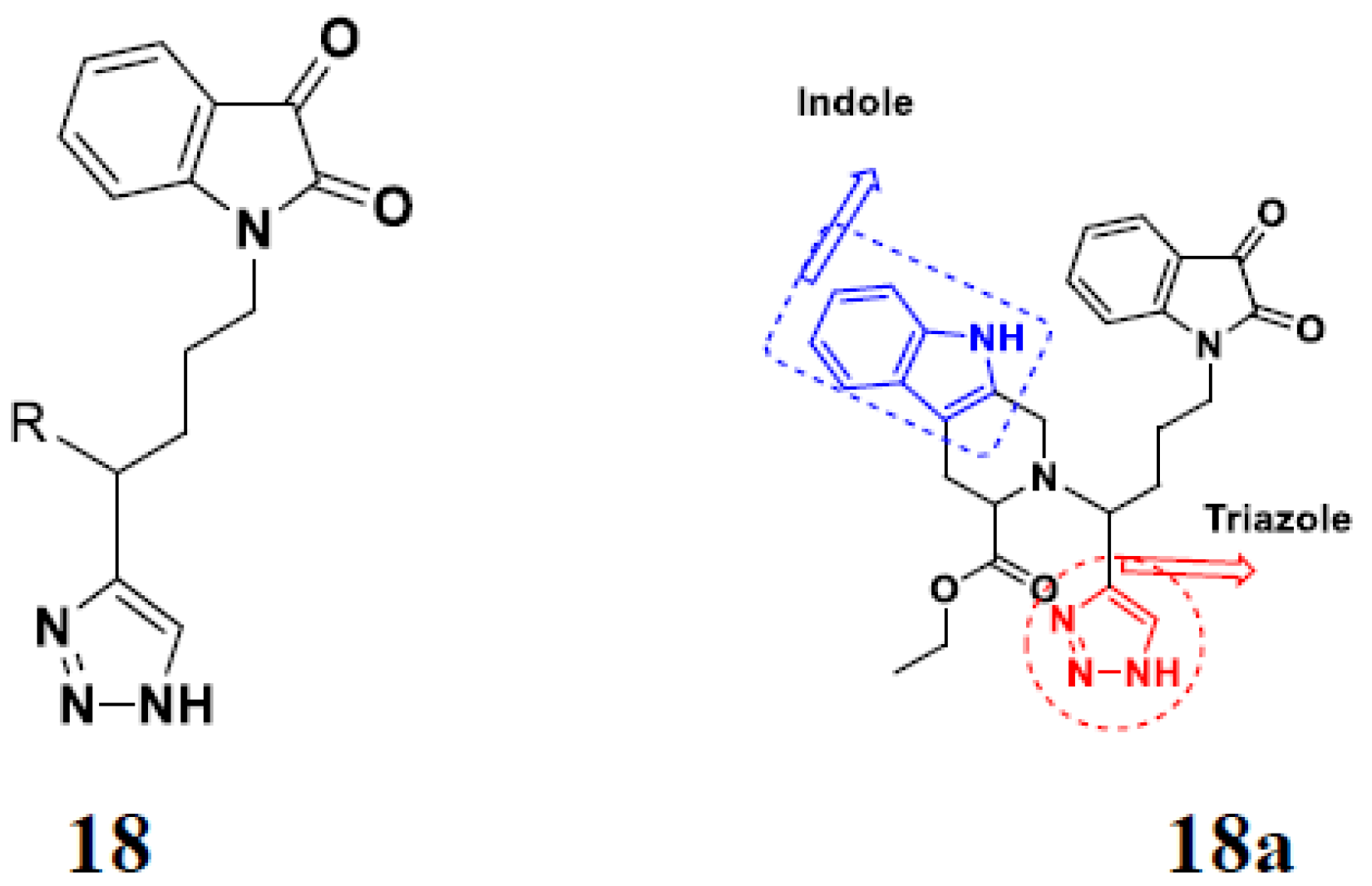{kind=link}
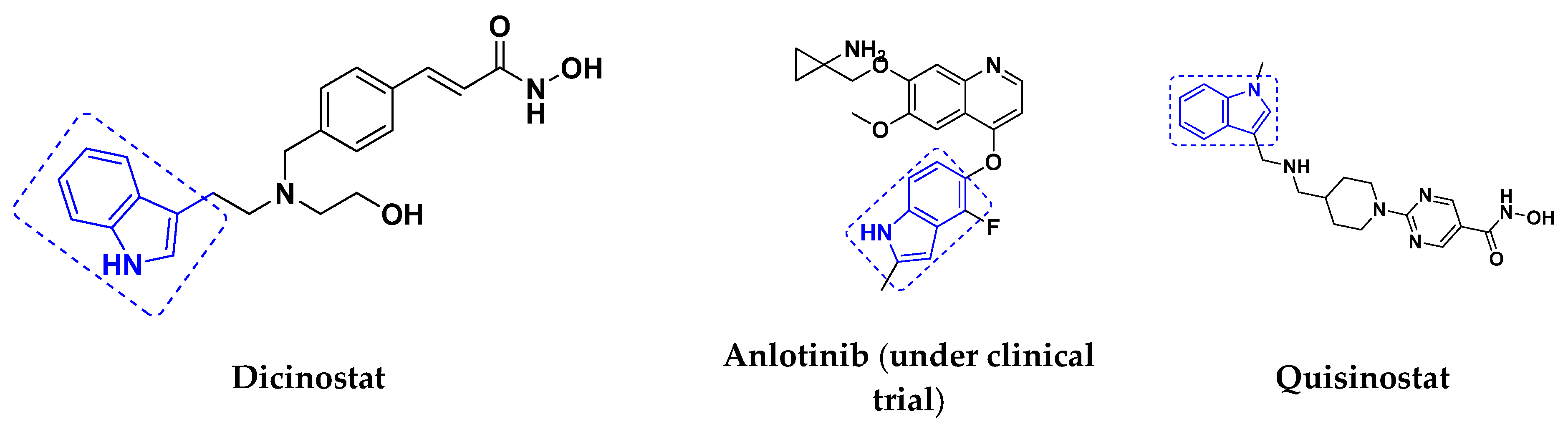{kind=link}
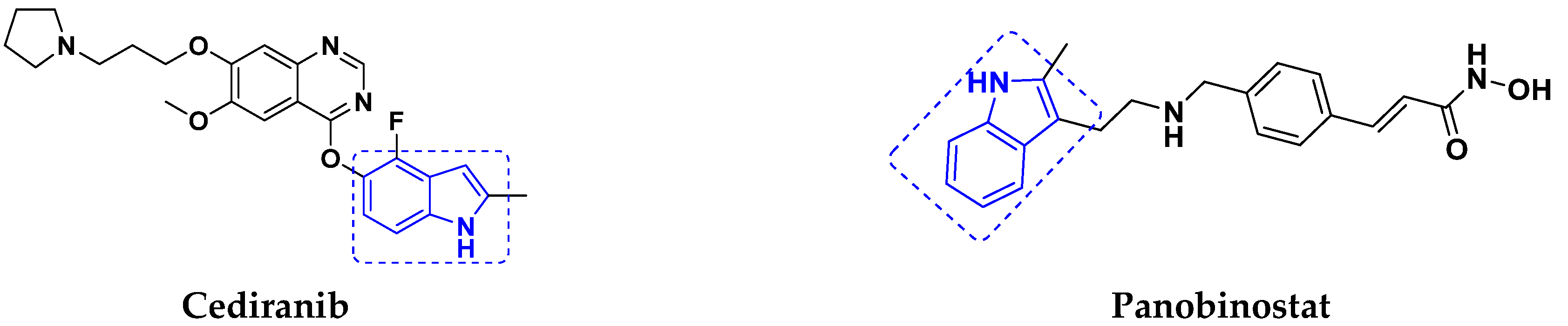{kind=link}
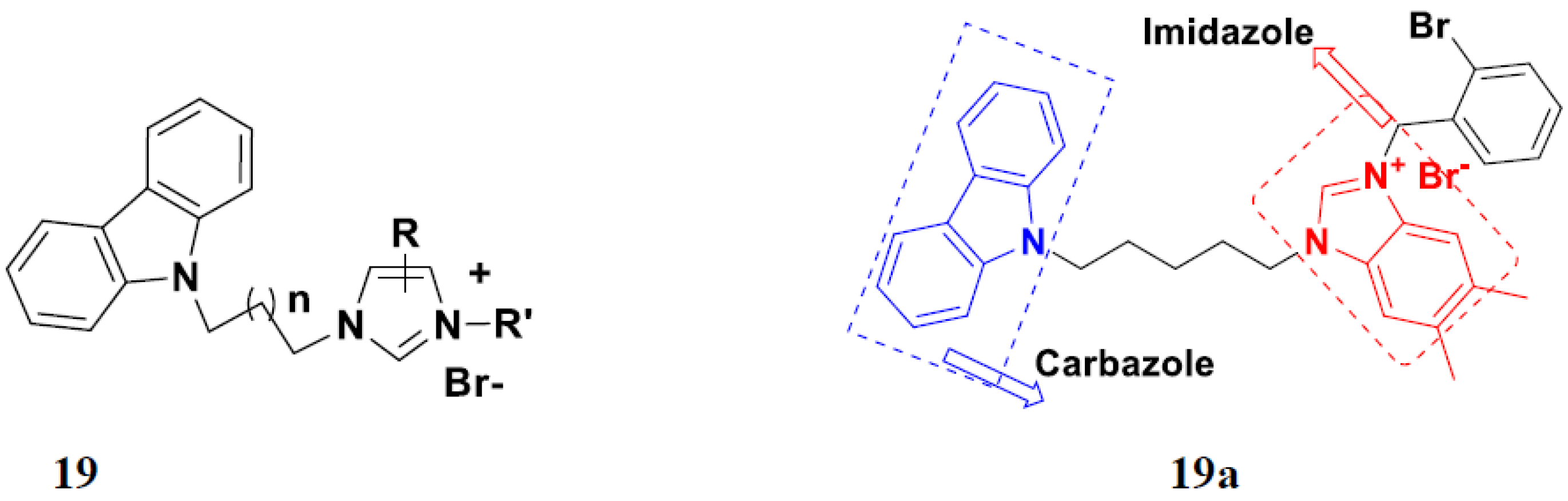{kind=link}
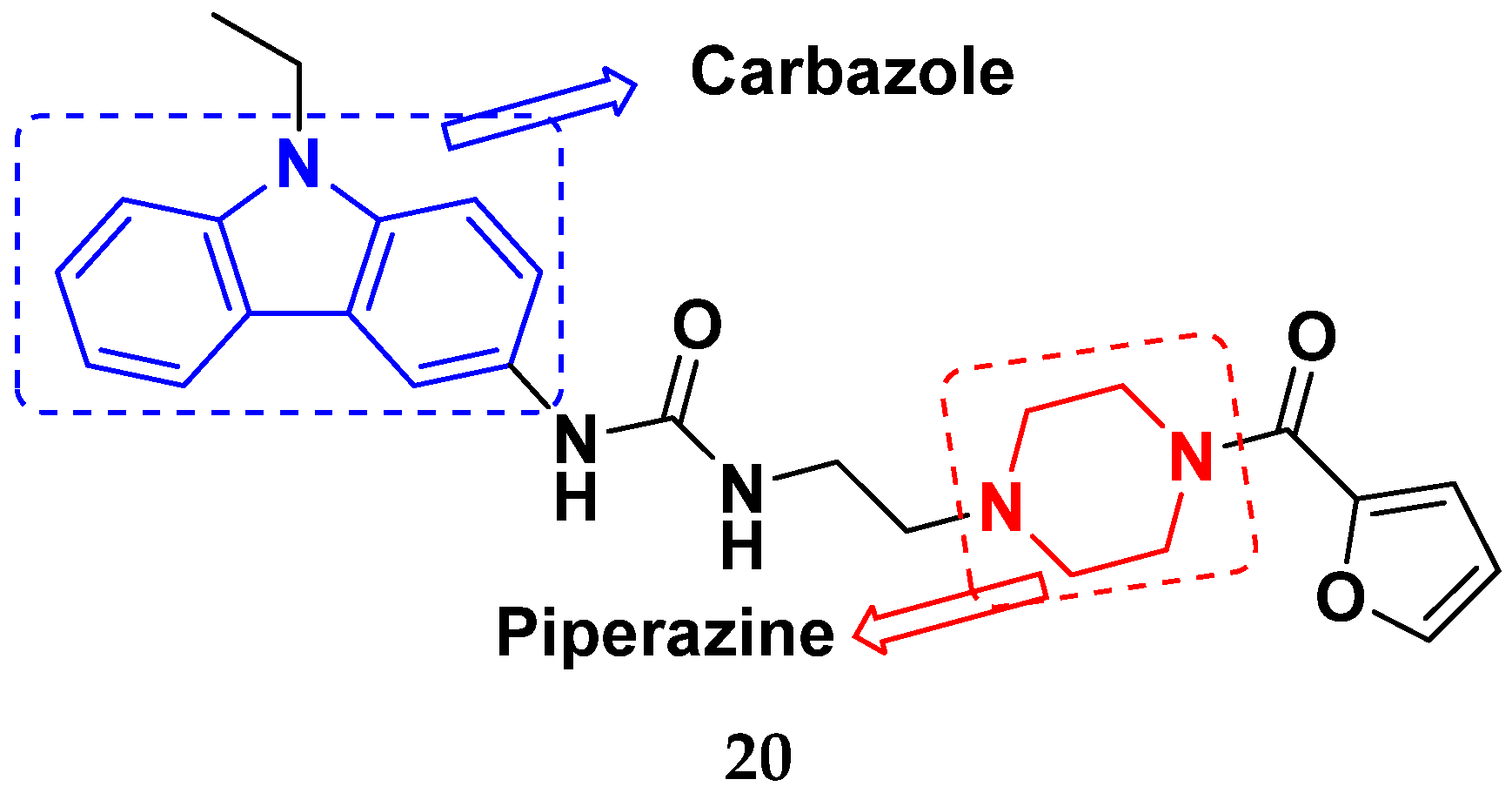{kind=link}
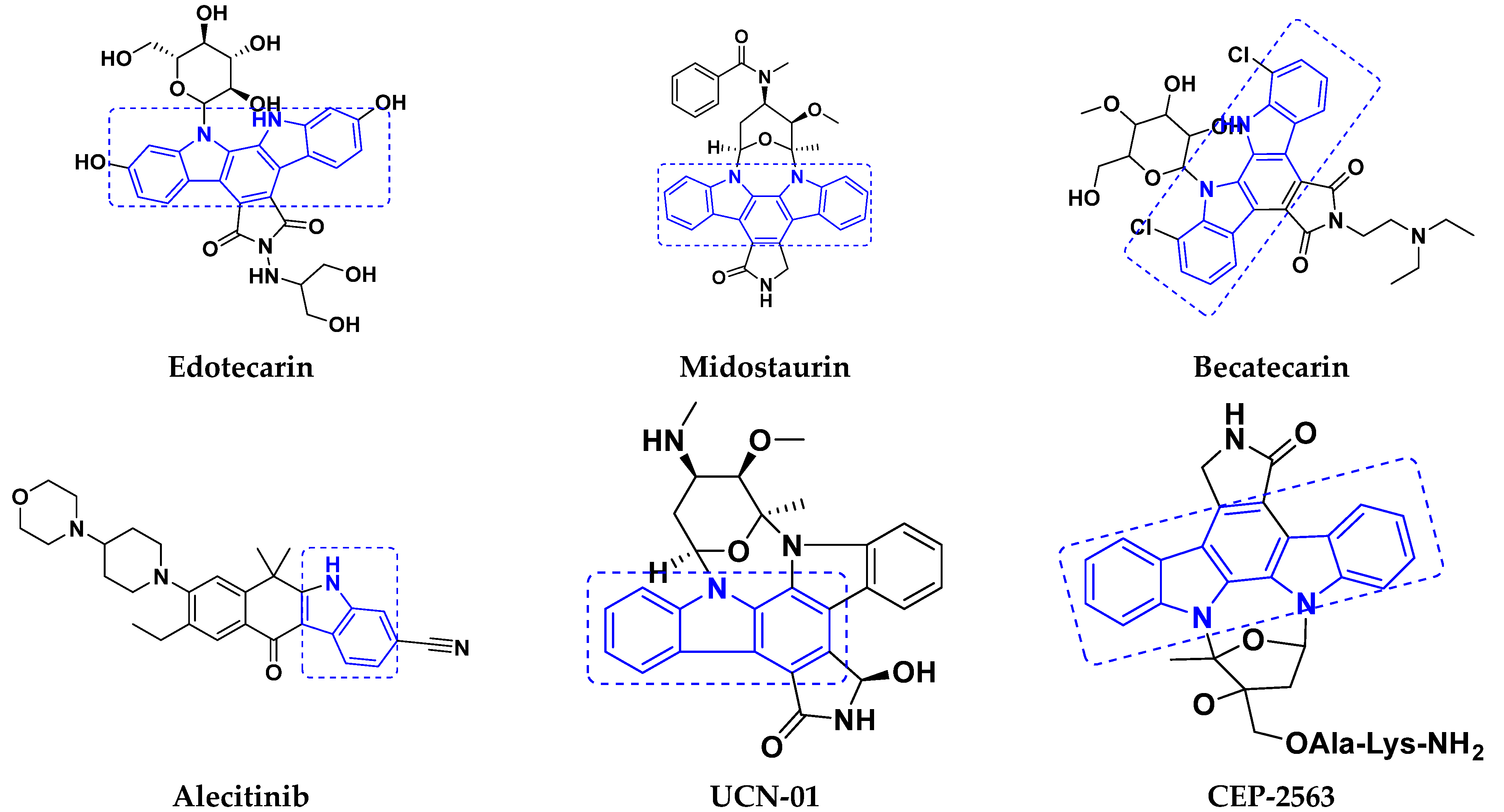{kind=link}
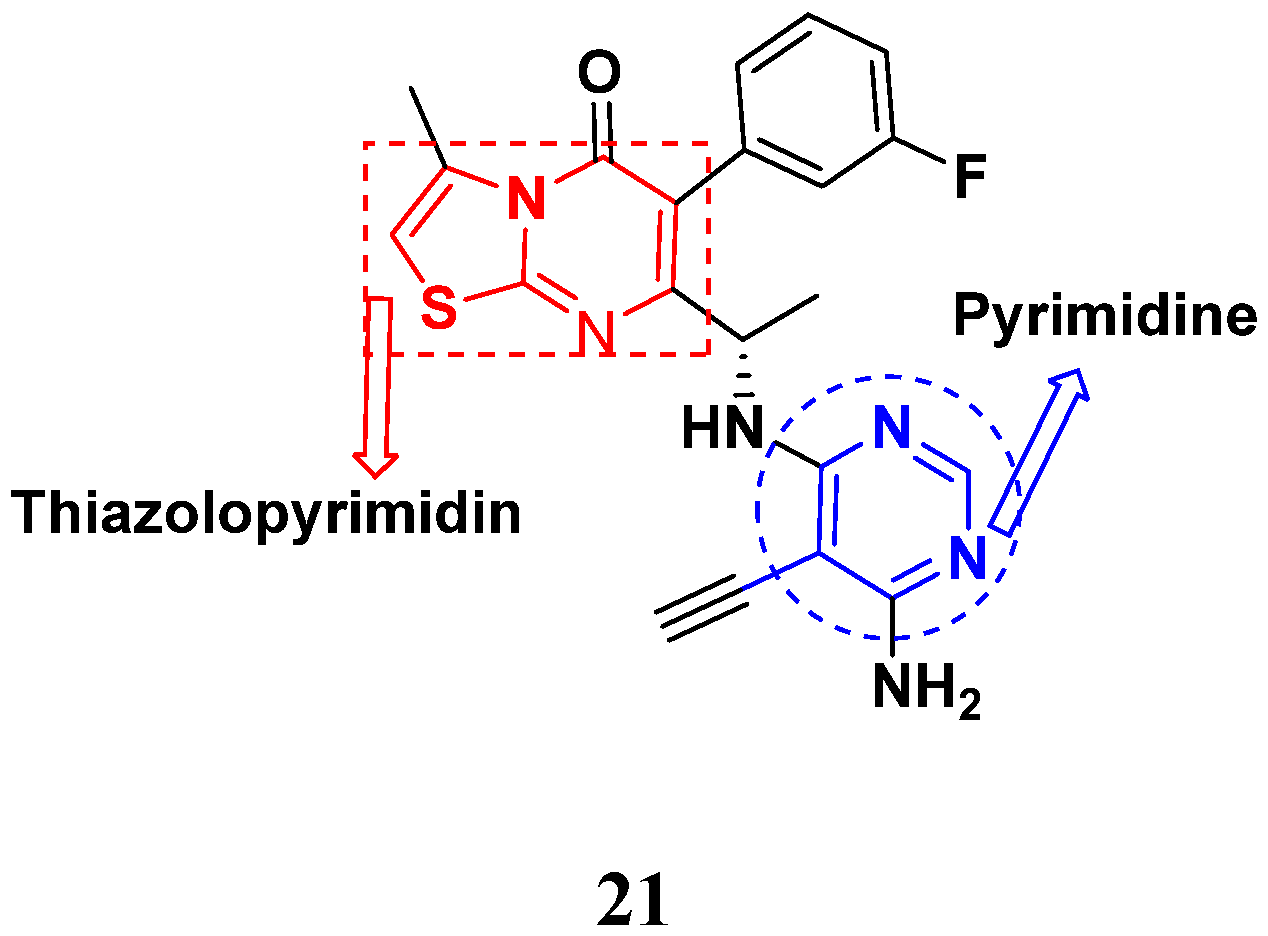{kind=link}
{kind=link}
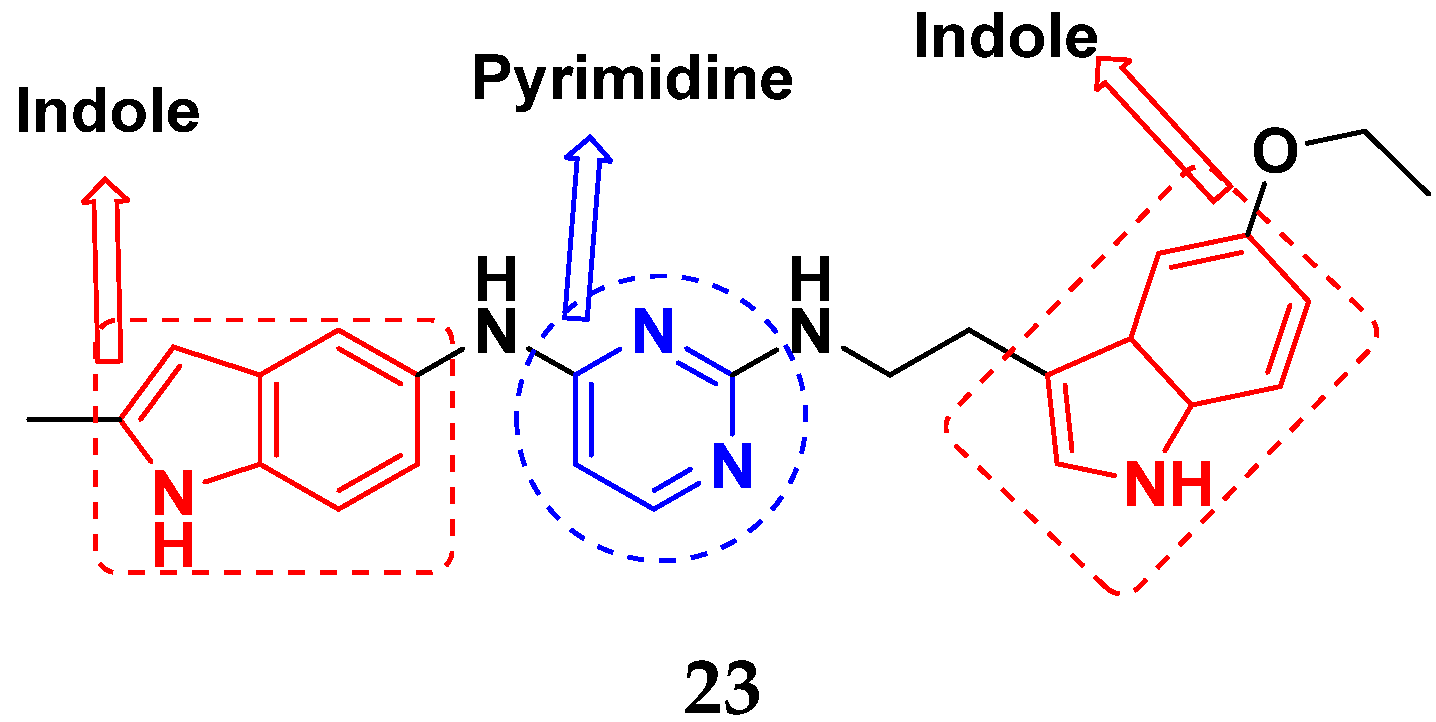{kind=link}
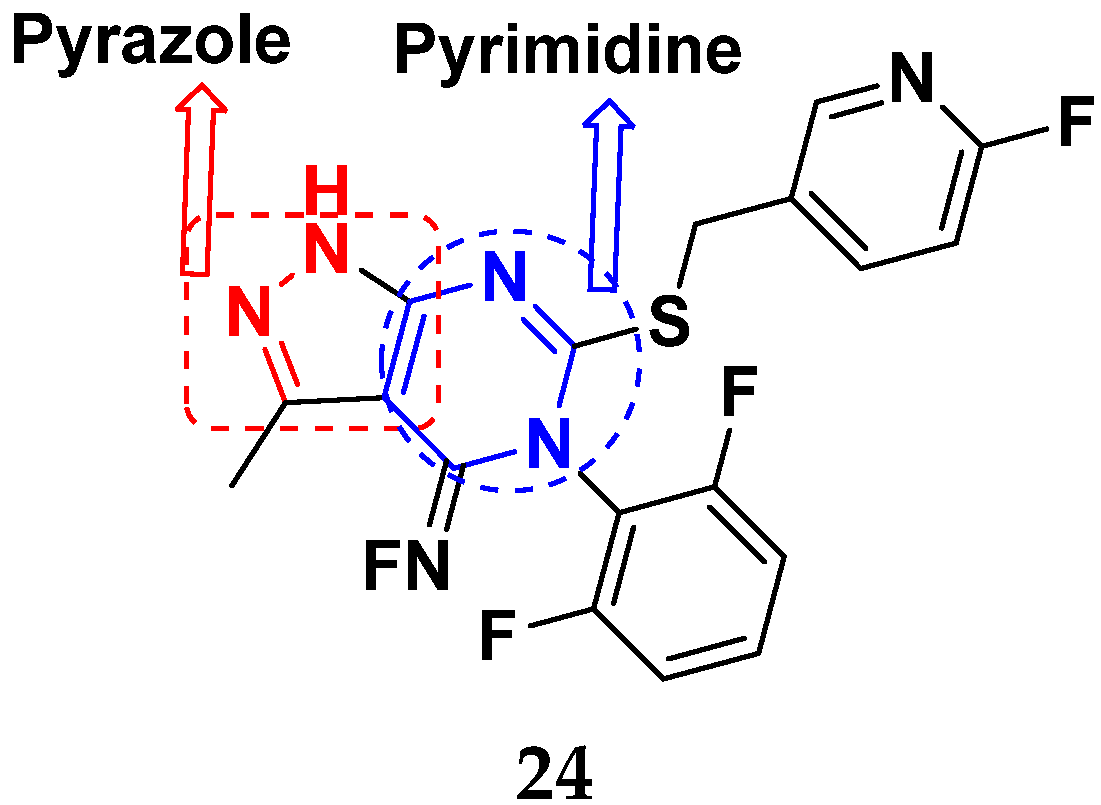{kind=link}
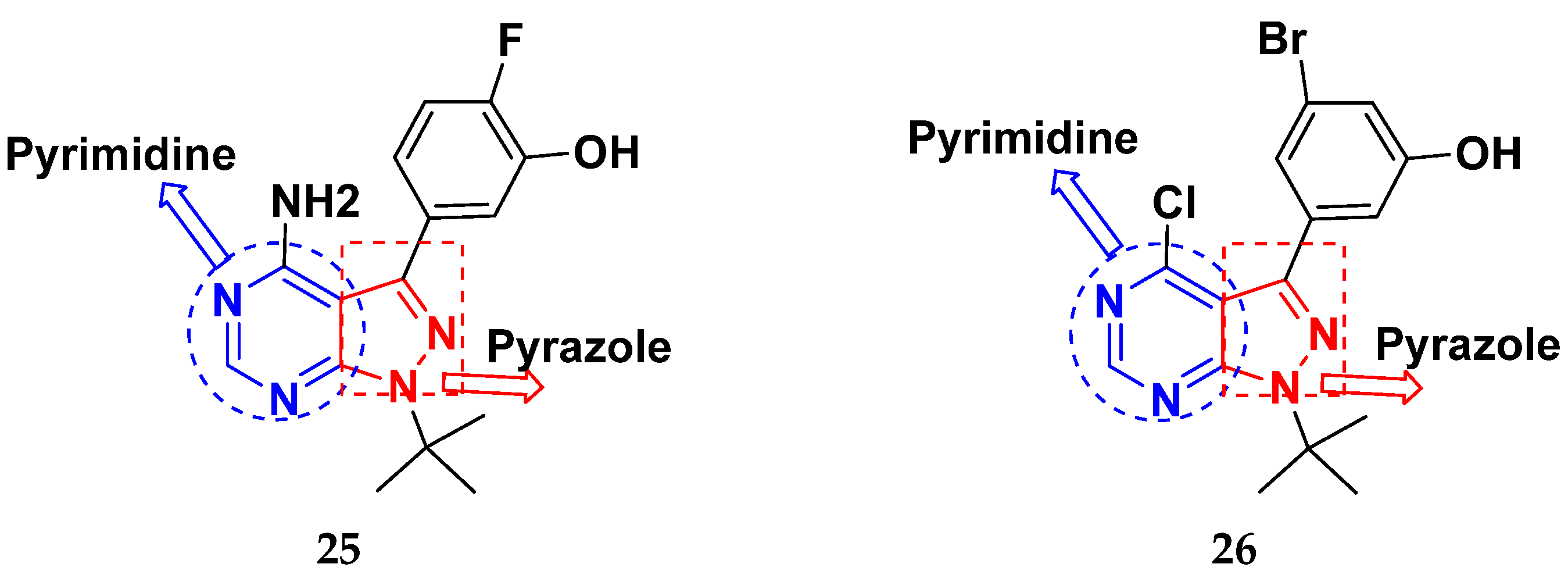{kind=link}
{kind=link}
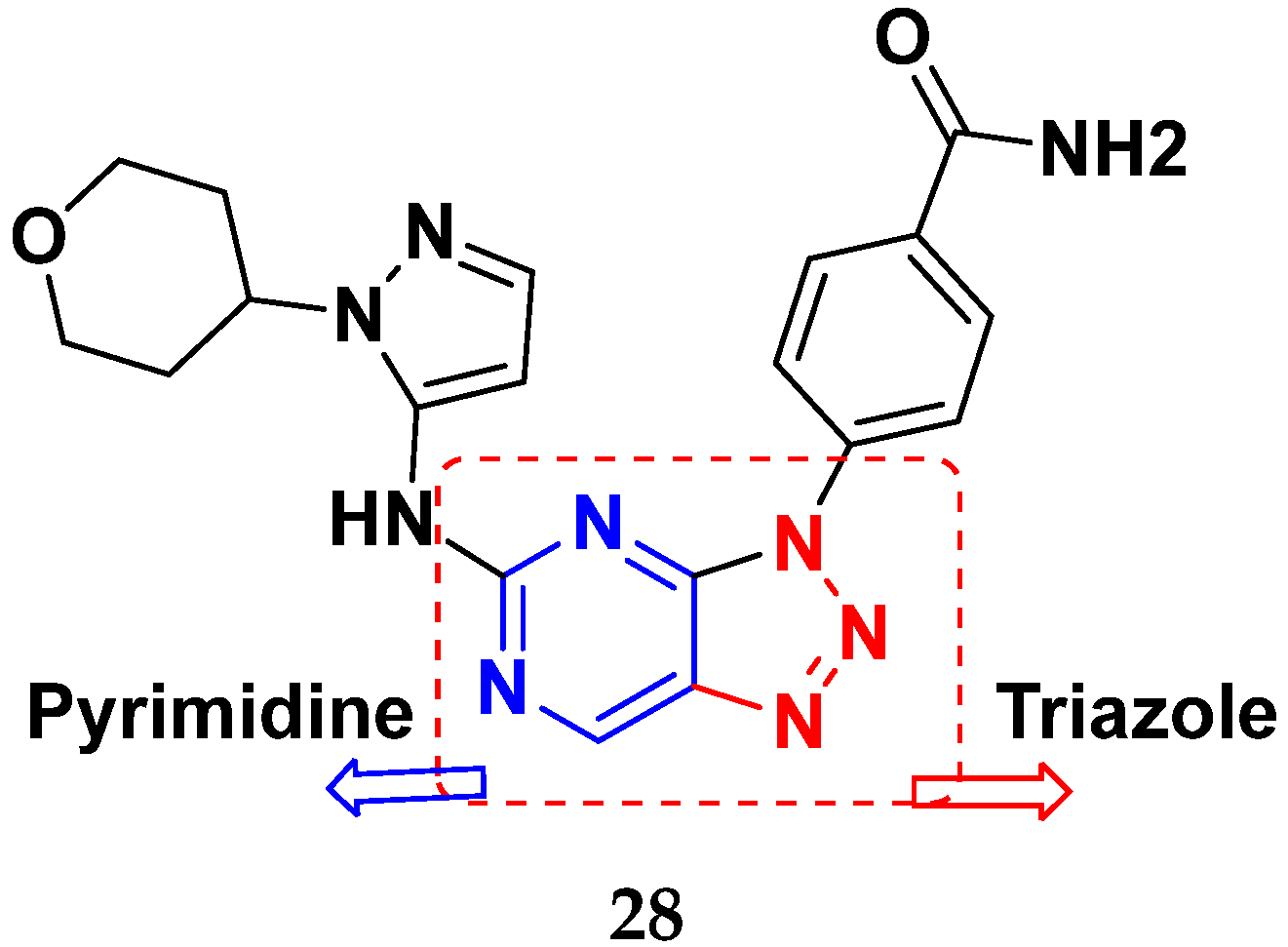{kind=link}
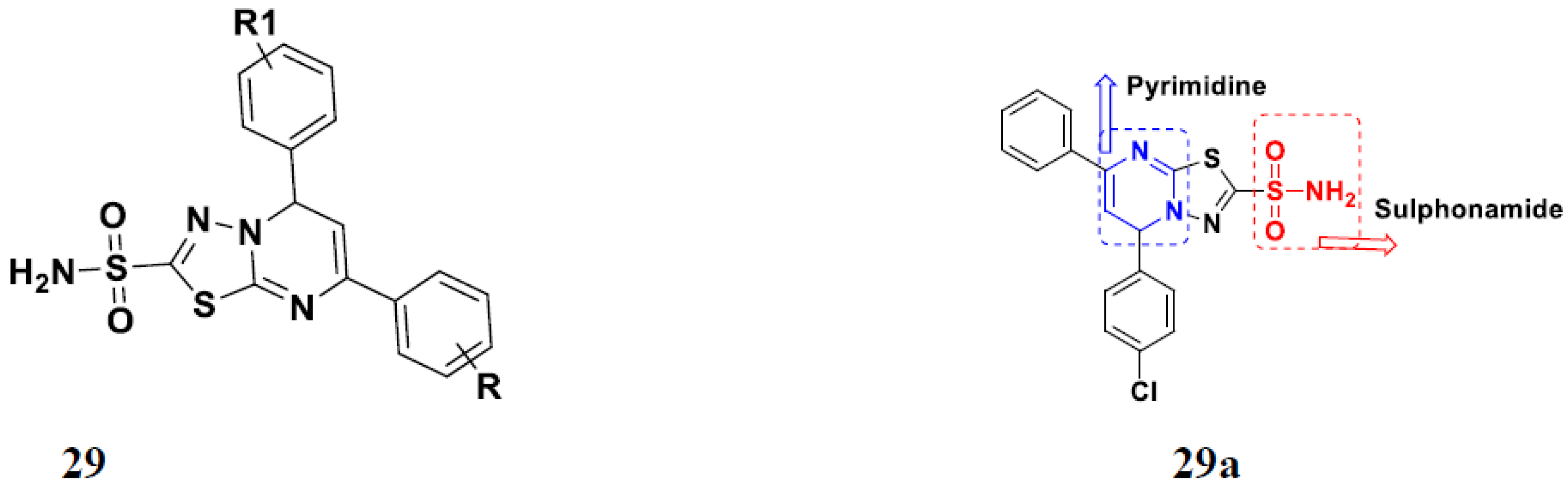{kind=link}
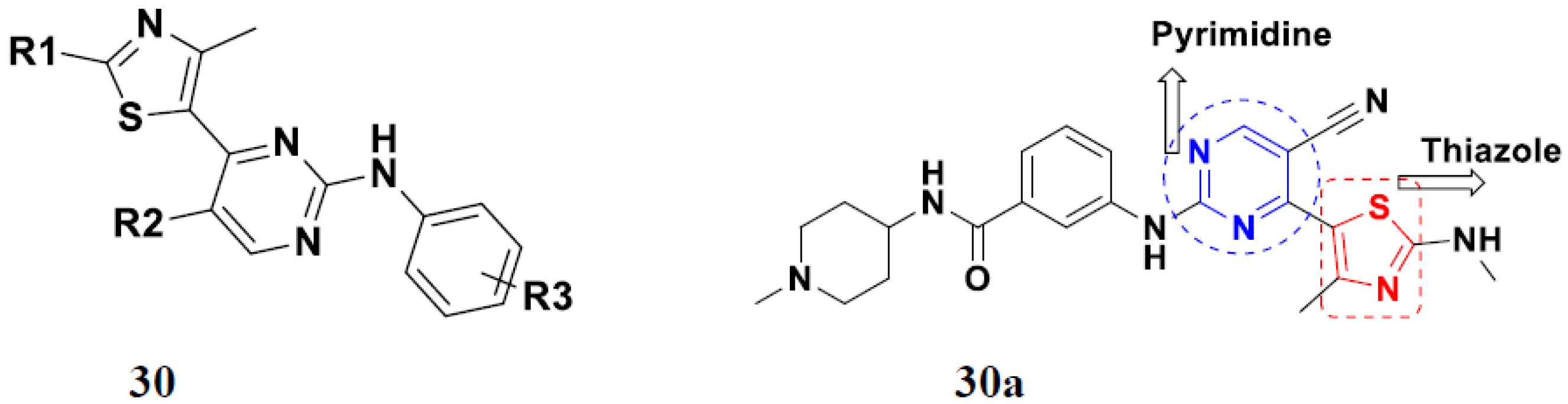{kind=link}
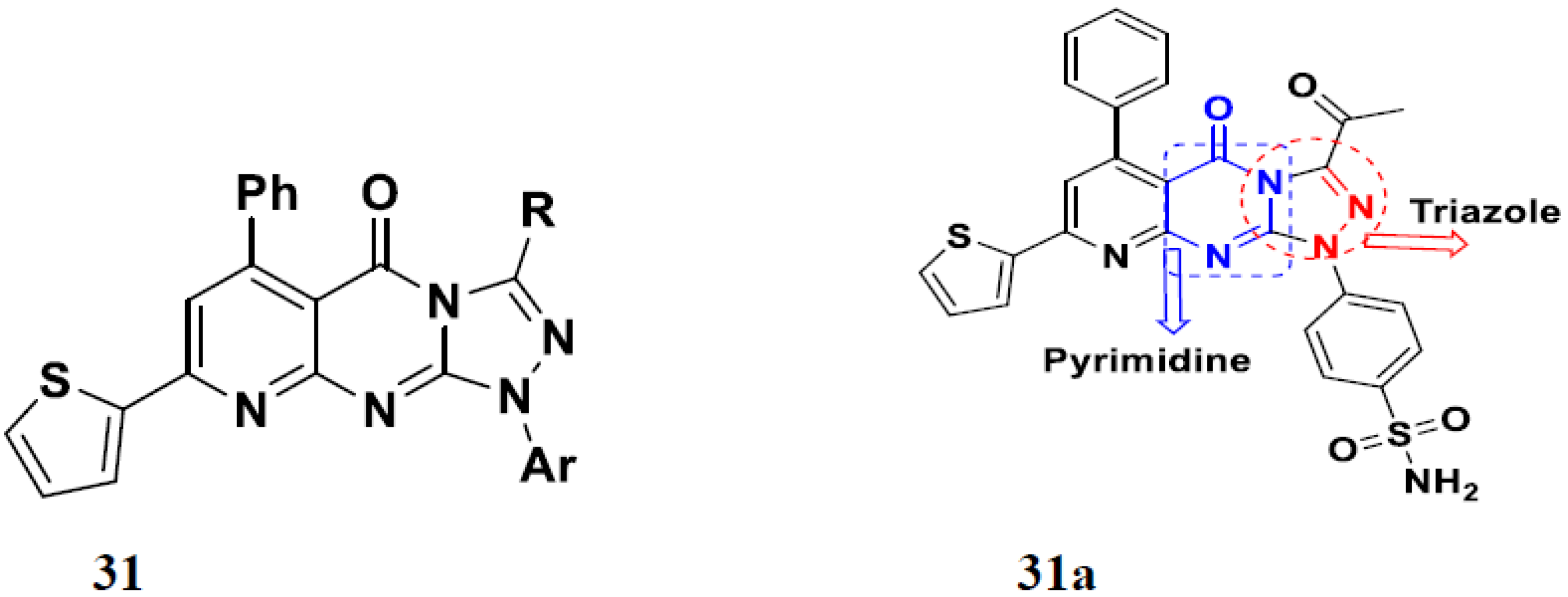{kind=link}
{kind=link}
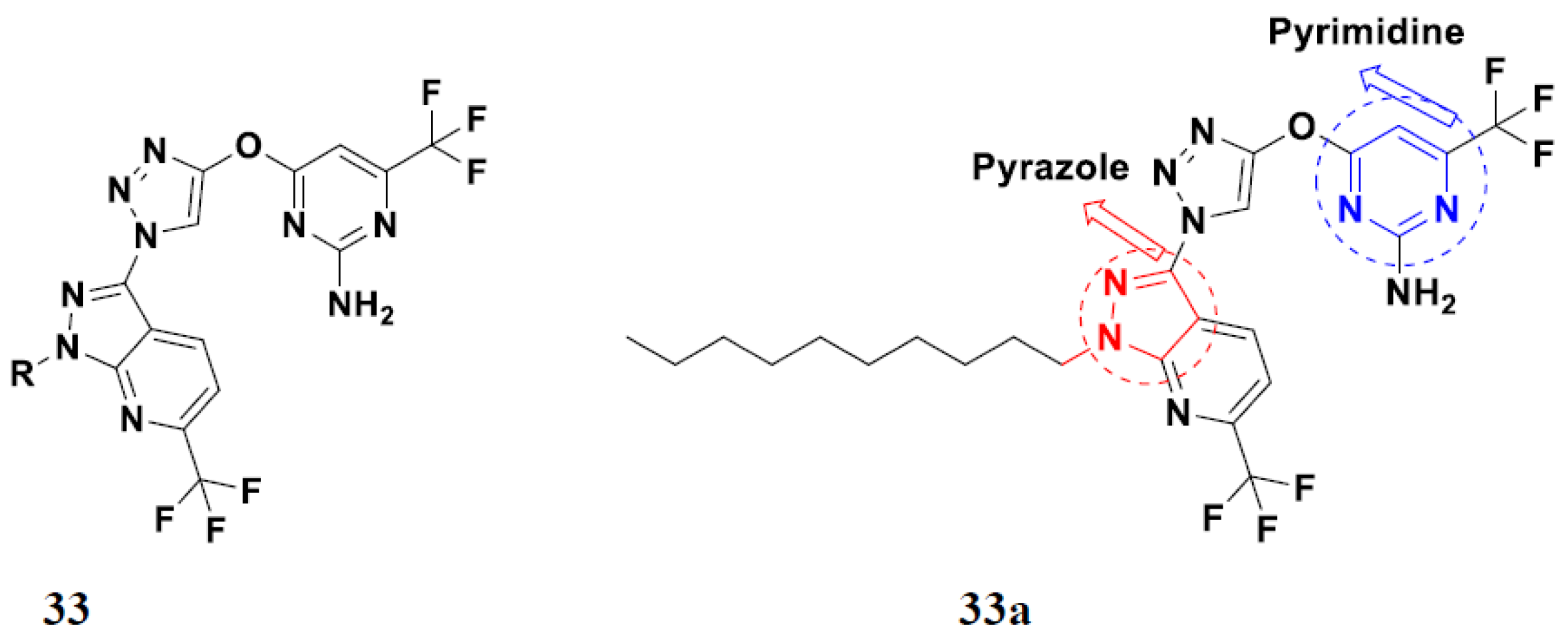{kind=link}
{kind=link}
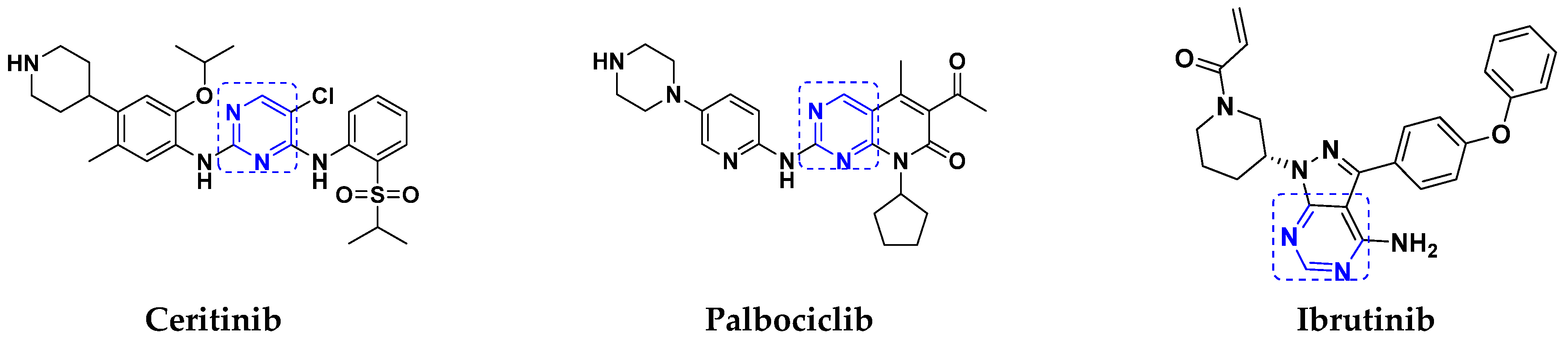{kind=link}
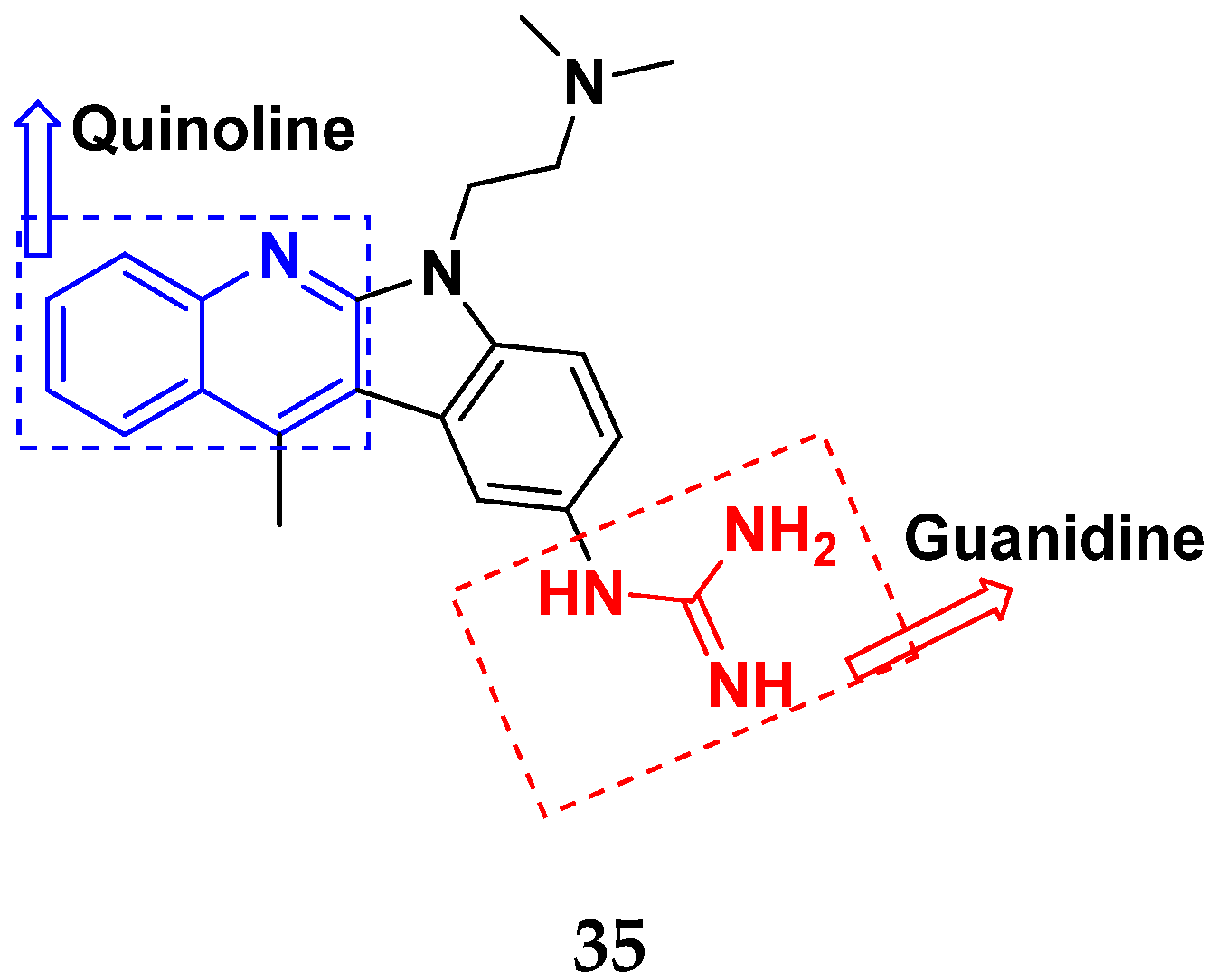{kind=link}
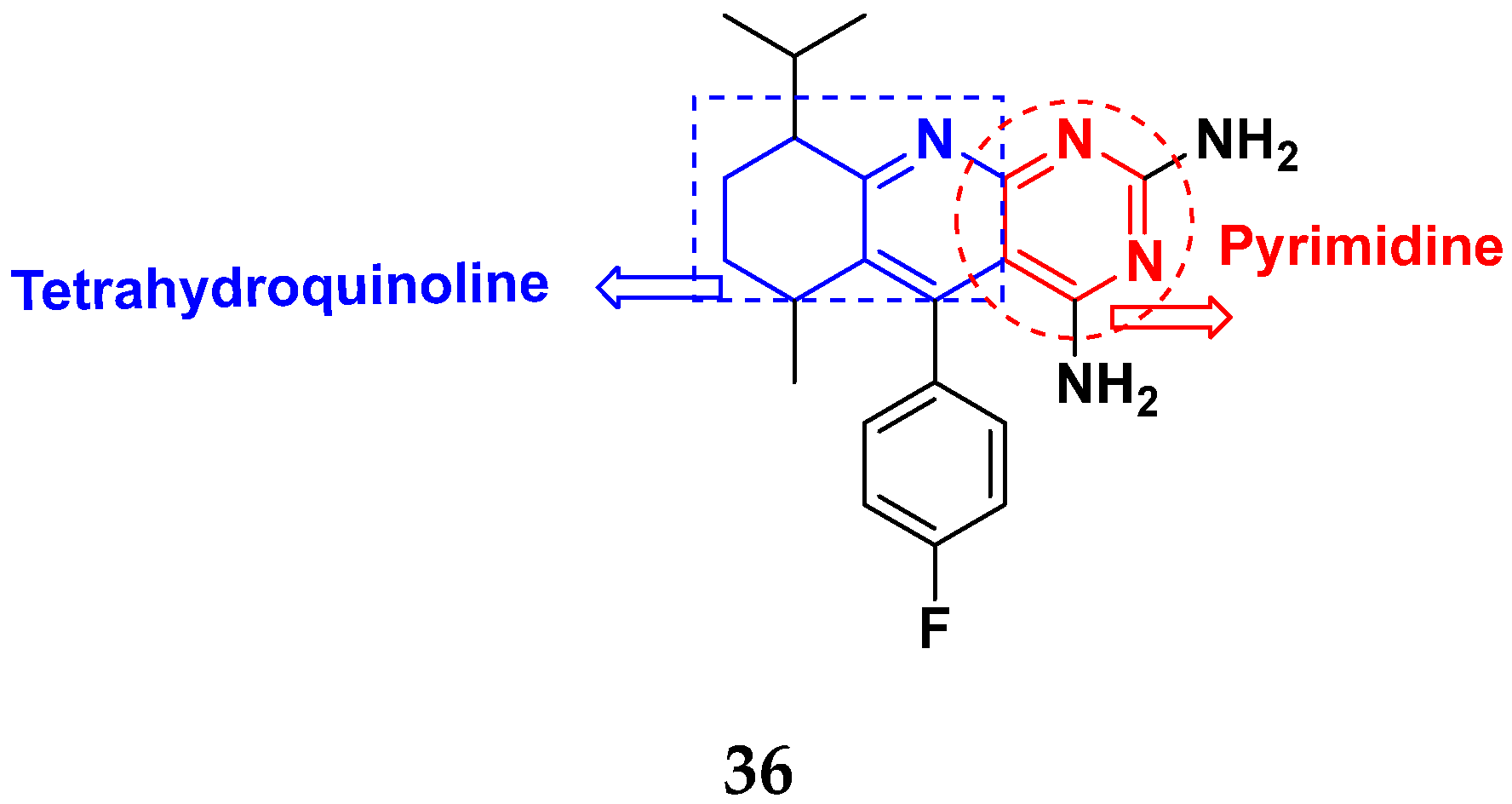{kind=link}
{kind=link}
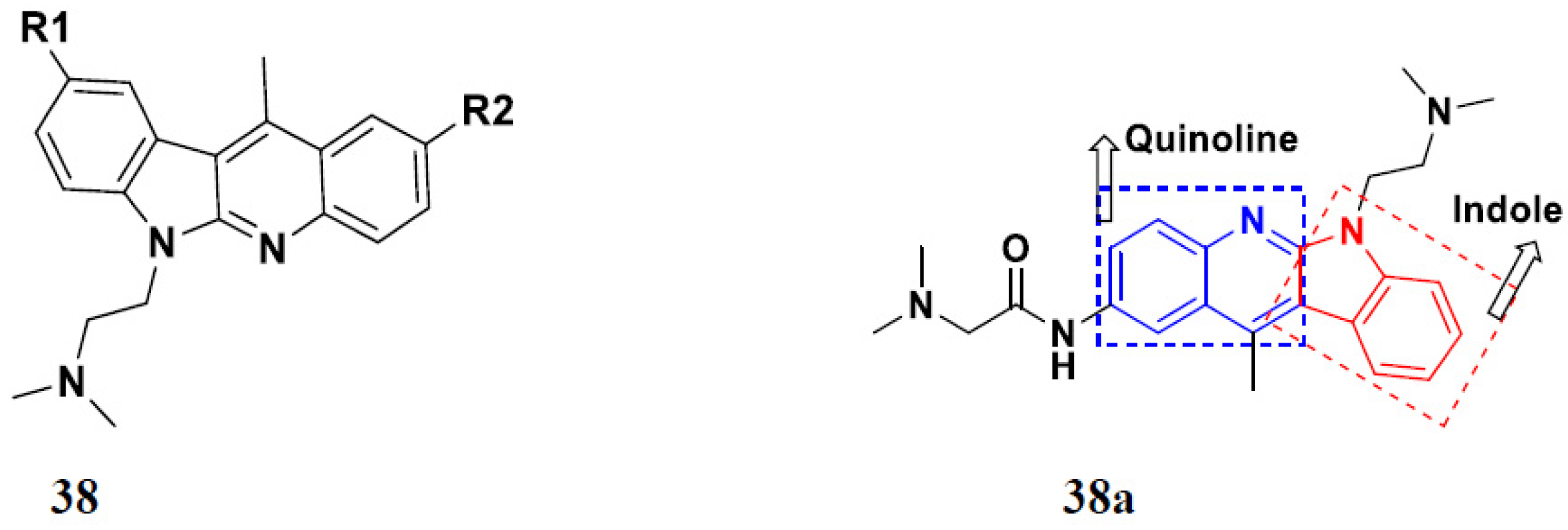{kind=link}
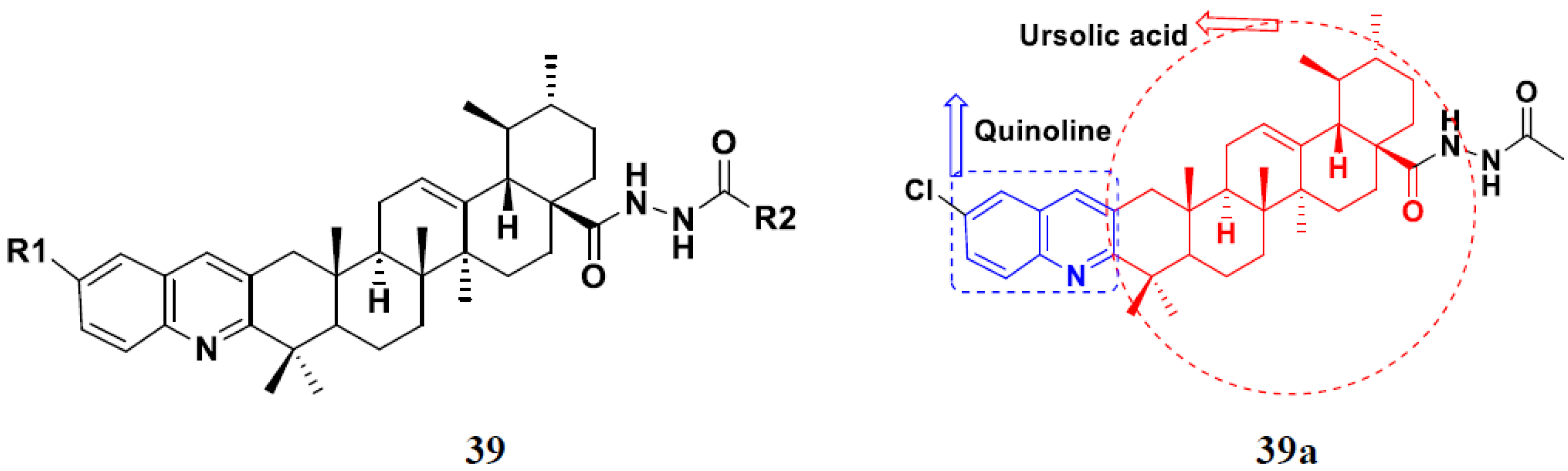{kind=link}
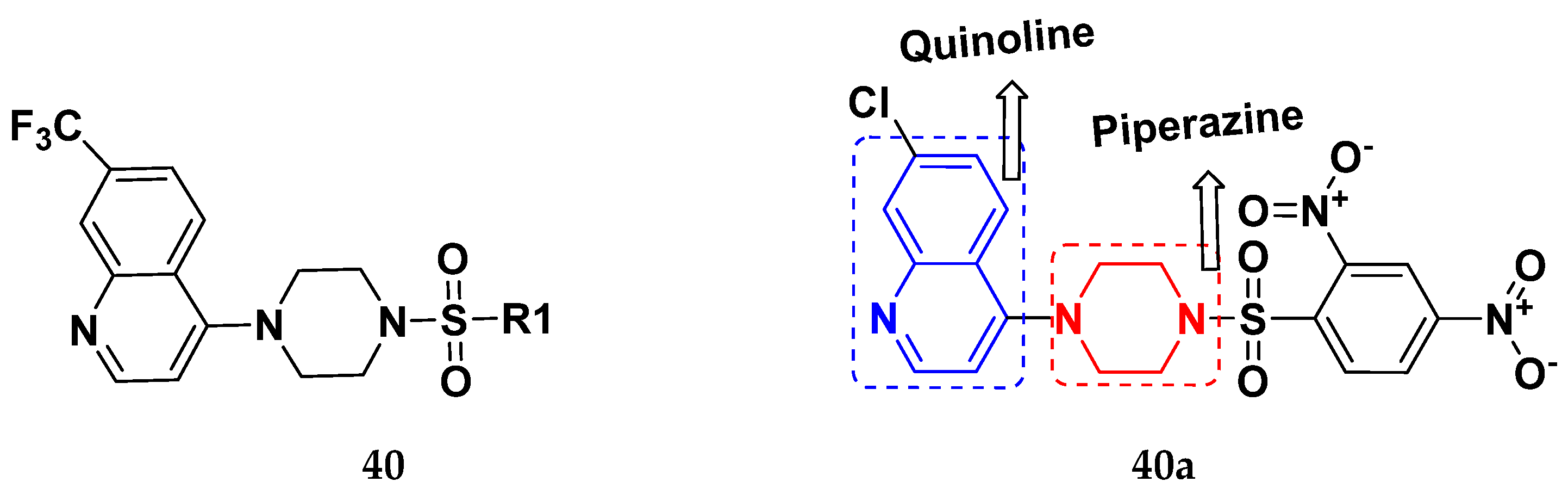{kind=link}
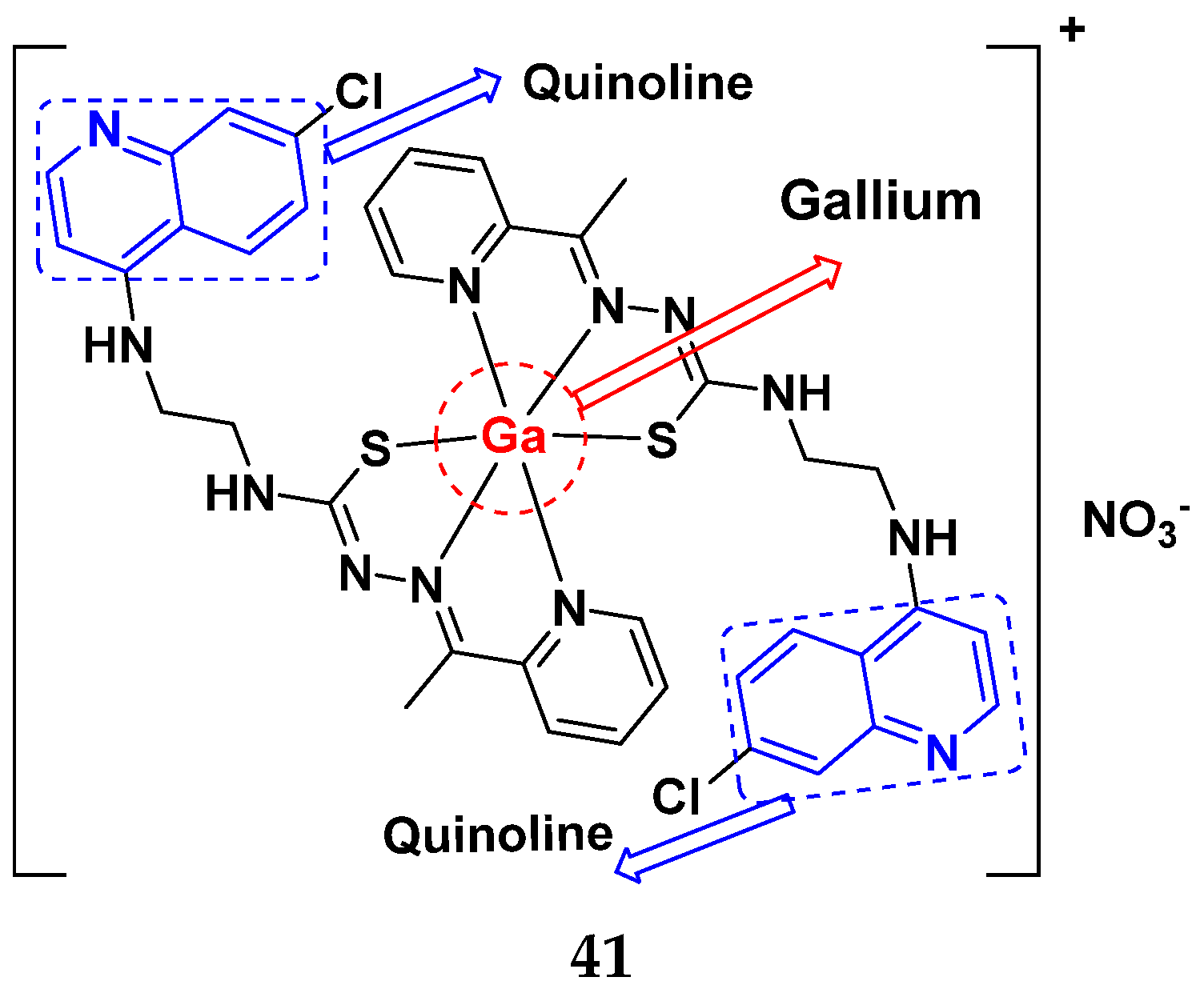{kind=link}
{kind=link}
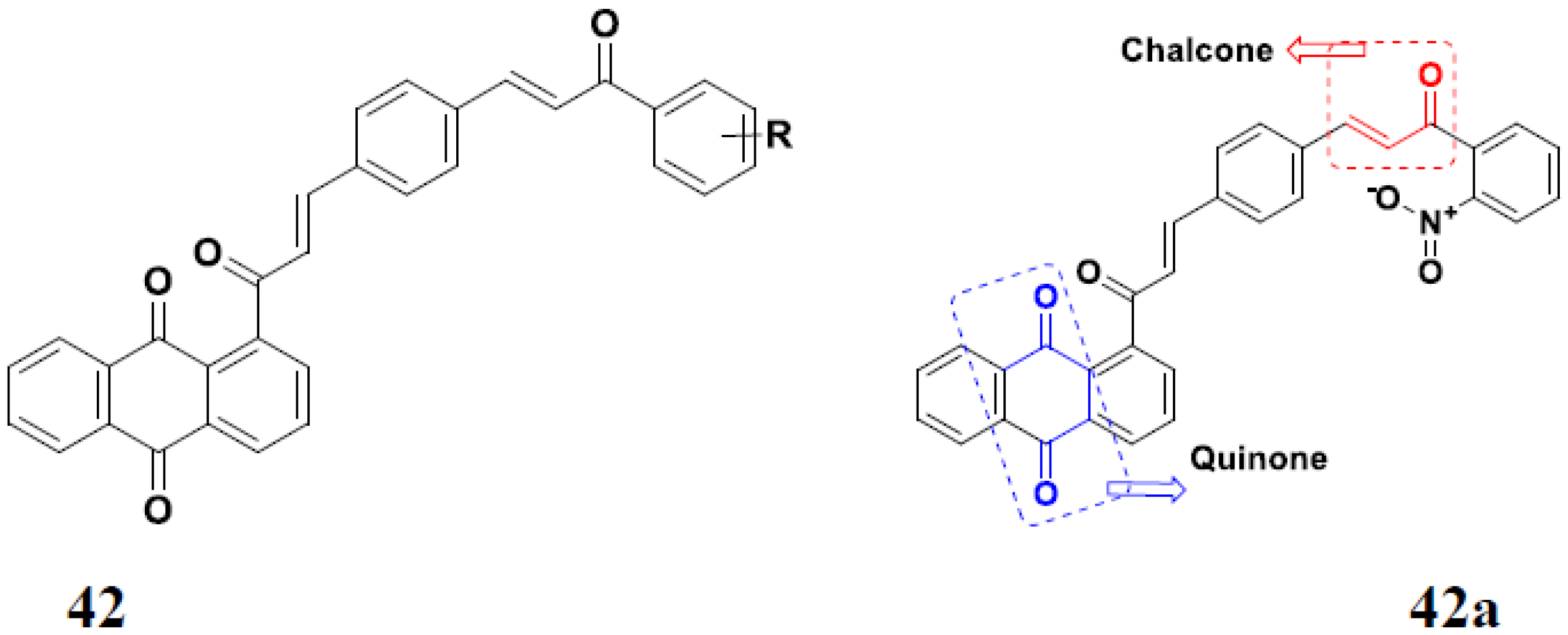{kind=link}
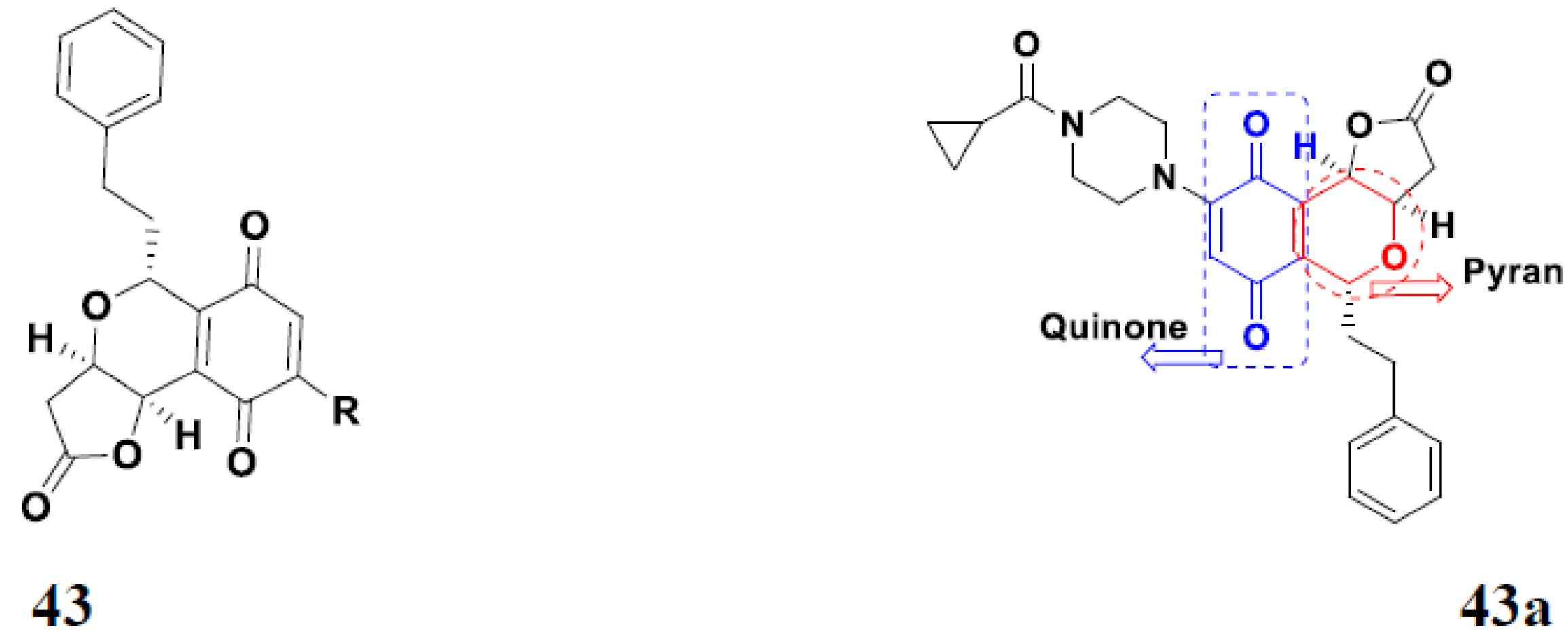{kind=link}
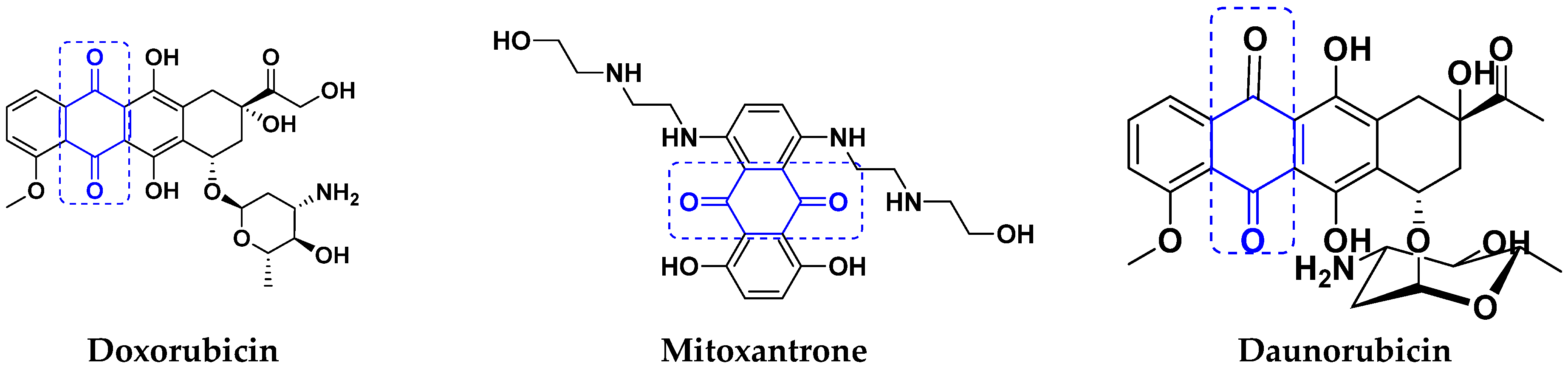{kind=link}
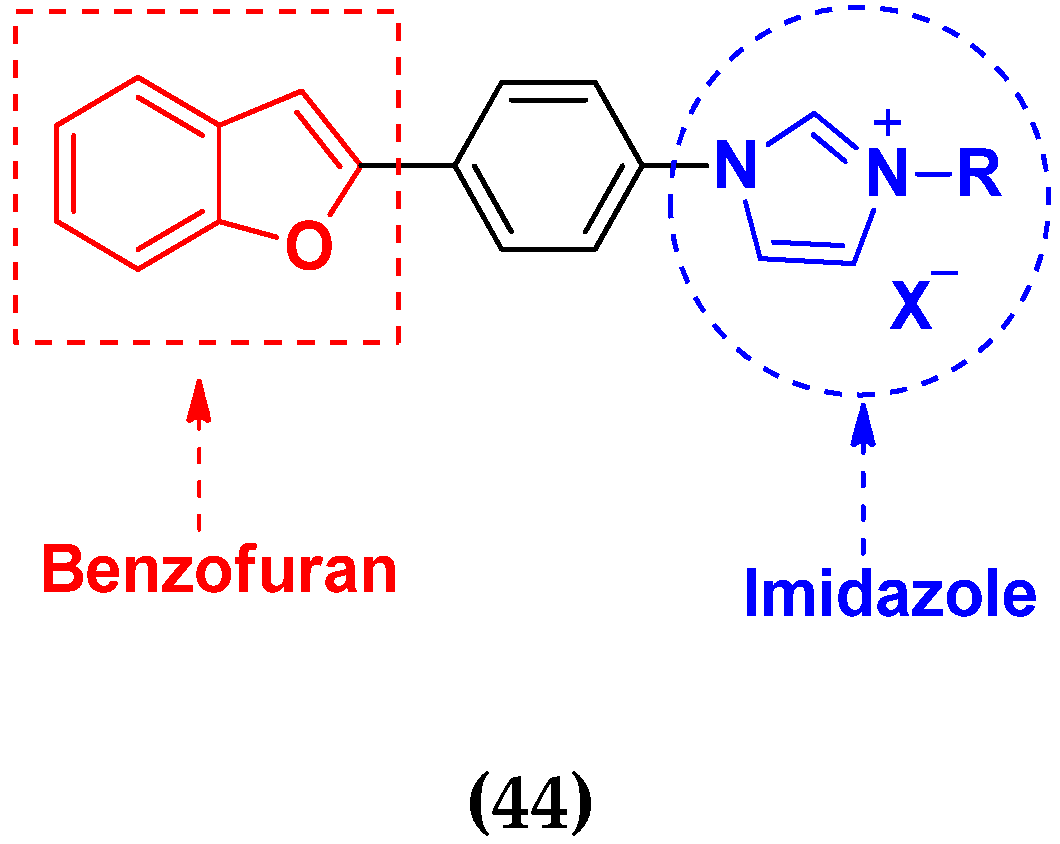{kind=link}
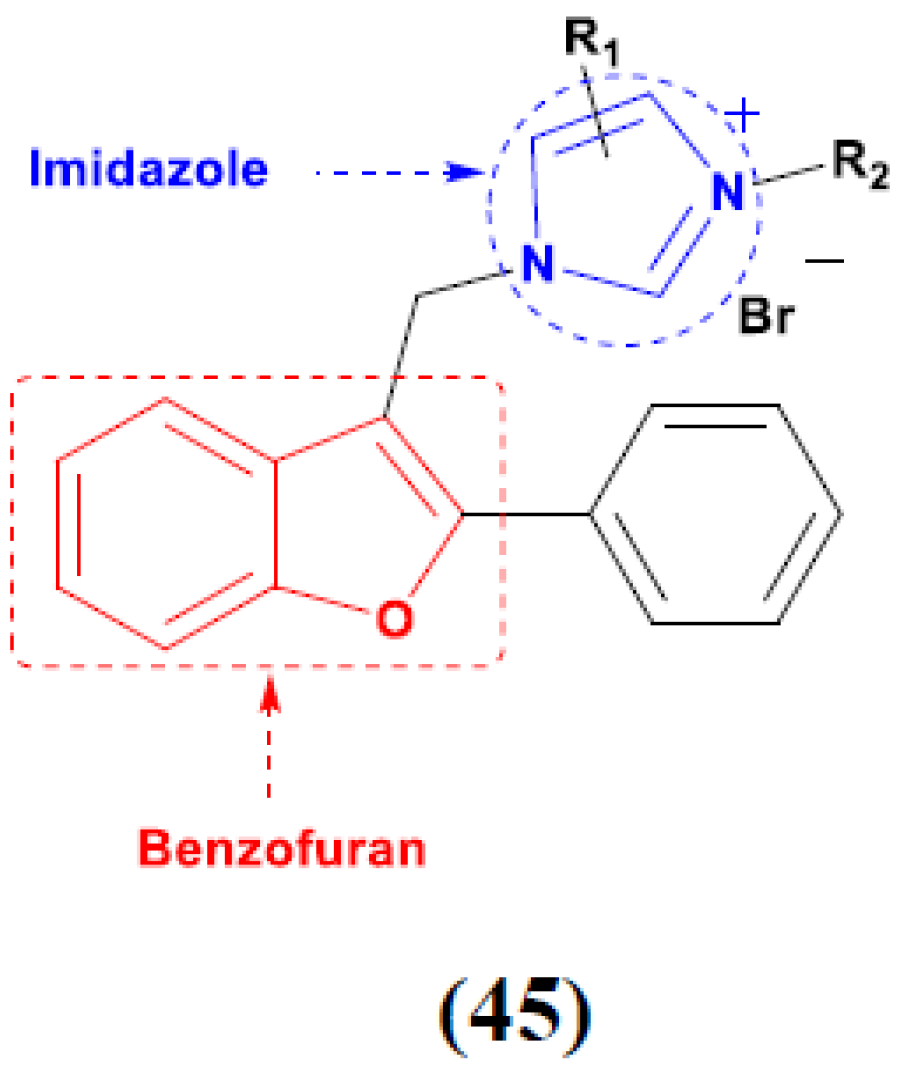{kind=link}
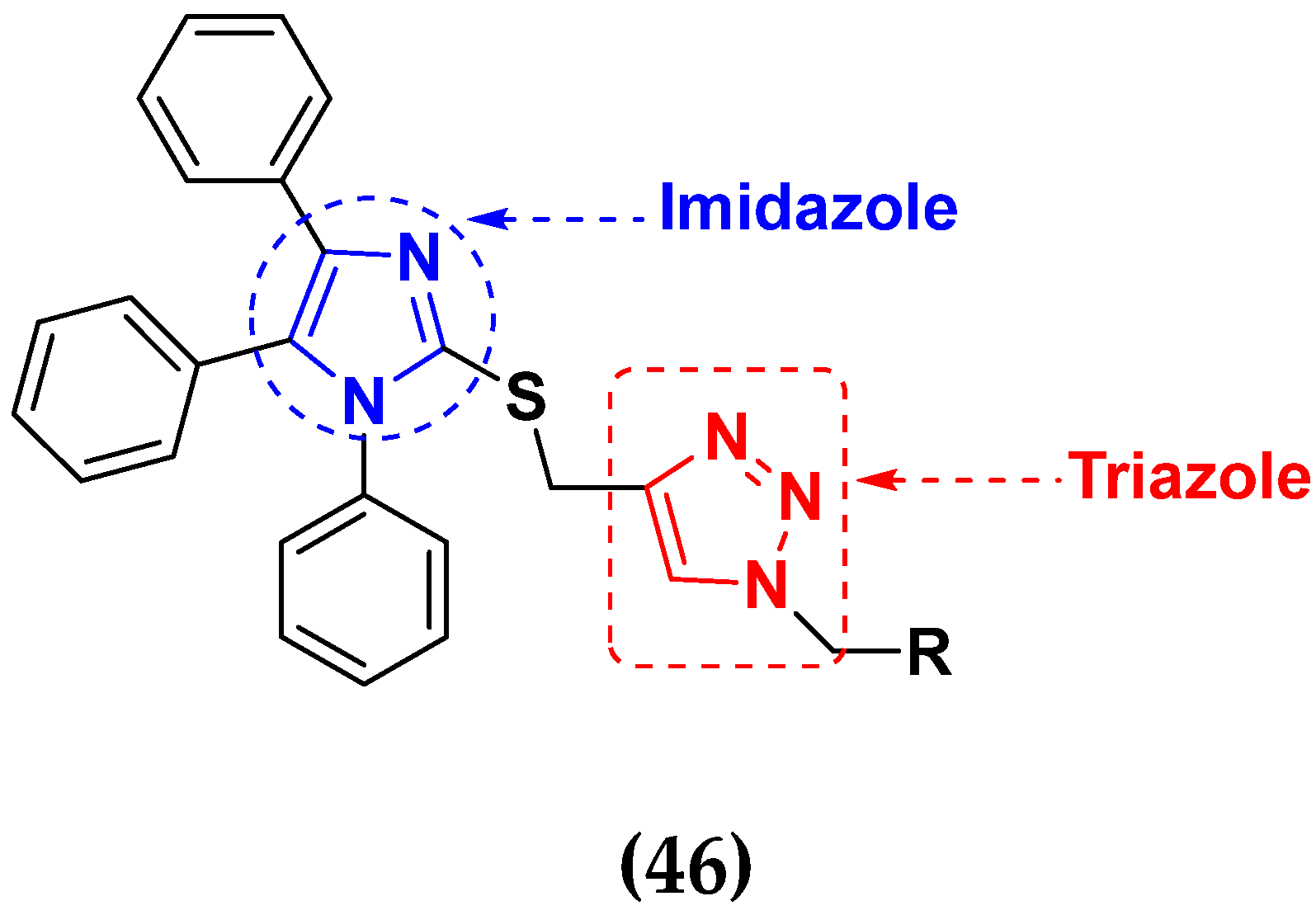{kind=link}
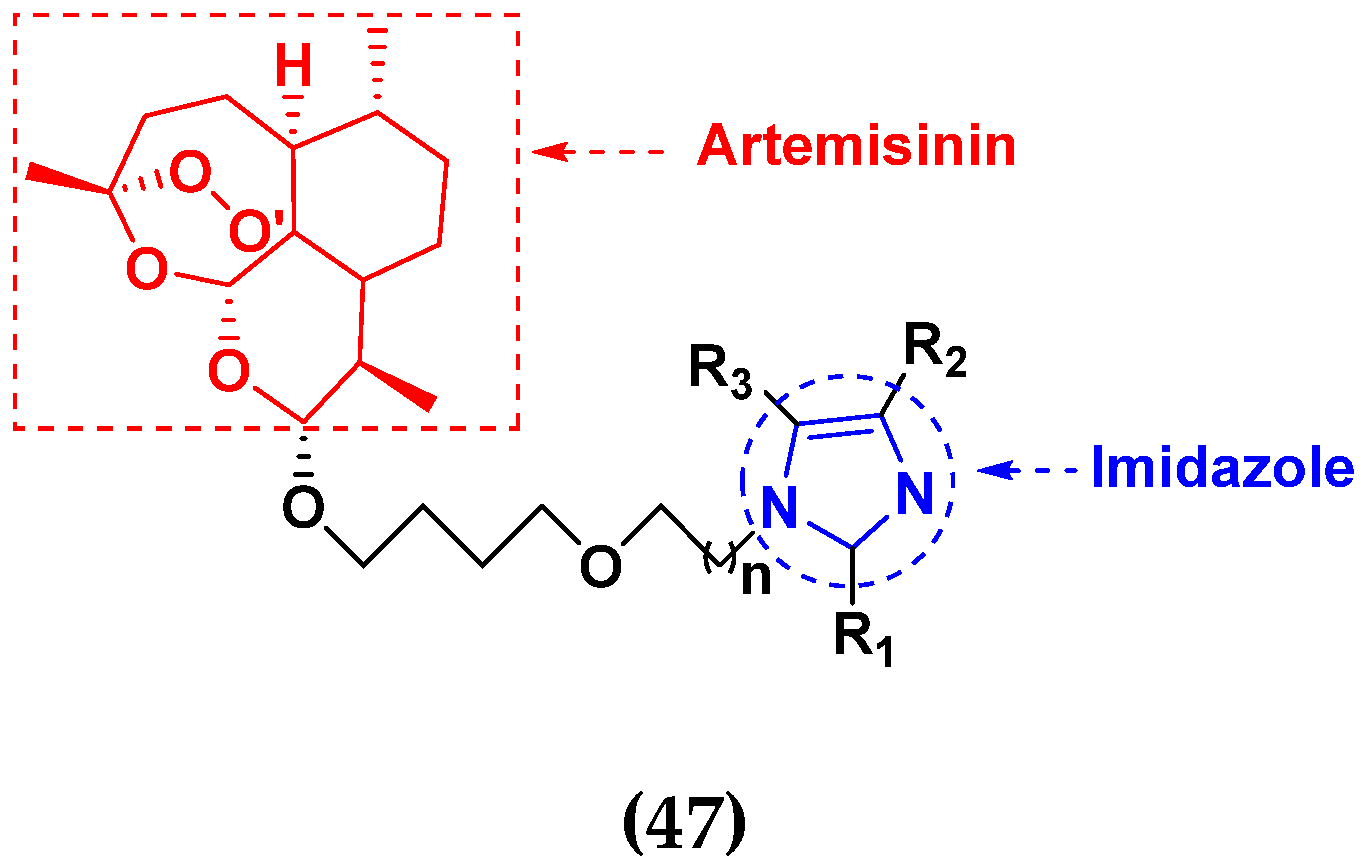{kind=link}
{kind=link}
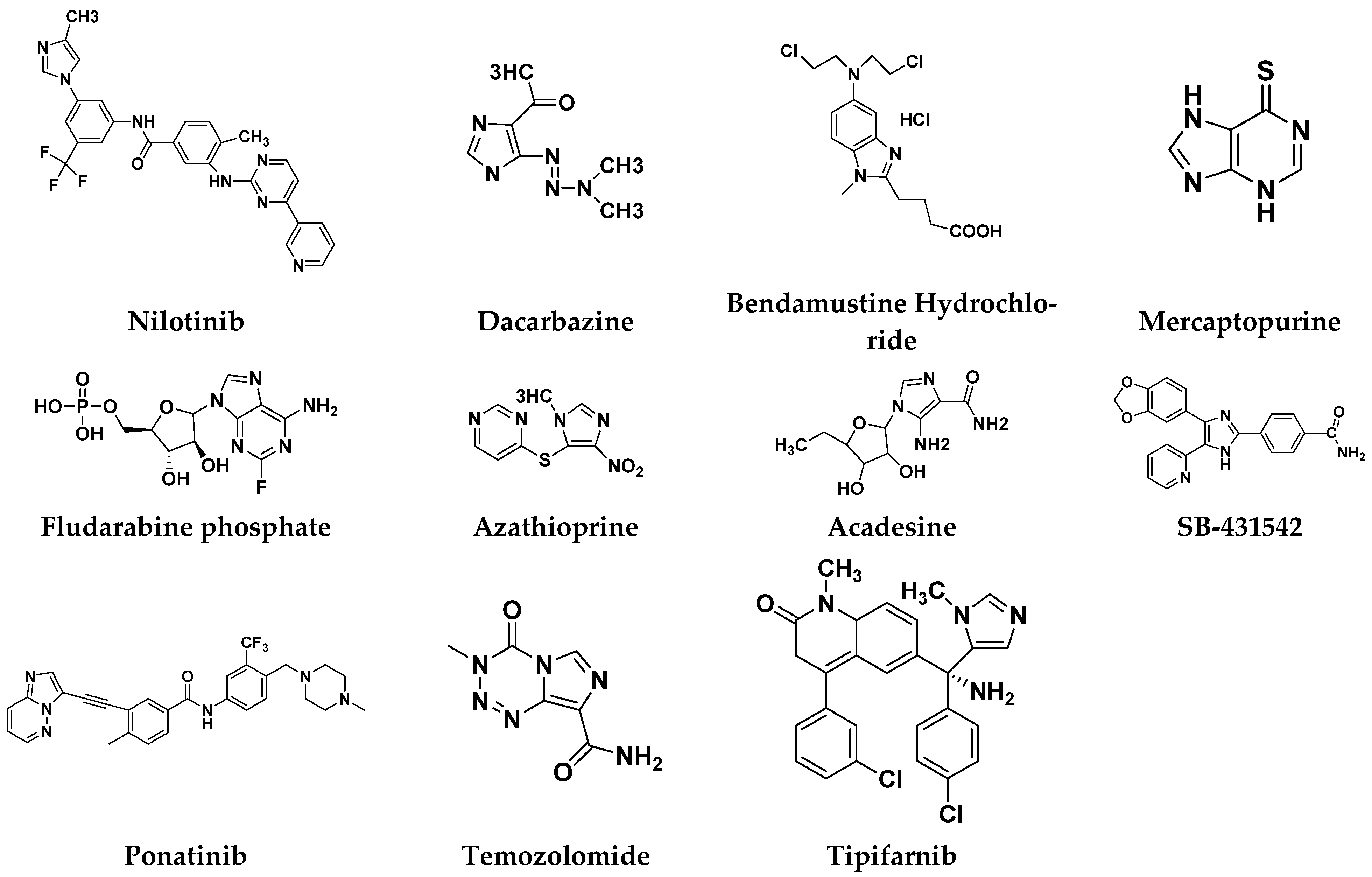{kind=link}
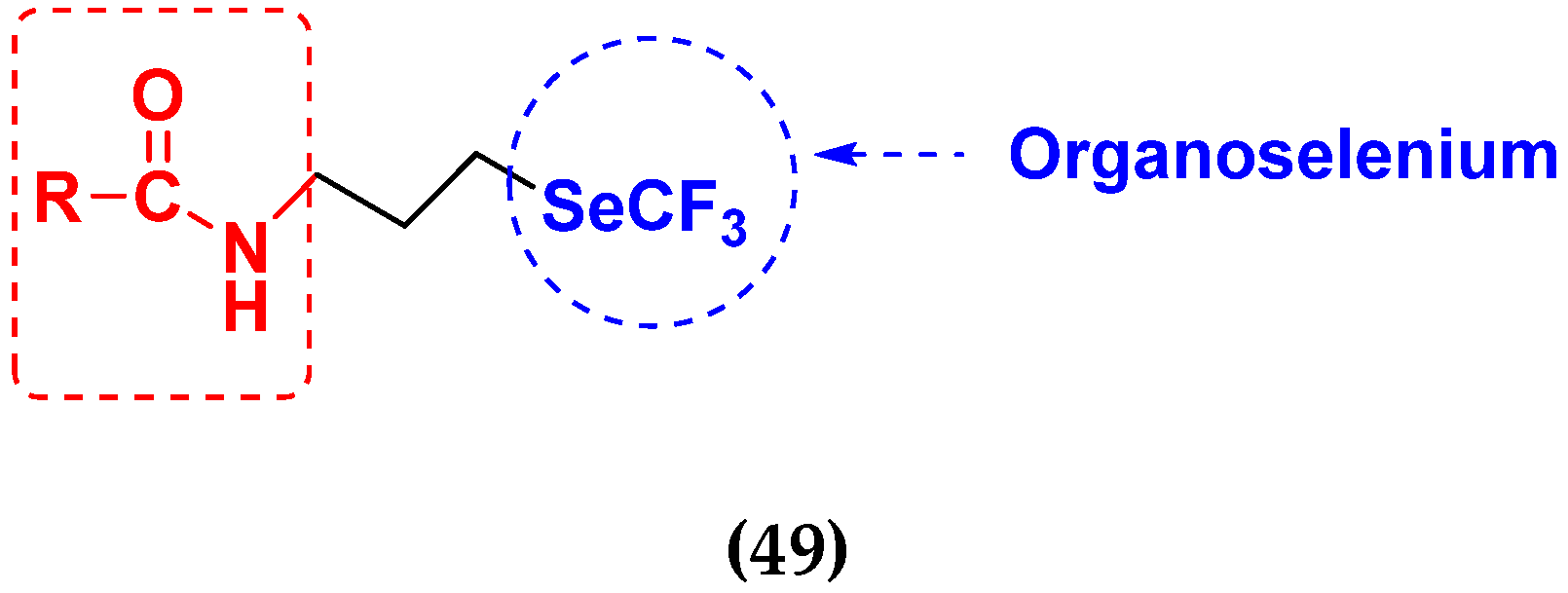{kind=link}
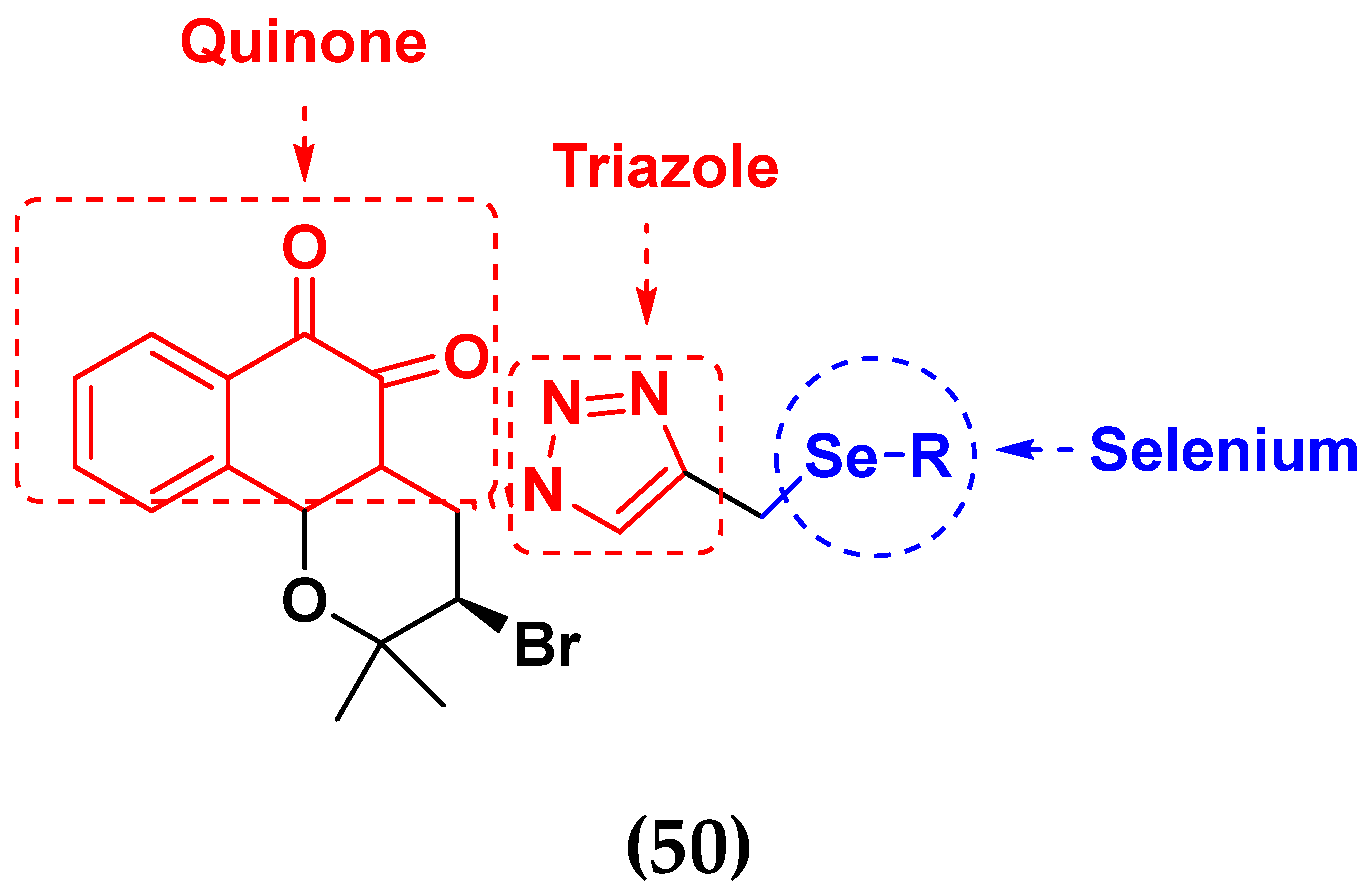{kind=link}
{kind=link}
{kind=link}
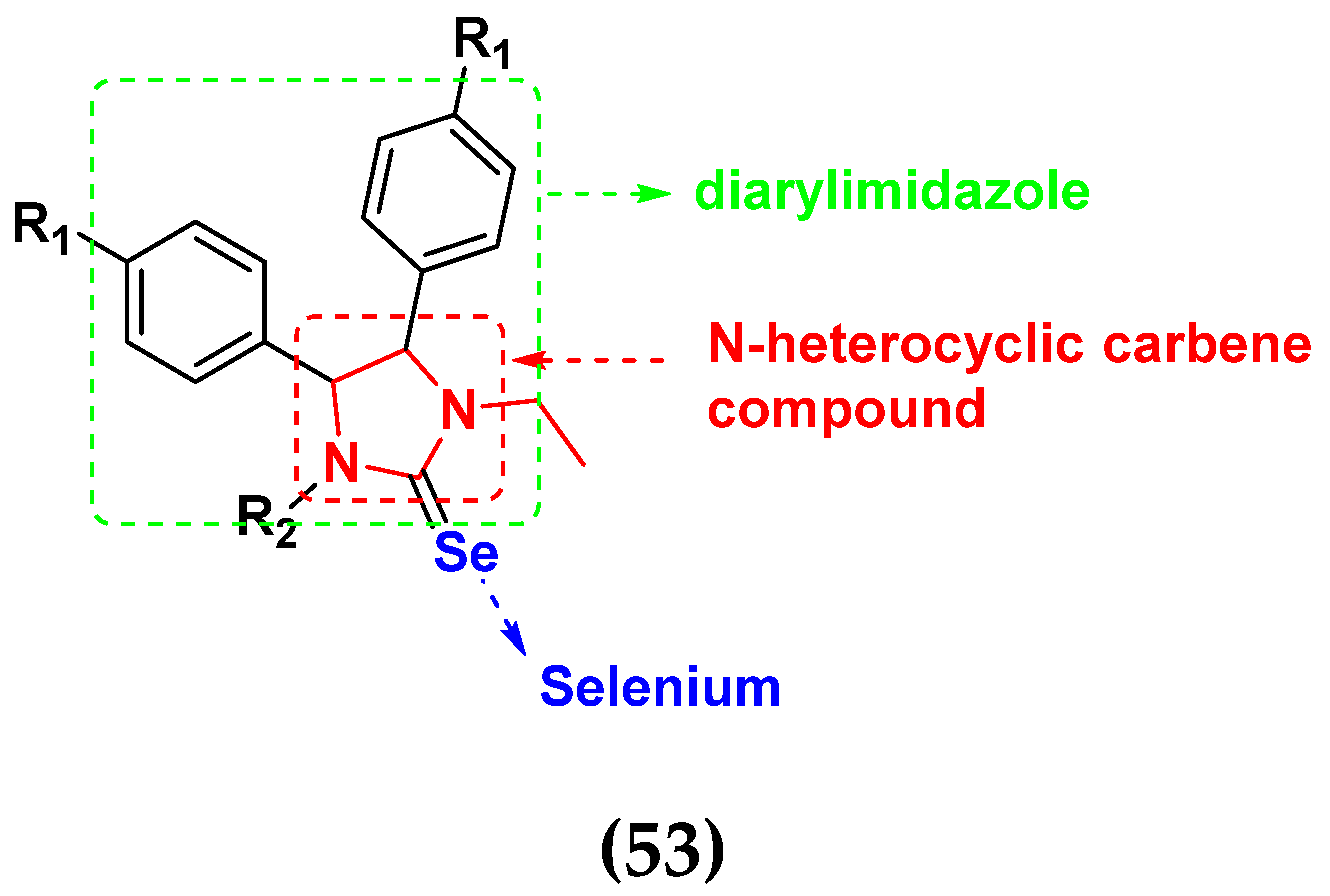{kind=link}
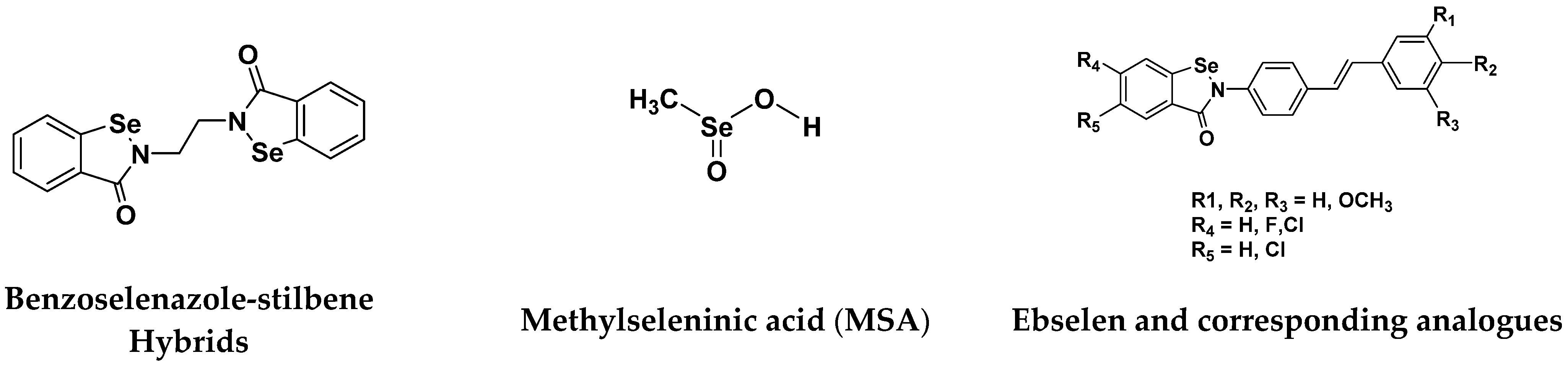{kind=link}
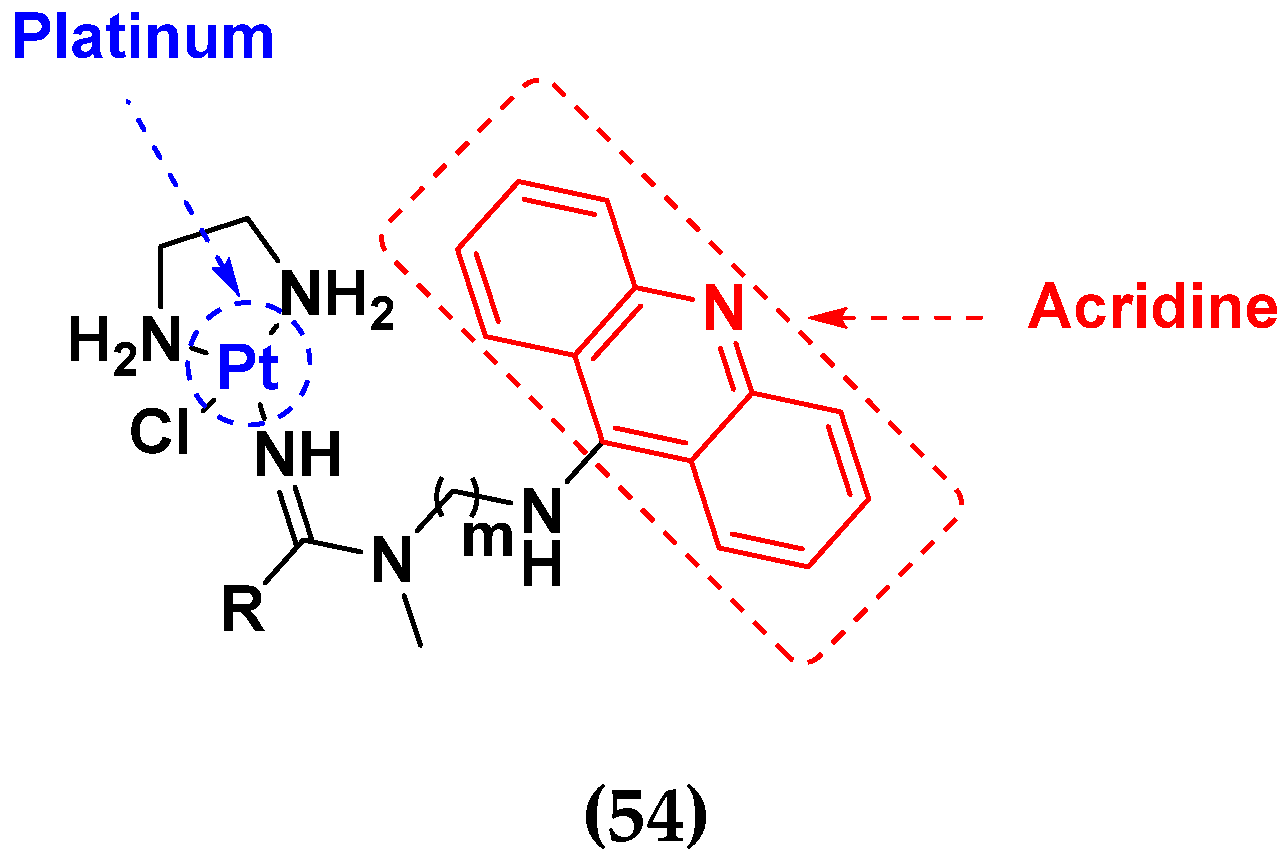{kind=link}
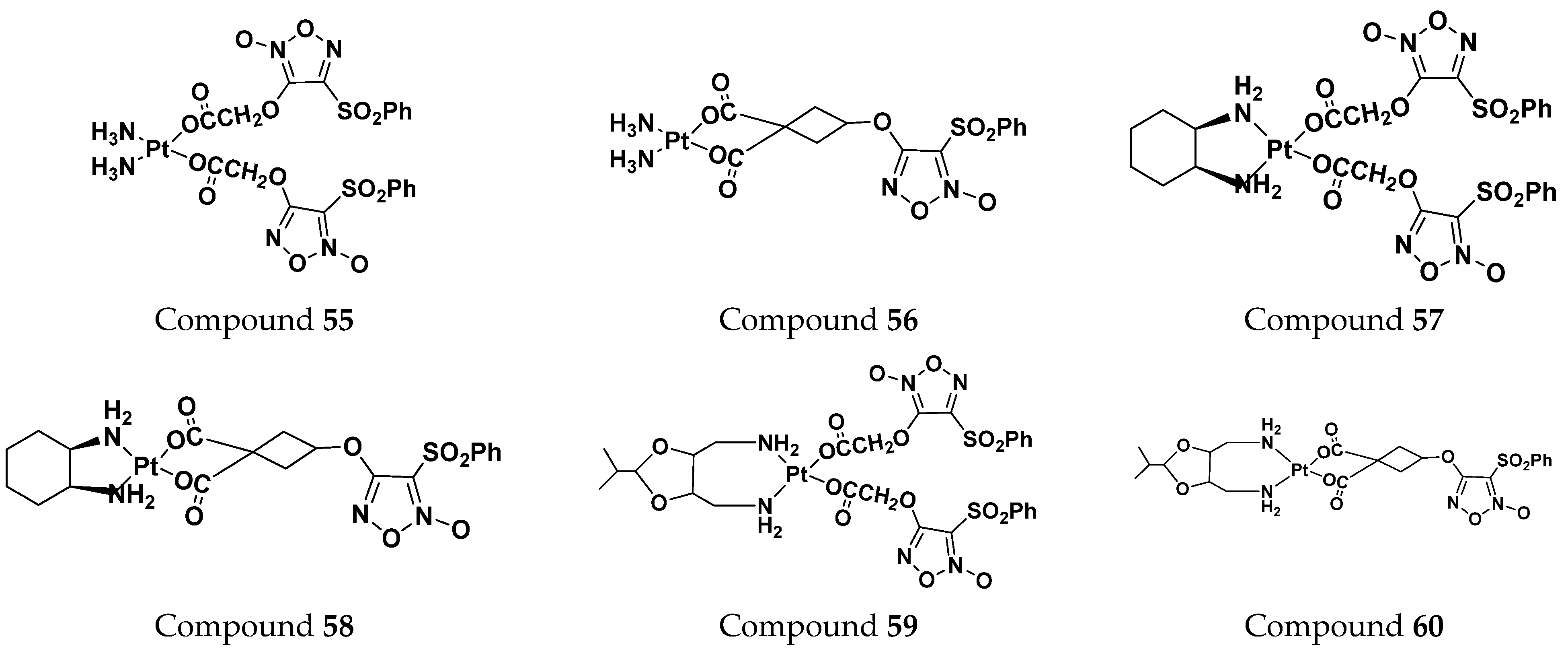{kind=link}
{kind=link}
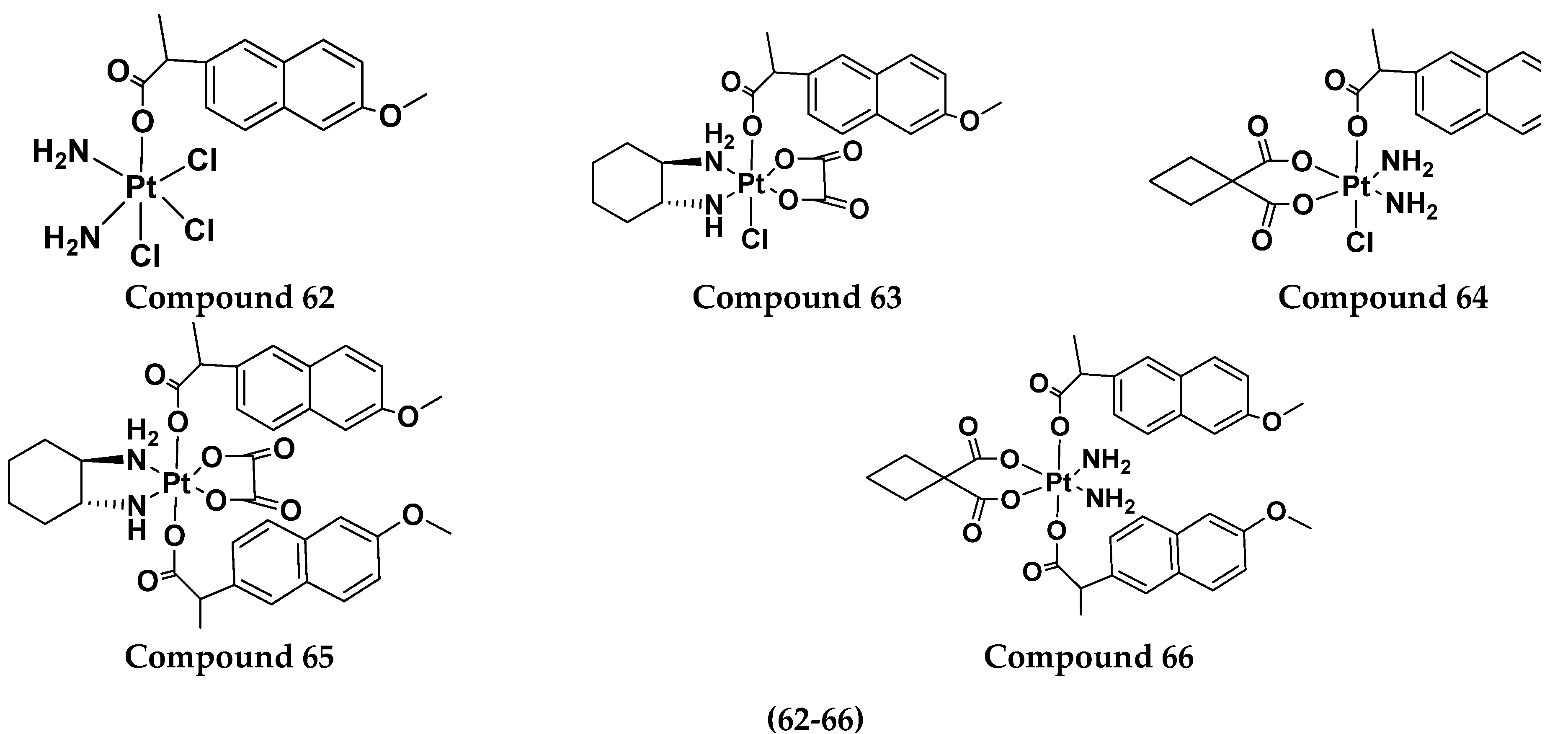{kind=link}
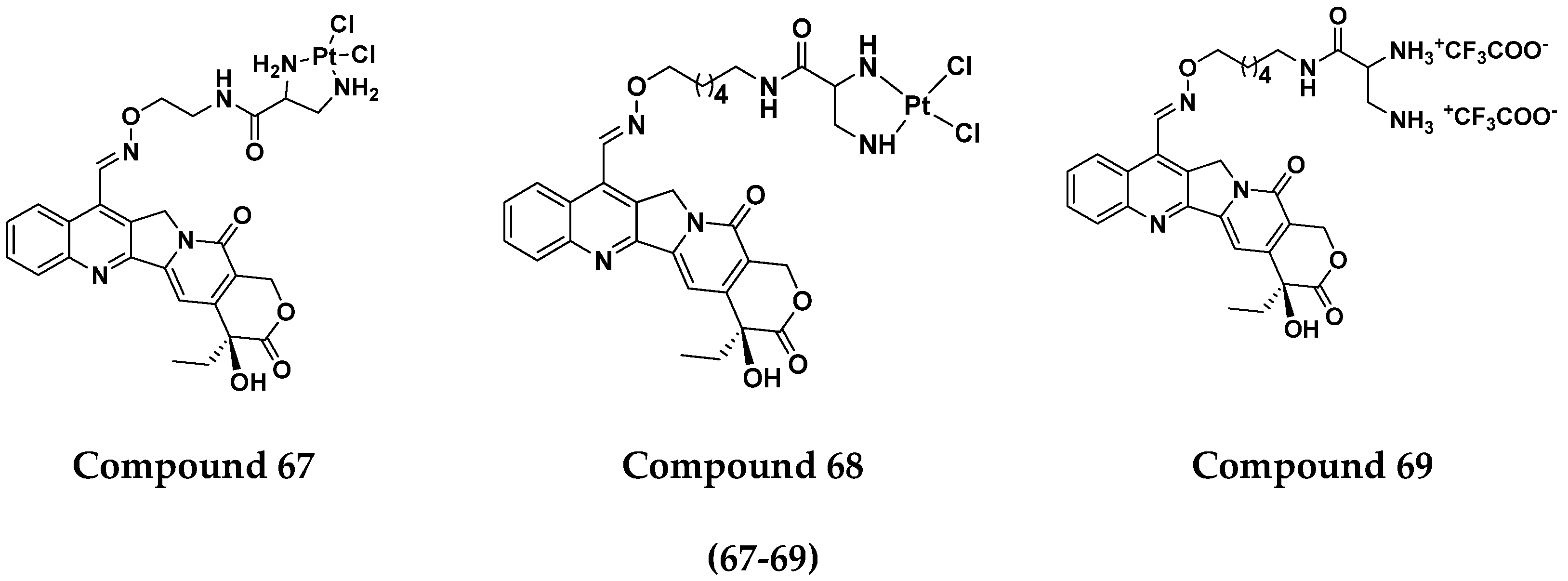{kind=link}
{kind=link}
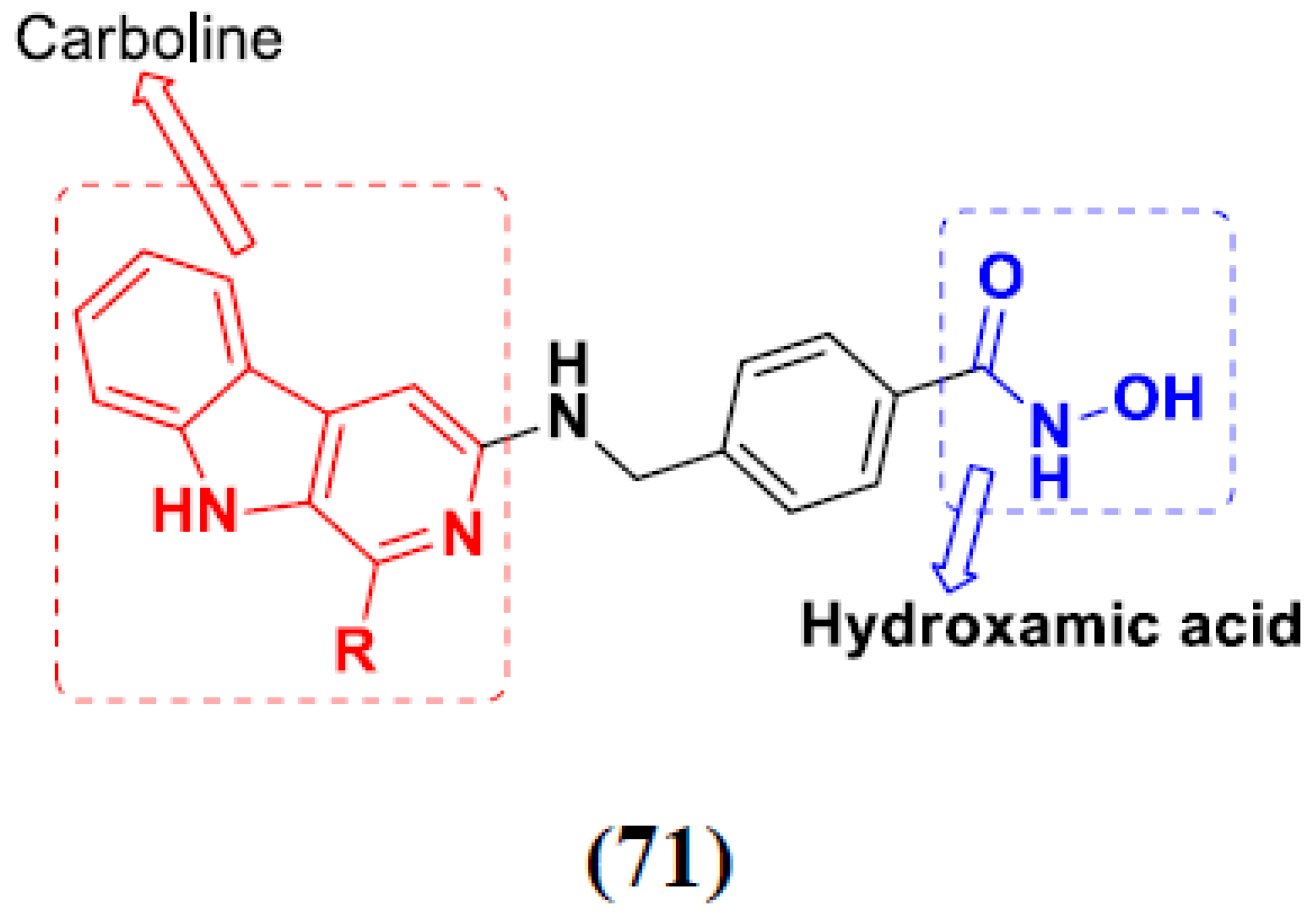{kind=link}
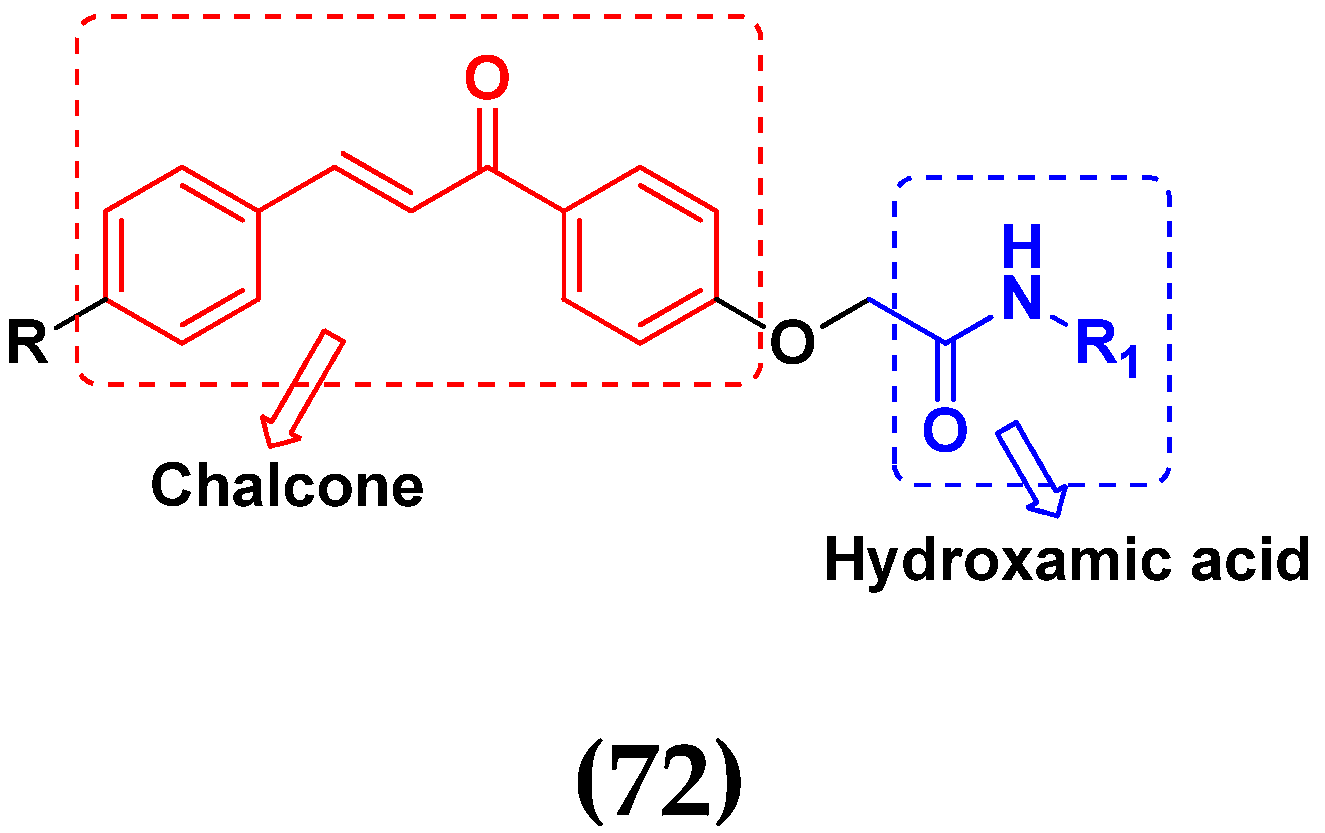{kind=link}
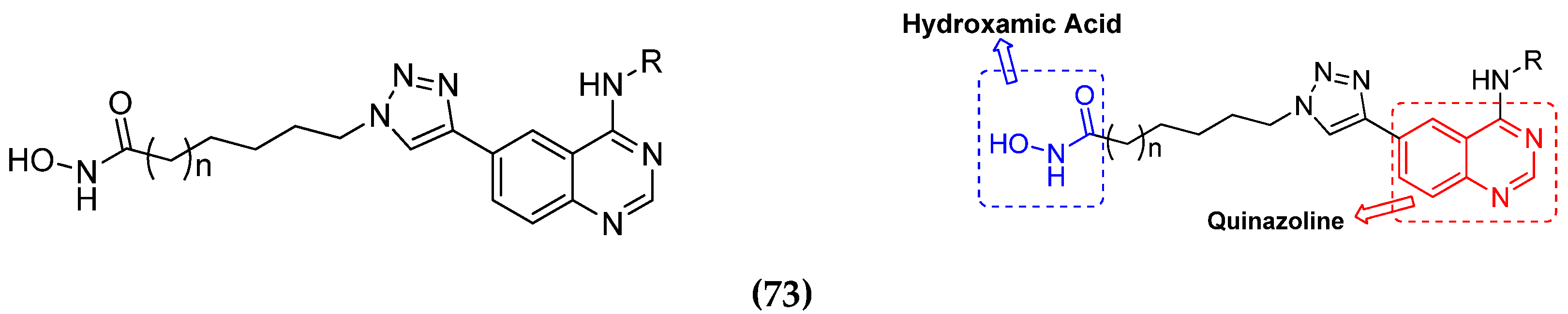{kind=link}
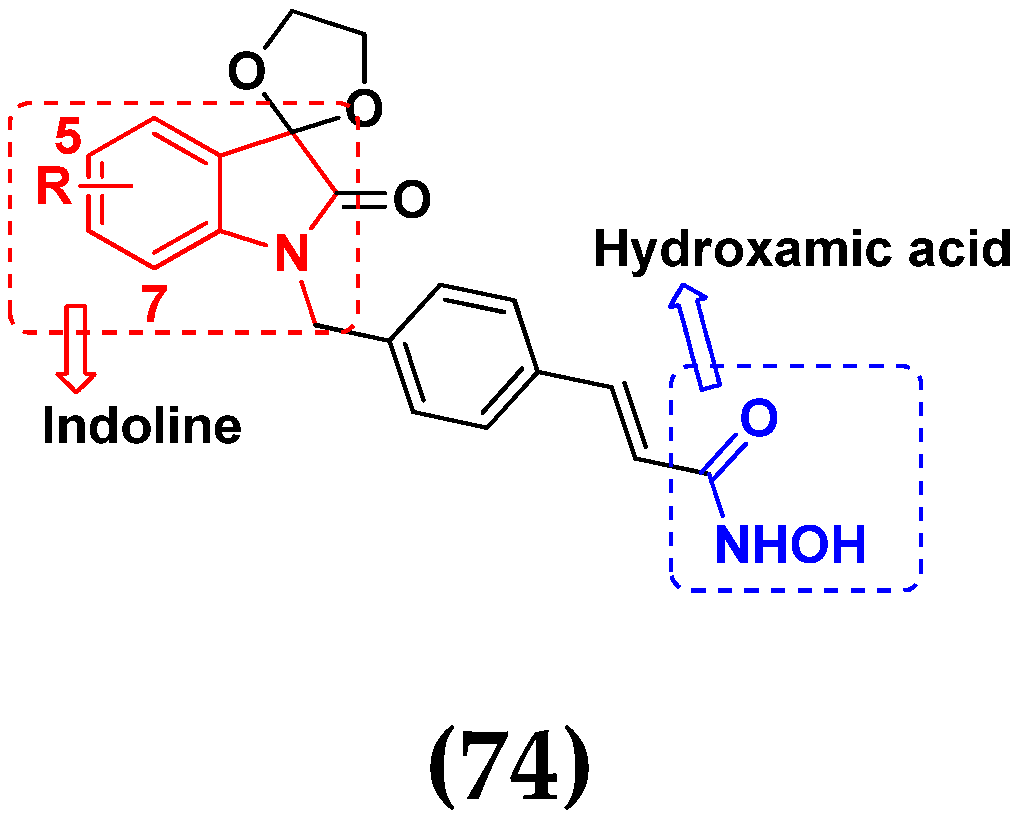{kind=link}
{kind=link}
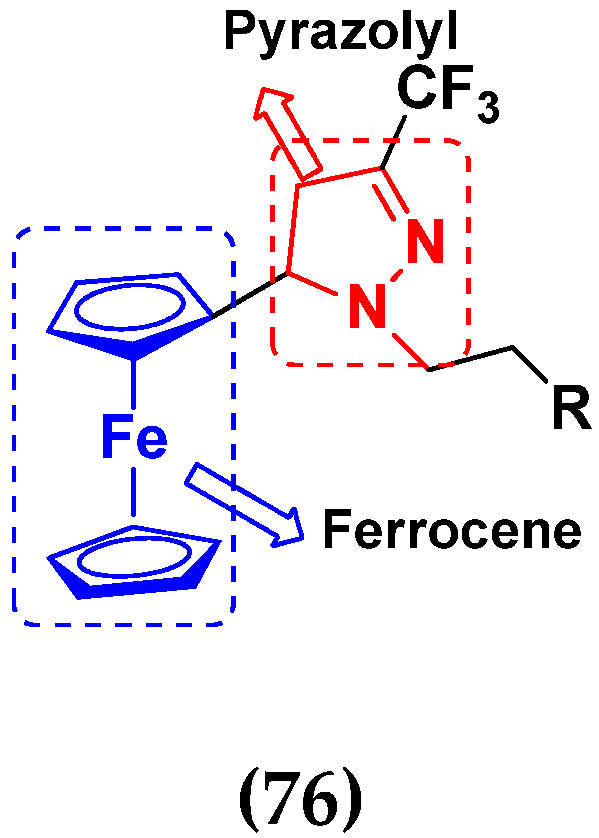{kind=link}
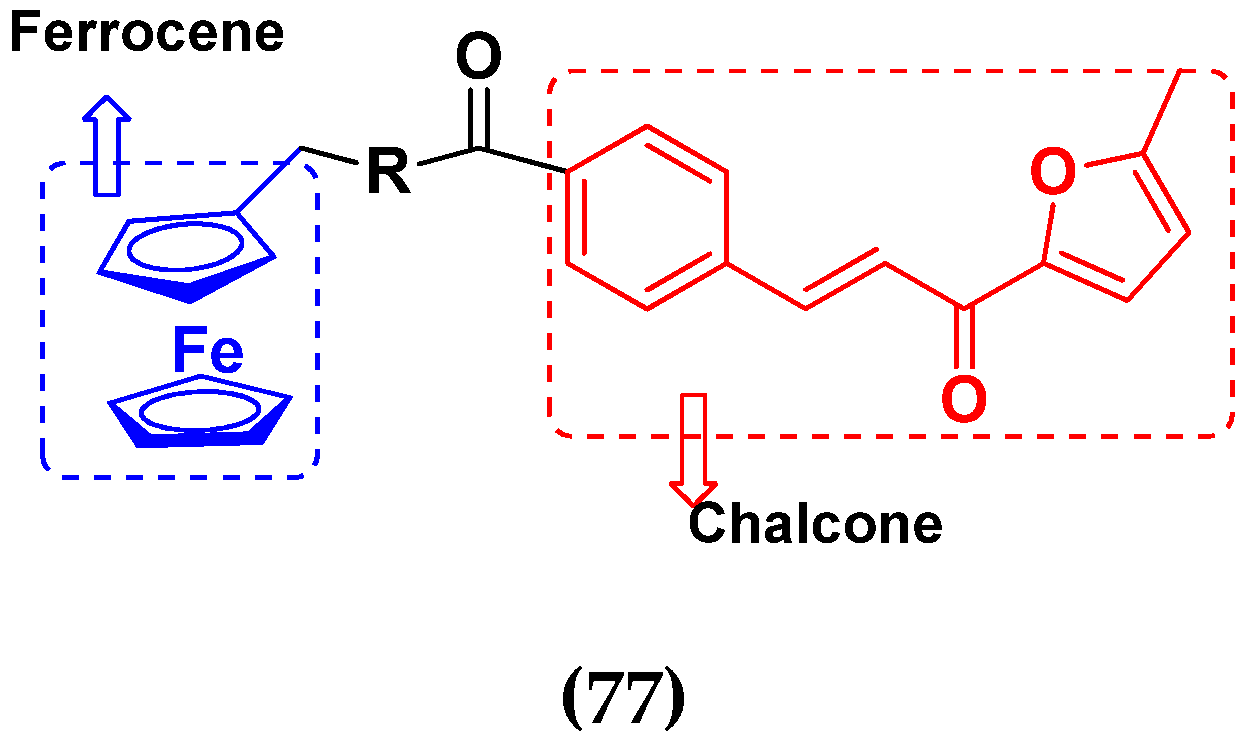{kind=link}
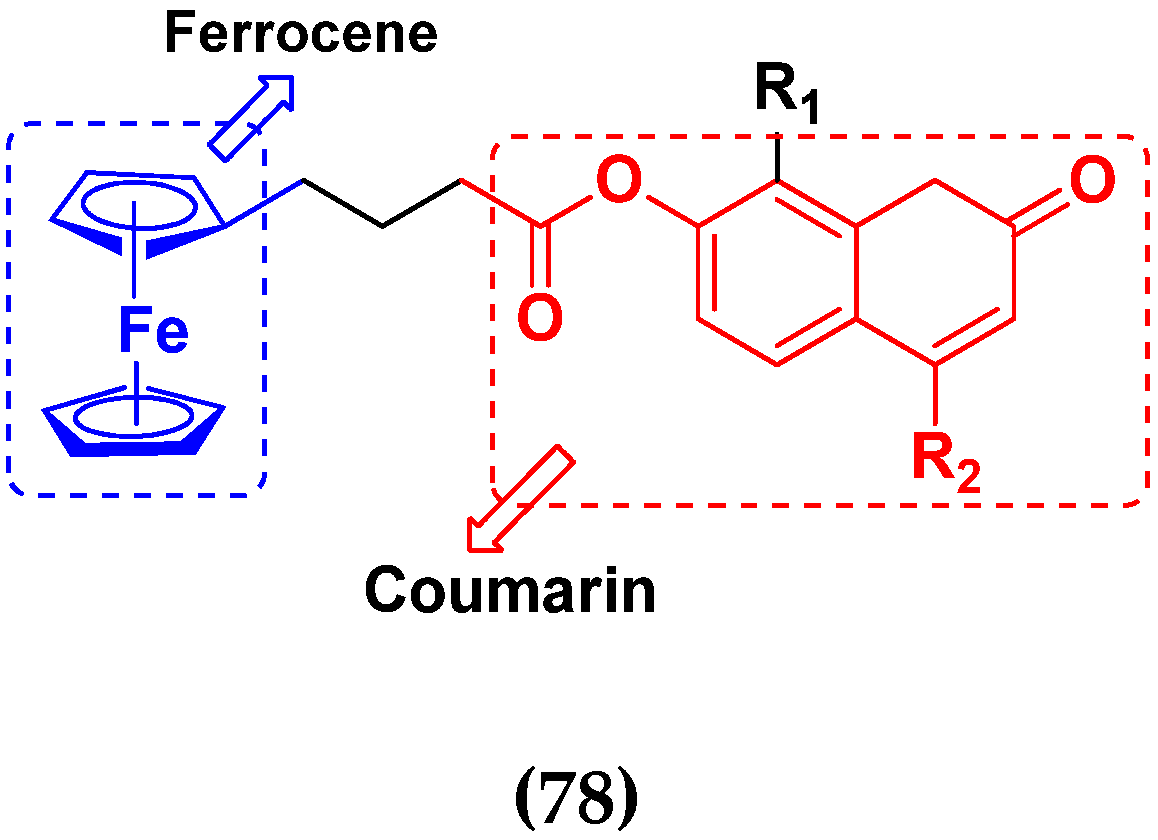{kind=link}
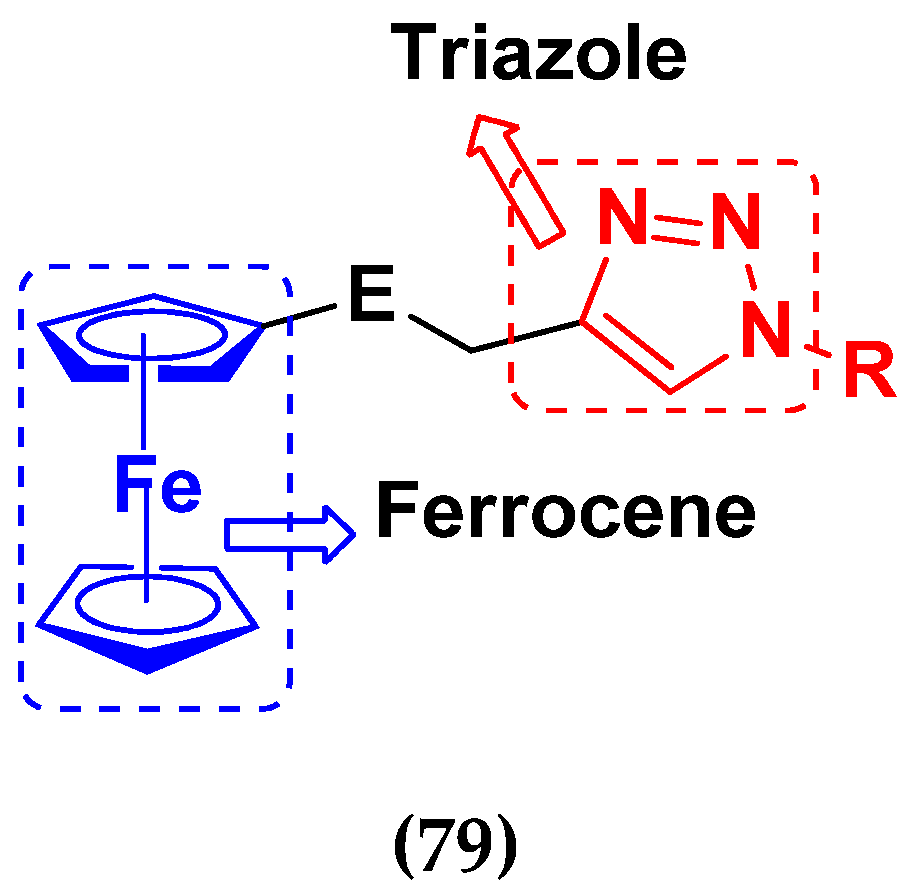{kind=link}
{kind=link}
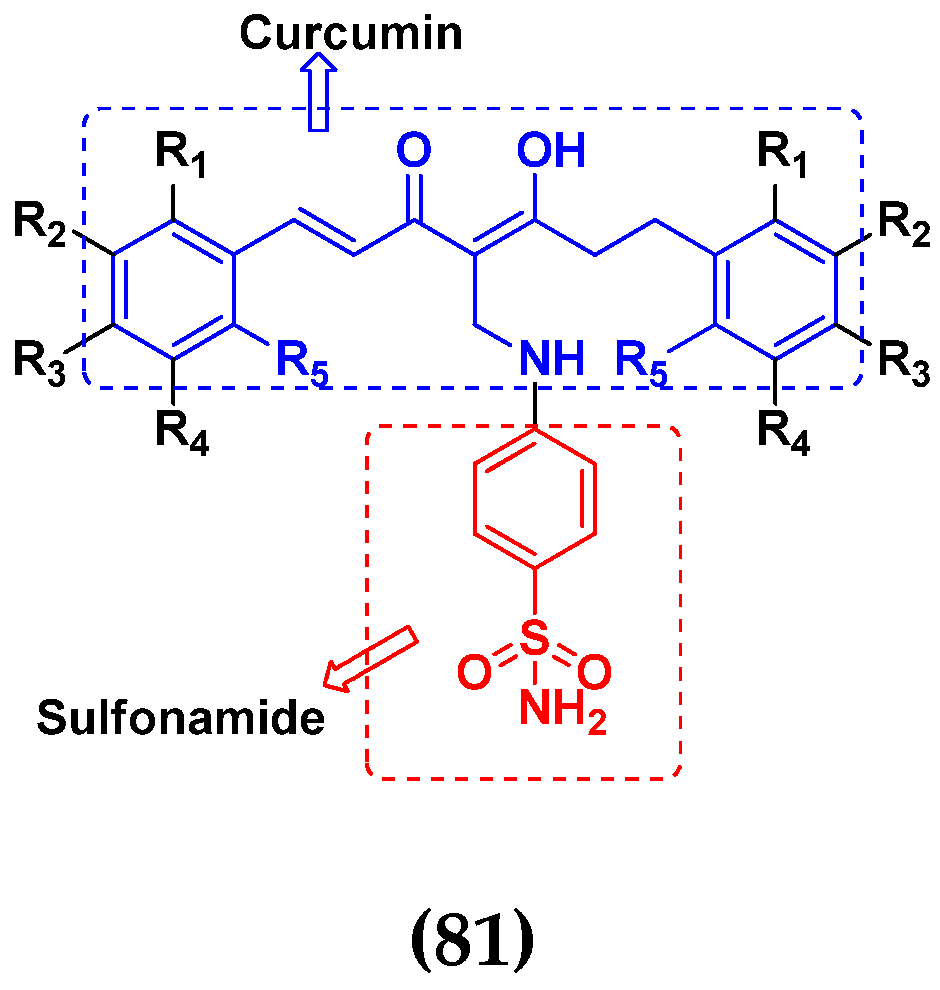{kind=link}
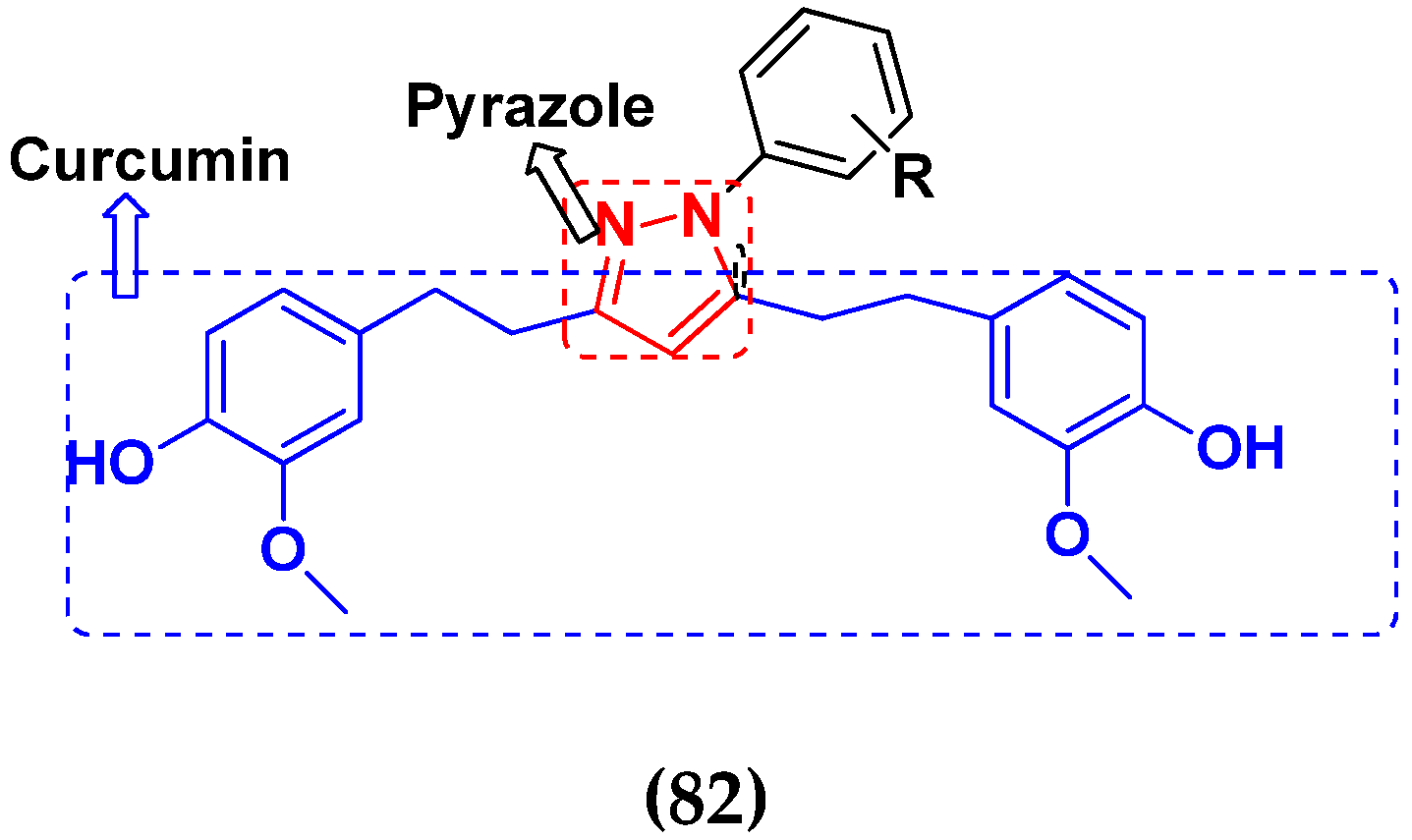{kind=link}
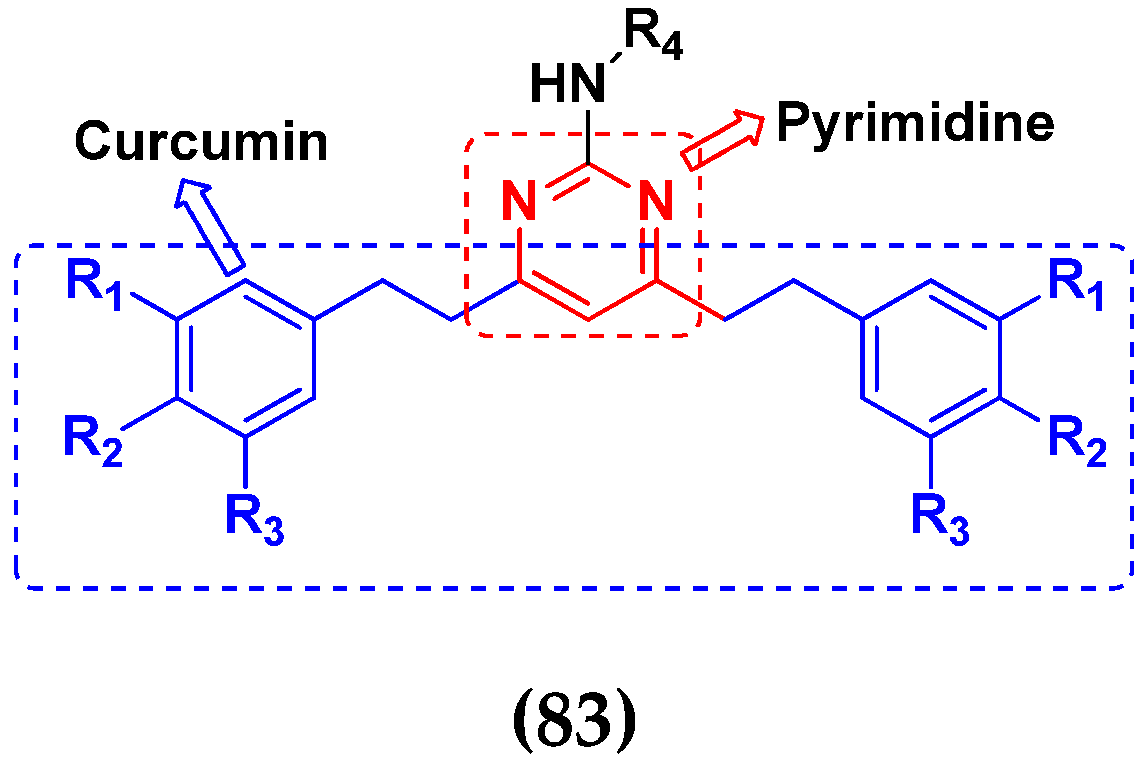{kind=link}
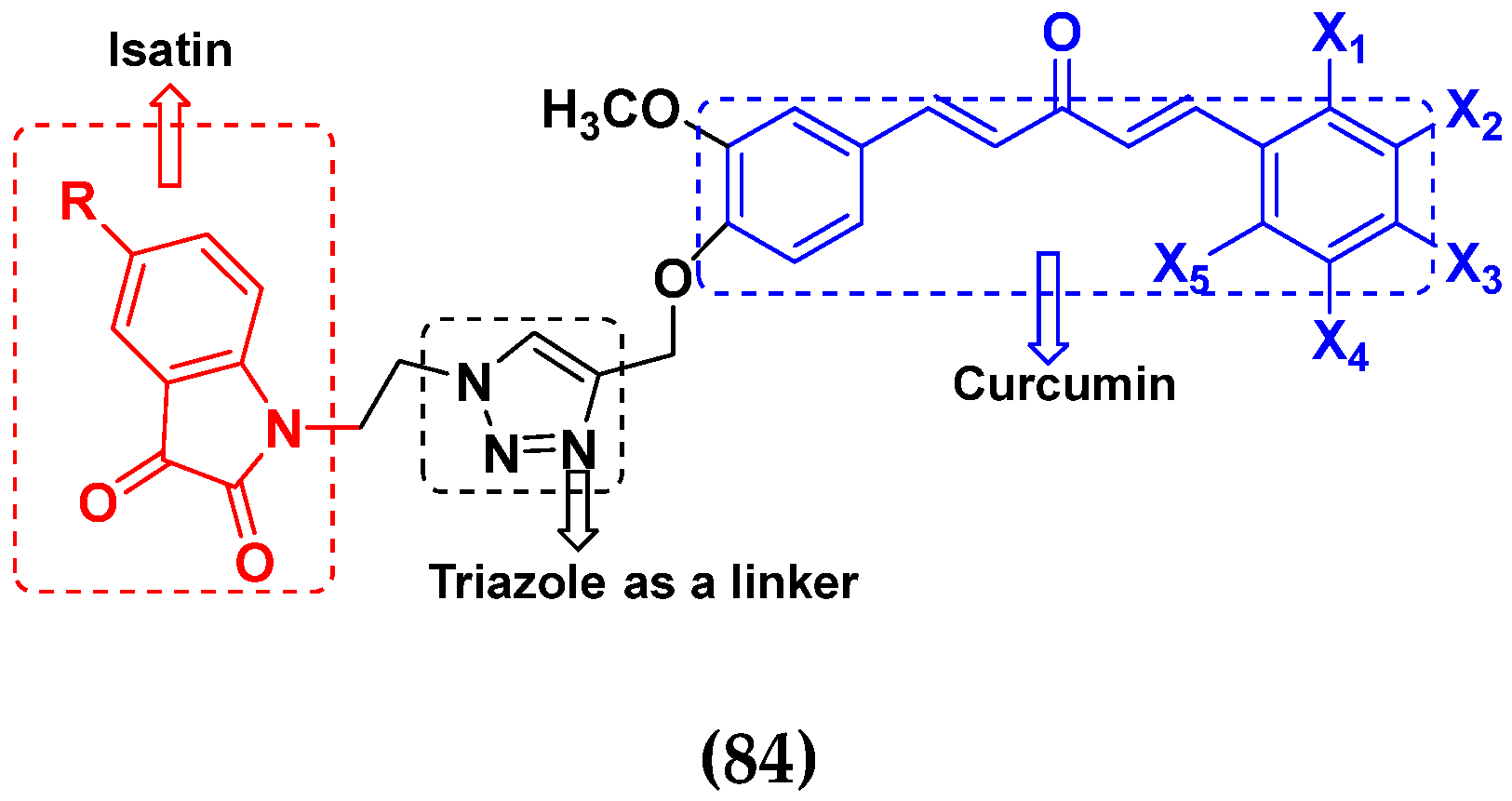{kind=link}
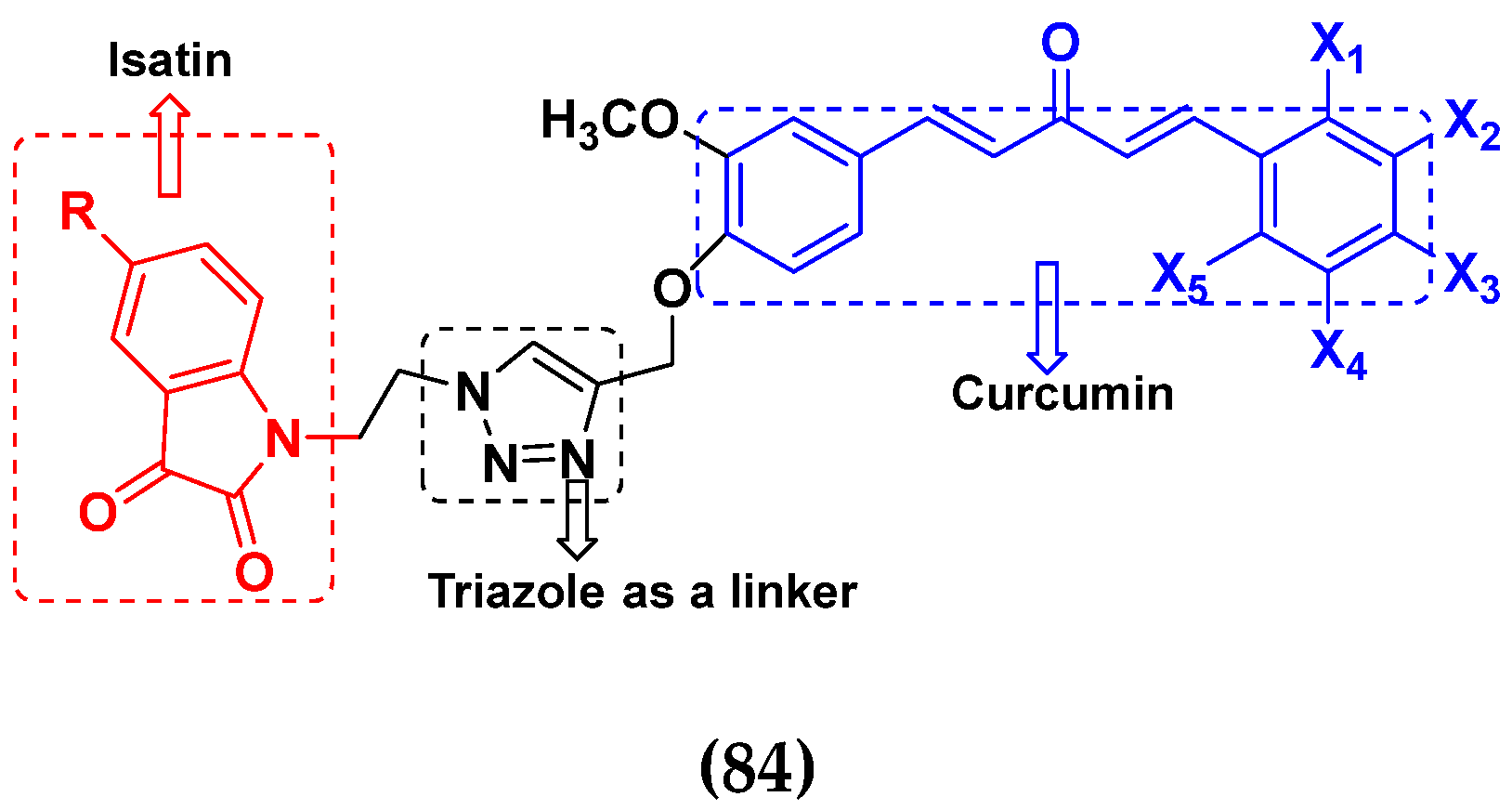{kind=link}
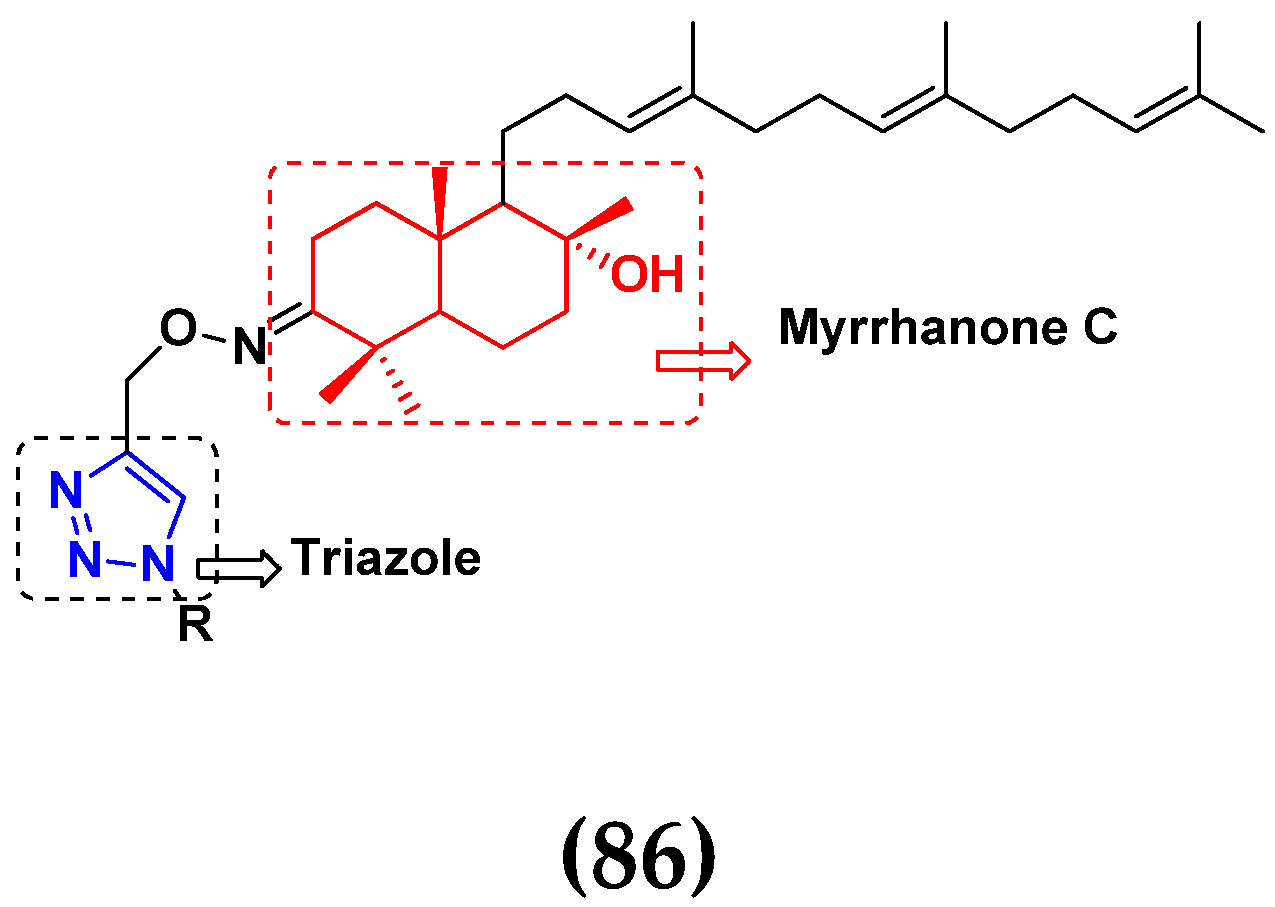{kind=link}
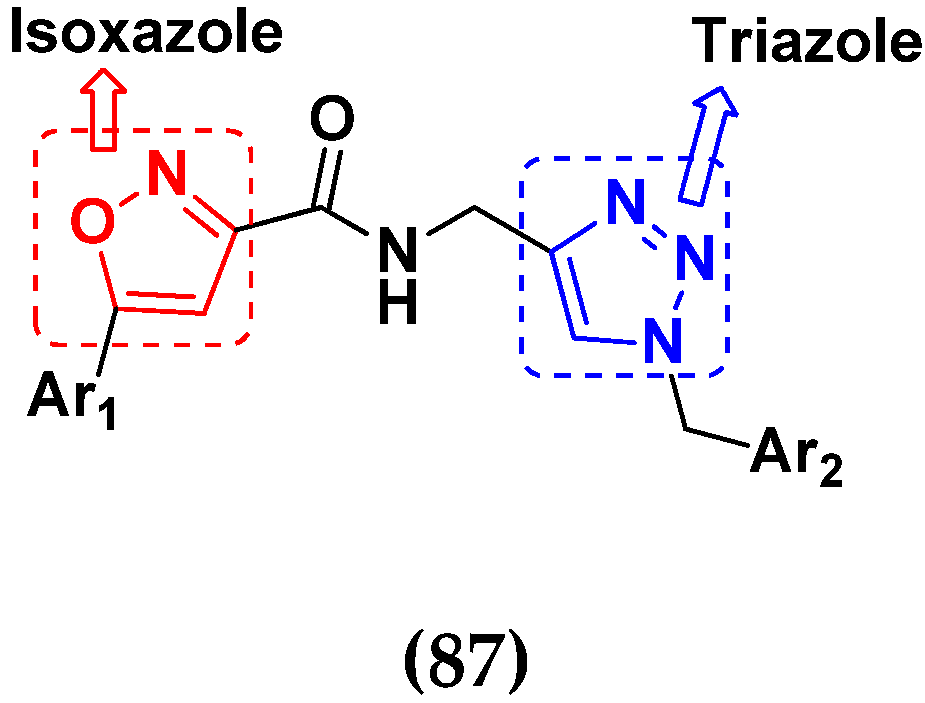{kind=link}
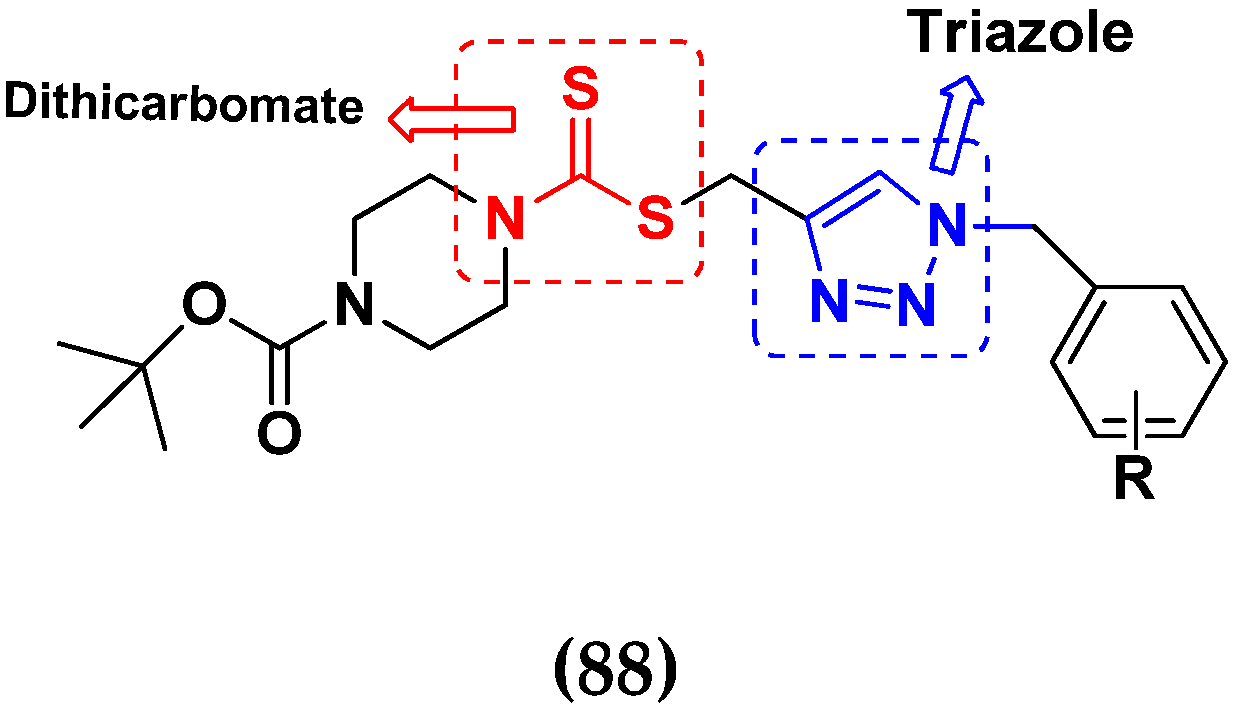{kind=link}
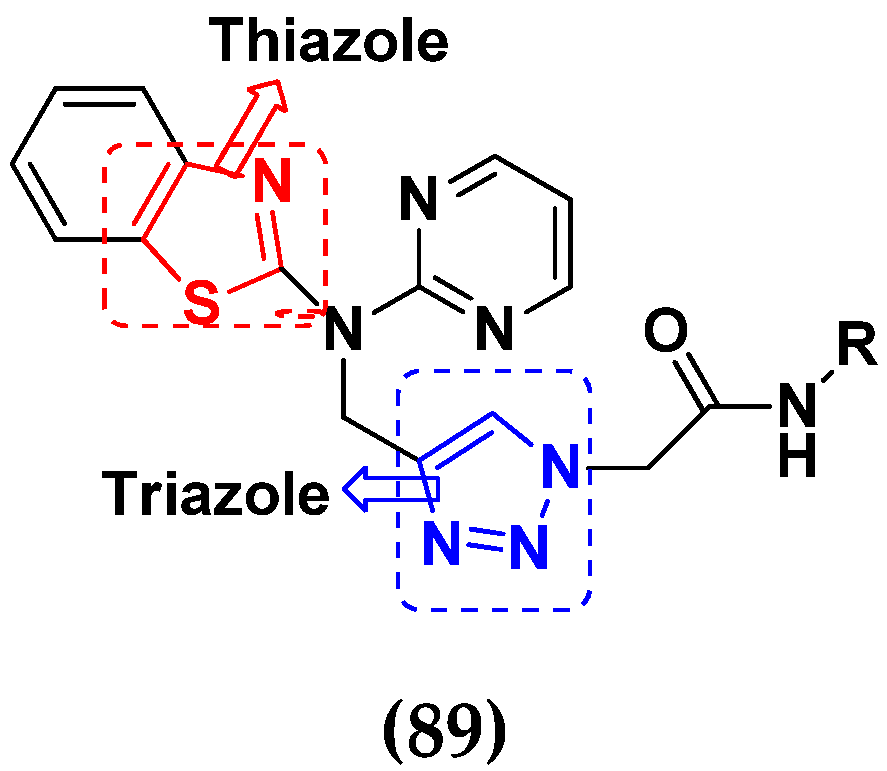{kind=link}
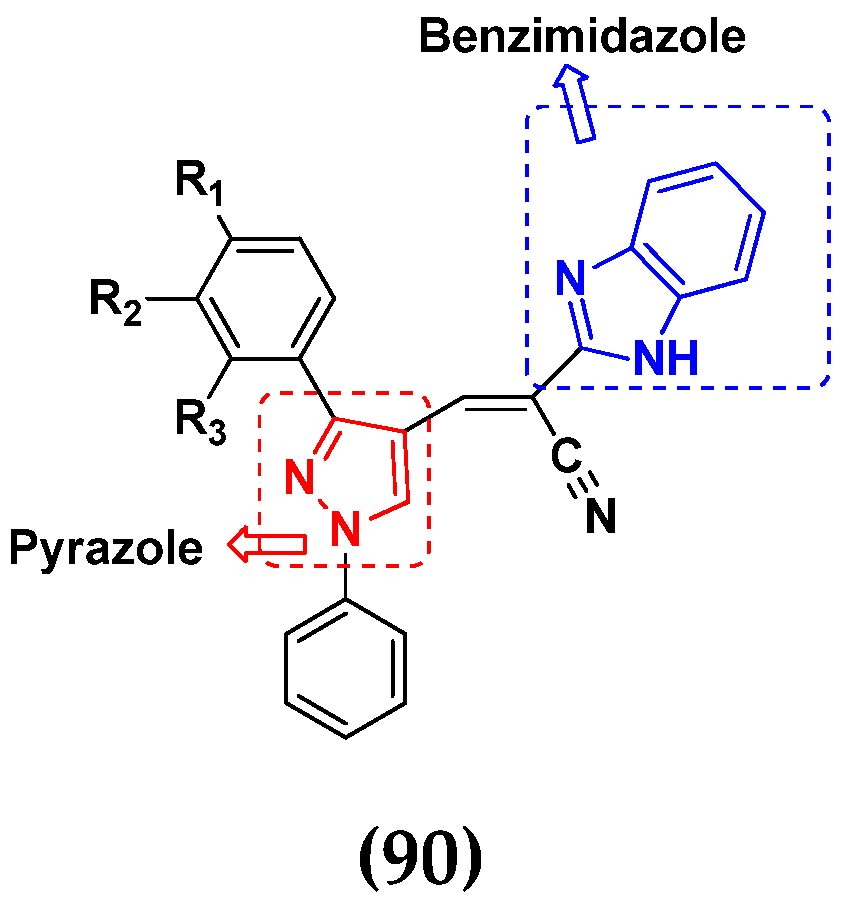{kind=link}
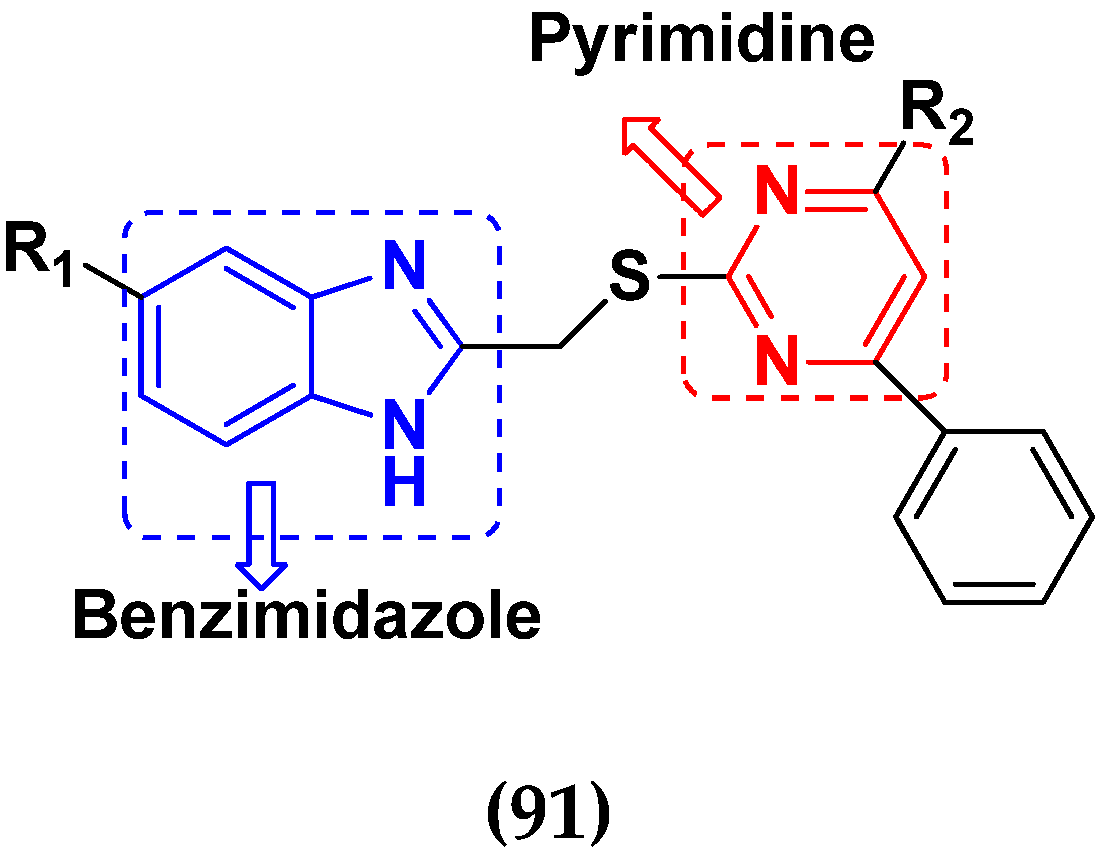{kind=link}
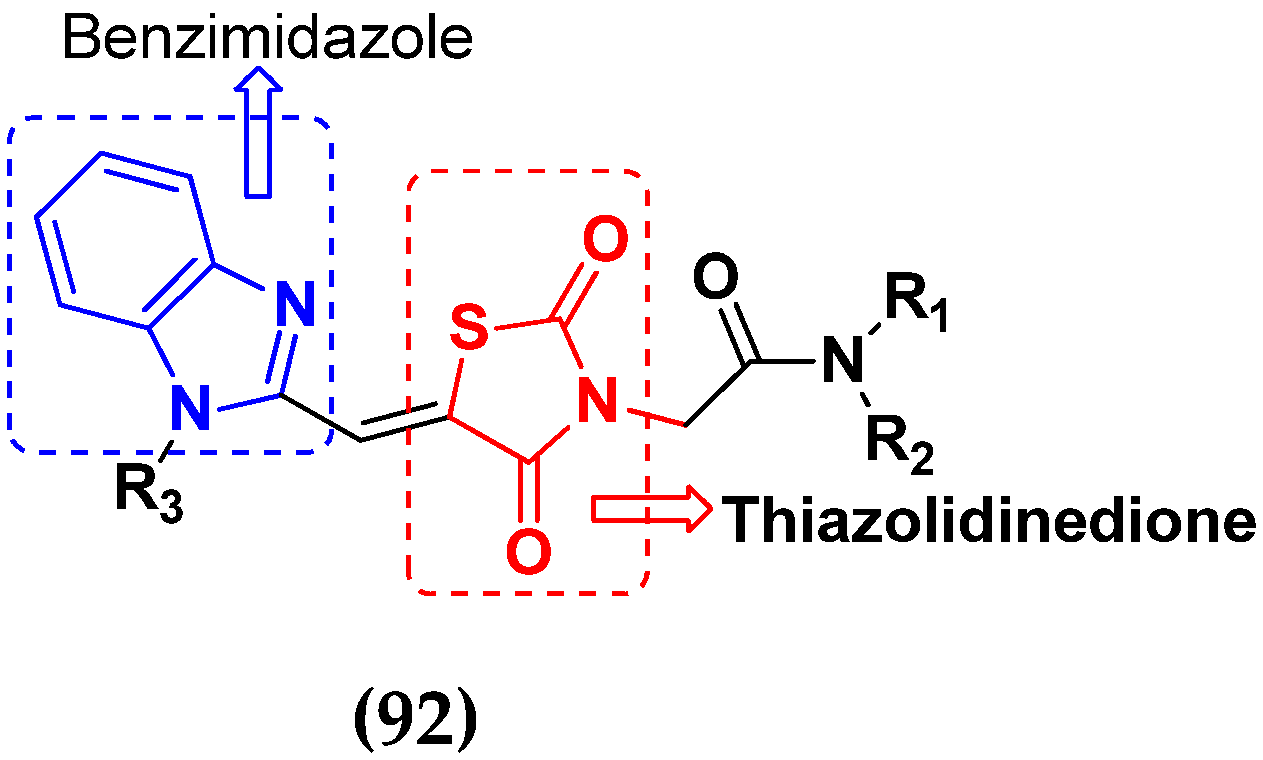{kind=link}
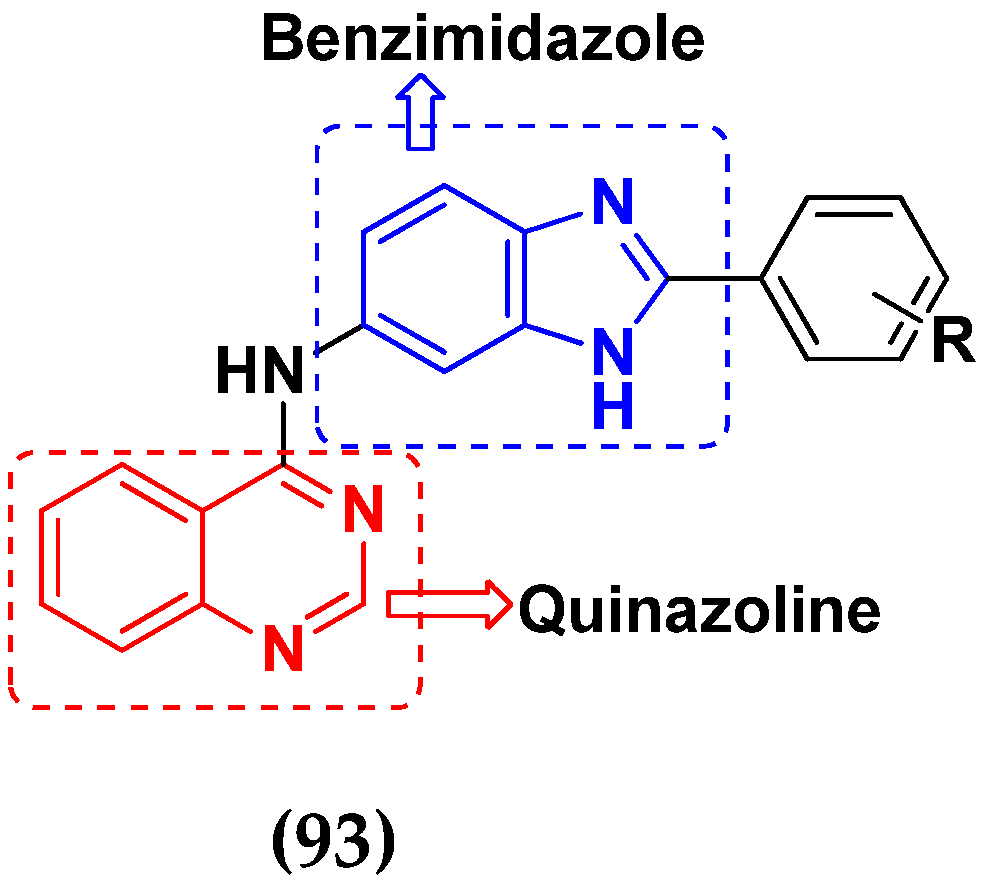{kind=link}
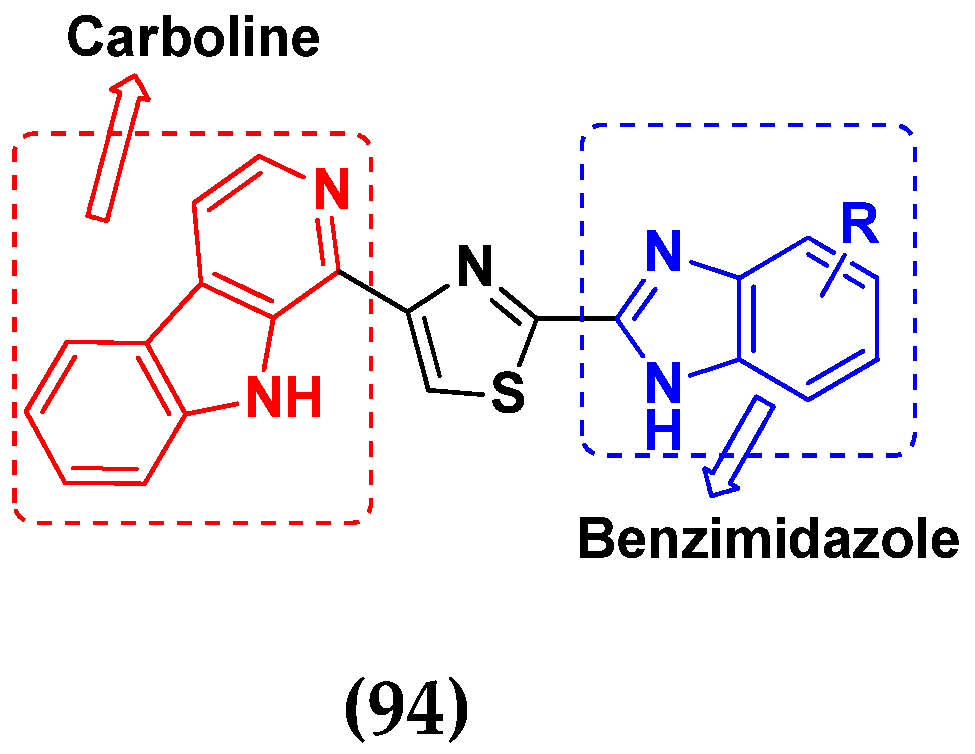{kind=link}
{kind=link}
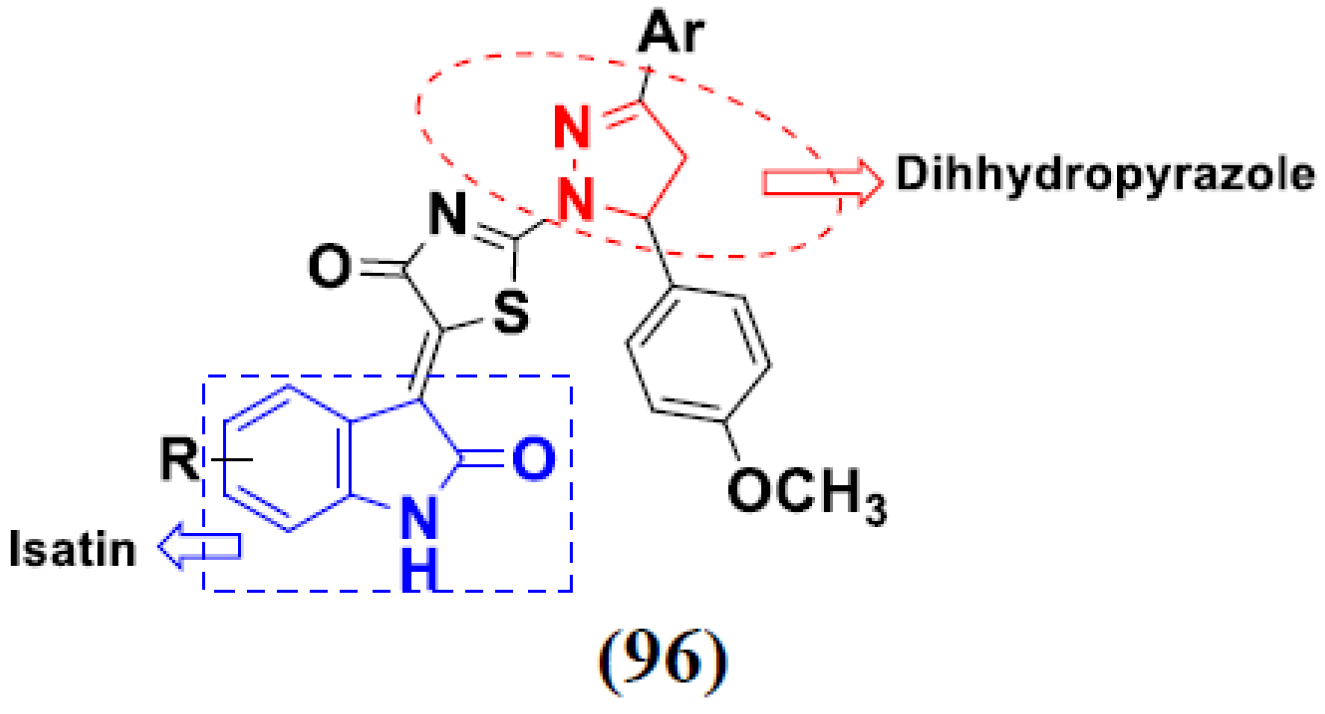{kind=link}
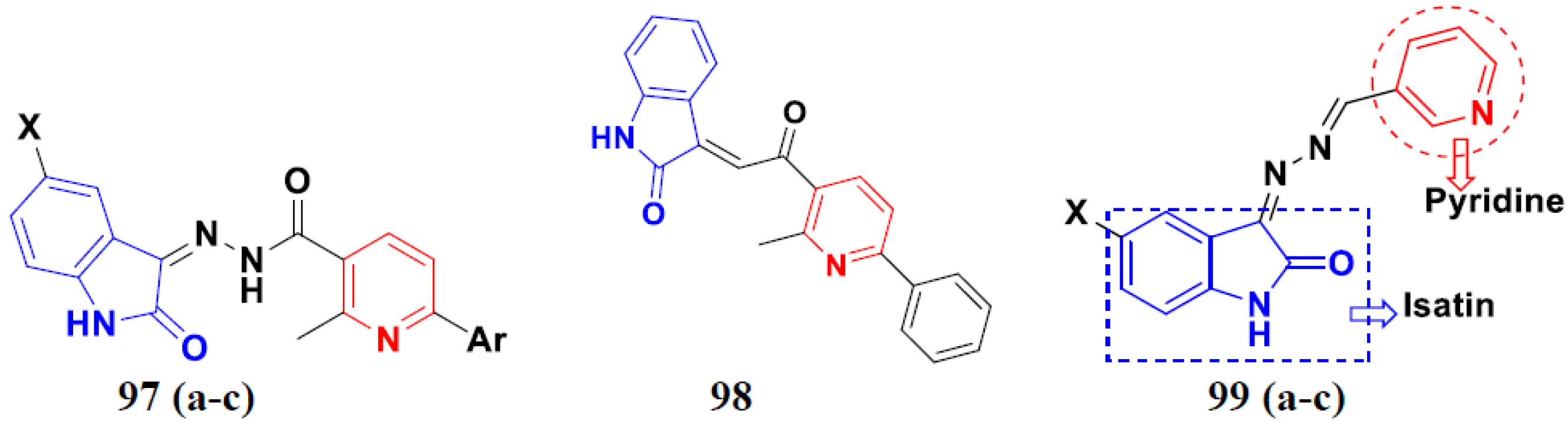{kind=link}
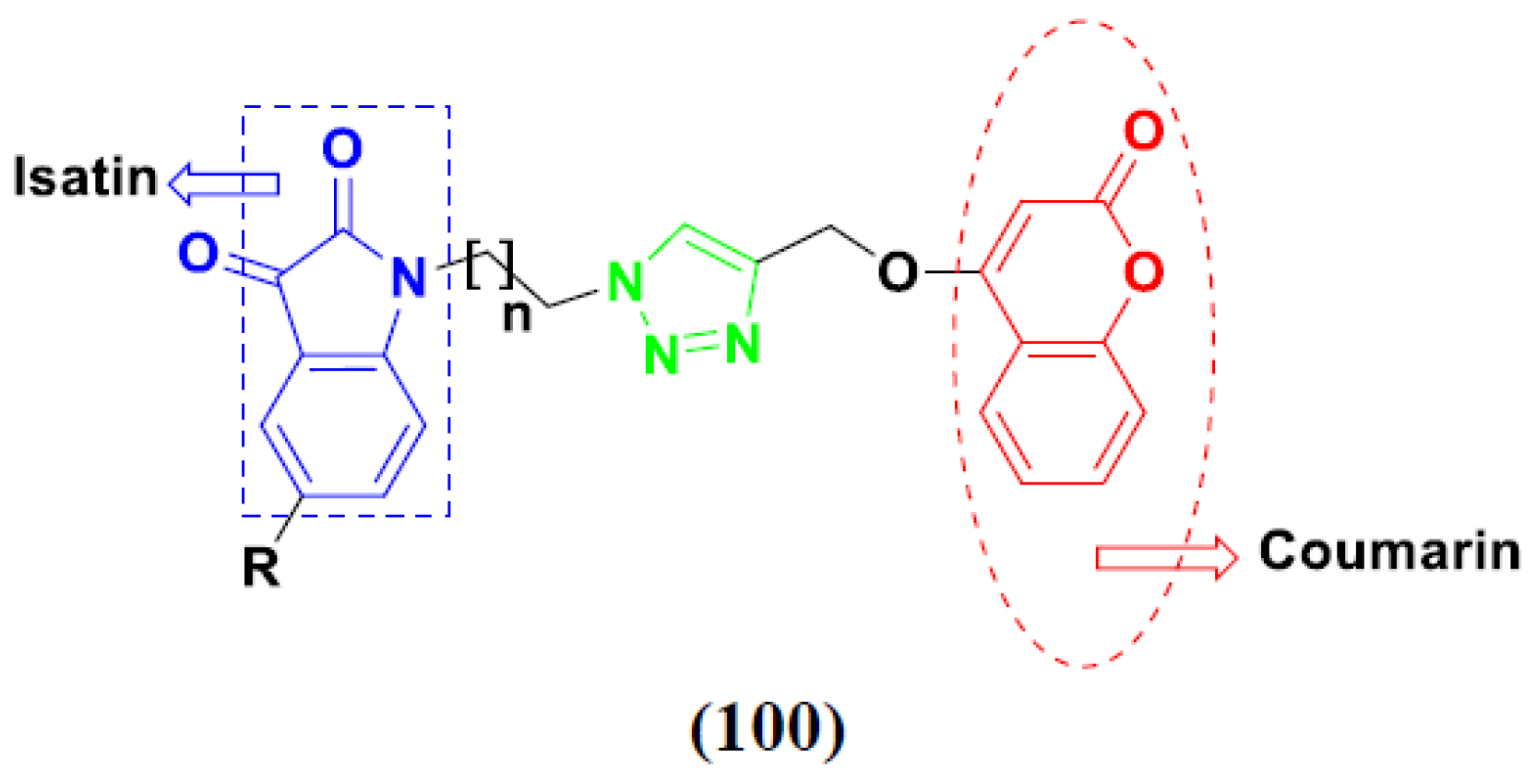{kind=link}
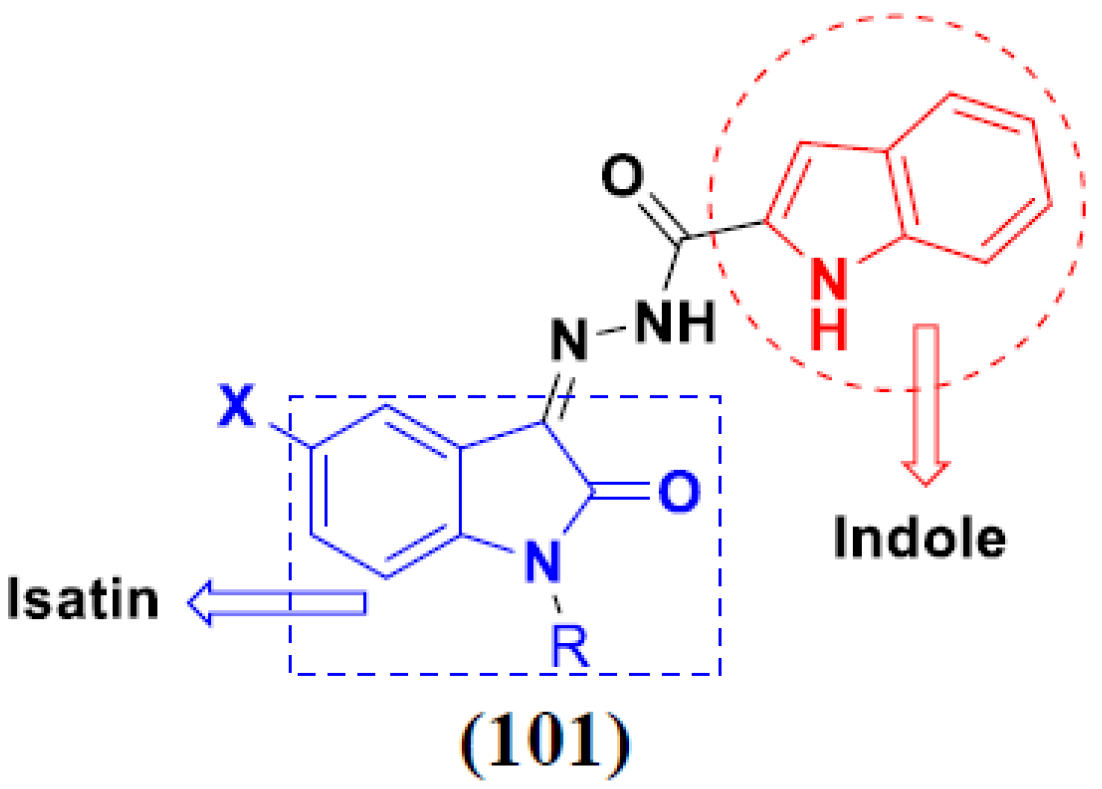{kind=link}
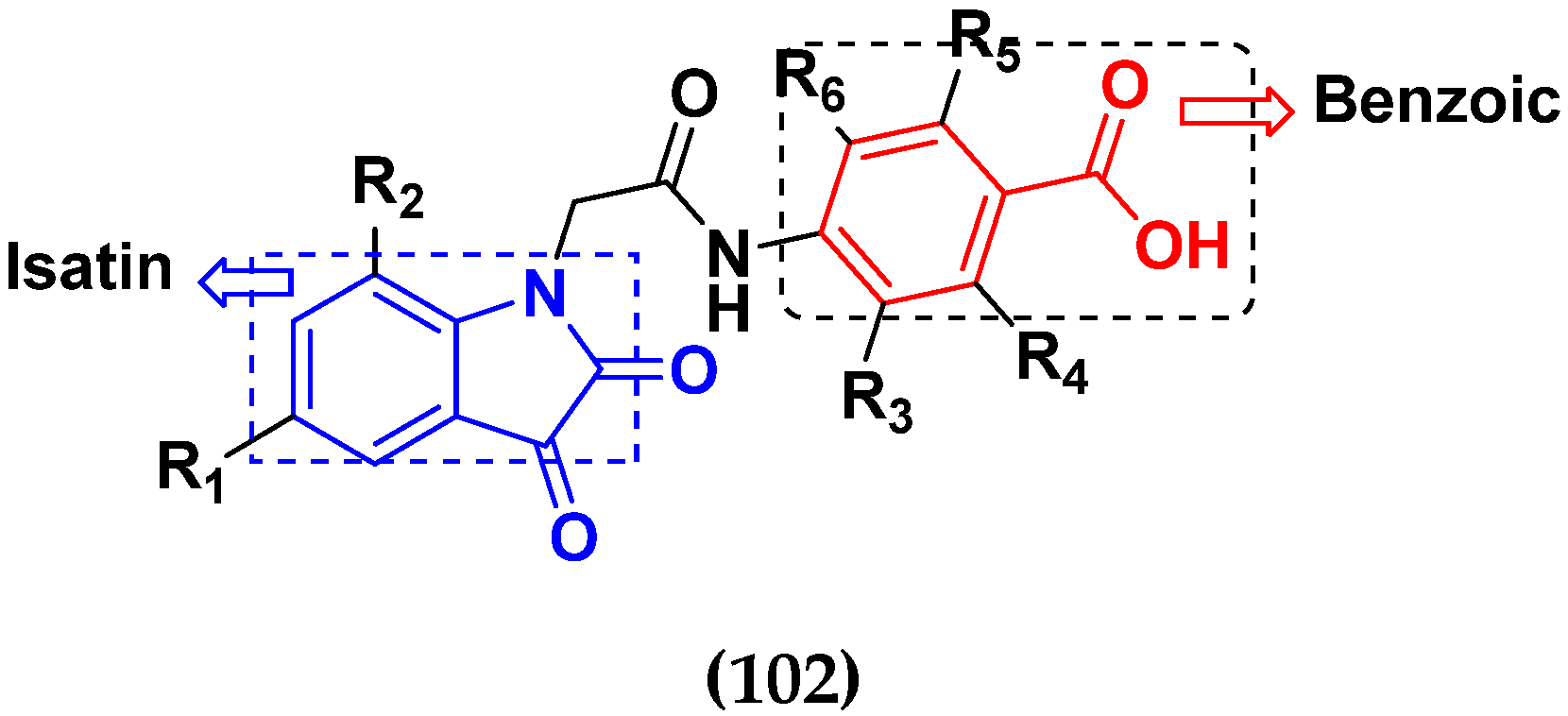{kind=link}
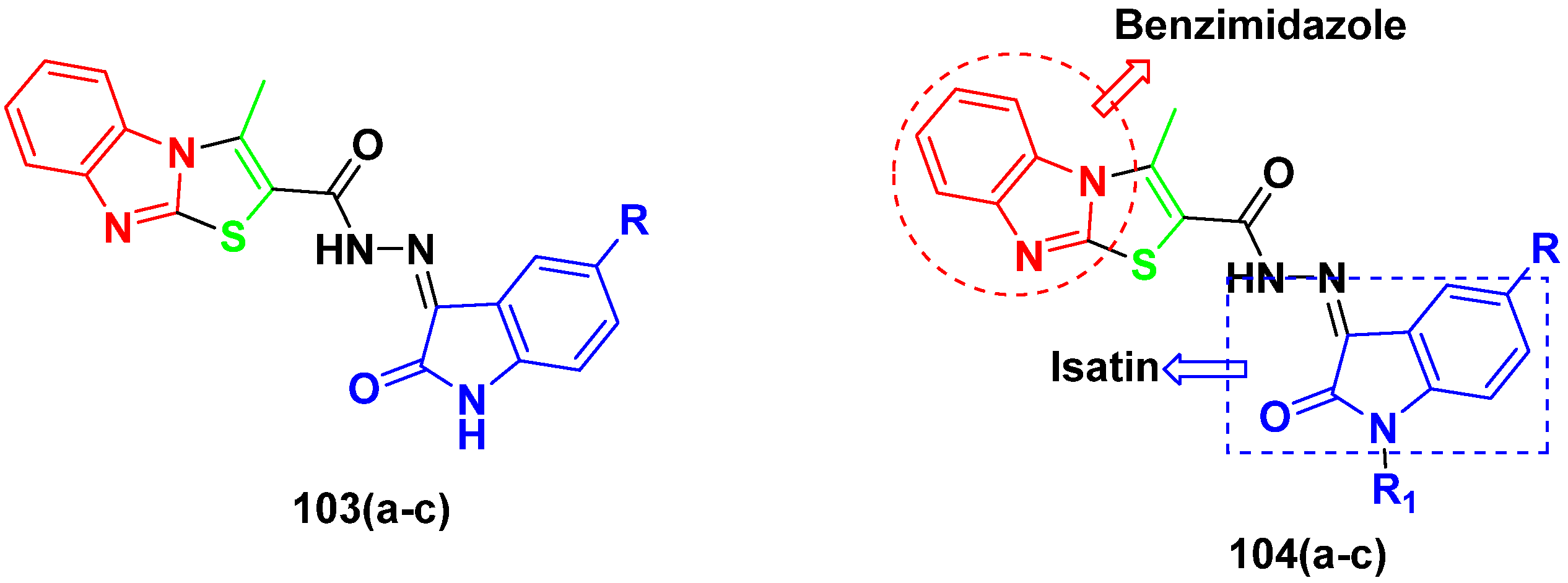{kind=link}
{kind=link}
{kind=link}
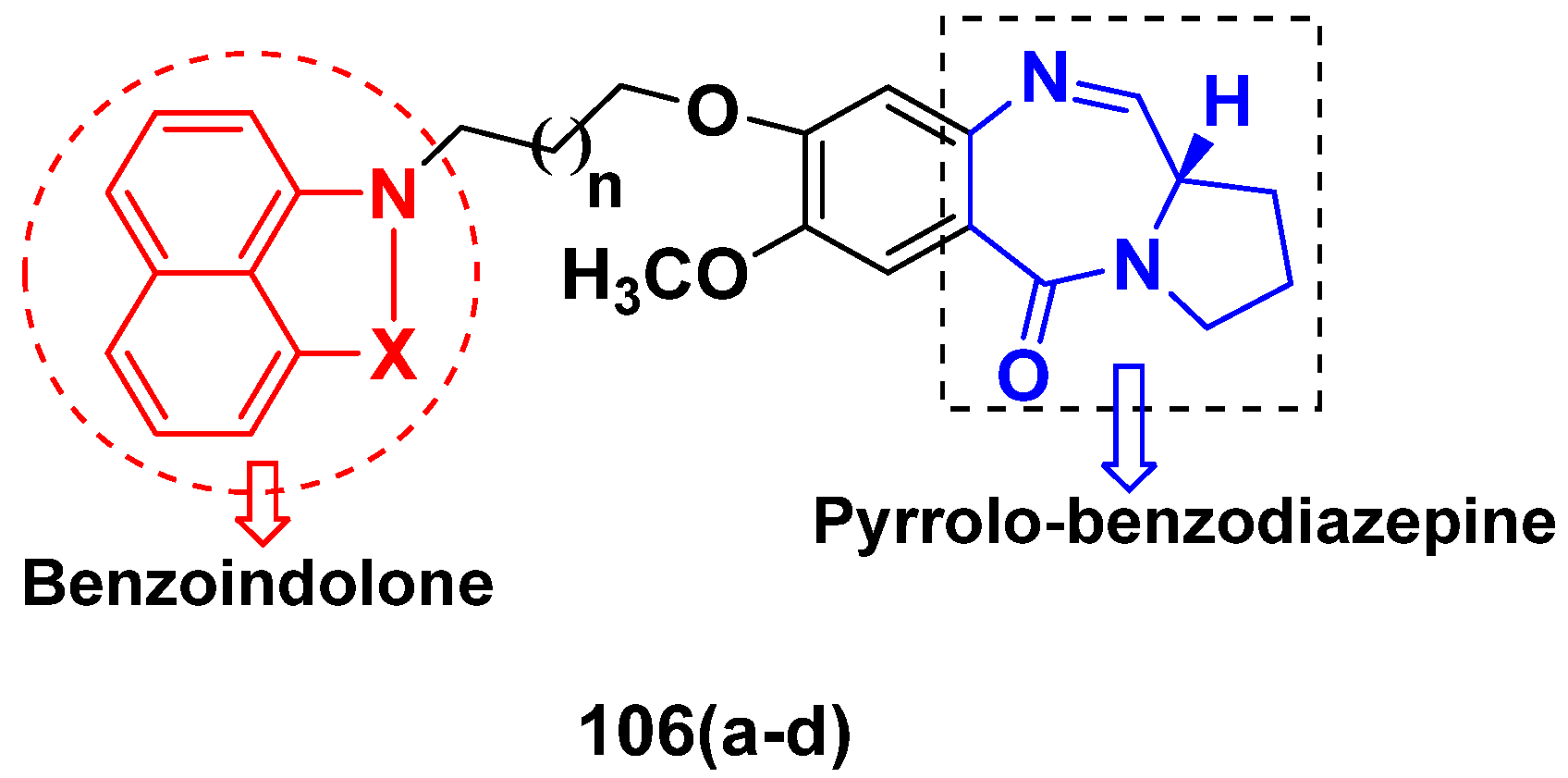{kind=link}
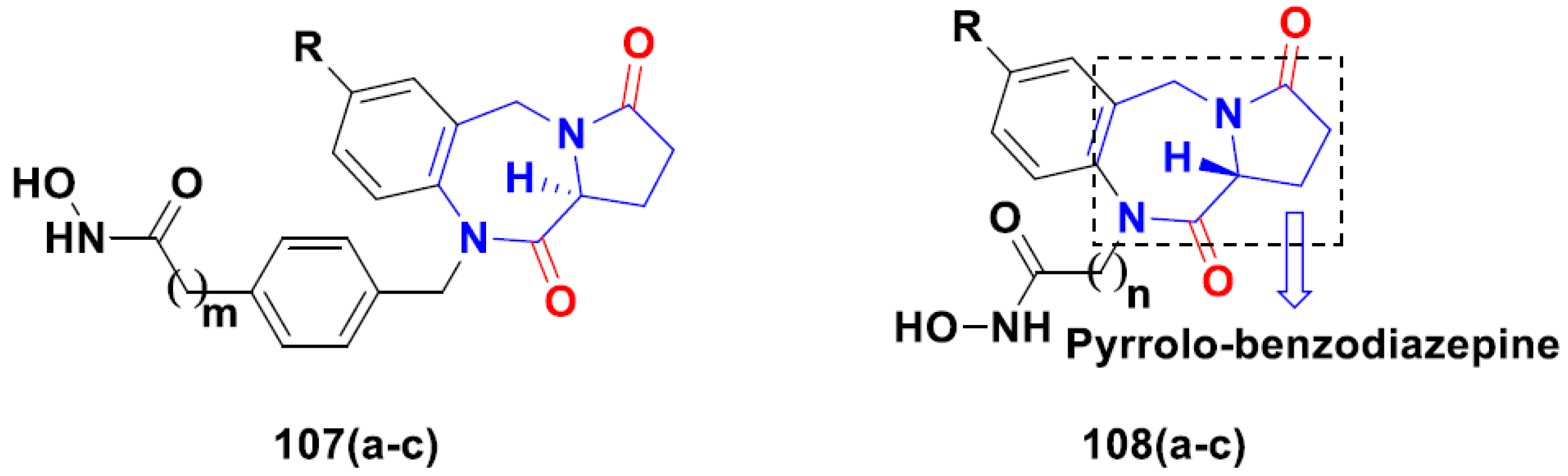{kind=link}
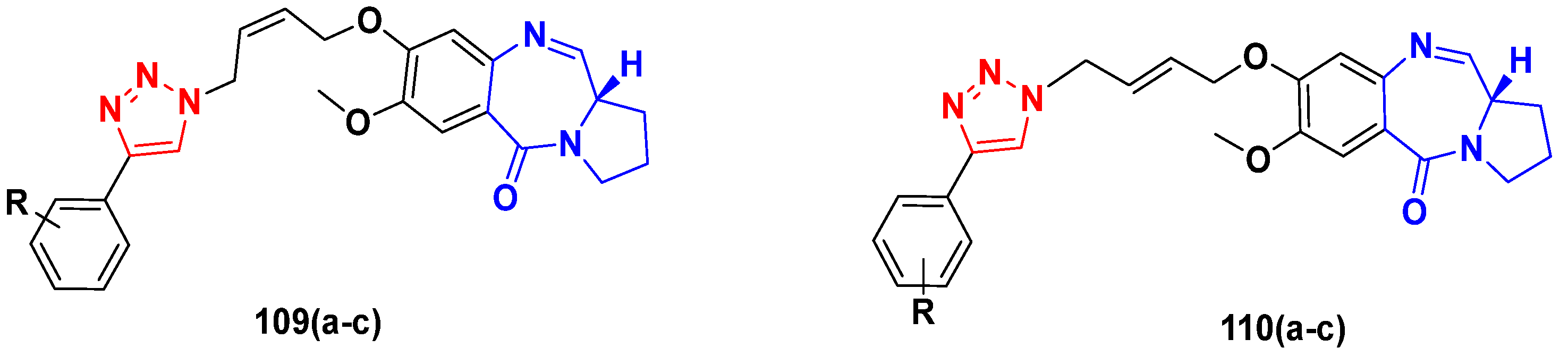{kind=link}
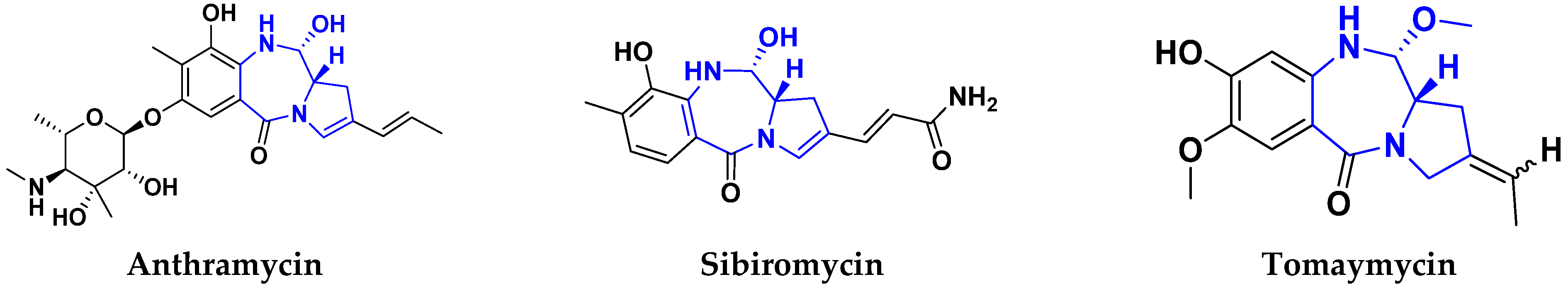{kind=link}
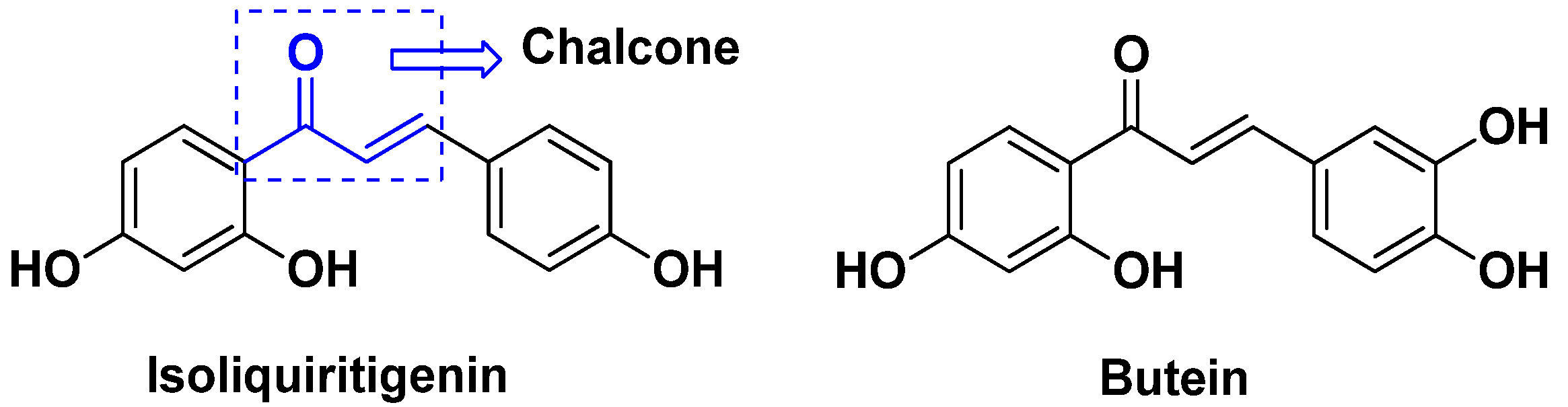{kind=link}
{kind=link}
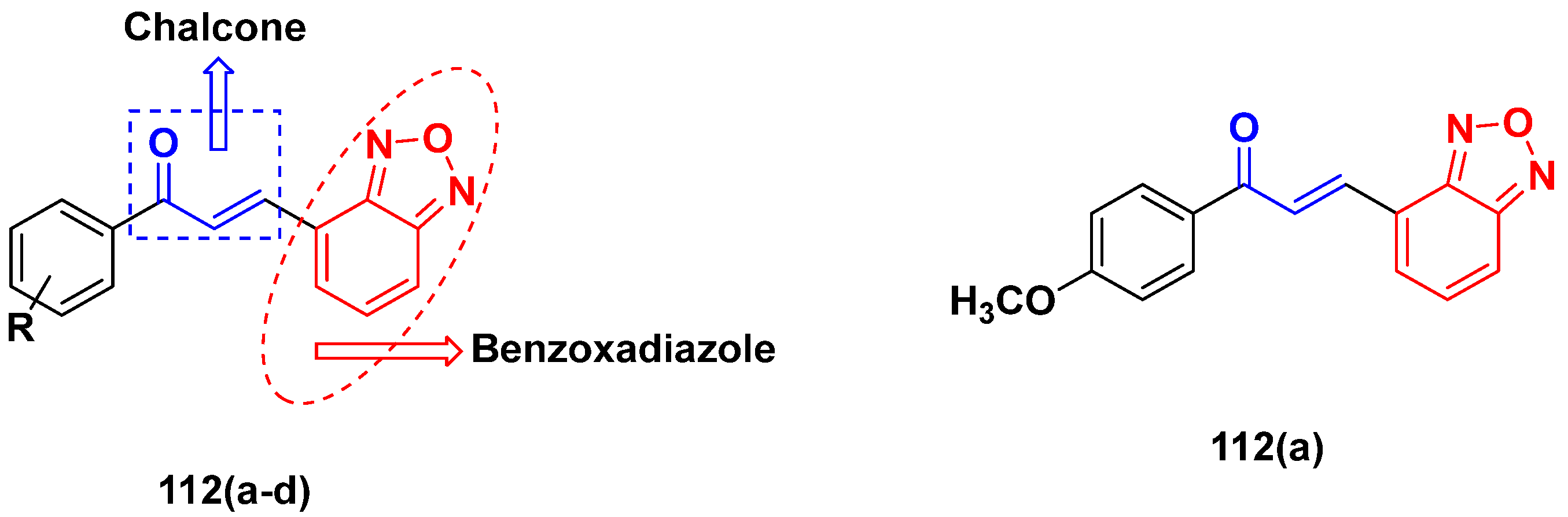{kind=link}
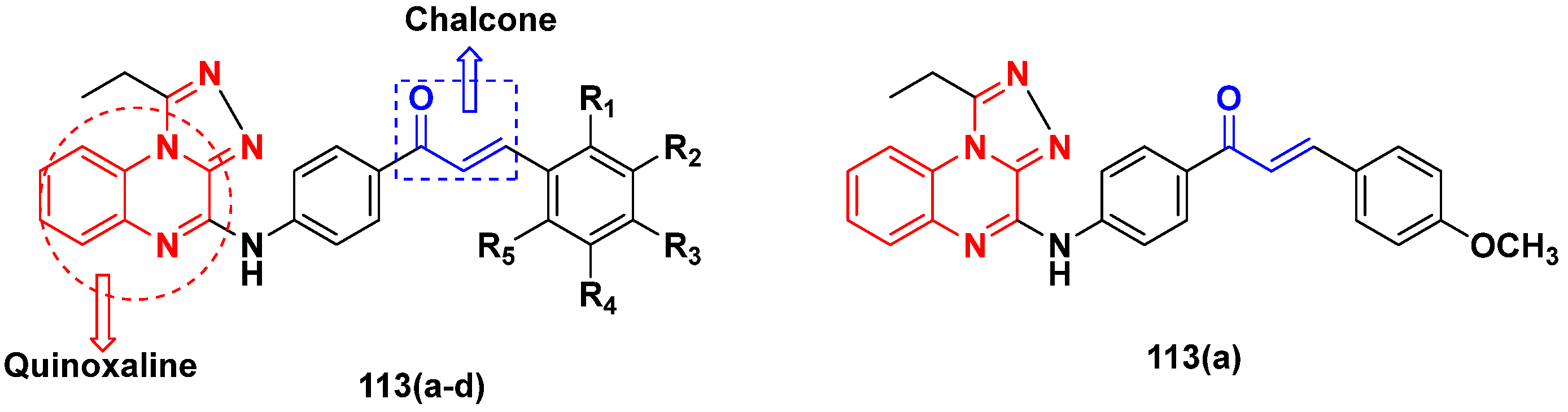{kind=link}
{kind=link}
{kind=link}
{kind=link}
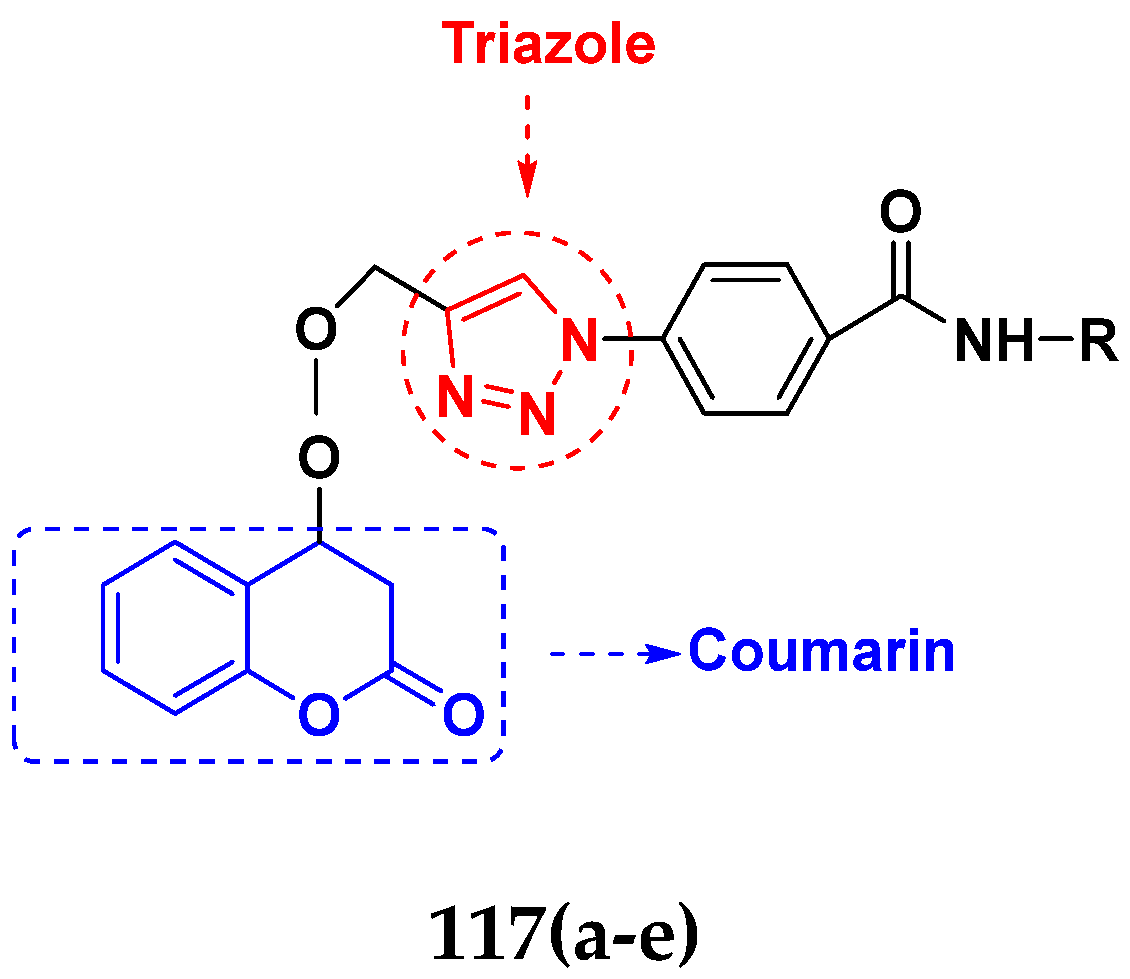{kind=link}
{kind=link}
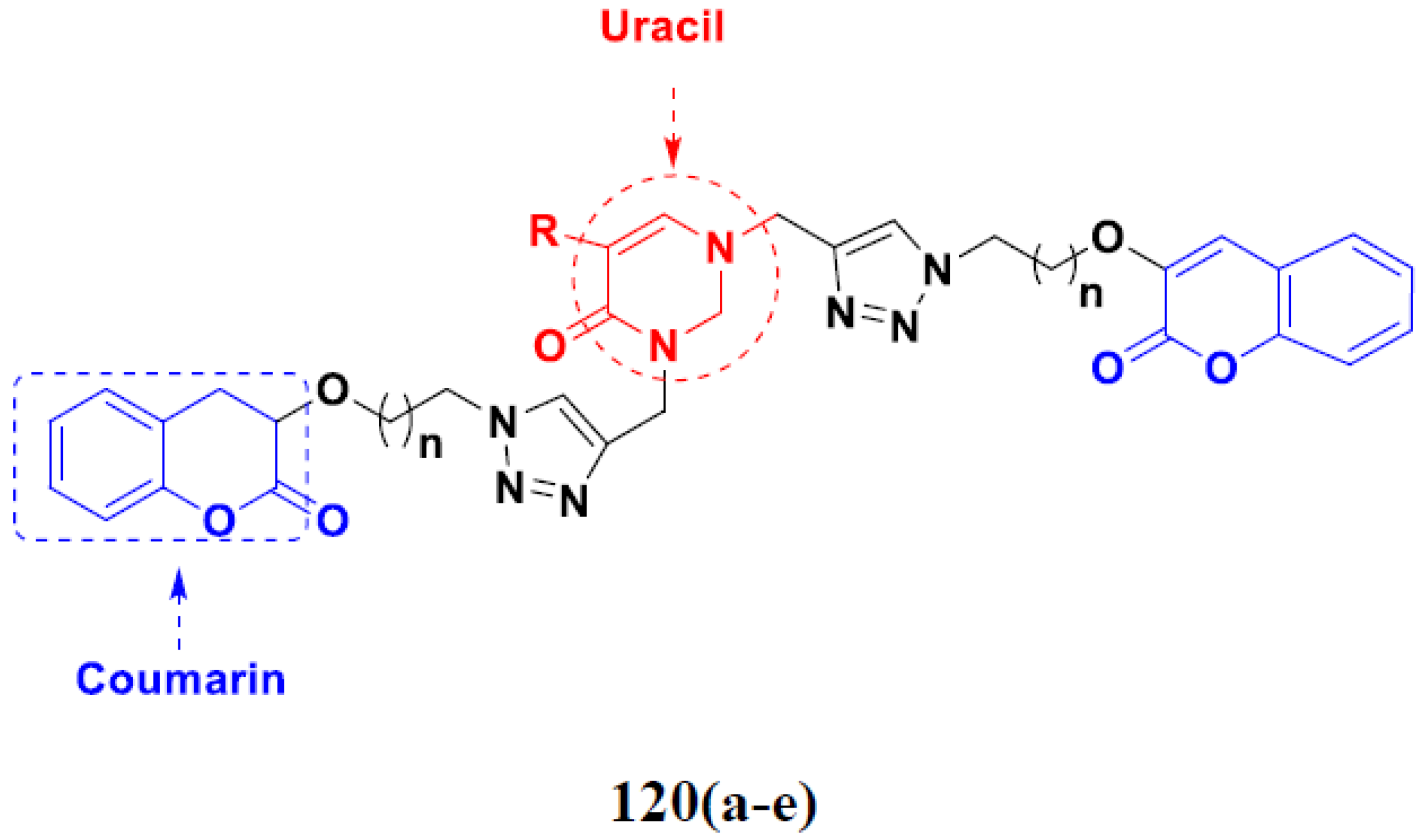{kind=link}
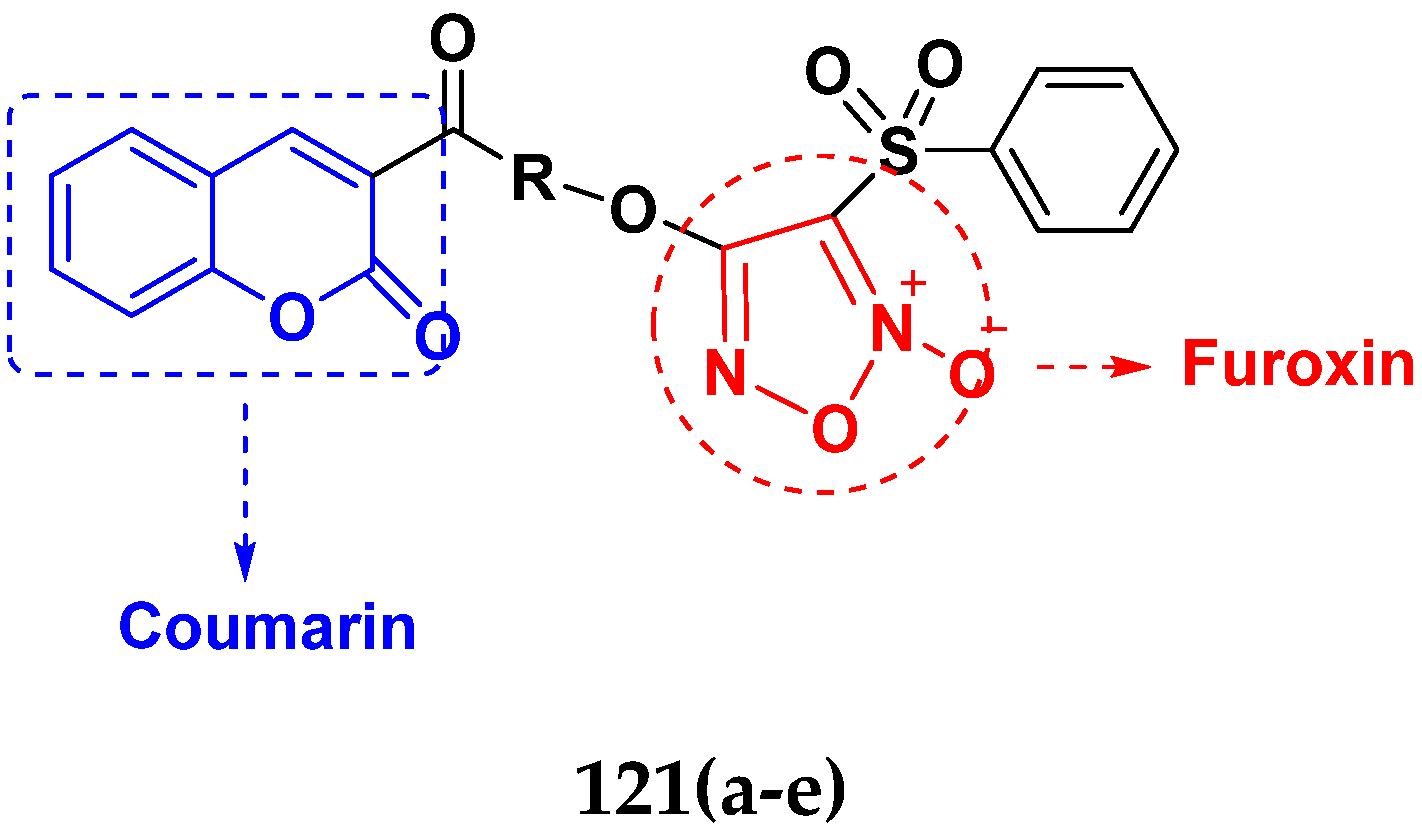{kind=link}
{kind=link}
{kind=link}
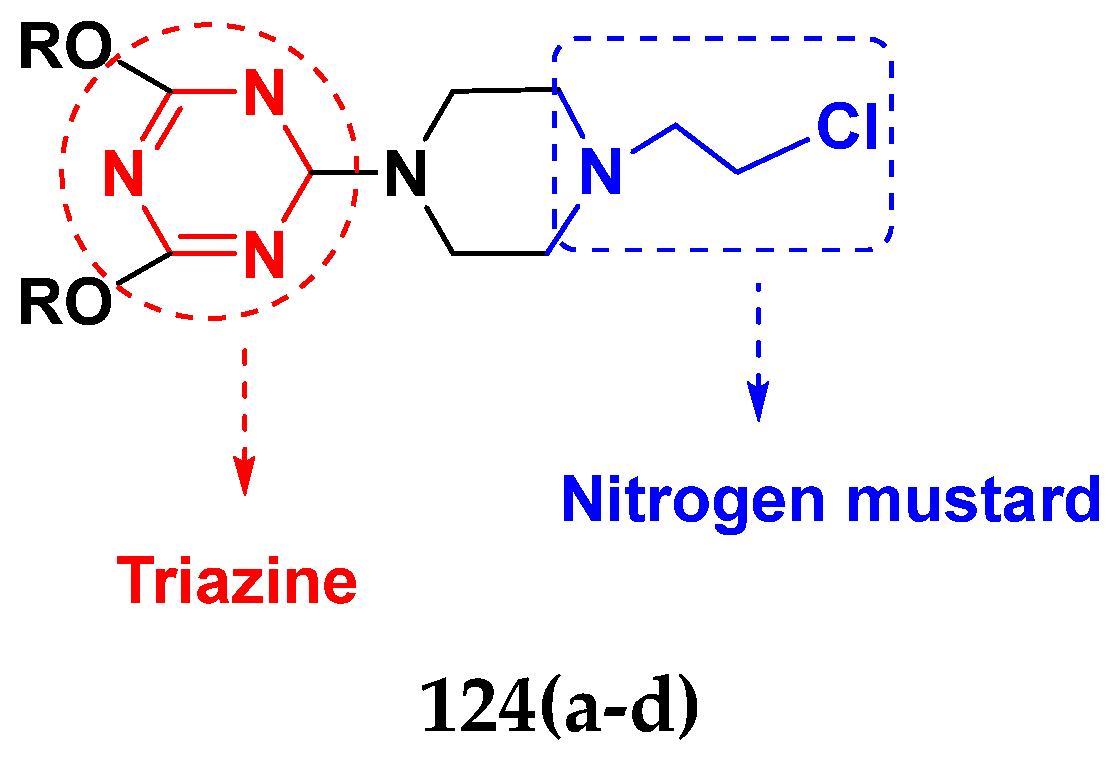{kind=link}
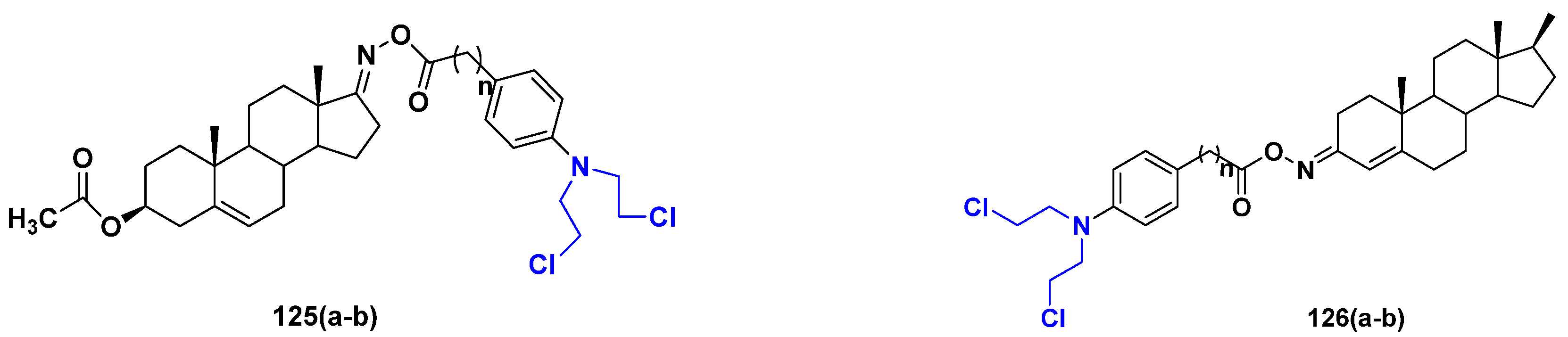{kind=link}
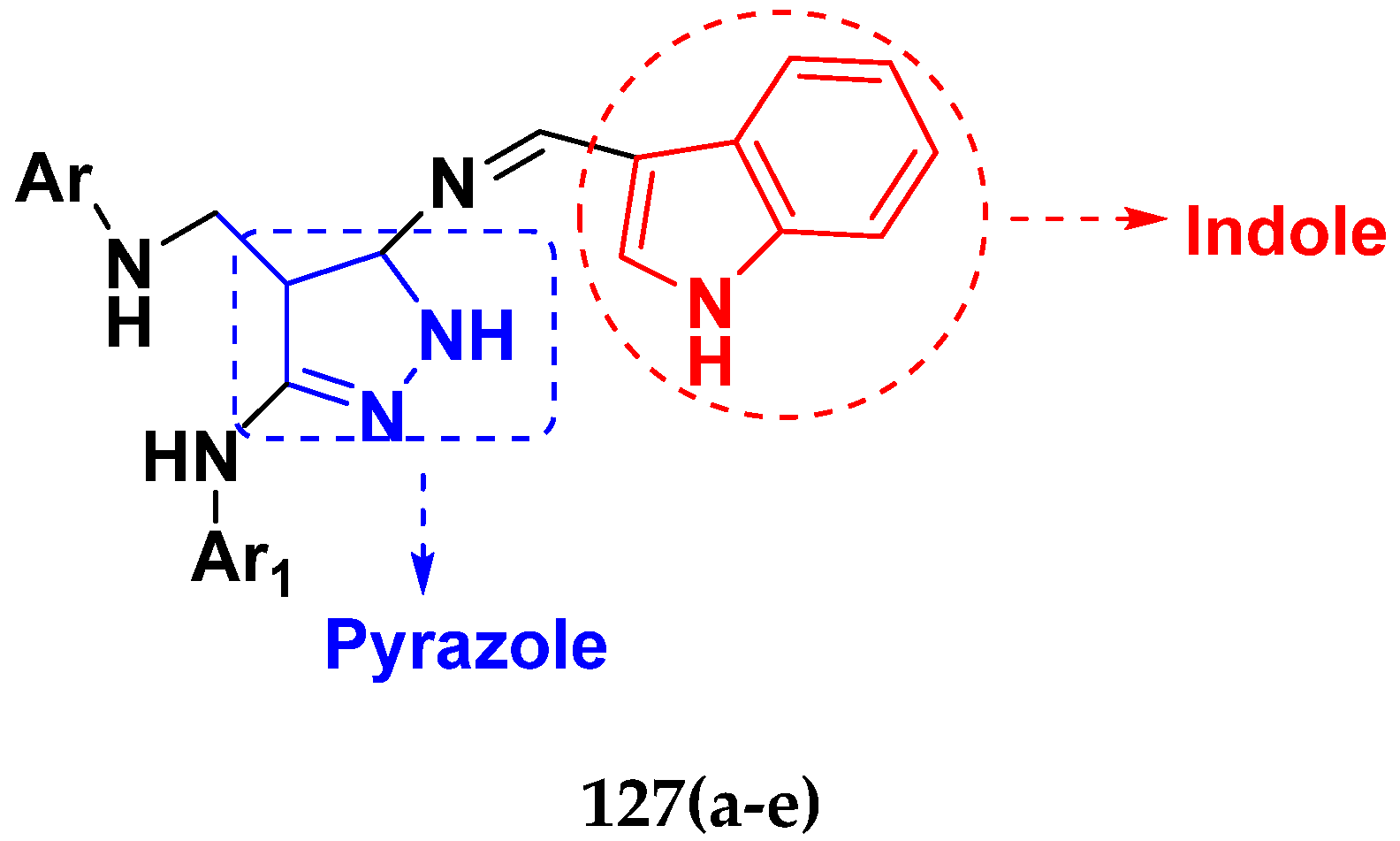{kind=link}
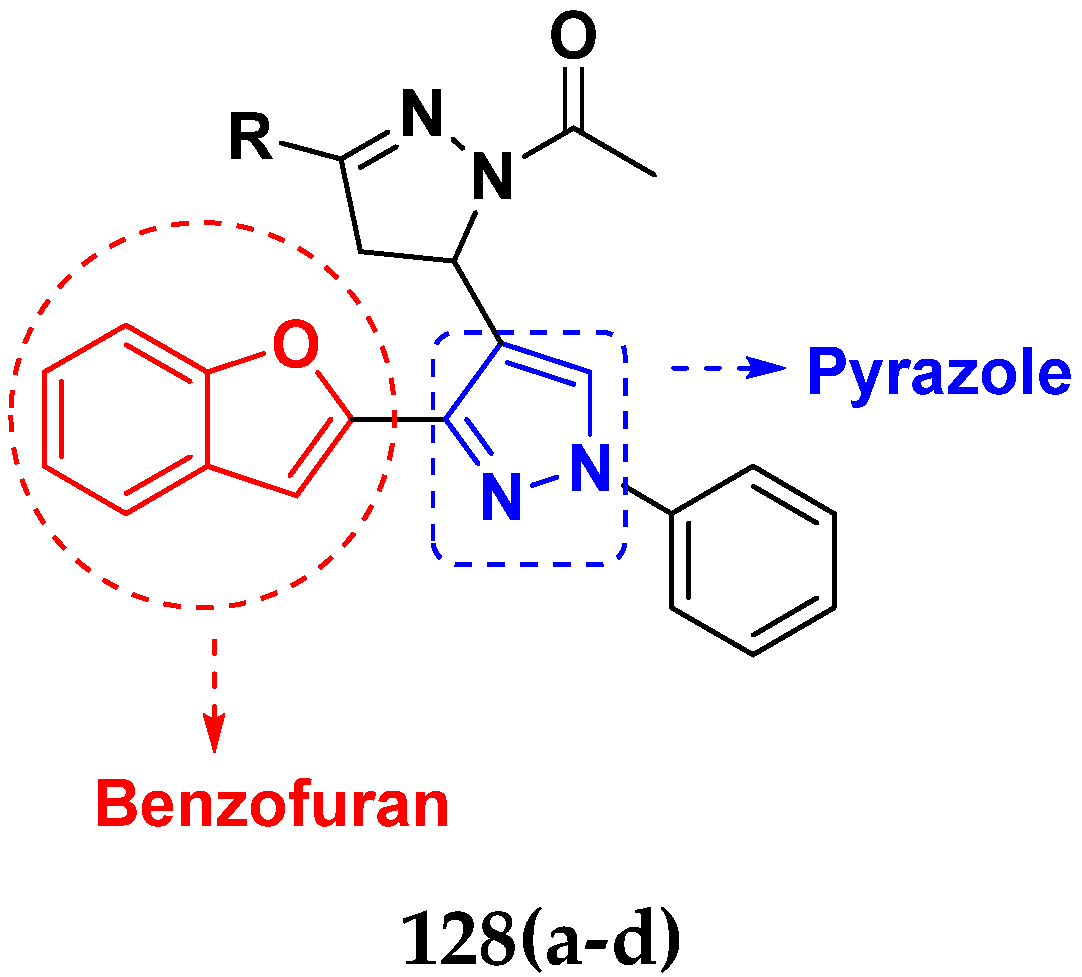{kind=link}
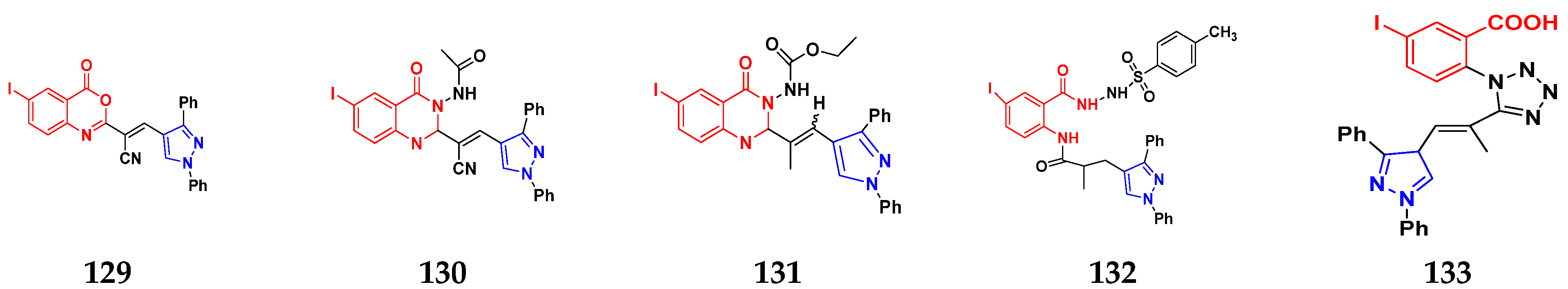{kind=link}
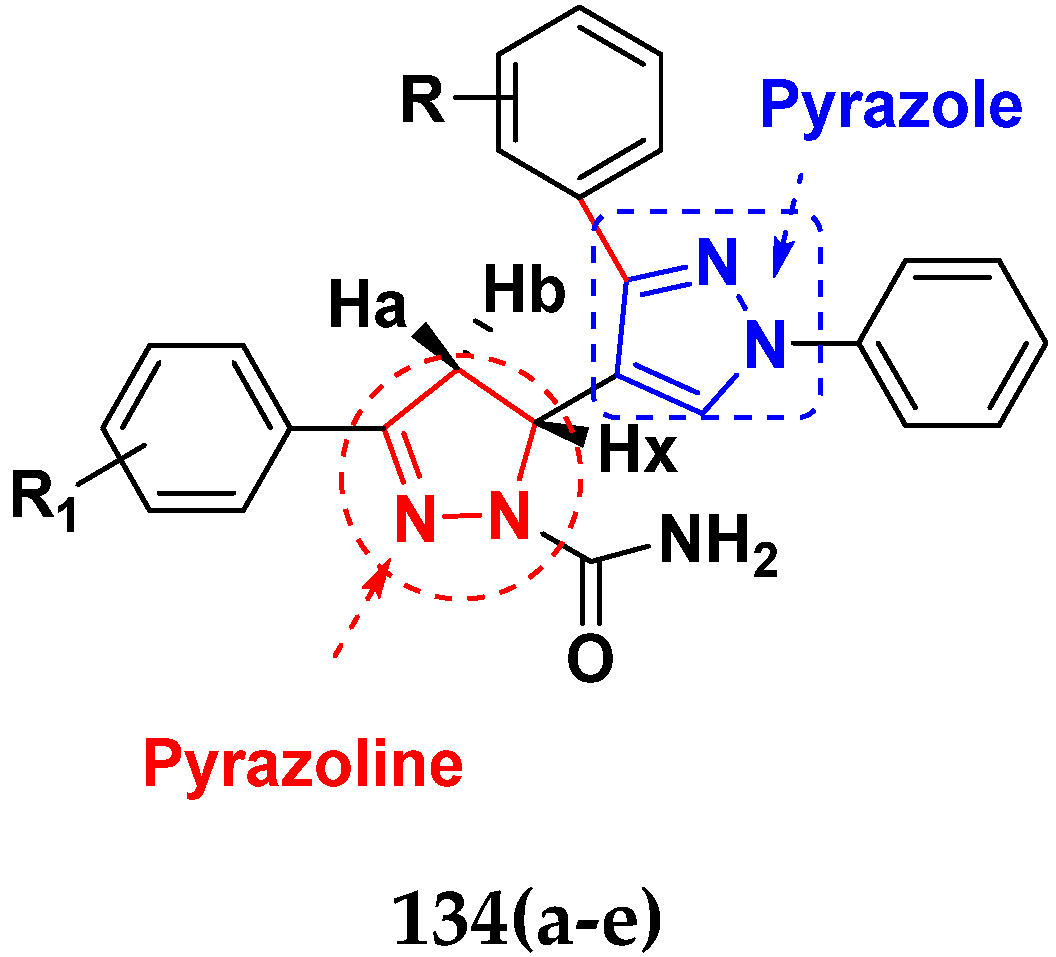{kind=link}
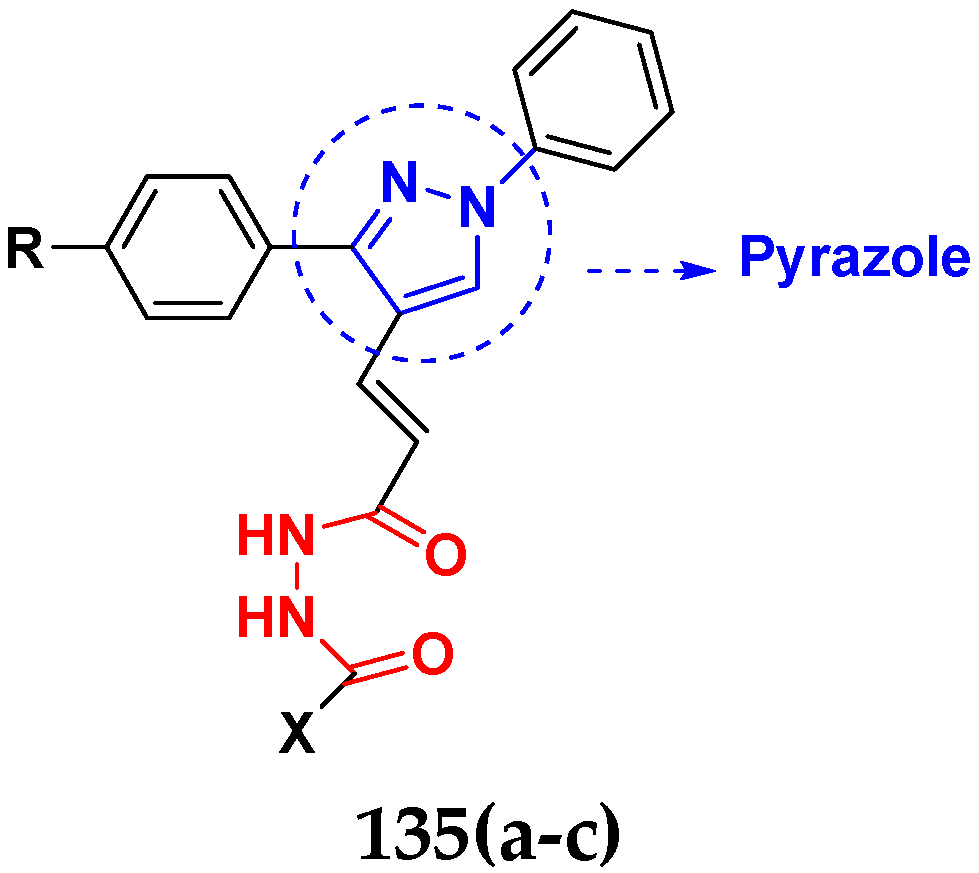{kind=link}
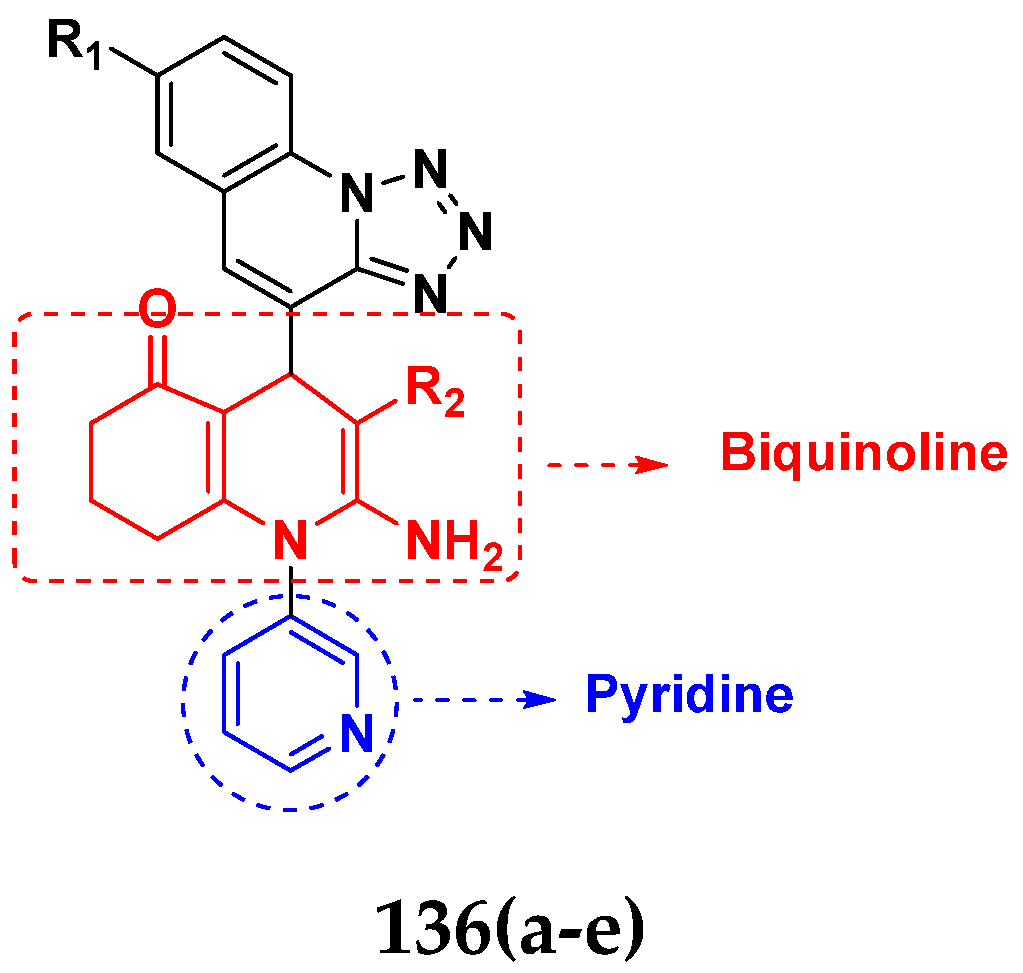{kind=link}
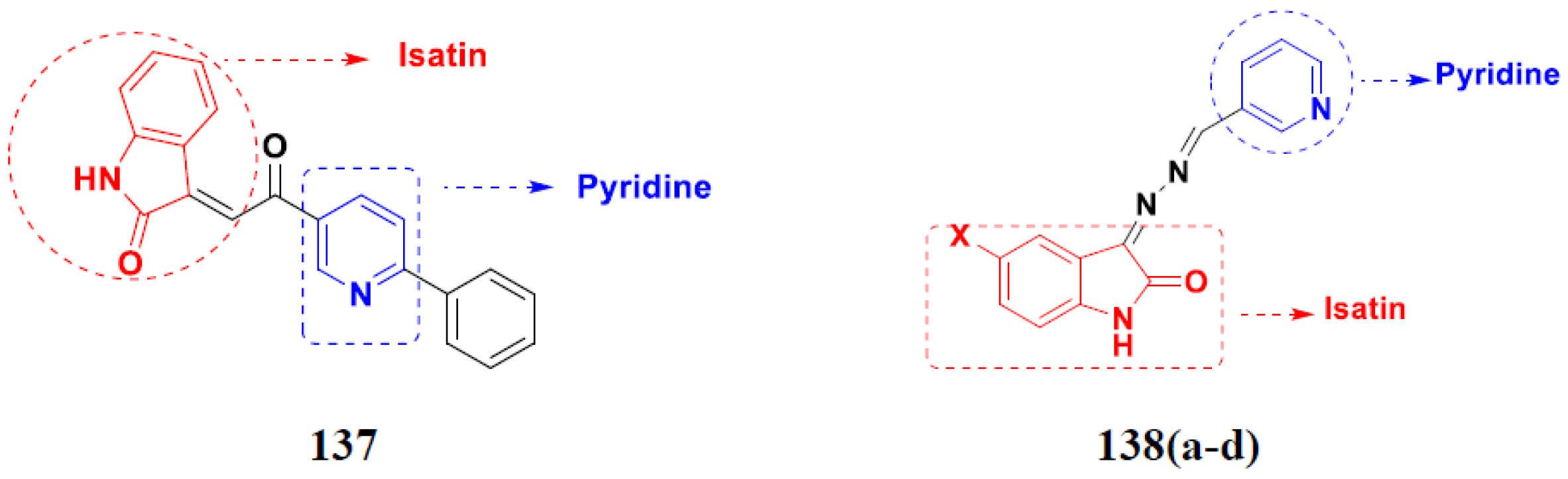{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, A.K.; Kumar, A.; Singh, H.; Sonawane, P.; Paliwal, H.; Thareja, S.; Pathak, P.; Grishina, M.; Jaremko, M.; Emwas, A.-H.; et al. Concept of Hybrid Drugs and Recent Advancements in Anticancer Hybrids. Pharmaceuticals 2022, 15, 1071. https://doi.org/10.3390/ph15091071
Singh AK, Kumar A, Singh H, Sonawane P, Paliwal H, Thareja S, Pathak P, Grishina M, Jaremko M, Emwas A-H, et al. Concept of Hybrid Drugs and Recent Advancements in Anticancer Hybrids. Pharmaceuticals. 2022; 15(9):1071. https://doi.org/10.3390/ph15091071
Chicago/Turabian StyleSingh, Ankit Kumar, Adarsh Kumar, Harshwardhan Singh, Pankaj Sonawane, Harshali Paliwal, Suresh Thareja, Prateek Pathak, Maria Grishina, Mariusz Jaremko, Abdul-Hamid Emwas, and et al. 2022. "Concept of Hybrid Drugs and Recent Advancements in Anticancer Hybrids" Pharmaceuticals 15, no. 9: 1071. https://doi.org/10.3390/ph15091071