
Synthesis and Biological Evaluation of Indole-2-Carboxamides with Potent Apoptotic Antiproliferative Activity as EGFR/CDK2 Dual Inhibitors

 ,
,
Abstract
:
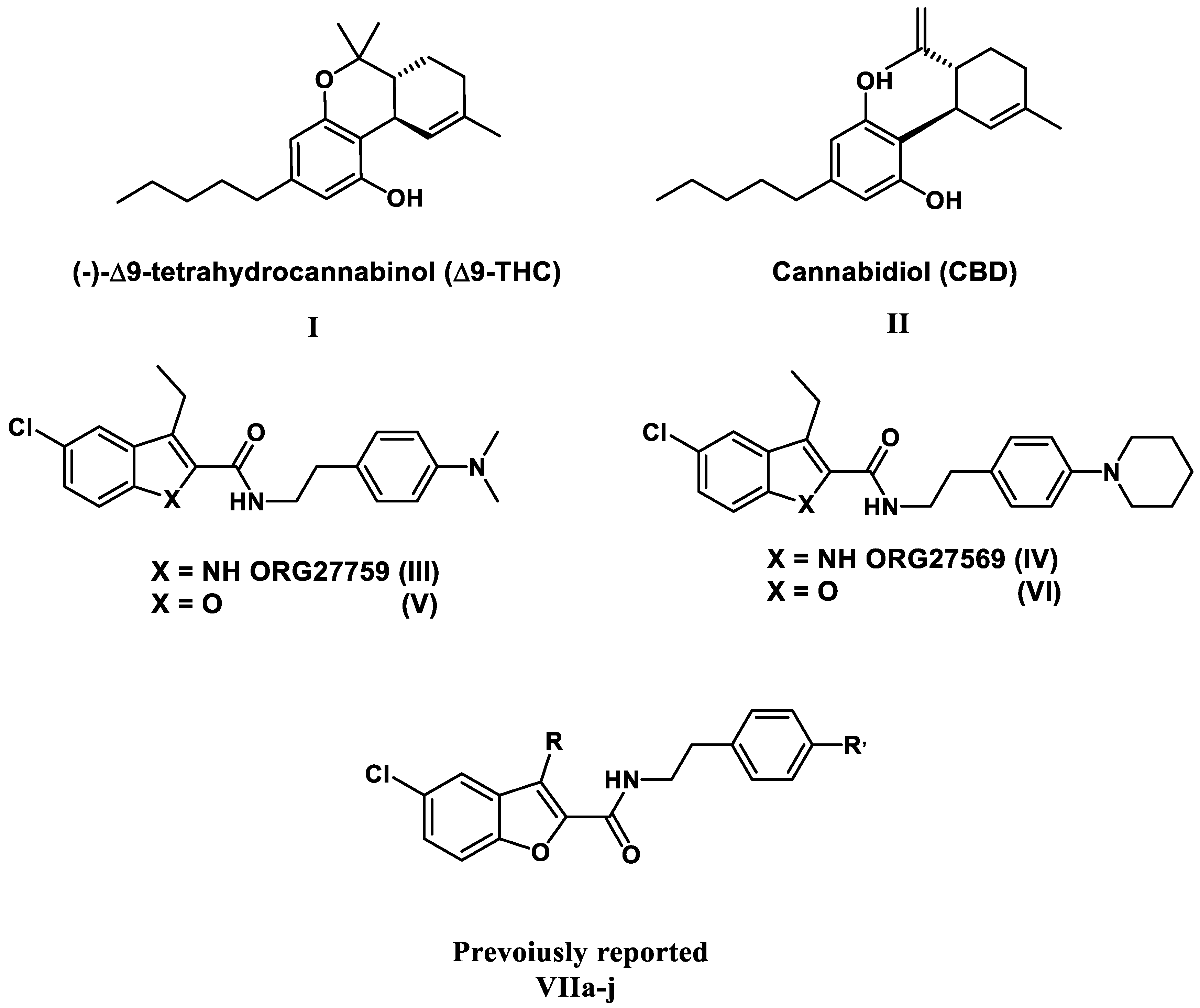
1. Introduction
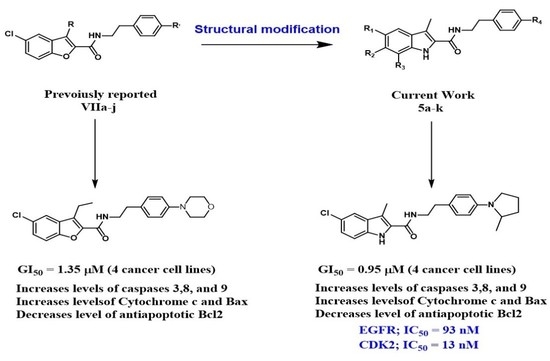
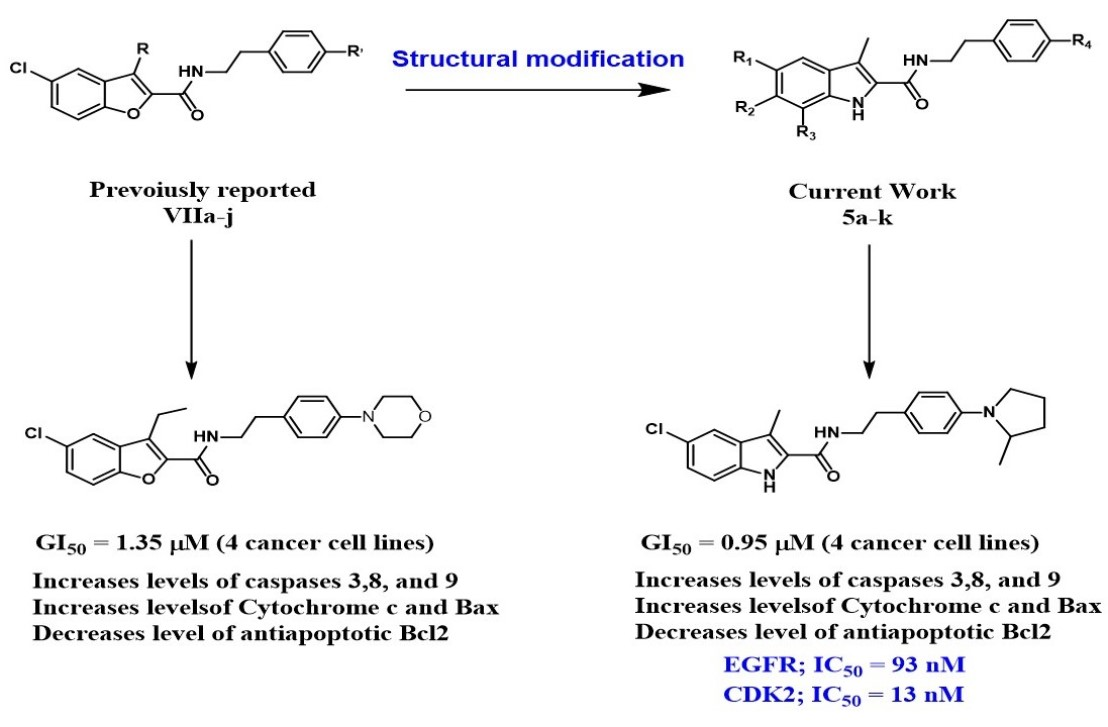
2. Results and Discussion
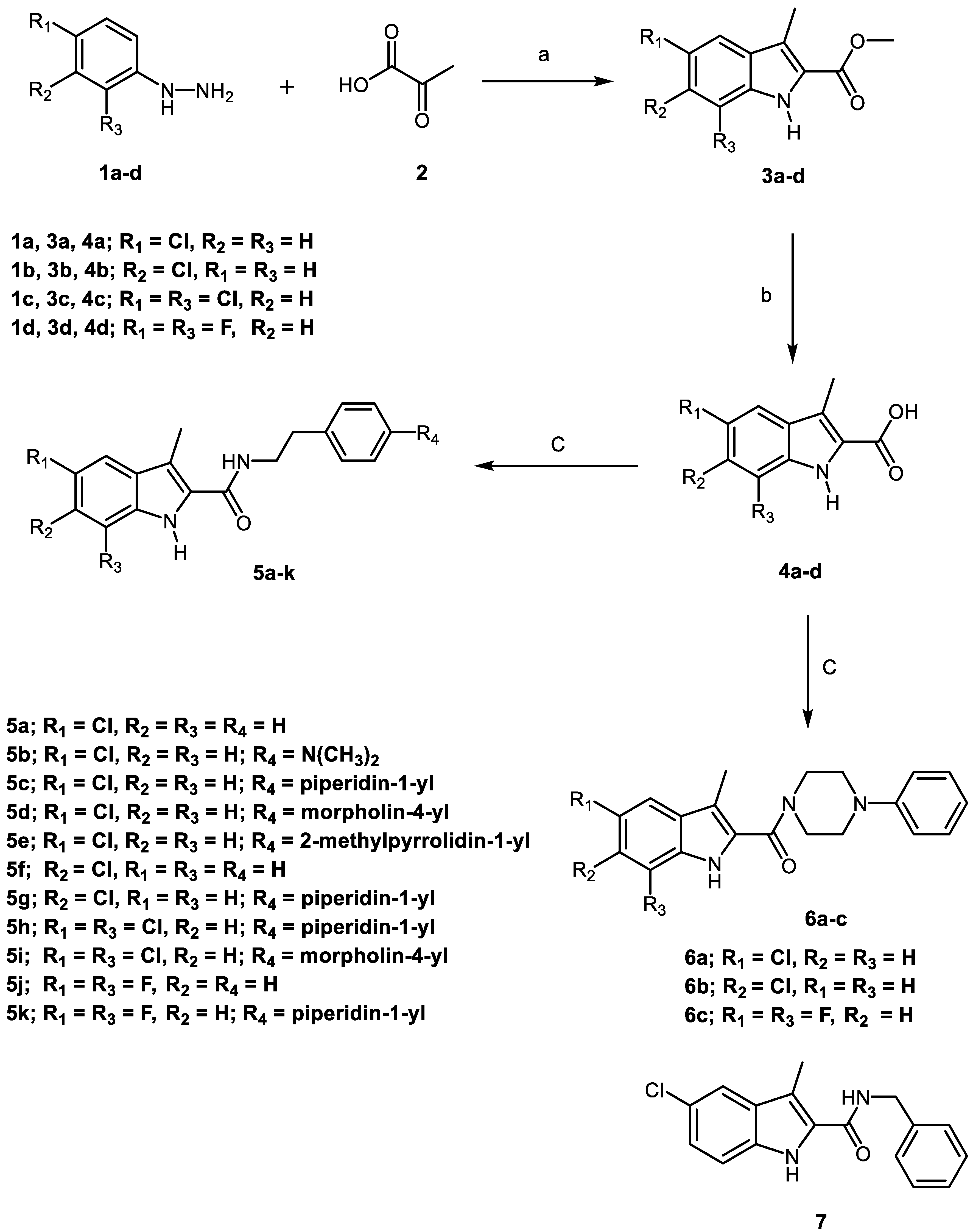
2.1. Chemistry
2.2. Evaluation of Biological Activities
2.2.1. In Vitro Anticancer Activity
Cell Viability Assay
Antiproliferative Activity
2.2.2. EGFR Inhibitory Activity
2.2.3. CDK2 Inhibitory Assay
2.2.4. Apoptosis Assay
Activation of Proteolytic Caspases Cascade
Cytochrome C Assay
Bax and Bcl-2 Levels Assay
Effect of Compounds 5d and 5e on p53 Transcription in MCF-7
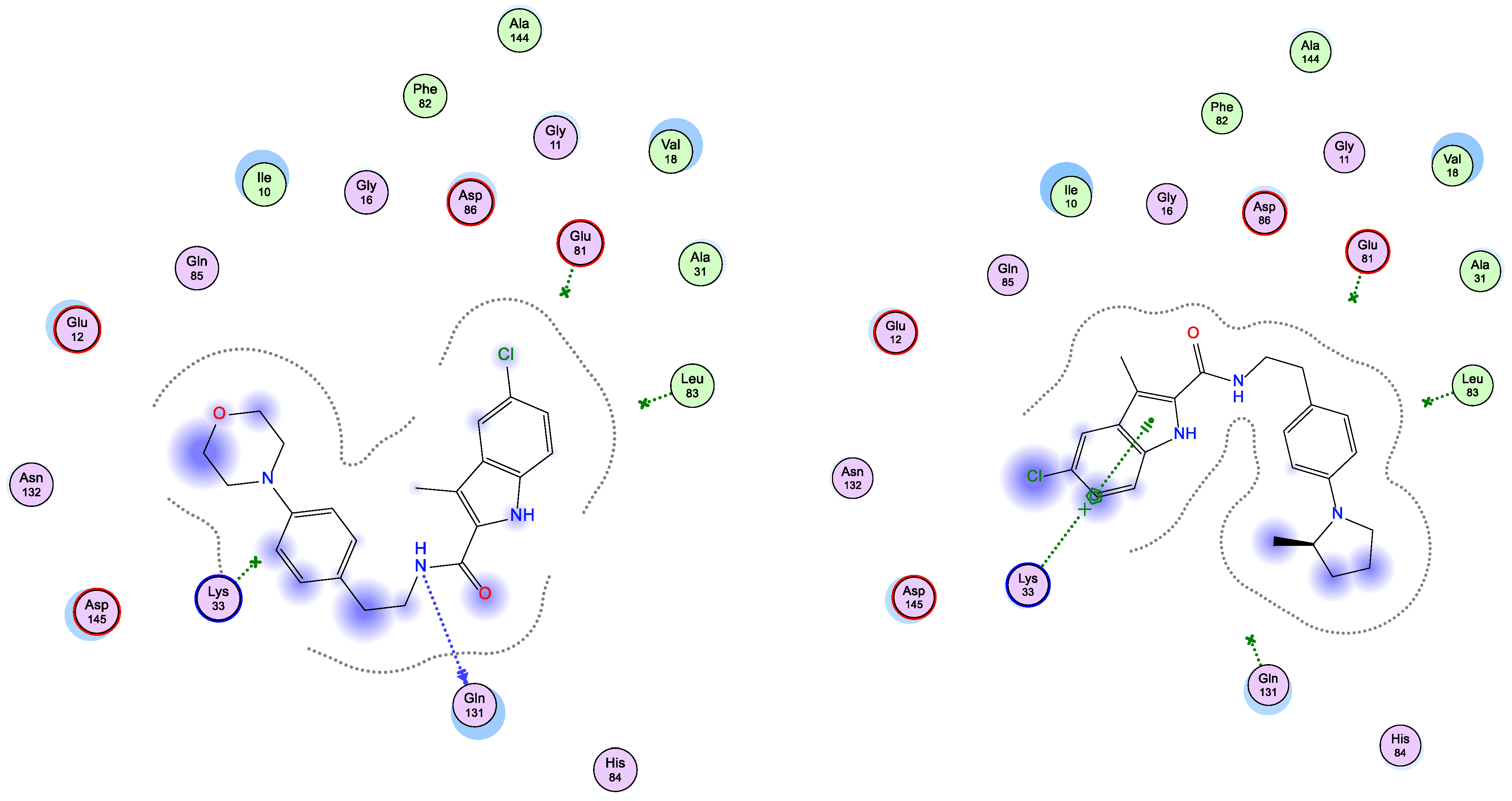
2.3. Docking Study
3. Materials and Methods
3.1. Chemistry
3.1.1. 5-Chloro-3-methyl-N-phenethyl-1H-indole-2-carboxamide (5a)
3.1.2. 5-Chloro-N-(4-(dimethylamino)phenethyl)-3-methyl-1H-indole-2-carboxamide (5b)
3.1.3. 5-Chloro-3-methyl-N-(4-(piperidin-1-yl)phenethyl)-1H-indole-2-carboxamide (5c)
3.1.4. 5-Chloro-3-methyl-N-(4-morpholinophenethyl)-1H-indole-2-carboxamide (5d)
3.1.5. 5-Chloro-3-methyl-N-(4-(2-methylpyrrolidin-1-yl)phenethyl)-1H-indole-2-carboxamide (5e)
3.1.6. 6-Chloro-3-methyl-N-phenethyl-1H-indole-2-carboxamide (5f)
3.1.7. 6-Chloro-3-methyl-N-(4-(piperidin-1-yl)phenethyl)-1H-indole-2-carboxamide (5g)
3.1.8. 5,7-Dichloro-3-methyl-N-(4-(piperidin-1-yl)phenethyl)-1H-indole-2-carboxamide (5h)
3.1.9. 5,7-Dichloro-3-methyl-N-(4-morpholinophenethyl)-1H-indole-2-carboxamide (5i)
3.1.10. 5,7-Difluoro-3-methyl-N-phenethyl-1H-indole-2-carboxamide (5j)
3.1.11. 5,7-Difluoro-3-methyl-N-(4-(piperidin-1-yl)phenethyl)-1H-indole-2-carboxamide (5k)
3.1.12. (5-Chloro-3-methyl-1H-indol-2-yl)(4-phenylpiperazin-1-yl)methanone (6a)
3.1.13. (6-Chloro-3-methyl-1H-indol-2-yl)(4-phenylpiperazin-1-yl)methanone (6b)
3.1.14. (5,7-Difluoro-3-methyl-1H-indol-2-yl)(4-phenylpiperazin-1-yl)methanone (6c)
3.1.15. N-Benzyl-5-chloro-3-methyl-1H-indole-2-carboxamide (7)
3.2. Biology
3.2.1. In Vitro Anticancer Activity
Cell Viability Assay
Antiproliferative Activity
3.2.2. EGFR Inhibitory Activity
3.2.3. CDK2 Inhibitory Assay
3.2.4. Apoptosis Assay
Activation of Proteolytic Caspases Cascade
Cytochrome C Assay
Bax and Bcl-2 Levels Assay
Effect of Compounds 5d and 5e on p53 Transcription in MCF-7
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Medina-Franco, J.L.; Giulianotti, M.A.; Welmaker, G.S.; Houghten, R.A. Shifting from the single to the multitarget paradigm in drug discovery. Drug Discov. Today 2013, 18, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.R.; Popovic-Nikolic, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Msomi, N.Z.; Shode, F.O.; Pooe, O.J.; Mazibuko-Mbeje, S.; Simelane, M.B.C. Iso-Mukaadial Acetate from Warburgia salutaris Enhances Glucose Uptake in the L6 Rat Myoblast Cell Line. Biomolecules 2019, 9, 520. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhang, M.; Hu, M.; Guo, H.-F.; Li, J.; Yu, Y.-L.; Jin, S.; Wang, X.-T.; Liu, L.; Liu, X.-D. Increased glucagon-like peptide-1 secretion may be involved in antidiabetic effects of ginsenosides. J. Endocrinol. 2013, 217, 185–196. [Google Scholar] [CrossRef]
- Zhou, P.; Xie, W.; He, S.; Sun, Y.; Meng, X.; Sun, G.; Sun, X. Ginsenoside Rb1 as an anti-diabetic agent and its underlying mechanism analysis. Cells 2019, 8, 204. [Google Scholar] [CrossRef]
- Qaseem, A.; Barry, M.J.; Humphrey, M.J.; Forciea, M.A.; Fitterman, N.; Horwitch, C.; Kansagara, D.; McLean, R.M.; Wilt, T.J. Oral pharmacologic treatment of type 2 diabetes mellitus: A clinical practice guideline update from the American college of physicians. Ann. Intern. Med. 2017, 166, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Garber, A.J.; Abrahamson, M.J.; Barzilay, J.I.; Blonde, L.; Bloomgarden, Z.T.; Bush, M.A.; Dagogo-Jack, S.; Davidson, M.B.; Einhorn, D.; Garvey, W.T.; et al. American Association of Clinical Endocrinologists’ comprehensive diabetes management algorithm 2013 consensus statement—Executive summary. Endocr. Pract. 2013, 19, 536–557. [Google Scholar] [CrossRef]
- Hussein, Z.; Wentworth, J.M.; Nankervis, A.J.; Proietto, J.; Colman, P.G. Effectiveness, and side effects of thiazolidinediones for type 2 diabetes: Real-life experience from a tertiary hospital. Med. J. Aust. 2004, 181, 536–539. [Google Scholar] [CrossRef]
- Elkady, A.I.; Abuzinadah, O.A.; Baeshen, N.A.; Rahmy, T.R. Differential control of growth, apoptotic activity, and gene expression in human breast cancer cells by extracts derived from medicinal herbs Zingiber officinale. J. Biomed. Biotechnol. 2012, 2012, 614356. [Google Scholar] [CrossRef]
- Ahmad, J.; Ahamed, M.; Akhtar, M.J.; Alrokayan, S.A.; Siddiqui, M.A.; Musarrat, J.; Al-Khedhairy, A.A. Apoptosis induction by silica nanoparticles mediated through reactive oxygen species in human liver cell in eHepG2. Toxicol. Appl. Pharmacol. 2012, 259, 160–168. [Google Scholar] [CrossRef]
- Teiten, M.H.; Eifes, S.; Dicato, M.; Diederich, M. Curcumin–the paradigm of a multi-target natural compound with applications in cancer prevention and treatment. Toxins 2010, 2, 128–162. [Google Scholar] [CrossRef] [PubMed]
- Pourhassanali, N.; Roshan-Milani, S.; Kheradmand, F.; Motazakker, M.; Bagheri, M.; Saboory, E. Zinc attenuates ethanol-induced Sertoli cell toxicity and apoptosis through caspase-3 mediated pathways. Reprod. Toxicol. 2016, 61, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.; Zhivotovsky, B. Caspases and cancer. Cell Death Differ. 2011, 8, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Overman, M.; Chatterjee, D.; Rashid, A.; Shroff, S.; Wang, H.; Katz, M.H.; Fleming, J.B.; Varadhachary, G.R.; Abbruzzese, J.L.; et al. Aberrant expression of p53, p21, cyclinD1, and BCL2 and their clinicopathological correlation in ampullary adenocarcinoma. Hum. Pathol. 2014, 45, 1015–1023. [Google Scholar] [CrossRef]
- Shi, L.; Teng, H.; Zhu, M.; Li, C.; Huang, K.; Chen, B.; Dai, Y.; Wang, J. Paeoniflorin inhibits nucleus pulposus cell apoptosis by regulating the expression of BCL2 family proteins and caspase-9 in a rabbit model of intervertebral disc degeneration. Exp. Ther. Med. 2015, 10, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Lu, H.; An, L.; Wang, C.; Zhou, Z.; Feng, F.; Ma, H.; Xu, Y.; Zhao, Q. Effect of active fraction of Eriocaulon sieboldianum on human leukemia K562 cells via proliferation inhibition, cell cycle arrest and apoptosis induction. Environ. Toxicol. Pharmacol. 2016, 43, 13–20. [Google Scholar] [CrossRef]
- Lohrum, M.A.E.; Vousden, K.H. Regulation and function of the p53-related proteins: Same family, different rules (Electronic version). Trends Cell Biol. 2000, 10, 197–202. [Google Scholar] [CrossRef]
- Shrivastava, A.; Kuzontkoski, P.M.; Groopman, J.E.; Prasad, A. Cannabidiol induces programmed cell death in breast cancer cells by coordinating the crosstalk between apoptosis and autophagy. Mol. Cancer Ther. 2011, 10, 1161–1172. [Google Scholar] [CrossRef]
- McAllister, S.D.; Murase, R.; Christian, R.T.; Lau, D.; Zielinski, A.J.; Allison, J.; Almanza, C.; Pakdel, A.; Lee, J.; Limbad, C.; et al. Pathways mediating the effects of cannabidiol on the reduction of breast cancer cell proliferation, invasion, and metastasis. Breast Cancer Res. Treat. 2011, 129, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Blazquez, C.; Casanova, M.L.; Planas, A.; Gomez Del Pulgar, T.; Villanueva, C.; Fernandez-Acenero, M.J.; Aragones, J.; Huffman, J.W.; Jorcano, J.L.; Guzman, M. Inhibition of tumor angiogenesis by cannabinoids. FASEB J. 2003, 17, 529–531. [Google Scholar] [CrossRef]
- Vaccani, A.; Massi, P.; Colombo, A.; Rubino, T.; Parolaro, D. Cannabidiol inhibits human glioma cell migration through a cannabinoid receptor-independent mechanism. Br. J. Pharmacol. 2005, 144, 1032–1036. [Google Scholar] [CrossRef] [PubMed]
- Ramer, R.; Bublitz, K.; Freimuth, N.; Merkord, J.; Rohde, H.; Haustein, M.; Borchert, P.; Schmuhl, E.; Linnebacher, M.; Hinz, B. Cannabidiol inhibits lung cancer cell invasion and metastasis via intercellular adhesion molecule 1. FASEB J. 2012, 26, 1535–1548. [Google Scholar] [CrossRef] [PubMed]
- Youssif, B.G.M.; Mohamed, A.M.; Osman, E.E.A.; Abou-Ghadir, O.F.; Elnaggar, D.H.; Abdelrahman, M.H.; Treamblu, L.; Gomaa, H.A. 5-Chlorobenzofuran-2-carboxamides: From allosteric CB1 modulators to potential apoptotic antitumor agents. Eur. J. Med. Chem. 2019, 177, 1–11. [Google Scholar] [CrossRef]
- Abdelrahman, M.H.; Aboraia, A.S.; Youssif, B.G.M.; Elsadek, B.E.M. Design, synthesis and pharmacophoric model building of new 3-alkoxymethyl/3-phenyl indole-2-carboxamides with potential antiproliferative activity. Chem. Biol. Drug Des. 2017, 90, 64–82. [Google Scholar] [CrossRef] [PubMed]
- Gomaa, H.A.M.; Shaker, M.E.; Alzarea, S.I.; Hendawy, O.M.; Mohamed, F.A.M.; Gouda, A.M.; Ali, A.T.; Morcoss, M.M.; Abdelrahman, M.H.; Trembleau, L.; et al. Optimization and SAR investigation of novel 2,3-dihydropyrazino[1,2-a]indole-1,4-dione derivatives as EGFR and BRAFV600E dual inhibitors with potent antiproliferative and antioxidant activities. Bioorg. Chem. 2022, 120, 105616. [Google Scholar] [CrossRef]
- Al-Wahaibi, L.H.; Gouda, A.M.; Abou-Ghadir, O.F.; Salem, O.I.A.; Ali, A.T.; Farghaly, H.S.; Abdelrahman, M.H.; Trembleau, L.; Abdu-Allah, H.H.M.; Youssif, B.G.M. Design, and synthesis of novel 2,3-dihydropyrazino[1,2-a]indole-1,4-dione derivatives as antiproliferative EGFR and BRAFV600E dual inhibitors. Bioorg. Chem. 2020, 104, 104260. [Google Scholar] [CrossRef]
- El-Sheref, E.M.; Elbastawesy, M.A.I.; Brown, A.B.; Shawky, A.M.; Gomaa, H.A.M.; Bräse, S.; Youssif, B.G.M. Design and Synthesis of (2-oxo-1, 2-Dihydroquinolin-4-yl)-1,2,3-triazole derivatives via Click Reaction: Potential Apoptotic Antiproliferative Agents. Molecules 2021, 26, 6798. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Aziz, S.A.; Taher, E.S.; Lan, P.; Asaad, G.F.; Gomaa, H.A.M.; El-Koussi, N.A.; Youssif, B.G.M. Design, synthesis, and biological evaluation of new pyrimidine-5-carbonitrile derivatives bearing 1, 3-thiazole moiety as novel anti-inflammatory EGFR inhibitors with cardiac safety profile. Bioorg. Chem. 2021, 111, 104890. [Google Scholar] [CrossRef]
- Mekheimer, R.A.; Allam, S.M.R.; Al-Sheikh, M.A.; Moustafa, M.S.; Al-Mousawi, S.M.; Mostafa, Y.A.; Youssif, B.G.M.; Gomaa, H.A.M.; Hayallah, A.M.; Abdel Aziz, M.; et al. Discovery of new pyrimido [5,4-c] quinolines as potential antiproliferative agents with multitarget actions: Rapid synthesis, docking, and ADME studies. Bioorg. Chem. 2022, 121, 105693. [Google Scholar] [CrossRef]
- Khurana, L.; Mackie, K.; Piomelli, D.; Kendall, D.A. Modulation of CB1 cannabinoid receptor by allosteric ligands: Pharmacology and therapeutic opportunities. Neuropharmacology 2017, 124, 3–12. [Google Scholar] [CrossRef]
- Nguyen, T.; Li, J.X.; Thomas, B.F.; Wiley, J.L.; Kenakin, T.P.; Zhang, Y. Allosteric Modulation: An Alternate Approach Targeting the Cannabinoid CB1 Receptor. Med. Res. Rev. 2017, 37, 441–474. [Google Scholar] [CrossRef] [PubMed]
- Cohen, G.M. Caspases: The executioners of apoptosis. Biochem. J. 1997, 236, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Abou-Zied, H.A.; Youssif, B.G.M.; Mohamed, M.F.A.; Hayallah, A.M.; Abdel-Aziz, M. EGFR inhibitors and apoptotic inducers: Design, synthesis, anticancer activity, and docking studies of novel xanthine derivatives carrying chalcone moiety as hybrid molecules. Bioorg. Chem. 2019, 89, 102997. [Google Scholar] [CrossRef] [PubMed]
- Slee, E.A.; Adrain, C.; Martin, S.J. Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J. Biol. Chem. 2001, 276, 7320–7326. [Google Scholar] [CrossRef]
- Hisham, M.; Youssif, B.G.M.; Osman, E.E.A.; Hayallah, A.M.; Abdel-Aziz, M. Synthesis and biological evaluation of novel xanthine derivatives as potential apoptotic antitumor agents. Eur. J. Med. Chem. 2019, 176, 117–128. [Google Scholar] [CrossRef]
- Mitupatum, T.; Aree, K.; Kittisenachai, S.; Roytrakul, S.; Puthong, S.; Kangsadalampai, S.; Rojpibulstit, P. mRNA Expression of Bax, Bcl-2, p53, Cathepsin B, Caspase-3 and Caspase-9 in the HepG2 Cell Line Following Induction by a Novel Monoclonal Ab Hep88 mAb: Cross-Talk for Paraptosis and Apoptosis. Asian Pac. J. Cancer Prev. 2016, 17, 703–712. [Google Scholar] [CrossRef]
- Gatza, C.; Moore, L.; Dumble, M.; Donehower, L.A. Tumor suppressor dosage regulates stem cell dynamics during aging. Cell Cycle 2007, 6, 52–55. [Google Scholar] [CrossRef]
- Godar, S.; Ince, T.A.; Bell, G.W.; Feldser, D.; Donaher, J.L.; Bergh, J.; Liu, A.; Miu, K.; Watnick, R.S.; Reinhardt, F.; et al. Growth-inhibitory and tumor- suppressive functions of p53 depend on its repression of CD44 expression. Cell 2008, 134, 62–73. [Google Scholar] [CrossRef]
- Finlay, C.A.; Hinds, P.W.; Levine, A.J. The p53 proto-oncogene can act as a suppressor of transformation. Cell 1989, 57, 1083–1093. [Google Scholar] [CrossRef]
- Levine, A.J.; Hu, W.; Feng, Z. The p53 pathway: What questions remain to be explored? Cell Death Differ. 2006, 13, 1027–1036. [Google Scholar] [CrossRef]
- Kim, N.H.; Kim, H.S.; Kim, N.-G.; Lee, I.; Choi, H.-S.; Li, X.-Y.; Kang, S.E.; Cha, S.Y.; Ryu, J.K.; Na, J.M.; et al. p53 and MicroRNA-34 are suppressors of canonical Wnt signaling. Sci. Signal. 2011, 4, 197. [Google Scholar] [CrossRef] [PubMed]
- Junttila, M.R.; Evan, G.I. p53 a Jack of all trades but master of none. Nat. Rev. Cancer 2009, 9, 821–829. [Google Scholar] [CrossRef]
- Gomaa, H.A.M.; El-Sherief, H.A.M.; Hussein, S.; Gouda, A.M.; Salem, O.I.A.; Alharbi, K.S.; Hayallah, A.M.; Youssif, B.G.M. Novel 1, 2,4-triazole derivatives as apoptotic inducers targeting p53: Synthesis and antiproliferative activity. Bioorg. Chem. 2020, 105, 104369. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Compd. | Cell Viability % | Antiproliferative Activity IC50 ± SEM (µM) | ||||
| A-549 | MCF-7 | Panc-1 | HT-29 | Average | ||
| 5a | 89 | 3.70 ± 0.30 | 3.20 ± 0.30 | 3.90 ± 0.30 | 3.90 ± 0.30 | 3.70 |
| 5b | 87 | 3.20 ± 0.30 | 2.90 ± 0.30 | 3.50 ± 0.30 | 3.60 ± 0.30 | 3.30 |
| 5c | 87 | 1.70 ± 0.20 | 1.40 ± 0.20 | 1.80 ± 0.20 | 1.80 ± 0.20 | 1.70 |
| 5d | 89 | 1.05 ± 0.10 | 0.90 ± 0.10 | 1.10 ± 0.10 | 1.10 ± 0.10 | 1.05 |
| 5e | 93 | 0.95 ± 0.05 | 0.80 ± 0.05 | 1.00 ± 0.20 | 1.10 ± 0.10 | 0.95 |
| 5f | 90 | 1.90 ± 0.20 | 1.70 ± 0.20 | 2.10 ± 0.20 | 2.10 ± 0.20 | 1.95 |
| 5g | 89 | 4.90 ± 0.50 | 4.80 ± 0.50 | 5.20 ± 0.50 | 5.10 ± 0.50 | 5.00 |
| 5h | 87 | 1.00 ± 0.10 | 0.90 ± 0.10 | 1.20 ± 0.10 | 1.20 ± 0.10 | 1.10 |
| 5i | 90 | 1.55 ± 0.20 | 1.30 ± 0.10 | 1.60 ± 0.20 | 1.65 ± 0.20 | 1.50 |
| 5j | 83 | 1.20 ± 0.10 | 1.00 ± 0.10 | 1.30 ± 0.10 | 1.30 ± 0.10 | 1.20 |
| 5k | 87 | 1.40 ± 0.20 | 1.20 ± 0.10 | 1.50 ± 0.20 | 1.50 ± 0.20 | 1.40 |
| 6a | 90 | 2.90 ± 0.30 | 2.60 ± 0.20 | 2.80 ± 0.20 | 2.90 ± 0.20 | 2.80 |
| 6b | 91 | 2.50 ± 0.20 | 2.30 ± 0.20 | 2.65 ± 0.20 | 2.80 ± 0.20 | 2.60 |
| 6c | 89 | 2.20 ± 0.20 | 2.10 ± 0.20 | 2.40 ± 0.20 | 2.50 ± 0.20 | 2.30 |
| 7 | 91 | 4.10 ± 0.40 | 4.00 ± 0.40 | 4.40 ± 0.40 | 4.60 ± 0.40 | 4.30 |
| Doxorubicin | - | 1.20 ± 0.20 | 0.90 ± 0.10 | 1.40 ± 0.20 | 1.00 ± 0.10 | 1.10 |
 | ||||||
|---|---|---|---|---|---|---|
| Compd. | R1 | R2 | R3 | R4 | EGFR Inhibition IC50 ± SEM (nM) | CDK2 Inhibition IC50 ± SEM (nM) |
| 5d | Cl | H | H |  | 89 ± 6 | 23 ± 2 |
| 5e | Cl | H | H |  | 93 ± 8 | 13 ± 1 |
| 5h | Cl | H | Cl |  | 118 ± 10 | 11 ± 1 |
| 5i | Cl | H | Cl |  | 137 ± 12 | 27 ± 3 |
| 5j | F | H | F | H | 98 ± 8 | 34 ± 3 |
| 5k | F | H | F |  | 129 ± 11 | 19 ± 2 |
| Erlotinib | -- | -- | -- | -- | 80 ± 5 | ND |
| Dinaciclib | -- | -- | -- | -- | ND | 20 ± 2 |
| Compound Number | Caspase-3 | Caspase-8 | Caspase-9 | Cytochrome C | ||||
|---|---|---|---|---|---|---|---|---|
| Conc (pg/mL) | Fold Change | Conc (ng/mL) | Fold Change | Conc (ng/mL) | Fold Change | Conc (ng/mL) | Fold Change | |
| 5d | 570.00 ± 5.00 | 8.70 | 1.94 | 9.70 | 16.90 | 17.80 | 0.70 | 14 |
| 5e | 635.50 ± 5.50 | 9.70 | 2.17 | 10.90 | 17.25 | 18.15 | 0.80 | 16 |
| 5h | 537.50 ± 5.00 | 8.20 | 1.88 | 9.50 | 16.65 | 17.50 | 0.65 | 13 |
| Doxorubicin | 503.50 ± 4.50 | 7.70 | 1.80 | 9.00 | 16.25 | 17.00 | 0.60 | 12 |
| Control | 65.50 | 1 | 0.20 | 1 | 0.95 | 1 | 0.05 | 1 |
| Compd. No. | Bax | Bcl-2 | ||
|---|---|---|---|---|
| Conc (pg/mL) | Fold Change | Conc (ng/mL) | Fold Change | |
| 5d | 289.70 ± 2.50 | 35 | 0.89 | 5.70 |
| 5e | 296.50 ± 2.50 | 36 | 0.87 | 5.90 |
| Doxorubicin | 275.80 ± 2.50 | 33 | 0.98 | 5.20 |
| Cont. | 8.25 | 1 | 5.10 | 1.00 |
| Compd. No. | p53 | |
|---|---|---|
| Conc (pg/mL) | Fold Change | |
| 5d | 1375 ± 15 | 27 |
| 5e | 1435 ± 15 | 28 |
| Doxorubicin | 1265 ± 10 | 25 |
| Cont. | 51.50 | 1 |
| Compd. | EFGR (PDB ID: 1M17) | CDK2 (PDB ID: 1PYE) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| S a | RMSD (Å) | Binding Interactions | S | RMSD (Å) | Binding Interactions | |||||
| a.a. Residue | Type | Distance (Å) | a.a. Residue | Type | Distance (Å) | |||||
| 5d | −6.90 | 1.49 | THR 766 | H-acceptor | 2.91 | −6.03 | 1.77 | GLN 131 | H-donor | 3.60 |
| LEU 694 | pi-H | 3.99 | ||||||||
| 5e | −6.79 | 1.51 | LEU 694 | pi-H | 3.70 | −6.99 | 1.68 | LYS 33 | pi-cation | 4.65 |
| Ref | −7.3 b | 1.28 | GLN 767 | H-donor | 3.15 | −5.89 c | 1.84 | GLU 81 | H-donor | 3.05 |
| MET 769 | H-acceptor | 2.70 | LEU 83 | H-acceptor | 3.07 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Wahaibi, L.H.; Mostafa, Y.A.; Abdelrahman, M.H.; El-Bahrawy, A.H.; Trembleau, L.; Youssif, B.G.M. Synthesis and Biological Evaluation of Indole-2-Carboxamides with Potent Apoptotic Antiproliferative Activity as EGFR/CDK2 Dual Inhibitors. Pharmaceuticals 2022, 15, 1006. https://doi.org/10.3390/ph15081006
Al-Wahaibi LH, Mostafa YA, Abdelrahman MH, El-Bahrawy AH, Trembleau L, Youssif BGM. Synthesis and Biological Evaluation of Indole-2-Carboxamides with Potent Apoptotic Antiproliferative Activity as EGFR/CDK2 Dual Inhibitors. Pharmaceuticals. 2022; 15(8):1006. https://doi.org/10.3390/ph15081006
Chicago/Turabian StyleAl-Wahaibi, Lamya H., Yaser A. Mostafa, Mostafa H. Abdelrahman, Ali H. El-Bahrawy, Laurent Trembleau, and Bahaa G. M. Youssif. 2022. "Synthesis and Biological Evaluation of Indole-2-Carboxamides with Potent Apoptotic Antiproliferative Activity as EGFR/CDK2 Dual Inhibitors" Pharmaceuticals 15, no. 8: 1006. https://doi.org/10.3390/ph15081006