
Design and Synthesis of New Hydantoin Acetanilide Derivatives as Anti-NSCLC Targeting EGFRL858R/T790M Mutations

 , , , and
, , , and
Abstract
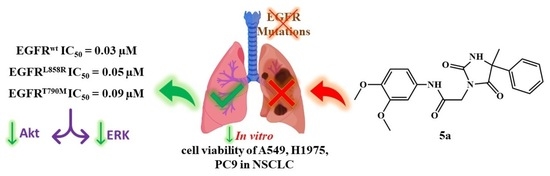
:
1. Introduction
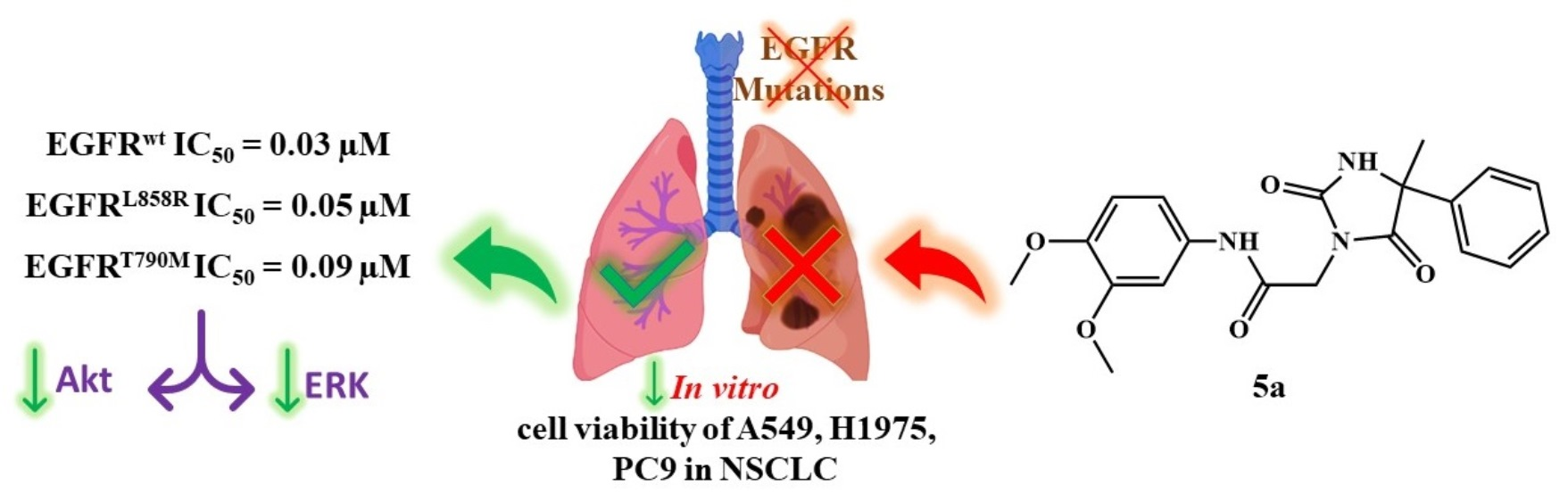
Rational Design of EGFR Inhibitors
2. Results and Discussion
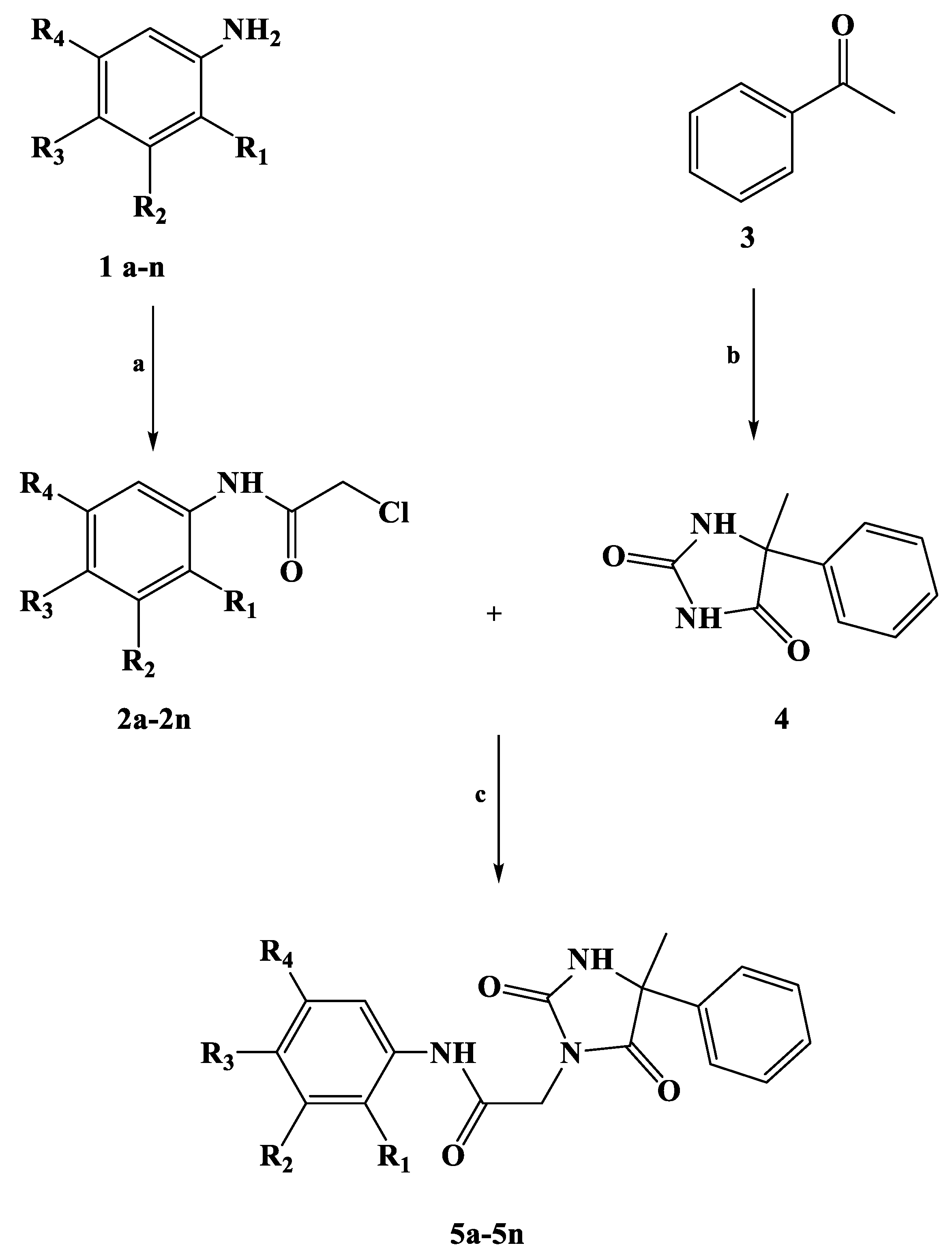
2.1. Chemistry
2.2. In Vitro Antiproliferative Activity
2.3. In Vitro EGFR Inhibition Assay
2.4. In Vitro Inhibition of the EGFR Downstream Pathway
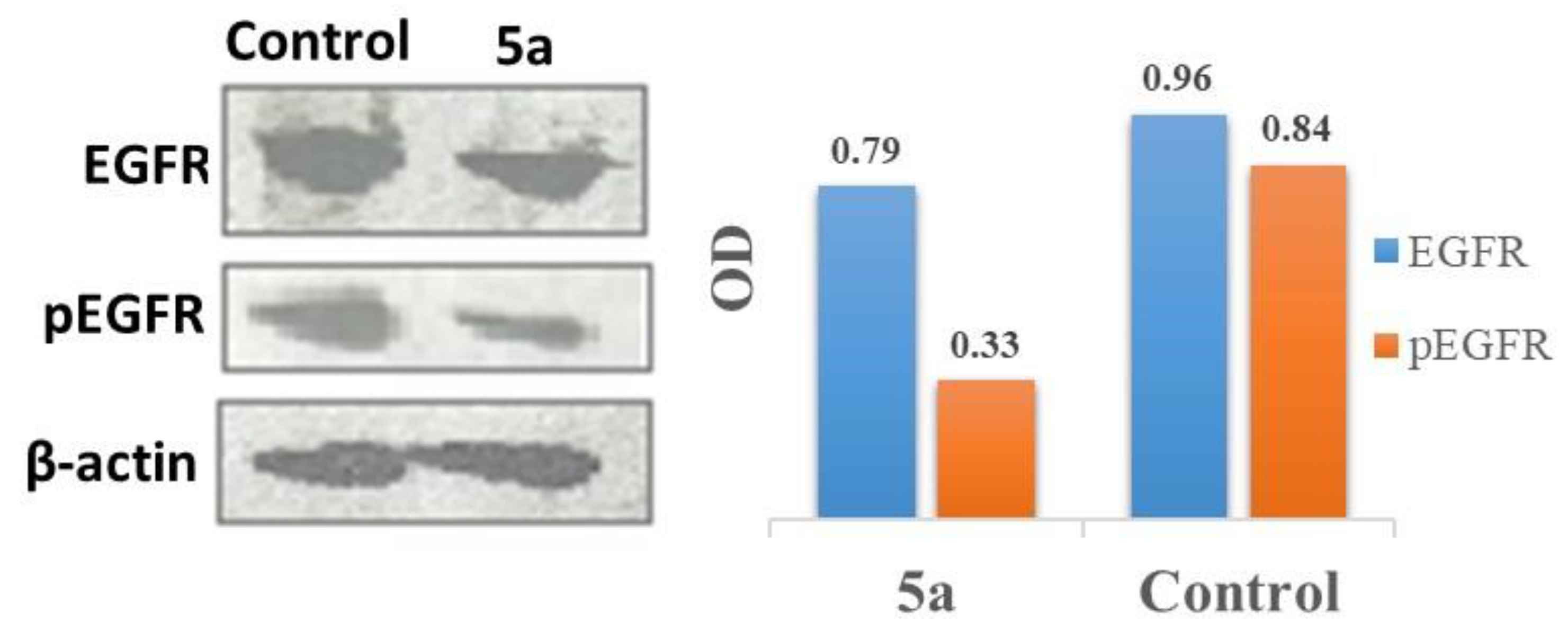
2.5. Inhibition of EGFR Phosphorylation Expression Using Western Blot Analysis
2.6. In Vitro Cytotoxic Activity towards Normal Fibroblast (WI-38) Cells
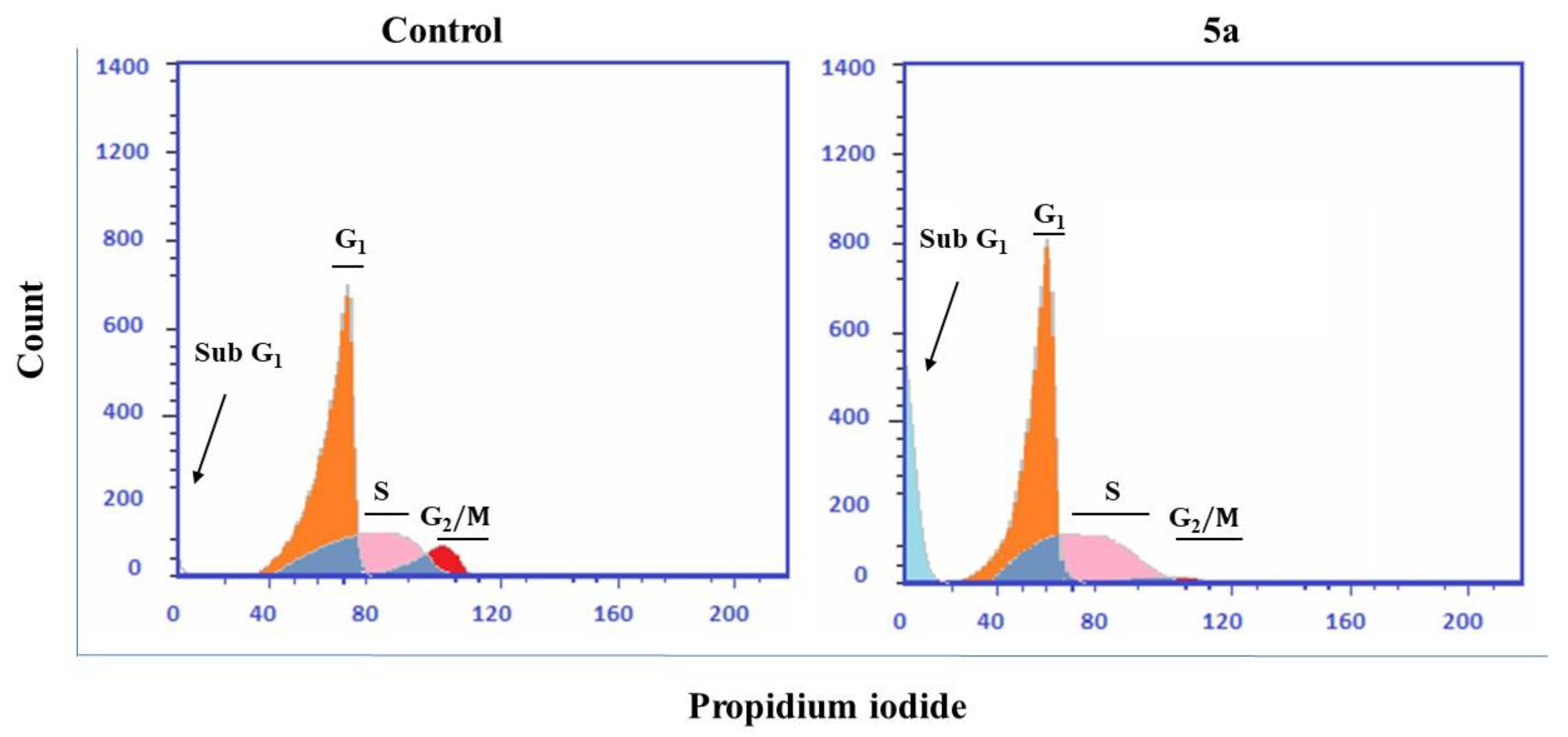
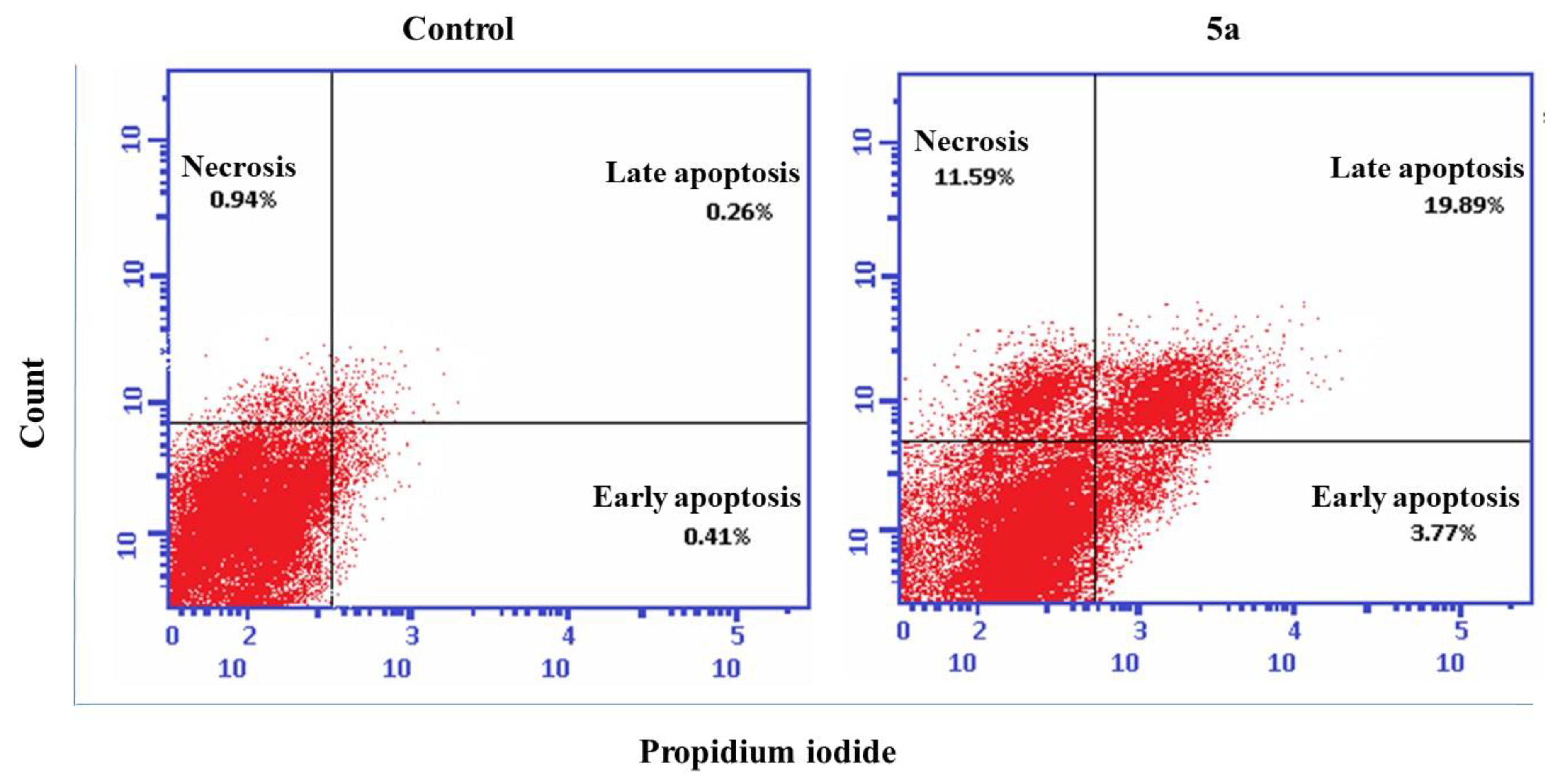
2.7. Cell Cycle Analysis and Detection of Apoptosis
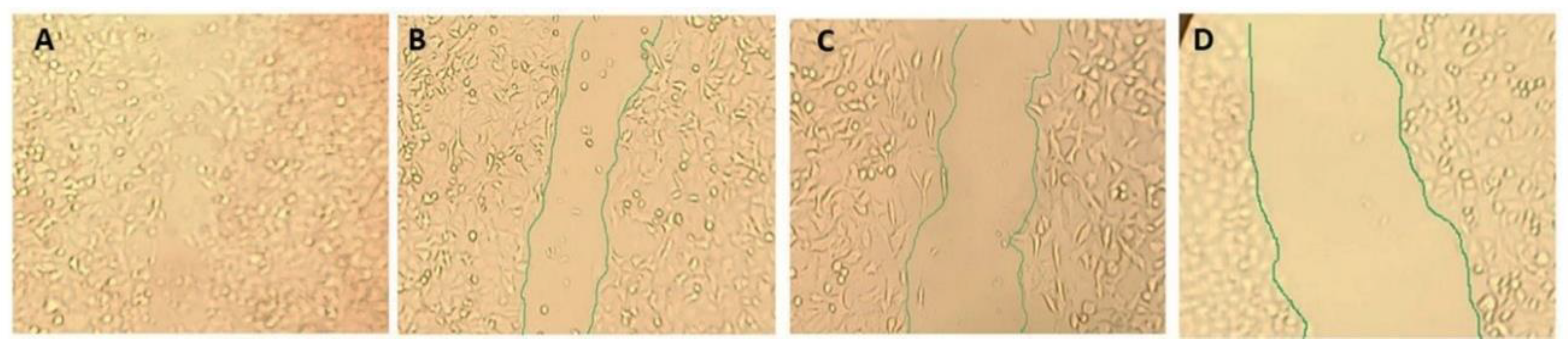
2.8. Wound Healing Assay Tau Aggregation Inhibition in a Cell Model of Tauopathy and Western Blot Analysis
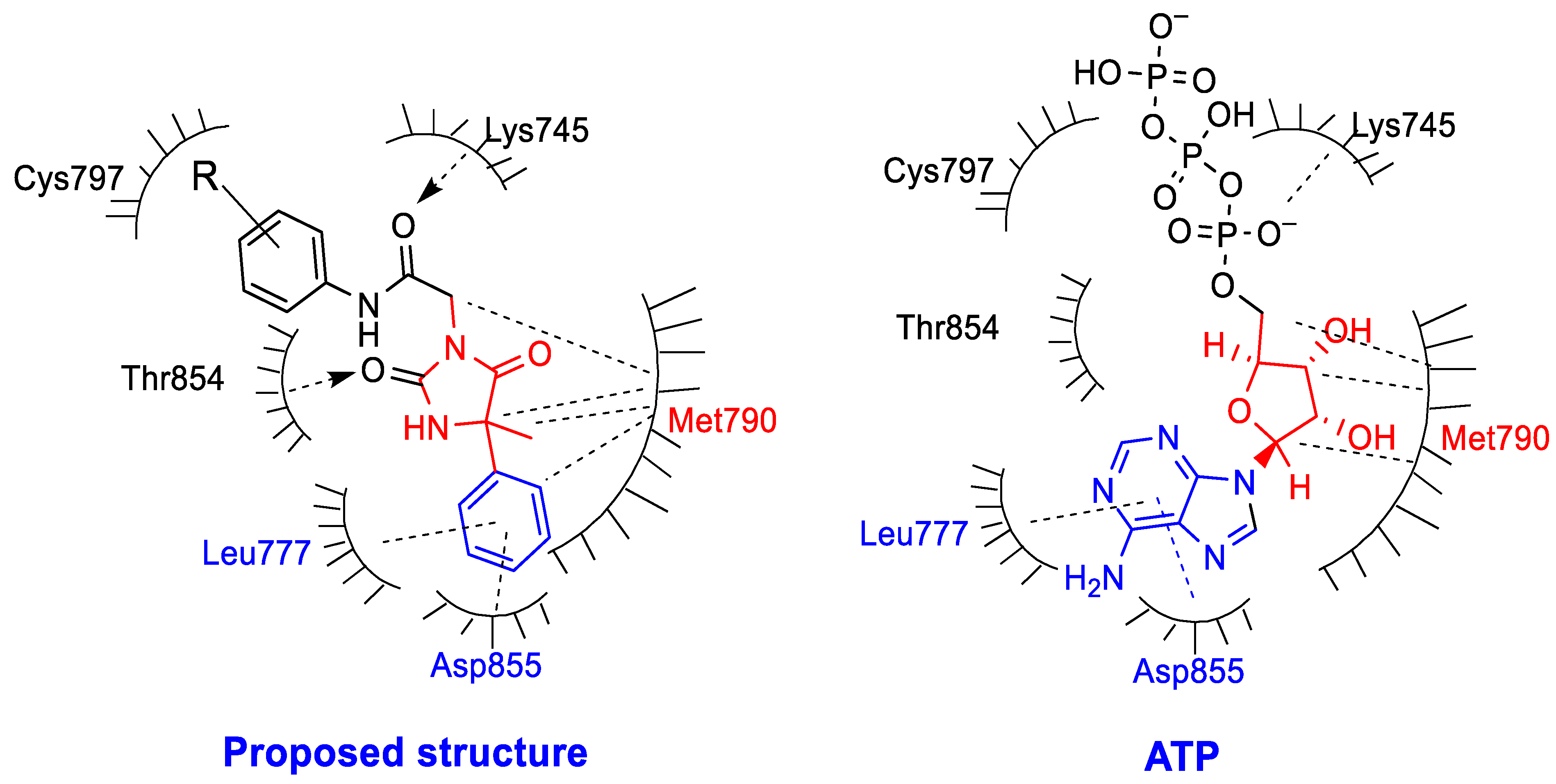
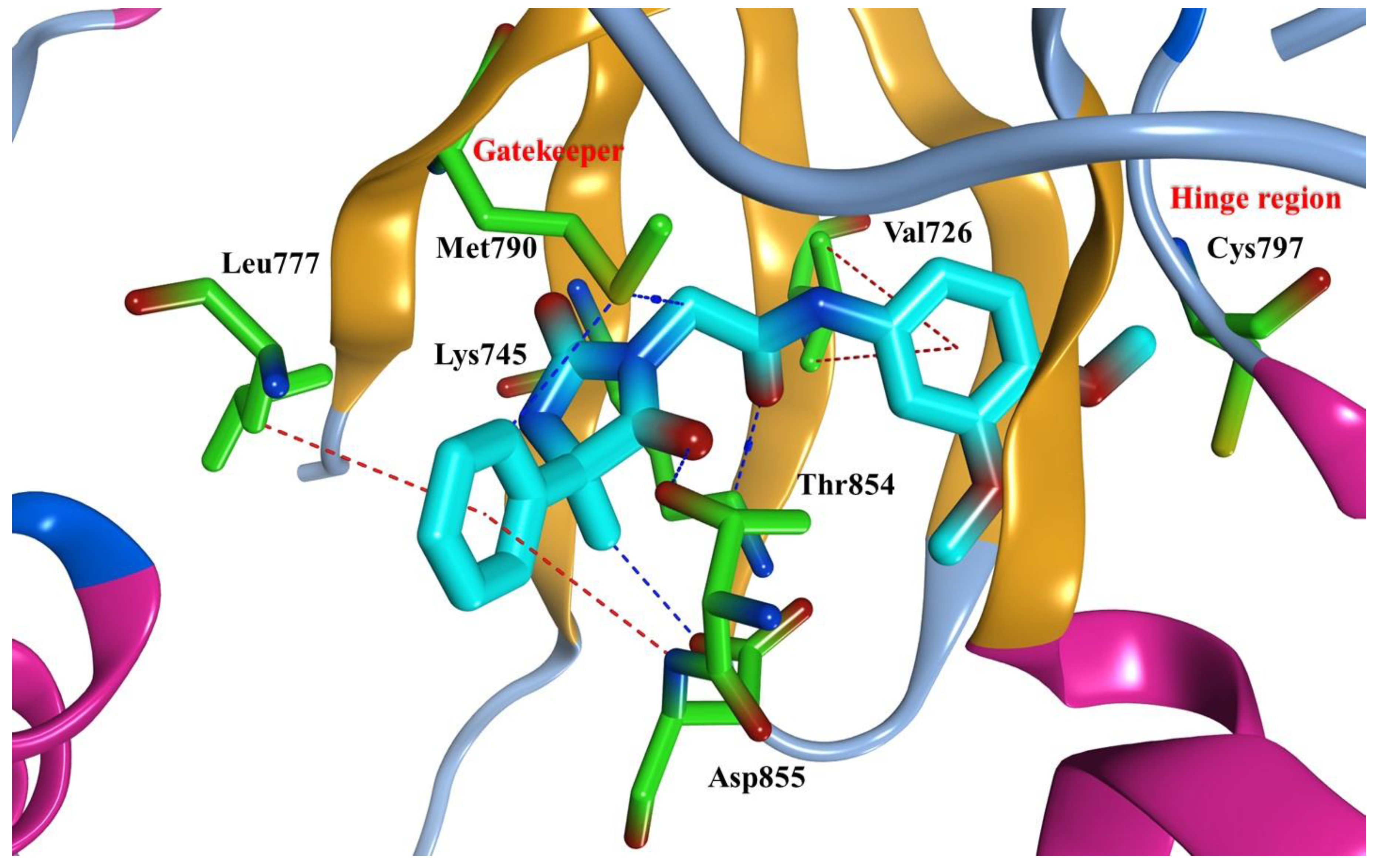
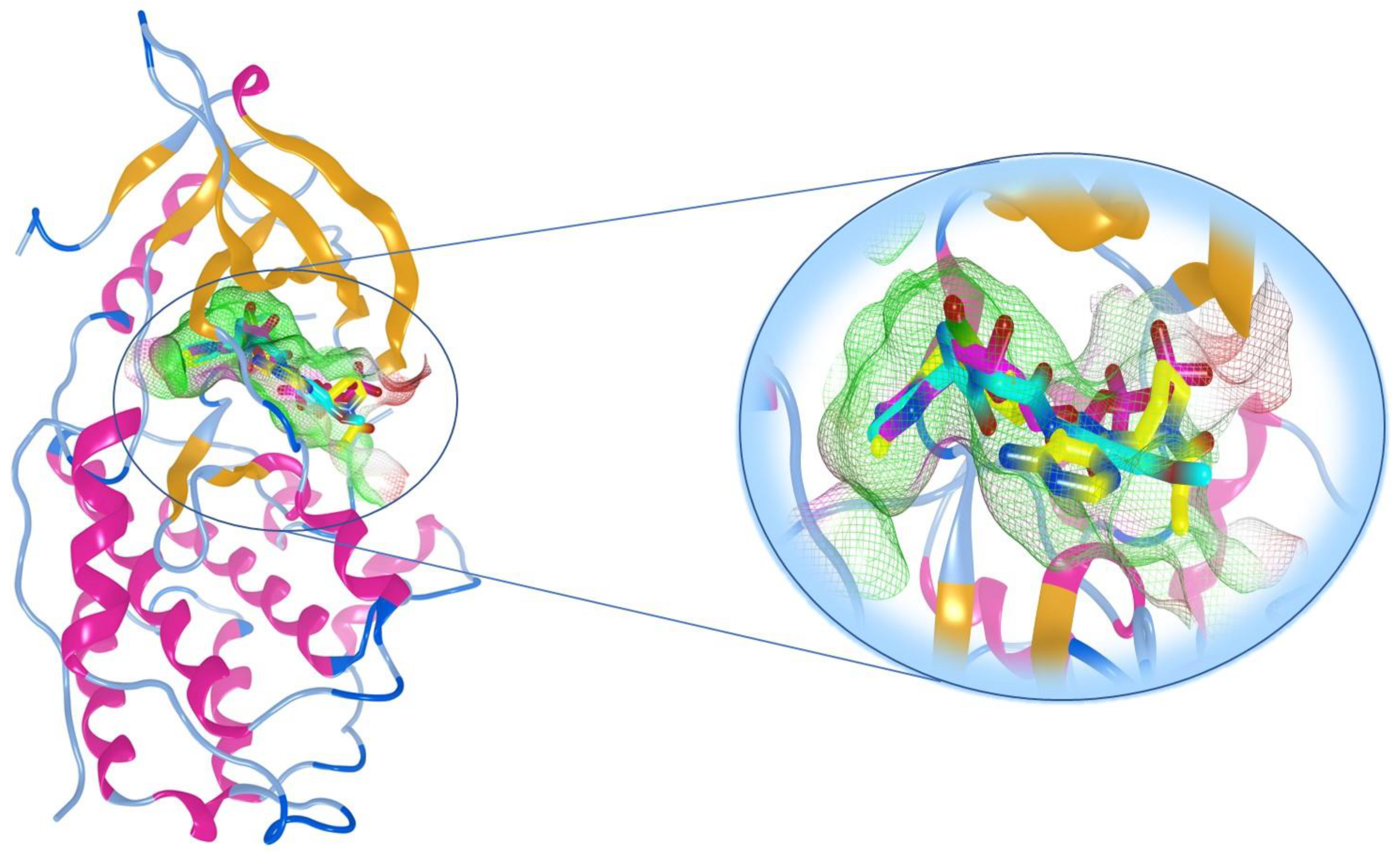
2.9. Molecular Docking Study
3. Materials and Methods
3.1. Chemistry
3.2. General Procedure for the Synthesis of Compounds 2a–2n
3.3. General Procedure for the Synthesis of Compound 4
3.4. Anti-Proliferative Activities against A549, H1975, PC9, WI-38 Cell Lines
3.5. EGFR Inhibition Assay
3.6. In Vitro Inhibition of EGFR Downstream Pathway
3.6.1. Akt Enzyme Assay
3.6.2. ERK Enzyme Assay
3.7. Inhibition of EGFR Phosphorylation Expression Using Western Blot Analysis
3.8. Cell Cycle Analysis and Detection of Apoptosis
3.9. Wound Healing Assay
3.10. Molecular Docking to EGFR Active Site
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Meng, Y.; Yu, B.; Huang, H.; Peng, Y.; Li, E.; Yao, Y.; Song, C.; Yu, W.; Zhu, K.; Wang, K.; et al. Discovery of dosimertinib, a highly potent, selective, and orally efficacious deuterated EGFR targeting clinical candidate for the treatment of non-small-cell lung cancer. J. Med. Chem. 2021, 64, 925–937. [Google Scholar] [CrossRef]
- An, B.; Pan, T.; Hu, J.; Pang, Y.; Huang, L.; Chan, A.S.; Li, X.; Yan, J. The discovery of a potent and selective third-generation EGFR kinase inhibitor as a therapy for EGFR L858R/T790M double mutant non-small cell lung cancer. Eur. J. Med. Chem. 2019, 183, 111709. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-Q.; Cui, J.-W. Non-small cell lung cancer patients with ex19del or exon 21 L858R mutation: Distinct mechanisms, different efficacies to treatments. J. Cancer Res. Clin. Oncol. 2020, 146, 2329–2338. [Google Scholar] [CrossRef] [PubMed]
- Pawara, R.; Ahmad, I.; Nayak, D.; Wagh, S.; Wadkar, A.; Ansari, A.; Belamkar, S.; Surana, S.; Kundu, C.N.; Patil, C.; et al. Novel, selective acrylamide linked quinazolines for the treatment of double mutant EGFR-L858R/T790M Non-Small-Cell lung cancer (NSCLC). Bioorg. Chem. 2021, 115, 105234. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lv, H.; Luo, L.; Xu, Y.; Pan, Y.; Wang, Y.; Lin, H.; Xiong, J.; Guo, P.; Zhang, J.; et al. Design, synthesis and pharmacological evaluation of N4,N6-disubstituted pyrimidine-4,6-diamine derivatives as potent EGFR inhibitors in non-small cell lung cancer. Eur. J. Med. Chem. 2018, 157, 1300–1325. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.M.; Pawara, R.; Ansari, A.; Noolvi, M.; Surana, S. Design and synthesis of quinazolinones as EGFR inhibitors to overcome EGFR resistance obstacle. Bioorg. Med. Chem. 2017, 25, 2713–2723. [Google Scholar] [CrossRef]
- Lee, J.Y.; Yang, H.; Kim, D.; Kyaw, K.Z.; Hu, R.; Fan, Y.; Lee, S.K. Antiproliferative Activity of a New Quinazolin-4(3H)-One Derivative via Targeting Aurora Kinase A in Non-Small Cell Lung Cancer. Pharmaceuticals 2022, 15, 698. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Dong, X.; Gao, Z.; Zheng, X.; Ji, J.; Zhang, M.; Liu, F.; Wu, S.; Li, M.; Song, W.; et al. Design, synthesis and biological evaluation of novel N-(3-amino-4-methoxyphenyl)acrylamide derivatives as selective EGFRL858R/T790M kinase inhibitors. Bioorg. Chem. 2021, 118, 105471. [Google Scholar] [CrossRef]
- Cohen, M.H.; Williams, G.A.; Sridhara, R.; Chen, G.; Pazdur, R. FDA Drug Approval Summary: Gefitinib (ZD1839) (Iressa®) Tablets. Oncologist 2003, 8, 303–306. [Google Scholar] [CrossRef]
- Sordella, R.; Bell, D.W.; Haber, D.A.; Settleman, J. Gefitinib-Sensitizing EGFR Mutations in Lung Cancer Activate Anti-Apoptotic Pathways. Science 2004, 305, 1163–1167. [Google Scholar] [CrossRef]
- Miyamoto, S.; Azuma, K.; Ishii, H.; Bessho, A.; Hosokawa, S.; Fukamatsu, N.; Kunitoh, H.; Ishii, M.; Tanaka, H.; Aono, H.; et al. Low-dose erlotinib treatment in elderly or frail patients with EGFR mutation–positive non–small cell lung cancer: A multicenter phase 2 trial. JAMA Oncol. 2020, 6, e201250. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.-H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.K.; Meyerson, M.; Eck, M.J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juchum, M.; Günther, M.; Döring, E.; Sievers-Engler, A.; Lämmerhofer, M.; Laufer, S. Trisubstituted imidazoles with a rigidized hinge binding motif act as single digit nM inhibitors of clinically relevant EGFR L858R/T790M and L858R/T790M/C797S mutants: An example of target hopping. J. Med. Chem. 2017, 60, 4636–4656. [Google Scholar] [CrossRef]
- Marquez-Medina, D.; Popat, S. Afatinib: A second-generation EGF receptor and ErbB tyrosine kinase inhibitor for the treatment of advanced non-small-cell lung cancer. Futur. Oncol. 2015, 11, 2525–2540. [Google Scholar] [CrossRef] [PubMed]
- Mitsudomi, T. Dacomitinib: Another option for EGFR-mutant lung cancer? Lancet Oncol. 2014, 15, 1408–1409. [Google Scholar] [CrossRef]
- Sequist, L.V.; Besse, B.; Lynch, T.J.; Miller, V.A.; Wong, K.K.; Gitlitz, B.; Eaton, K.; Zacharchuk, C.; Freyman, A.; Powell, C.; et al. Neratinib, an irreversible pan-ErbB receptor tyrosine kinase inhibitor: Results of a phase II trial in patients with advanced non–small-cell lung cancer. J. Clin. Oncol. 2010, 28, 3076–3083. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; John, T.; Grohe, C.; Majem, M.; Goldman, J.W.; Kim, S.W.; Kato, T.; Laktionov, K.; Vu, H.V.; Wang, Z.; et al. Postoperative Chemotherapy Use and Outcomes From ADAURA: Osimertinib as Adjuvant Therapy for Resected EGFR-Mutated NSCLC. J. Thorac. Oncol. 2021, 17, 423–433. [Google Scholar] [CrossRef]
- Yan, X.E.; Ayaz, P.; Zhu, S.J.; Zhao, P.; Liang, L.; Zhang, C.H.; Wu, Y.C.; Li, J.L.; Choi, H.G.; Huang, X.; et al. Structural basis of AZD9291 selectivity for EGFR T790M. J. Med. Chem. 2020, 63, 8502–8511. [Google Scholar] [CrossRef]
- Shen, J.; Zhang, T.; Zhu, S.J.; Sun, M.; Tong, L.; Lai, M.; Zhang, R.; Xu, W.; Wu, R.; Ding, J.; et al. Structure-based design of 5-methylpyrimidopyridone derivatives as new wild-type sparing inhibitors of the epidermal growth factor receptor triple mutant (EGFRL858R/T790M/C797S). J. Med. Chem. 2019, 62, 7302–7308. [Google Scholar] [CrossRef]
- Cheng, H.; Nair, S.K.; Murray, B.W.; Almaden, C.; Bailey, S.; Baxi, S.; Behenna, D.; Cho-Schultz, S.; Dalvie, D.; Dinh, D.M.; et al. Discovery of 1-{(3 R, 4 R)-3-[({5-Chloro-2-[(1-methyl-1 H-pyrazol-4-yl) amino]-7 H-pyrrolo [2, 3-d] pyrimidin-4-yl} oxy) methyl]-4-methoxypyrrolidin-1-yl} prop-2-en-1-one (PF-06459988), a Potent, WT Sparing, Irreversible Inhibitor of T790M-Containing EGFR Mutants. J. Med. Chem. 2016, 59, 2005–2024. [Google Scholar]
- Azizmohammadi, M.; Khoobi, M.; Ramazani, A.; Emami, S.; Zarrin, A.; Firuzi, O.; Miri, R.; Shafiee, A. 2H-chromene derivatives bearing thiazolidine-2,4-dione, rhodanine or hydantoin moieties as potential anticancer agents. Eur. J. Med. Chem. 2013, 59, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Alkahtani, H.M.; Alanazi, M.M.; Aleanizy, F.S.; Alqahtani, F.Y.; Alhoshani, A.; Alanazi, F.E.; Almehizia, A.A.; Abdalla, A.N.; Alanazi, M.G.; El-Azab, A.S.; et al. Synthesis, anticancer, apoptosis-inducing activities and EGFR and VEGFR2 assay mechanistic studies of 5,5-diphenylimidazolidine-2,4-dione derivatives: Molecular docking studies. Saudi Pharm. J. 2019, 27, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Mourad, A.A.; Farouk, N.; El-Sayed, E.-S.H.; Mahdy, A.R. EGFR/VEGFR-2 dual inhibitor and apoptotic inducer: Design, synthesis, anticancer activity and docking study of new 2-thioxoimidazolidin-4one derivatives. Life Sci. 2021, 277, 119531. [Google Scholar] [CrossRef]
- Kucwaj-Brysz, K.; Kurczab, R.; Jastrzębska-Więsek, M.; Żesławska, E.; Satała, G.; Nitek, W.; Partyka, A.; Siwek, A.; Jankowska, A.; Wesołowska, A.; et al. Computer-aided insights into receptor-ligand interaction for novel 5-arylhydantoin derivatives as serotonin 5-HT 7 receptor agents with antidepressant activity. Eur. J. Med. Chem. 2018, 147, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chang, Y.; Fu, J.; Ding, R.; Zhang, L.; Liang, T.; Liu, Y.; Liu, Y.; Hu, J. Design, synthesis and biological evaluation of aminopyrimidine derivatives bearing a 4,5,6,7-tetrahydrothieno [3,2-c]pyridine as potent EGFR inhibitors. Eur. J. Med. Chem. 2021, 226, 113845. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Mustafa, M.; Anwar, S.; Elgamal, F.; Ahmed, E.R.; Aly, O.M. Potent combretastatin A-4 analogs containing 1,2,4-triazole: Synthesis, antiproliferative, anti-tubulin activity, and docking study. Eur. J. Med. Chem. 2019, 183, 111697. [Google Scholar] [CrossRef]
- Mghwary, A.E.-S.; Gedawy, E.M.; Kamal, A.M.; Abuel-Maaty, S.M. Novel thienopyrimidine derivatives as dual EGFR and VEGFR-2 inhibitors: Design, synthesis, anticancer activity and effect on cell cycle profile. J. Enzym. Inhib. Med. Chem. 2019, 34, 838–852. [Google Scholar] [CrossRef] [Green Version]
- El-Deen, E.M.M.; Anwar, M.M.; El-Gwaad, A.A.A.; Karam, E.A.; El-Ashrey, M.K.; Kassab, R.R. Design and synthesis of some novel pyridothienopyrimidine derivatives and their biological evaluation as antimicrobial and anticancer agents targeting EGFR enzyme. Arab. J. Chem. 2022, 15, 103751. [Google Scholar] [CrossRef]
- Mustafa, M.; Abuo-Rahma, G.E.; Abd El-Hafeez, A.A.; Ahmed, E.R.; Abdelhamid, D.; Ghosh, P.; Hayallah, A.M. Discovery of antiproliferative and anti-FAK inhibitory activity of 1, 2, 4-triazole derivatives containing acetamido carboxylic acid skeleton. Bioorg. Med. Chem. Lett. 2021, 40, 127965. [Google Scholar] [CrossRef]
- Hu, C.; Wang, A.; Wu, H.; Qi, Z.; Li, X.; Yan, X.-E.; Chen, C.; Yu, K.; Zou, F.; Wang, W.; et al. Discovery and characterization of a novel irreversible EGFR mutants selective and potent kinase inhibitor CHMFL-EGFR-26 with a distinct binding mode. Oncotarget 2017, 8, 18359–18372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mustafa, M.; Abd El-Hafeez, A.A.; Abdelhamid, D.; Katkar, G.D.; Mostafa, Y.A.; Ghosh, P.; Hayallah, A.M.; Abuo-Rahma, G.E. A first-in-class anticancer dual HDAC2/FAK inhibitors bearing hydroxamates/benzamides capped by pyridinyl-1,2,4-triazoles. Eur. J. Med. Chem. 2021, 222, 113569. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cpd | R1 | R2 | R3 | R4 | Cpd | R1 | R2 | R3 | R4 |
|---|---|---|---|---|---|---|---|---|---|
| 5a | H | OCH3 | OCH3 | H | 5h | H | F | F | H |
| 5b | H | SO2NH2 | H | H | 5i | F | F | F | H |
| 5c | H | Cl | OCH3 | H | 5j | CH3 | H | OCH3 | H |
| 5d | OCH3 | H | H | CH3 | 5k | H | Cl | F | H |
| 5e | H | H | F | H | 5l | H | H | OC2H5 | H |
| 5f | F | H | H | H | 5m | OCH3 | H | H | H |
| 5g | H | F | H | H | 5n | H | OCH3 | H | H |
| Cpd | IC50 (µM) | Cpd | IC50 (µM) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| A549 | H1975 | PC9 | Average | A549 | H1975 | PC9 | Average | ||
| 5a | 2.26 ± 0.12 | 1.94 ± 0.25 | 10.19 ± 0.53 | 4.79 | 5i | 0.73 ± 0.05 | 14.00 ± 0.76 | 6.06 ± 0.35 | 6.93 |
| 5b | 14.54 ± 0.92 | 8.20 ± 0.45 | 5.12 ± 0.56 | 9.29 | 5j | 4.11 ± 0.26 | 30.90 ± 1.68 | 9.97 ± 1.08 | 14.99 |
| 5c | 91.39 ± 5.75 | 55.30 ± 3.01 | 21.34 ± 2.31 | 56.01 | 5k | 68.57 ± 4.32 | 5.84 ± 0.32 | 3.25 ± 0.35 | 25.89 |
| 5d | 35.82 ± 2.26 | 11.90 ± 0.65 | 7.50 ± 0.81 | 18.41 | 5l | 2.37 ± 0.15 | 43.80 ± 2.39 | 6.69 ± 0.73 | 17.62 |
| 5e | 7.19 ± 0.45 | 9.34 ± 0.51 | 2.52 ± 0.27 | 6.35 | 5m | 12.33 ± 0.78 | 2.27 ± 0.12 | 28.34 ± 3.08 | 14.31 |
| 5f | 1.94 ± 0.12 | 1.38 ± 0.07 | 4.40 ± 0.48 | 2.57 | 5n | 0.42 ± 0.03 | 18.80 ± 1.02 | 6.87 ± 0.75 | 8.70 |
| 5g | 37.65 ± 2.37 | 6.84 ± 0.37 | 11.50 ± 1.25 | 18.66 | Erloinib | 7.81 ± 0.43 | 9.70 ± 0.53 | 16.26 ± 0.84 | 11.26 |
| 5h | 11.94 ± 0.75 | 21.30 ± 1.16 | 15.41 ± 1.67 | 16.22 | |||||
| Cpd | EGFR Enzymatic Inhibition, IC50, (µM) | ||
|---|---|---|---|
| WT | L858R | T790M | |
| 5a | 0.03 ± 0.005 | 0.05 ± 0.001 | 0.09 ± 0.002 |
| 5f | 0.06 ± 0.011 | 0.17 ± 0.003 | 0.26 ± 0.005 |
| Erlotinib | 0.09 ± 0.002 | 0.03 ± 0.001 | 0.07 ± 0.002 |
| Cpd | Enzymatic Inhibition, IC50, (nM) | |
|---|---|---|
| Akt | ERK | |
| 5a | 33.75 ± 0.55 | 25.76 ± 41 |
| Erlotinib | 22.97 ± 0.20 | 17.46 ± 0.18 |
| Compound | WI-38, IC50 (µM) |
|---|---|
| 5a | 49.30 ± 2.75 |
| 5f | 42.45 ± 2.32 |
| Erlotinib | 18.62 ± 1.24 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassanin, M.A.; Mustafa, M.; Abourehab, M.A.S.; Hassan, H.A.; Aly, O.M.; Beshr, E.A.M. Design and Synthesis of New Hydantoin Acetanilide Derivatives as Anti-NSCLC Targeting EGFRL858R/T790M Mutations. Pharmaceuticals 2022, 15, 857. https://doi.org/10.3390/ph15070857
Hassanin MA, Mustafa M, Abourehab MAS, Hassan HA, Aly OM, Beshr EAM. Design and Synthesis of New Hydantoin Acetanilide Derivatives as Anti-NSCLC Targeting EGFRL858R/T790M Mutations. Pharmaceuticals. 2022; 15(7):857. https://doi.org/10.3390/ph15070857
Chicago/Turabian StyleHassanin, Moamen A., Muhamad Mustafa, Mohammed A. S. Abourehab, Heba A. Hassan, Omar M. Aly, and Eman A. M. Beshr. 2022. "Design and Synthesis of New Hydantoin Acetanilide Derivatives as Anti-NSCLC Targeting EGFRL858R/T790M Mutations" Pharmaceuticals 15, no. 7: 857. https://doi.org/10.3390/ph15070857