
High-Dose Vitamin C for Cancer Therapy

 ,
,  ,
,  , ,
, ,  , ,
, ,  , , , and
, , , and
Abstract
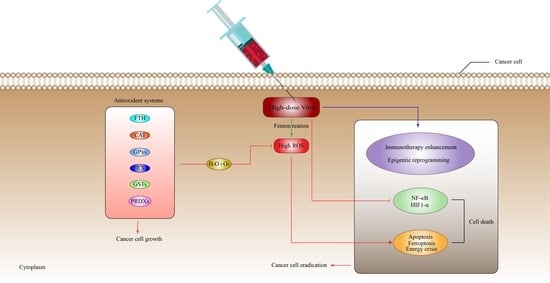
:
1. Introduction
2. Historical Background and Justification of High-Dose Vitamin-C in Cancer Management
3. Different Oxidized Forms of Vitamin-C
4. Enzymatic Activities of Vitamin-C
5. ROS
6. Anti-Cancer Mechanisms of Vitamin-C
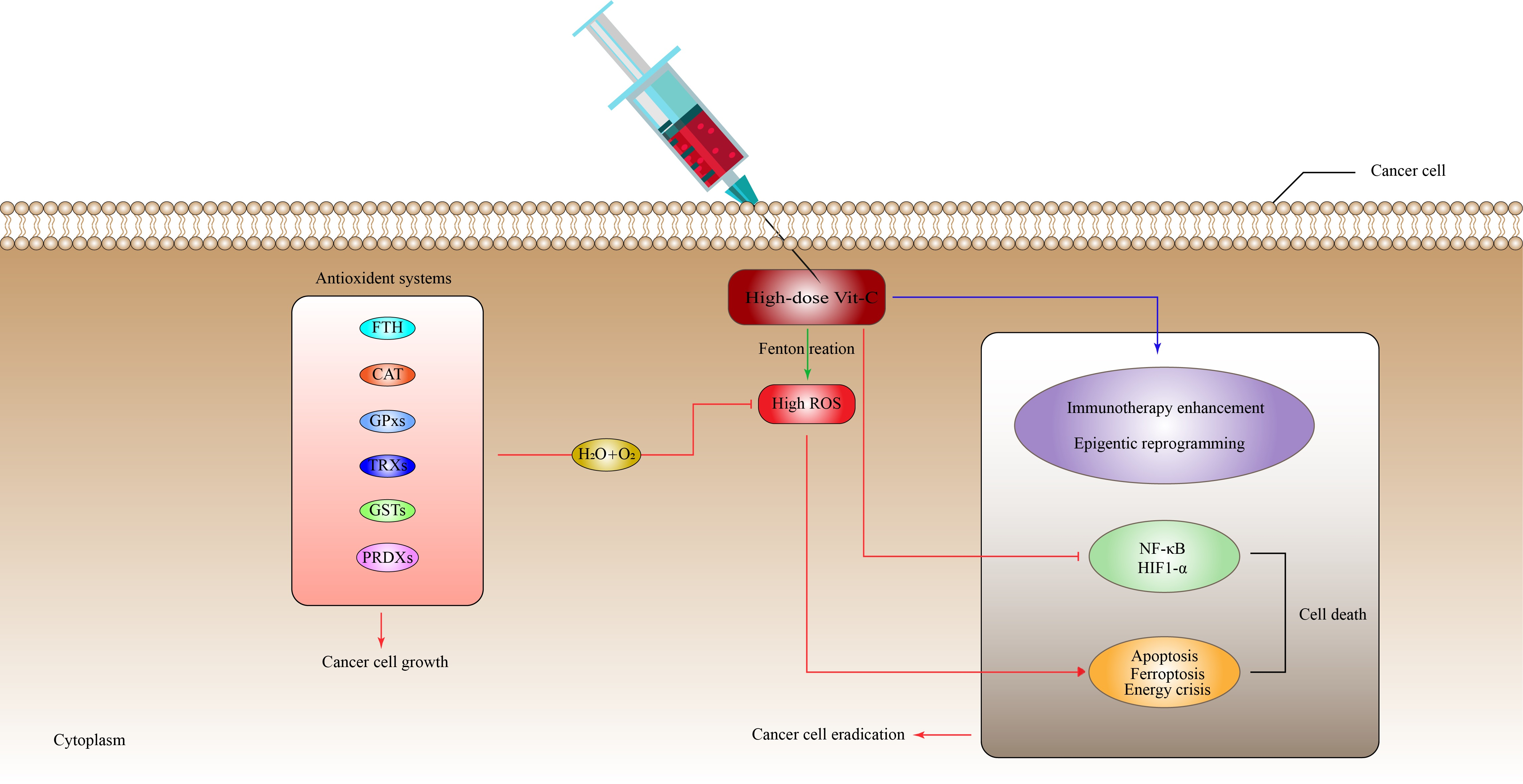
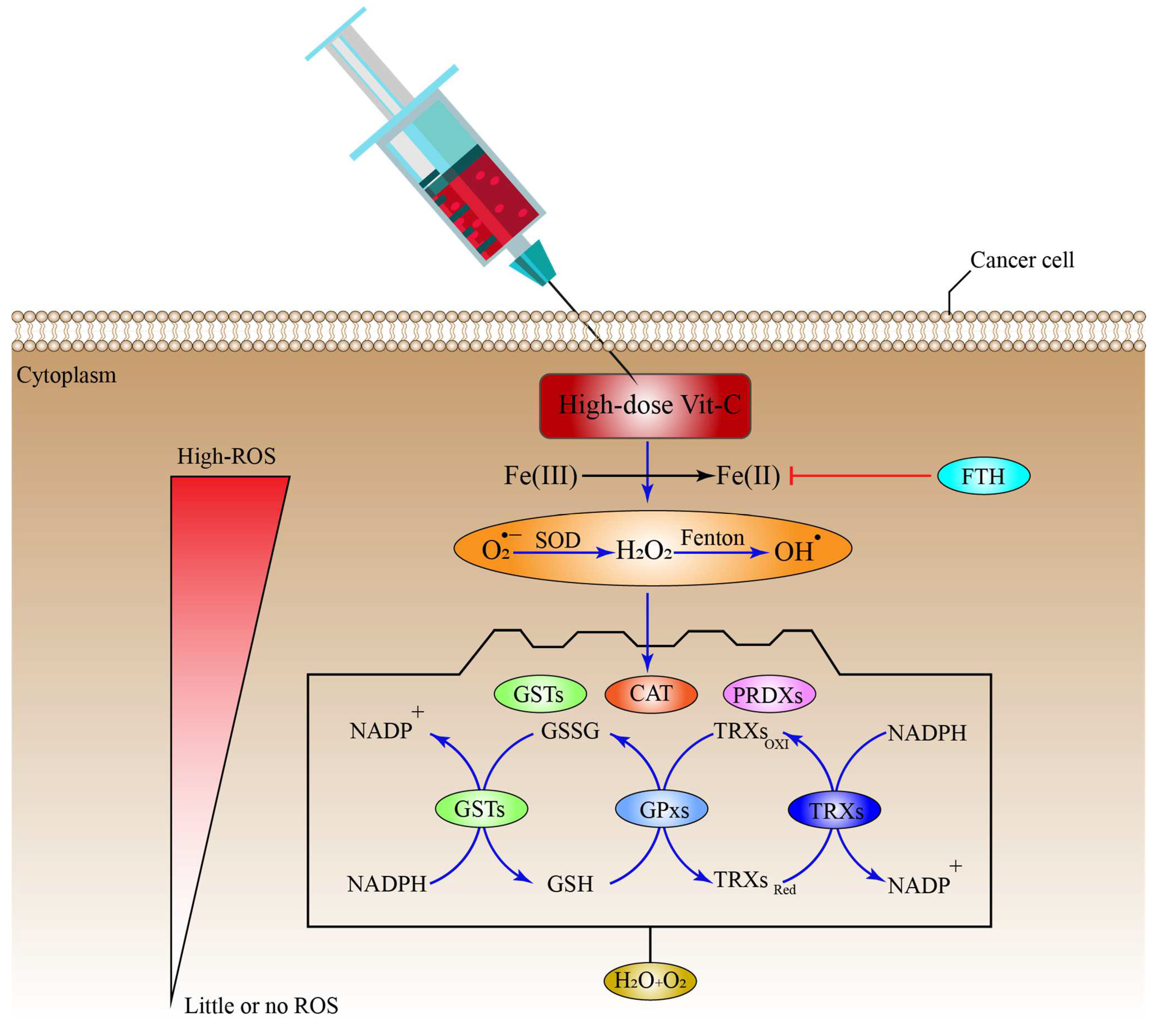
6.1. High-Dose Vitamin-C Targeted the Intracellular Labile Fe(II) Iron Ion Hemostasis

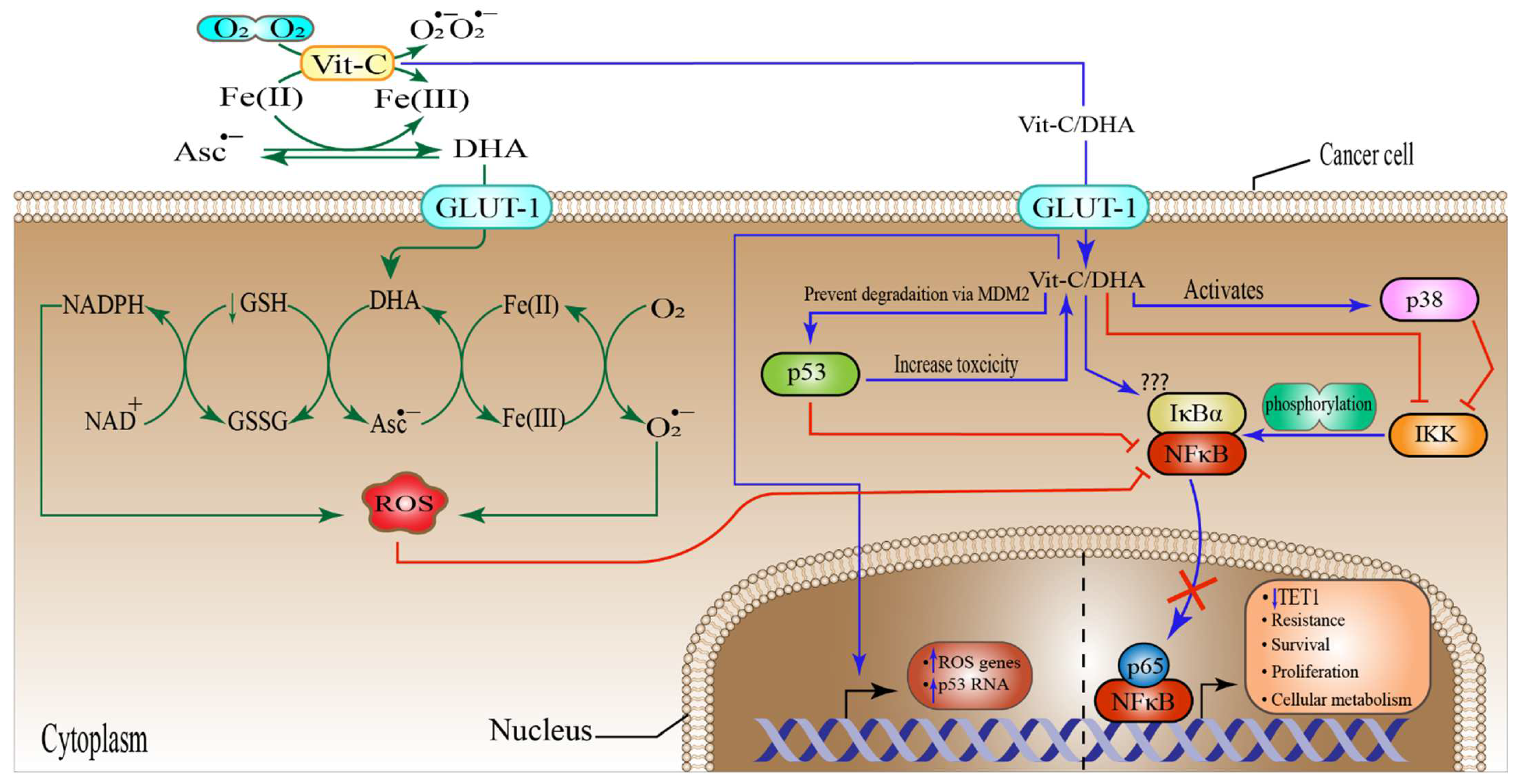
6.2. High-Dose Vitamin-C Targeted Glycolysis and GLUT-1
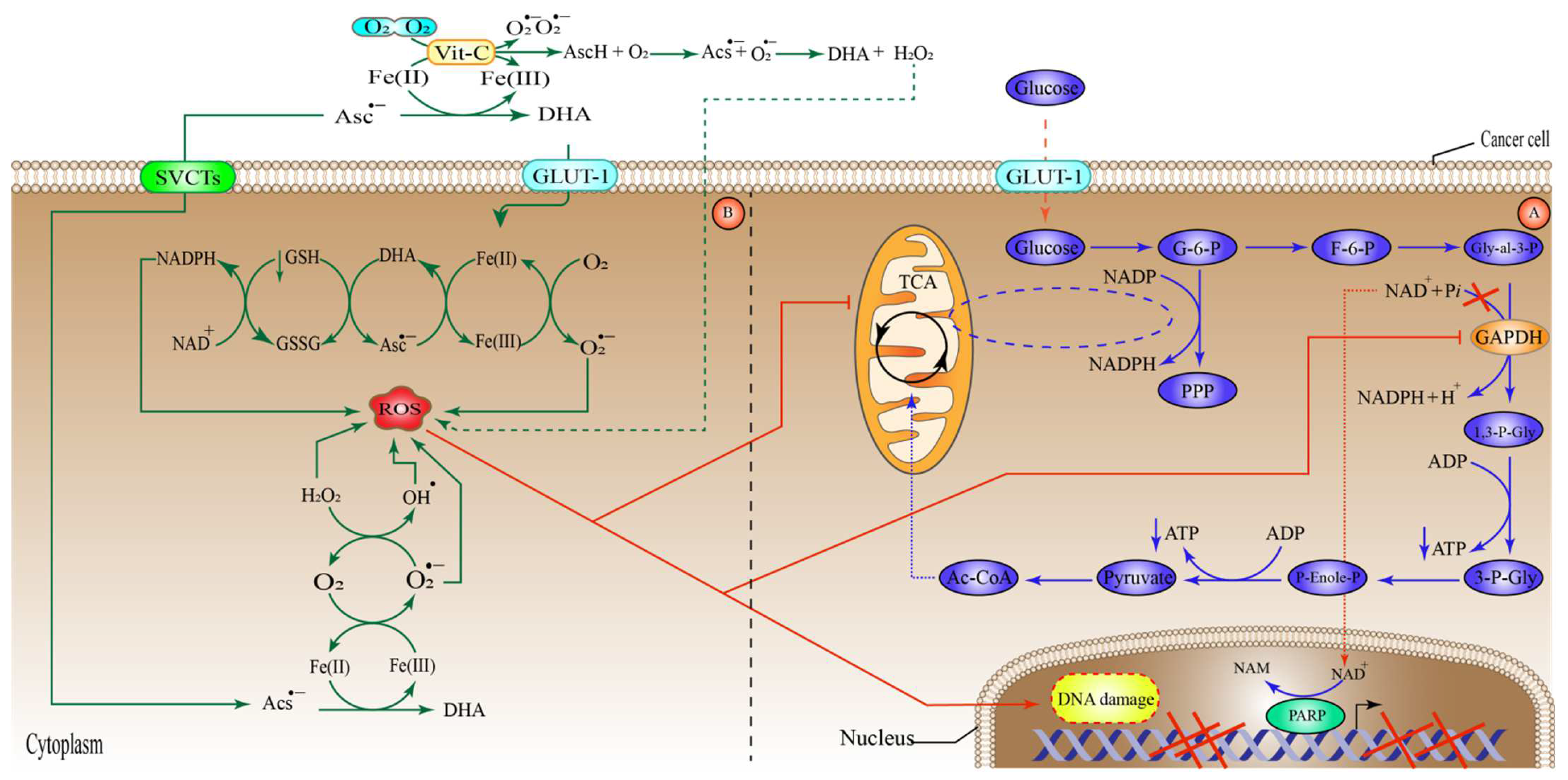
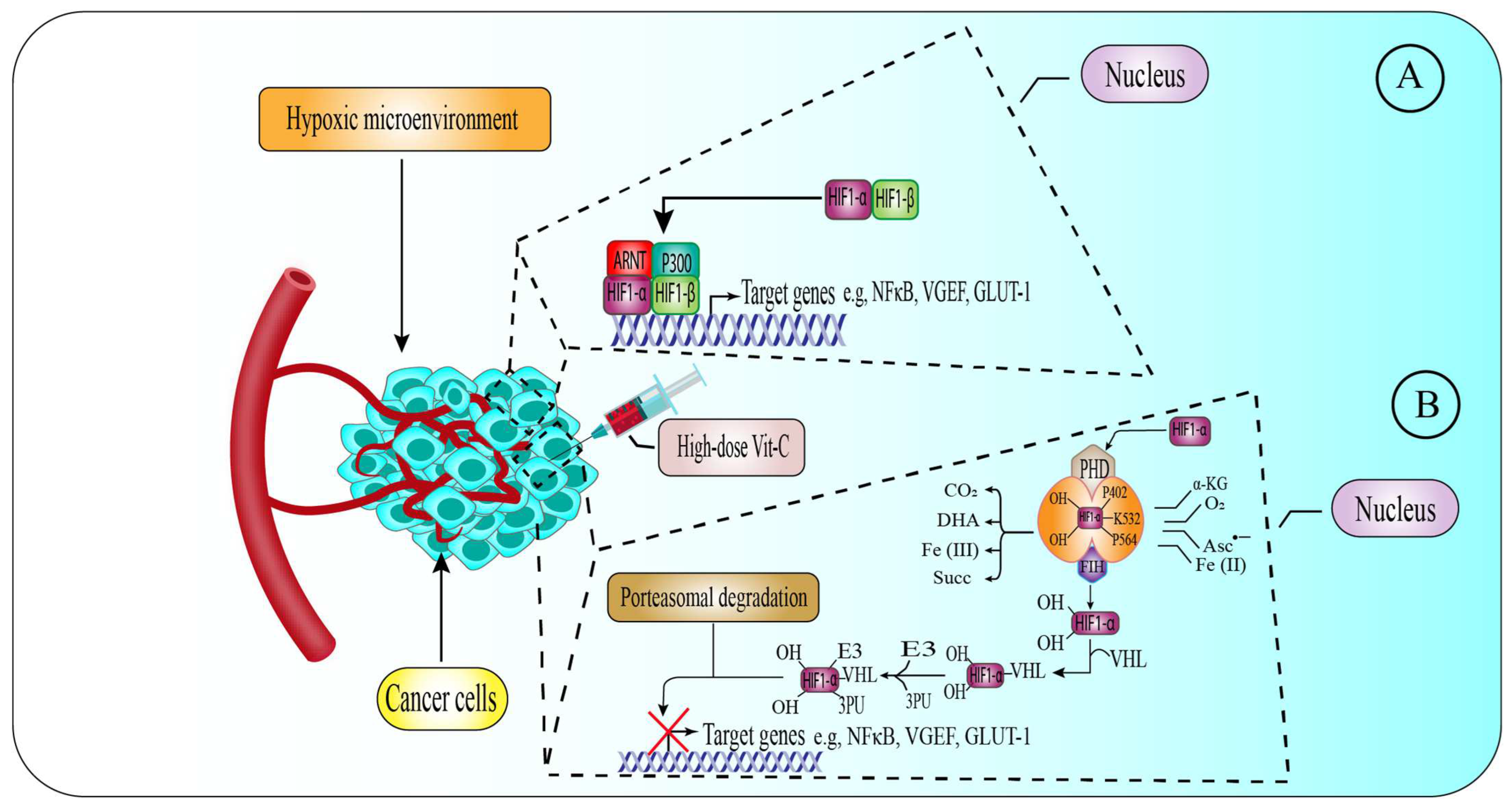
6.3. High-Dose Vitamin-C Targeted the Hypoxic Enfironment
6.4. High-Dose Vitamin-C Targeted NF-κB in the Tumor Microenvironment
6.5. High-Dose Vitamin-C Implies Epigenetic Regulation in Tumor Microenvironment
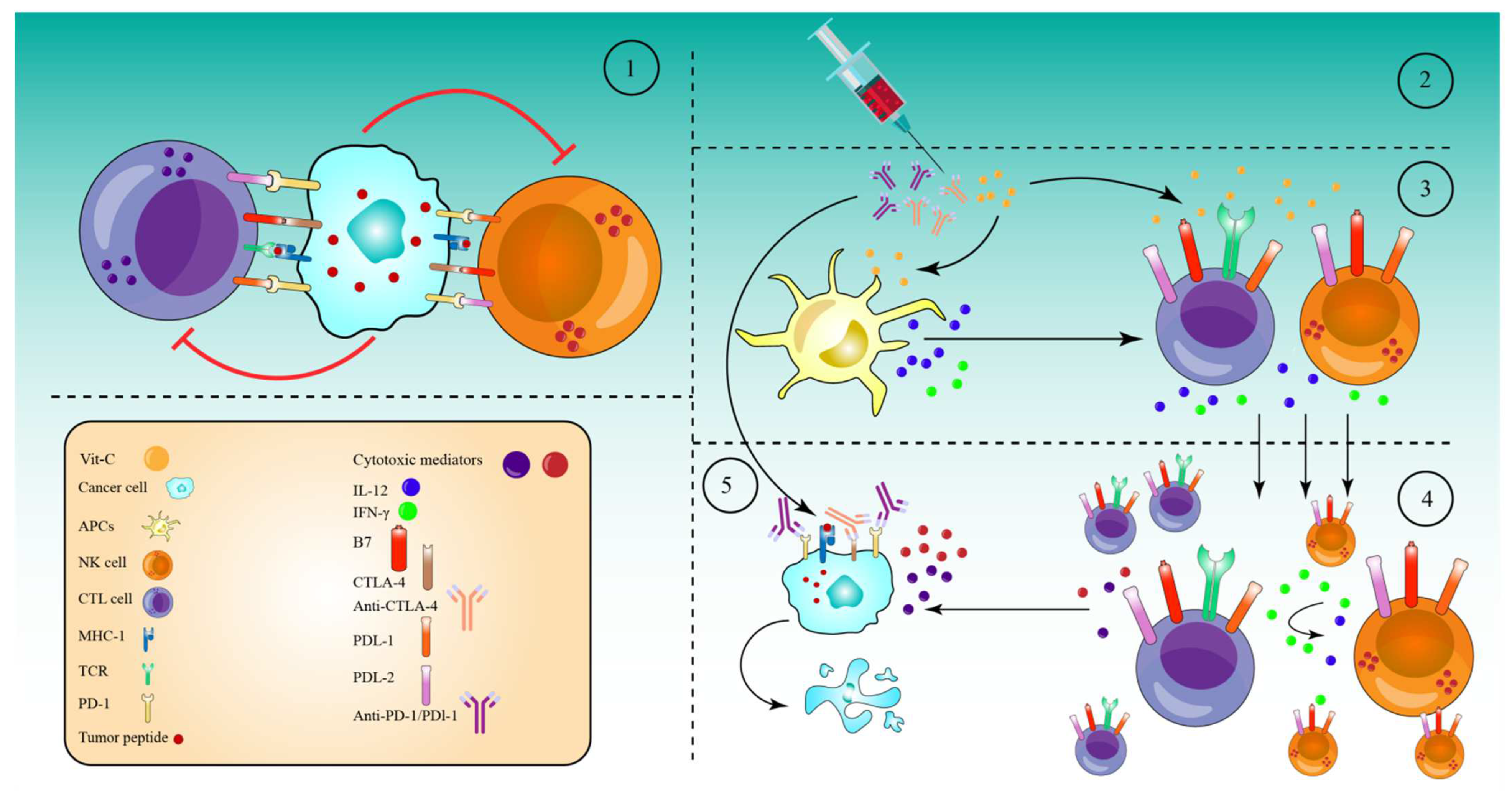
7. High-Dose Vitamin-C Enhances Cancer Immunotherapy
8. Antioxidant Systems That Might Inhibit the Effect of High-Dose Vitamin-C Therapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Defense System | Sub-Cellular Localization(s) | ROS Type and System Function | Ref. |
|---|---|---|---|
| SODs | Cytosol and Peroxisomes | O2•− to O2 and H2O2 | [202] |
| CAT | Peroxisomes | H2O2 to H2O and O2 | [202] |
| GPxs | Mitochondria and Cytosol | H2O2 to H2O lipid peroxides to alcohols | [202] |
| PRDXs | Cytosol, Mitochondria, Nucleus And Endoplasmic reticulum | H2O2 to H2O | [217] |
| TRXs | Cytosol, Mitochondria, Nucleus | H2O2 to H2O and O2 | [220,221] |
| GSH | Cytosol, Mitochondria, Nucleus And Endoplasmic reticulum | H2O2 to H2O | [227,229] |
| GSTs * | Cytosol, Membrane-bound | conjugates with reduced GSH) | [232] |
| FTH * | Nucleus, Lysosome, Cytoplasm | Sequester Fe(II) to inhibit ROS generation | [81] |
9. Clinical Studies of High-Dose Vitamin-C in Cancer Therapies
10. Conclusions and Future Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nishikimi, M.; Fukuyama, R.; Minoshima, S.; Shimizu, N.; Yagi, K. Cloning and chromosomal mapping of the human nonfunctional gene for L-gulono-gamma-lactone oxidase, the enzyme for L-ascorbic acid biosynthesis missing in man. J. Biol. Chem. 1994, 269, 13685–13688. [Google Scholar] [CrossRef]
- Drouin, G.; Godin, J.-R.; Pagé, B. The genetics of vitamin C loss in vertebrates. Curr. Genom. 2011, 12, 371–378. [Google Scholar] [CrossRef]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef]
- Carr, A.; Frei, B. Does vitamin C act as a pro-oxidant under physiological conditions? FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1999, 13, 1007–1024. [Google Scholar] [CrossRef]
- Englard, S.; Seifter, S. The biochemical functions of ascorbic acid. Annu. Rev. Nutr. 1986, 6, 365–406. [Google Scholar] [CrossRef]
- Kuiper, C.; Vissers, M.C.M. Ascorbate as a Co-Factor for Fe- and 2-Oxoglutarate Dependent Dioxygenases: Physiological Activity in Tumor Growth and Progression. Front. Oncol. 2014, 4, 359. [Google Scholar] [CrossRef]
- Young, J.I.; Züchner, S.; Wang, G. Regulation of the Epigenome by Vitamin C. Annu. Rev. Nutr. 2015, 35, 545–564. [Google Scholar] [CrossRef]
- Carr, A.C.; McCall, C. The role of vitamin C in the treatment of pain: New insights. J. Transl. Med. 2017, 15, 77. [Google Scholar] [CrossRef]
- Ting, H.H.; Timimi, F.K.; Boles, K.S.; Creager, S.J.; Ganz, P.; Creager, M.A. Vitamin C improves endothelium-dependent vasodilation in patients with non-insulin-dependent diabetes mellitus. J. Clin. Investig. 1996, 97, 22–28. [Google Scholar] [CrossRef]
- Salonen, R.M.; Nyyssönen, K.; Kaikkonen, J.; Porkkala-Sarataho, E.; Voutilainen, S.; Rissanen, T.H.; Tuomainen, T.P.; Valkonen, V.P.; Ristonmaa, U.; Lakka, H.M.; et al. Six-year effect of combined vitamin C and E supplementation on atherosclerotic progression: The Antioxidant Supplementation in Atherosclerosis Prevention (ASAP) Study. Circulation 2003, 107, 947–953. [Google Scholar] [CrossRef]
- Van Straten, M.; Josling, P. Preventing the common cold with a vitamin C supplement: A double-blind, placebo-controlled survey. Adv. Ther. 2002, 19, 151–159. [Google Scholar] [CrossRef]
- Valero, M.P.; Fletcher, A.E.; De Stavola, B.L.; Vioque, J.; Alepuz, V.C. Vitamin C is associated with reduced risk of cataract in a Mediterranean population. J. Nutr. 2002, 132, 1299–1306. [Google Scholar] [CrossRef]
- Ramdas, W.D.; Schouten, J.; Webers, C.A.B. The Effect of Vitamins on Glaucoma: A Systematic Review and Meta-Analysis. Nutrients 2018, 10, 359. [Google Scholar] [CrossRef]
- Seddon, J.M.; Ajani, U.A.; Sperduto, R.D.; Hiller, R.; Blair, N.; Burton, T.C.; Farber, M.D.; Gragoudas, E.S.; Haller, J.; Miller, D.T.; et al. Dietary carotenoids, vitamins A, C, and E, and advanced age-related macular degeneration. Eye Disease Case-Control Study Group. Jama 1994, 272, 1413–1420. [Google Scholar] [CrossRef]
- Chen, G.C.; Lu, D.B.; Pang, Z.; Liu, Q.F. Vitamin C intake, circulating vitamin C and risk of stroke: A meta-analysis of prospective studies. J. Am. Heart Assoc. 2013, 2, e000329. [Google Scholar] [CrossRef]
- Losonczy, K.G.; Harris, T.B.; Havlik, R.J. Vitamin E and vitamin C supplement use and risk of all-cause and coronary heart disease mortality in older persons: The Established Populations for Epidemiologic Studies of the Elderly. Am. J. Clin. Nutr. 1996, 64, 190–196. [Google Scholar] [CrossRef]
- Zhang, J.; Rao, X.; Li, Y.; Zhu, Y.; Liu, F.; Guo, G.; Luo, G.; Meng, Z.; De Backer, D.; Xiang, H.; et al. Pilot trial of high-dose vitamin C in critically ill COVID-19 patients. Ann. Intensive Care 2021, 11, 5. [Google Scholar] [CrossRef]
- Cameron, E.; Campbell, A. The orthomolecular treatment of cancer. II. Clinical trial of high-dose ascorbic acid supplements in advanced human cancer. Chem. Biol. Interact. 1974, 9, 285–315. [Google Scholar] [CrossRef]
- Cameron, E.; Pauling, L. Supplemental ascorbate in the supportive treatment of cancer: Prolongation of survival times in terminal human cancer. Proc. Natl. Acad. Sci. USA 1976, 73, 3685–3689. [Google Scholar] [CrossRef]
- Cameron, E.; Pauling, L. Supplemental ascorbate in the supportive treatment of cancer: Reevaluation of prolongation of survival times in terminal human cancer. Proc. Natl. Acad. Sci. USA 1978, 75, 4538–4542. [Google Scholar] [CrossRef]
- Murata, A.; Morishige, F.; Yamaguchi, H. Prolongation of survival times of terminal cancer patients by administration of large doses of ascorbate. Int. J. Vitam. Nutr. Res. 1982, 23, 103–113. [Google Scholar]
- Creagan, E.T.; Moertel, C.G.; O’Fallon, J.R.; Schutt, A.J.; O’Connell, M.J.; Rubin, J.; Frytak, S. Failure of High-Dose Vitamin C (Ascorbic Acid) Therapy to Benefit Patients with Advanced Cancer. N. Engl. J. Med. 1979, 301, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Moertel, C.G.; Fleming, T.R.; Creagan, E.T.; Rubin, J.; O’Connell, M.J.; Ames, M.M. High-dose vitamin C versus placebo in the treatment of patients with advanced cancer who have had no prior chemotherapy. A randomized double-blind comparison. N. Engl. J. Med. 1985, 312, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Block, G. Vitamin C and cancer prevention: The epidemiologic evidence. Am. J. Clin. Nutr. 1991, 53 (Suppl. 1), 270s–282s. [Google Scholar] [CrossRef]
- Block, G. Epidemiologic evidence regarding vitamin C and cancer. Am. J. Clin. Nutr. 1991, 54 (Suppl. 6), 1310s–1314s. [Google Scholar] [CrossRef]
- Head, K.A. Ascorbic acid in the prevention and treatment of cancer. Altern. Med. Rev. A J. Clin. Ther. 1998, 3, 174–186. [Google Scholar]
- Block, G.; Patterson, B.; Subar, A. Fruit, vegetables, and cancer prevention: A review of the epidemiological evidence. Nutr. Cancer 1992, 18, 1–29. [Google Scholar] [CrossRef]
- Steinmetz, K.A.; Potter, J.D. Vegetables, fruit, and cancer prevention: A review. J. Am. Diet. Assoc. 1996, 96, 1027–1039. [Google Scholar] [CrossRef]
- Loria, C.M.; Klag, M.J.; Caulfield, L.E.; Whelton, P.K. Vitamin C status and mortality in US adults. Am. J. Clin. Nutr. 2000, 72, 139–145. [Google Scholar] [CrossRef]
- Padayatty, S.J.; Riordan, H.D.; Hewitt, S.M.; Katz, A.; Hoffer, L.J.; Levine, M. Intravenously administered vitamin C as cancer therapy: Three cases. CMAJ 2006, 174, 937–942. [Google Scholar] [CrossRef]
- Dachs, G.U.; Gandhi, J.; Wohlrab, C.; Carr, A.C.; Morrin, H.R.; Pullar, J.M.; Bayer, S.B.; Eglinton, T.W.; Robinson, B.A.; Vissers, M.C.M. Vitamin C Administration by Intravenous Infusion Increases Tumor Ascorbate Content in Patients With Colon Cancer: A Clinical Intervention Study. Front. Oncol. 2021, 10, 600715. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Krishna, M.C.; Mitchell, J.B.; Corpe, C.P.; Buettner, G.R.; Shacter, E.; Levine, M. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: Action as a pro-drug to deliver hydrogen peroxide to tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 13604–13609. [Google Scholar] [CrossRef] [PubMed]
- Cabanillas, F. Vitamin C and cancer: What can we conclude—1609 patients and 33 years later? Puerto Rico Health Sci. J. 2010, 29, 215–217. [Google Scholar]
- Cha, J.; Roomi, M.W.; Ivanov, V.; Kalinovsky, T.; Niedzwiecki, A.; Rath, M. Ascorbate depletion increases growth and metastasis of melanoma cells in vitamin C deficient mice. Exp. Oncol. 2011, 33, 226–230. [Google Scholar] [PubMed]
- Cha, J.; Roomi, M.W.; Ivanov, V.; Kalinovsky, T.; Niedzwiecki, A.; Rath, M. Ascorbate supplementation inhibits growth and metastasis of B16FO melanoma and 4T1 breast cancer cells in vitamin C-deficient mice. Int. J. Oncol. 2013, 42, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Magrì, A.; Germano, G.; Lorenzato, A.; Lamba, S.; Chilà, R.; Montone, M.; Amodio, V.; Ceruti, T.; Sassi, F.; Arena, S.; et al. High-dose vitamin C enhances cancer immunotherapy. Sci. Transl. Med. 2020, 12, eaay8707. [Google Scholar] [CrossRef] [PubMed]
- Padayatty, S.J.; Sun, H.; Wang, Y.; Riordan, H.D.; Hewitt, S.M.; Katz, A.; Wesley, R.A.; Levine, M. Vitamin C pharmacokinetics: Implications for oral and intravenous use. Ann. Intern. Med. 2004, 140, 533–537. [Google Scholar] [CrossRef]
- Kushi, L.H.; Fee, R.M.; Sellers, T.A.; Zheng, W.; Folsom, A.R. Intake of vitamins A, C, and E and postmenopausal breast cancer. The Iowa Women’s Health Study. Am. J. Epidemiol. 1996, 144, 165–174. [Google Scholar] [CrossRef]
- Roa, F.J.; Pena, E.; Gatica, M.; Escobar-Acuna, K.; Saavedra, P.; Maldonado, M.; Cuevas, M.; Moraga-Cid, G.; Rivas, C.I.; Munoz-Montesino, C. Therapeutic Use of Vitamin C in Cancer: Physiological Considerations. Front. Pharmacol. 2020, 11, 211. [Google Scholar] [CrossRef]
- Assouline, S.; Miller, W.H. High-dose vitamin C therapy: Renewed hope or false promise? CMAJ 2006, 174, 956–957. [Google Scholar] [CrossRef]
- Yeom, C.H.; Jung, G.C.; Song, K.J. Changes of terminal cancer patients’ health-related quality of life after high dose vitamin C administration. J. Korean Med. Sci. 2007, 22, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Corti, A.; Casini, A.F.; Pompella, A. Cellular pathways for transport and efflux of ascorbate and dehydroascorbate. Arch. Biochem. Biophys. 2010, 500, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Glorieux, C.; Buc Calderon, P. Vitamin C (Ascorbate) and Redox Topics in Cancer. Antioxid Redox Signal 2021, 35, 1157–1175. [Google Scholar] [CrossRef] [PubMed]
- Vera, J.C.; Rivas, C.I.; Zhang, R.H.; Farber, C.M.; Golde, D.W. Human HL-60 myeloid leukemia cells transport dehydroascorbic acid via the glucose transporters and accumulate reduced ascorbic acid. Blood 1994, 84, 1628–1634. [Google Scholar] [CrossRef] [PubMed]
- Padayatty, S.J.; Levine, M. Vitamin C: The known and the unknown and Goldilocks. Oral Dis. 2016, 22, 463–493. [Google Scholar] [CrossRef]
- Wilson, J.X. The physiological role of dehydroascorbic acid. FEBS Lett. 2002, 527, 5–9. [Google Scholar] [CrossRef]
- Aguilera, O.; Muñoz-Sagastibelza, M.; Torrejón, B.; Borrero-Palacios, A.; Del Puerto-Nevado, L.; Martínez-Useros, J.; Rodriguez-Remirez, M.; Zazo, S.; García, E.; Fraga, M.; et al. Vitamin C uncouples the Warburg metabolic switch in KRAS mutant colon cancer. Oncotarget 2016, 7, 47954–47965. [Google Scholar] [CrossRef]
- Buettner, G.R. The pecking order of free radicals and antioxidants: Lipid peroxidation, alpha-tocopherol, and ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543. [Google Scholar] [CrossRef]
- Podmore, I.D.; Griffiths, H.R.; Herbert, K.E.; Mistry, N.; Mistry, P.; Lunec, J. Vitamin C exhibits pro-oxidant properties. Nature 1998, 392, 559. [Google Scholar] [CrossRef]
- Flashman, E.; Davies, S.L.; Yeoh, K.K.; Schofield, C.J. Investigating the dependence of the hypoxia-inducible factor hydroxylases (factor inhibiting HIF and prolyl hydroxylase domain 2) on ascorbate and other reducing agents. Biochem. J. 2010, 427, 135–142. [Google Scholar] [CrossRef]
- Dao, J.H.; Kurzeja, R.J.; Morachis, J.M.; Veith, H.; Lewis, J.; Yu, V.; Tegley, C.M.; Tagari, P. Kinetic characterization and identification of a novel inhibitor of hypoxia-inducible factor prolyl hydroxylase 2 using a time-resolved fluorescence resonance energy transfer-based assay technology. Anal. Biochem. 2009, 384, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; DesMarais, T.L.; Tong, Z.; Yao, Y.; Costa, M. Oxidative stress alters global histone modification and DNA methylation. Free. Radic. Biol. Med. 2015, 82, 22–28. [Google Scholar] [CrossRef] [PubMed]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Biol. Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef]
- Muthukumar, K.; Nachiappan, V. Cadmium-induced oxidative stress in Saccharomyces cerevisiae. Indian J. Biochem. Biophys. 2010, 47, 383–387. [Google Scholar]
- Tominaga, H.; Kodama, S.; Matsuda, N.; Suzuki, K.; Watanabe, M. Involvement of reactive oxygen species (ROS) in the induction of genetic instability by radiation. J. Radiat. Res. 2004, 45, 181–188. [Google Scholar] [CrossRef]
- van der Toorn, M.; Rezayat, D.; Kauffman, H.F.; Bakker, S.J.L.; Gans, R.O.B.; Koëter, G.H.; Choi, A.M.K.; van Oosterhout, A.J.M.; Slebos, D.-J. Lipid-soluble components in cigarette smoke induce mitochondrial production of reactive oxygen species in lung epithelial cells. Am. J. Physiol. Lung. Cell Mol. Physiol. 2009, 297, L109–L114. [Google Scholar] [CrossRef]
- Deavall, D.G.; Martin, E.A.; Horner, J.M.; Roberts, R. Drug-Induced Oxidative Stress and Toxicity. J. Toxicol. 2012, 2012, 645460. [Google Scholar] [CrossRef]
- Klotz, L.-O.; Steinbrenner, H. Cellular adaptation to xenobiotics: Interplay between xenosensors, reactive oxygen species and FOXO transcription factors. Redox Biol. 2017, 13, 646–654. [Google Scholar] [CrossRef]
- Dickinson, B.C.; Chang, C.J. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat. Chem. Biol. 2011, 7, 504–511. [Google Scholar] [CrossRef]
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free. Radic Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Schumacker, P.T. Reactive oxygen species in cancer cells: Live by the sword, die by the sword. Cancer Cell 2006, 10, 175–176. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. CB 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Chio, I.I.C.; Tuveson, D.A. ROS in Cancer: The Burning Question. Trends Mol. Med. 2017, 23, 411–429. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef]
- Thyagarajan, A.; Sahu, R.P. Potential Contributions of Antioxidants to Cancer Therapy: Immunomodulation and Radiosensitization. Integr. Cancer Ther. 2018, 17, 210–216. [Google Scholar] [CrossRef]
- Sayin, V.I.; Ibrahim, M.X.; Larsson, E.; Nilsson, J.A.; Lindahl, P.; Bergo, M.O. Antioxidants accelerate lung cancer progression in mice. Sci. Transl. Med. 2014, 6, 221ra215. [Google Scholar] [CrossRef]
- Zou, Z.V.; Le Gal, K.; El Zowalaty, A.E.; Pehlivanoglu, L.E.; Garellick, V.; Gul, N.; Ibrahim, M.X.; Bergh, P.-O.; Henricsson, M.; Wiel, C.; et al. Antioxidants Promote Intestinal Tumor Progression in Mice. Antioxidants 2021, 10, 241. [Google Scholar] [CrossRef]
- Klein, E.A.; Thompson, I.M., Jr.; Tangen, C.M.; Crowley, J.J.; Lucia, M.S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; et al. Vitamin E and the risk of prostate cancer: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2011, 306, 1549–1556. [Google Scholar] [CrossRef]
- Omenn, G.S.; Goodman, G.E.; Thornquist, M.D.; Balmes, J.; Cullen, M.R.; Glass, A.; Keogh, J.P.; Meyskens, F.L.; Valanis, B.; Williams, J.H.; et al. Effects of a combination of beta carotene and vitamin A on lung cancer and cardiovascular disease. N. Engl. J. Med. 1996, 334, 1150–1155. [Google Scholar] [CrossRef] [PubMed]
- Wondrak, G.T. Redox-directed cancer therapeutics: Molecular mechanisms and opportunities. Antioxid Redox Signal 2009, 11, 3013–3069. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Pantopoulos, K. Regulation of cellular iron metabolism. Biochem. J. 2011, 434, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.R.; Merlot, A.M.; Huang, M.L.H.; Bae, D.H.; Jansson, P.J.; Sahni, S.; Kalinowski, D.S.; Richardson, D.R. Cellular iron uptake, trafficking and metabolism: Key molecules and mechanisms and their roles in disease. Biochim. Biophys. Acta 2015, 1853, 1130–1144. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol. Lett. 1995, 82, 969–974. [Google Scholar] [CrossRef]
- Du, J.; Cullen, J.J.; Buettner, G.R. Ascorbic acid: Chemistry, biology and the treatment of cancer. Biochim. Biophys. Acta 2012, 1826, 443–457. [Google Scholar] [CrossRef]
- Di Tano, M.; Raucci, F.; Vernieri, C.; Caffa, I.; Buono, R.; Fanti, M.; Brandhorst, S.; Curigliano, G.; Nencioni, A.; de Braud, F.; et al. Synergistic effect of fasting-mimicking diet and vitamin C against KRAS mutated cancers. Nat. Commun. 2020, 11, 2332. [Google Scholar] [CrossRef]
- Ha, Y.M.; Park, M.K.; Kim, H.J.; Seo, H.G.; Lee, J.H.; Chang, K.C. High concentrations of ascorbic acid induces apoptosis of human gastric cancer cell by p38-MAP kinase-dependent up-regulation of transferrin receptor. Cancer Lett. 2009, 277, 48–54. [Google Scholar] [CrossRef]
- Lane, D.J.R.; Richardson, D.R. The active role of vitamin C in mammalian iron metabolism: Much more than just enhanced iron absorption! Free. Radic. Biol. Med. 2014, 75, 69–83. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Zhang, K.H.; Tian, H.Y.; Gao, X.; Lei, W.W.; Hu, Y.; Wang, D.M.; Pan, X.C.; Yu, M.L.; Xu, G.J.; Zhao, F.K.; et al. Ferritin heavy chain-mediated iron homeostasis and subsequent increased reactive oxygen species production are essential for epithelial-mesenchymal transition. Cancer Res. 2009, 69, 5340–5348. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, J.D.; Sibenaller, Z.A.; Mapuskar, K.A.; Wagner, B.A.; Cramer-Morales, K.L.; Furqan, M.; Sandhu, S.; Carlisle, T.L.; Smith, M.C.; Abu Hejleh, T.; et al. O2—and H2O2-Mediated Disruption of Fe Metabolism Causes the Differential Susceptibility of NSCLC and GBM Cancer Cells to Pharmacological Ascorbate. Cancer Cell 2017, 31, 487–500.e488. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Pooput, C.; Kirk, K.L.; Krishna, M.C.; Khosh, D.B.; Drisko, J.; Levine, M. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 11105–11109. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Martin, S.M.; Levine, M.; Wagner, B.A.; Buettner, G.R.; Wang, S.H.; Taghiyev, A.F.; Du, C.; Knudson, C.M.; Cullen, J.J. Mechanisms of ascorbate-induced cytotoxicity in pancreatic cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2010, 16, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Rawal, M.; Schroeder, S.R.; Wagner, B.A.; Cushing, C.M.; Welsh, J.L.; Button, A.M.; Du, J.; Sibenaller, Z.A.; Buettner, G.R.; Cullen, J.J. Manganoporphyrins increase ascorbate-induced cytotoxicity by enhancing H2O2 generation. Cancer Res. 2013, 73, 5232–5241. [Google Scholar] [CrossRef] [PubMed]
- Duarte, T.L.; Almeida, G.M.; Jones, G.D. Investigation of the role of extracellular H2O2 and transition metal ions in the genotoxic action of ascorbic acid in cell culture models. Toxicol Lett. 2007, 170, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Sakagami, H.; Satoh, K.; Fukuchi, K.; Gomi, K.; Takeda, M. Effect on an iron-chelator on ascorbate-induced cytotoxicity. Free. Radic. Biol. Med. 1997, 23, 260–270. [Google Scholar] [CrossRef]
- Buettner, G.R.; Jurkiewicz, B.A. Catalytic metals, ascorbate and free radicals: Combinations to avoid. Radiat. Res. 1996, 145, 532–541. [Google Scholar] [CrossRef]
- Clément, M.V.; Ramalingam, J.; Long, L.H.; Halliwell, B. The in vitro cytotoxicity of ascorbate depends on the culture medium used to perform the assay and involves hydrogen peroxide. Antioxid Redox Signal 2001, 3, 157–163. [Google Scholar] [CrossRef]
- Chen, Q.; Espey, M.G.; Sun, A.Y.; Lee, J.H.; Krishna, M.C.; Shacter, E.; Choyke, P.L.; Pooput, C.; Kirk, K.L.; Buettner, G.R.; et al. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 8749–8754. [Google Scholar] [CrossRef]
- Ma, E.; Chen, P.; Wilkins, H.M.; Wang, T.; Swerdlow, R.H.; Chen, Q. Pharmacologic ascorbate induces neuroblastoma cell death by hydrogen peroxide mediated DNA damage and reduction in cancer cell glycolysis. Free. Radic. Biol. Med. 2017, 113, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Chapman, J.; Levine, M.; Polireddy, K.; Drisko, J.; Chen, Q. High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci. Transl. Med. 2014, 6, 222ra218. [Google Scholar] [CrossRef] [PubMed]
- Manz, D.H.; Blanchette, N.L.; Paul, B.T.; Torti, F.M.; Torti, S.V. Iron and cancer: Recent insights. Ann. New York Acad. Sci. 2016, 1368, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Rychtarcikova, Z.; Lettlova, S.; Tomkova, V.; Korenkova, V.; Langerova, L.; Simonova, E.; Zjablovskaja, P.; Alberich-Jorda, M.; Neuzil, J.; Truksa, J. Tumor-initiating cells of breast and prostate origin show alterations in the expression of genes related to iron metabolism. Oncotarget 2017, 8, 6376–6398. [Google Scholar] [CrossRef]
- Pinnix, Z.K.; Miller, L.D.; Wang, W.; D’Agostino, R., Jr.; Kute, T.; Willingham, M.C.; Hatcher, H.; Tesfay, L.; Sui, G.; Di, X.; et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci. Transl. Med. 2010, 2, 43ra56. [Google Scholar] [CrossRef]
- Corna, G.; Campana, L.; Pignatti, E.; Castiglioni, A.; Tagliafico, E.; Bosurgi, L.; Campanella, A.; Brunelli, S.; Manfredi, A.A.; Apostoli, P.; et al. Polarization dictates iron handling by inflammatory and alternatively activated macrophages. Haematologica 2010, 95, 1814–1822. [Google Scholar] [CrossRef]
- Recalcati, S.; Locati, M.; Marini, A.; Santambrogio, P.; Zaninotto, F.; De Pizzol, M.; Zammataro, L.; Girelli, D.; Cairo, G. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur. J. Immunol. 2010, 40, 824–835. [Google Scholar] [CrossRef]
- Panis, C.; Victorino, V.J.; Herrera, A.C.; Freitas, L.F.; De Rossi, T.; Campos, F.C.; Simão, A.N.; Barbosa, D.S.; Pinge-Filho, P.; Cecchini, R.; et al. Differential oxidative status and immune characterization of the early and advanced stages of human breast cancer. Breast Cancer Res. Treat. 2012, 133, 881–888. [Google Scholar] [CrossRef]
- Zhang, W.; Wu, Y.; Dong, H.J.; Yin, J.J.; Zhang, H.; Wu, H.A.; Song, L.N.; Chong, Y.; Li, Z.X.; Gu, N.; et al. Sparks fly between ascorbic acid and iron-based nanozymes: A study on Prussian blue nanoparticles. Colloids Surfaces B Biointerfaces 2018, 163, 379–384. [Google Scholar] [CrossRef]
- Xia, J.; Xu, H.; Zhang, X.; Allamargot, C.; Coleman, K.L.; Nessler, R.; Frech, I.; Tricot, G.; Zhan, F. Multiple Myeloma Tumor Cells are Selectively Killed by Pharmacologically-dosed Ascorbic Acid. EBioMedicine 2017, 18, 41–49. [Google Scholar] [CrossRef]
- Franqui-Machin, R.; Xu, H.; Yethava, Y.; Frech, I.; Tricot, G.J.; Zhan, F. Multiple Myeloma Tumor Cells Are Selectively Killed By Pharmacologically-Dosed Ascorbic Acid. Blood 2017, 130, 5391. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.; Markowitz, S.; Zhou, S.; et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009, 325, 1555–1559. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Montesino, C.; Peña, E.; Roa, F.J.; Sotomayor, K.; Escobar, E.; Rivas, C.I. Transport of Vitamin C in Cancer. Antioxid Redox Signal 2021, 35, 61–74. [Google Scholar] [CrossRef]
- May, J.M.; Qu, Z.C.; Whitesell, R.R.; Cobb, C.E. Ascorbate recycling in human erythrocytes: Role of GSH in reducing dehydroascorbate. Free. Radic. Biol. Med. 1996, 20, 543–551. [Google Scholar] [CrossRef]
- Ngo, B.; Van Riper, J.M.; Cantley, L.C.; Yun, J. Targeting cancer vulnerabilities with high-dose vitamin C. Nature reviews. Cancer 2019, 19, 271–282. [Google Scholar] [CrossRef]
- Ghanem, A.; Melzer, A.M.; Zaal, E.; Neises, L.; Baltissen, D.; Matar, O.; Glennemeier-Marke, H.; Almouhanna, F.; Theobald, J.; Abu El Maaty, M.A.; et al. Ascorbate kills breast cancer cells by rewiring metabolism via redox imbalance and energy crisis. Free. Radic. Biol. Med. 2021, 163, 196–209. [Google Scholar] [CrossRef]
- Uetaki, M.; Tabata, S.; Nakasuka, F.; Soga, T.; Tomita, M. Metabolomic alterations in human cancer cells by vitamin C-induced oxidative stress. Sci. Rep. 2015, 5, 13896. [Google Scholar] [CrossRef]
- Lu, Y.-X.; Wu, Q.-N.; Chen, D.-l.; Chen, L.-Z.; Wang, Z.-X.; Ren, C.; Mo, H.-y.; Chen, Y.; Sheng, H.; Wang, Y.-N.; et al. Pharmacological Ascorbate Suppresses Growth of Gastric Cancer Cells with GLUT1 Overexpression and Enhances the Efficacy of Oxaliplatin Through Redox Modulation. Theranostics 2018, 8, 1312–1326. [Google Scholar] [CrossRef]
- Tian, W.; Wang, Y.; Xu, Y.; Guo, X.; Wang, B.; Sun, L.; Liu, L.; Cui, F.; Zhuang, Q.; Bao, X.; et al. The Hypoxia-inducible Factor Renders Cancer Cells More Sensitive to Vitamin C-induced Toxicity *. J. Biol. Chem. 2014, 289, 3339–3351. [Google Scholar] [CrossRef] [PubMed]
- Vera, J.C.; Rivas, C.I.; Velásquez, F.V.; Zhang, R.H.; Concha, I.I.; Golde, D.W. Resolution of the facilitated transport of dehydroascorbic acid from its intracellular accumulation as ascorbic acid. J. Biol. Chem. 1995, 270, 23706–23712. [Google Scholar] [CrossRef] [PubMed]
- Szatrowski, T.P.; Nathan, C.F. Production of Large Amounts of Hydrogen Peroxide by Human Tumor Cells. Cancer Res. 1991, 51, 794. [Google Scholar] [PubMed]
- Policastro, L.L.; Ibañez, I.L.; Notcovich, C.; Duran, H.A.; Podhajcer, O.L. The Tumor Microenvironment: Characterization, Redox Considerations, and Novel Approaches for Reactive Oxygen Species-Targeted Gene Therapy. Antioxid. Redox Signal 2012, 19, 854–895. [Google Scholar] [CrossRef]
- Rees, D.C.; Kelsey, H.; Richards, J.D. Acute haemolysis induced by high dose ascorbic acid in glucose-6-phosphate dehydrogenase deficiency. BMJ 1993, 306, 841–842. [Google Scholar] [CrossRef]
- Quinn, J.; Gerber, B.; Fouche, R.; Kenyon, K.; Blom, Z.; Muthukanagaraj, P. Effect of High-Dose Vitamin C Infusion in a Glucose-6-Phosphate Dehydrogenase-Deficient Patient. Case Rep. Med. 2017, 2017, 5202606. [Google Scholar] [CrossRef]
- Rakitzis, E.T.; Papandreou, P.T. Ascorbate-induced generation of free radical species in normal and glucose-6-phosphate dehydrogenase-deficient erythrocytes. Biochem. Soc. Trans. 1989, 17, 371–372. [Google Scholar] [CrossRef]
- Mehta, J.; Singhal, S.; Mehta, B. Ascorbic-acid-induced haemolysis in G-6-PD deficiency. Lancet 1990, 336, 944. [Google Scholar] [CrossRef]
- Gaetani, G.; Ferraris, A.; Rolfo, M.; Mangerini, R.; Arena, S.; Kirkman, H. Predominant role of catalase in the disposal of hydrogen peroxide within human erythrocytes. Blood 1996, 87, 1595–1599. [Google Scholar] [CrossRef]
- May, J.M.; Qu, Z.-C.; Morrow, J.D. Mechanisms of ascorbic acid recycling in human erythrocytes. Biochim. Biophys. Acta (BBA)—Gen. Subj. 2001, 1528, 159–166. [Google Scholar] [CrossRef]
- Bissinger, R.; Bhuyan, A.A.M.; Qadri, S.M.; Lang, F. Oxidative stress, eryptosis and anemia: A pivotal mechanistic nexus in systemic diseases. FEBS J. 2019, 286, 826–854. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhong, Z.-F.; Wang, S.-P.; Vong, C.-T.; Yu, B.; Wang, Y.-T. HIF-1: Structure, biology and natural modulators. Chin. J. Nat. Med. 2021, 19, 521–527. [Google Scholar] [CrossRef]
- Xu, D.; Li, C. Regulation of the SIAH2-HIF-1 Axis by Protein Kinases and Its Implication in Cancer Therapy. Front. Cell Dev Biol. 2021, 9, 646687. [Google Scholar] [CrossRef]
- Dengler, V.L.; Galbraith, M.D.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef]
- Koivunen, P.; Hirsilä, M.; Günzler, V.; Kivirikko, K.I.; Myllyharju, J. Catalytic Properties of the Asparaginyl Hydroxylase (FIH) in the Oxygen Sensing Pathway Are Distinct from Those of Its Prolyl 4-Hydroxylases *. J. Biol. Chem. 2004, 279, 9899–9904. [Google Scholar] [CrossRef]
- Lee, G.; Won, H.-S.; Lee, Y.-M.; Choi, J.-W.; Oh, T.-I.; Jang, J.-H.; Choi, D.-K.; Lim, B.-O.; Kim, Y.J.; Park, J.-W.; et al. Oxidative Dimerization of PHD2 is Responsible for its Inactivation and Contributes to Metabolic Reprogramming via HIF-1α Activation. Sci. Rep. 2016, 6, 18928. [Google Scholar] [CrossRef]
- Metzen, E.; Stiehl, D.P.; Marxsen, J.H.; Hellwig-Burgel, T.; Jelkmann, W. Regulation of the prolyl hydroxylase domain protein 2 (phd2/egln-1) gene: Identification of a functional hypoxia-responsive element. Biochemical J. 2005, 387, 711–717. [Google Scholar] [CrossRef]
- Lando, D.; Peet, D.J.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine Hydroxylation of the HIF Transactivation Domain: A Hypoxic Switch. Science 2002, 295, 858. [Google Scholar] [CrossRef]
- Mahon, P.C.; Hirota, K.; Semenza, G.L. FIH-1: A novel protein that interacts with HIF-1α and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001, 15, 2675–2686. [Google Scholar]
- Cavadas, M.A.; Nguyen, L.K.; Cheong, A. Hypoxia-inducible factor (HIF) network: Insights from mathematical models. Cell Commun. Signal. CCS 2013, 11, 42. [Google Scholar] [CrossRef] [PubMed]
- Vissers, M.C.M.; Das, A.B. Potential Mechanisms of Action for Vitamin C in Cancer: Reviewing the Evidence. Front. Physiol. 2018, 9, 809. [Google Scholar] [CrossRef] [PubMed]
- Osipyants, A.I.; Poloznikov, A.A.; Smirnova, N.A.; Hushpulian, D.M.; Khristichenko, A.Y.; Chubar, T.A.; Zakhariants, A.A.; Ahuja, M.; Gaisina, I.N.; Thomas, B.; et al. L-ascorbic acid: A true substrate for HIF prolyl hydroxylase? Biochimie 2018, 147, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Wilkes, J.G.; O’Leary, B.R.; Du, J.; Klinger, A.R.; Sibenaller, Z.A.; Doskey, C.M.; Gibson-Corley, K.N.; Alexander, M.S.; Tsai, S.; Buettner, G.R.; et al. Pharmacologic ascorbate (P-AscH(-)) suppresses hypoxia-inducible Factor-1α (HIF-1α) in pancreatic adenocarcinoma. Clin. Exp. Metastasis 2018, 35, 37–51. [Google Scholar] [CrossRef]
- Gao, P.; Zhang, H.; Dinavahi, R.; Li, F.; Xiang, Y.; Raman, V.; Bhujwalla, Z.M.; Felsher, D.W.; Cheng, L.; Pevsner, J.; et al. HIF-Dependent Antitumorigenic Effect of Antioxidants In Vivo. Cancer Cell 2007, 12, 230–238. [Google Scholar] [CrossRef]
- Jóźwiak, P.; Ciesielski, P.; Zaczek, A.; Lipińska, A.; Pomorski, L.; Wieczorek, M.; Bryś, M.; Forma, E.; Krześlak, A. Expression of hypoxia inducible factor 1α and 2α and its association with vitamin C level in thyroid lesions. J. Biomed. Sci. 2017, 24, 83. [Google Scholar] [CrossRef]
- Campbell, E.J.; Vissers, M.C.M.; Bozonet, S.; Dyer, A.; Robinson, B.A.; Dachs, G.U. Restoring physiological levels of ascorbate slows tumor growth and moderates HIF-1 pathway activity in Gulo−/− mice. Cancer Med. 2015, 4, 303–314. [Google Scholar] [CrossRef]
- Campbell, E.J.; Vissers, M.C.; Dachs, G.U. Ascorbate availability affects tumor implantation-take rate and increases tumor rejection in Gulo(-/-) mice. Hypoxia (Auckl) 2016, 4, 41–52. [Google Scholar] [CrossRef]
- Campbell, E.J.; Vissers, M.C.M.; Wohlrab, C.; Hicks, K.O.; Strother, R.M.; Bozonet, S.M.; Robinson, B.A.; Dachs, G.U. Pharmacokinetic and anti-cancer properties of high dose ascorbate in solid tumours of ascorbate-dependent mice. Free. Radic. Biol. Med. 2016, 99, 451–462. [Google Scholar] [CrossRef]
- Kuiper, C.; Dachs, G.; Munn, D.; Currie, M.; Robinson, B.; Pearson, J.; Vissers, M. Increased Tumor Ascorbate is Associated with Extended Disease-Free Survival and Decreased Hypoxia-Inducible Factor-1 Activation in Human Colorectal Cancer. Front. Oncol. 2014, 4, 10. [Google Scholar] [CrossRef]
- Kuiper, C.; Dachs, G.; Currie, M.; Pearson, J.; Munn, D.; Vissers, M. Abstract 494: Tumor ascorbate content is associated with extended disease-free survival and decreased hypoxia-inducible factor-1 activation in patients with colorectal cancer. Cancer Res. 2014, 74, 494. [Google Scholar] [CrossRef]
- Wohlrab, C.; Vissers, M.C.M.; Phillips, E.; Morrin, H.; Robinson, B.A.; Dachs, G.U. The Association Between Ascorbate and the Hypoxia-Inducible Factors in Human Renal Cell Carcinoma Requires a Functional Von Hippel-Lindau Protein. Front. Oncol. 2018, 8, 574. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, C.; Molenaar, I.G.M.; Dachs, G.U.; Currie, M.J.; Sykes, P.H.; Vissers, M.C.M. Low Ascorbate Levels Are Associated with Increased Hypoxia-Inducible Factor-1 Activity and an Aggressive Tumor Phenotype in Endometrial Cancer. Cancer Res. 2010, 70, 5749. [Google Scholar] [CrossRef] [PubMed]
- King, A.; Selak, M.A.; Gottlieb, E. Succinate dehydrogenase and fumarate hydratase: Linking mitochondrial dysfunction and cancer. Oncogene 2006, 25, 4675–4682. [Google Scholar] [CrossRef] [PubMed]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Koivunen, P.; Hirsilä, M.; Remes, A.M.; Hassinen, I.E.; Kivirikko, K.I.; Myllyharju, J. Inhibition of Hypoxia-inducible Factor (HIF) Hydroxylases by Citric Acid Cycle Intermediates: Possible Links between Cell Metabolism and Stabilization of HIF *. J. Biol. Chem. 2007, 282, 4524–4532. [Google Scholar] [CrossRef]
- Gill, A.J.; Pachter, N.S.; Clarkson, A.; Tucker, K.M.; Winship, I.M.; Benn, D.E.; Robinson, B.G.; Clifton-Bligh, R.J. Renal tumors and hereditary pheochromocytoma-paraganglioma syndrome type 4. N. Engl. J. Med. 2011, 364, 885–886. [Google Scholar] [CrossRef]
- Pasini, B.; Stratakis, C.A. SDH mutations in tumorigenesis and inherited endocrine tumours: Lesson from the phaeochromocytoma–paraganglioma syndromes. J. Intern. Med. 2009, 266, 19–42. [Google Scholar] [CrossRef]
- Ricketts, C.J.; Shuch, B.; Vocke, C.; Metwalli, A.; Bratslavsky, G.; Middleton, L.; Yang, Y.; Wei, M.; Pautler, S.; Peterson, J.; et al. Succinate dehydrogenase kidney cancer: An aggressive example of the Warburg effect in cancer. J. Urol. 2012, 188, 2063–2071. [Google Scholar] [CrossRef]
- Clark, G.R.; Sciacovelli, M.; Gaude, E.; Walsh, D.M.; Kirby, G.; Simpson, M.A.; Trembath, R.C.; Berg, J.N.; Woodward, E.R.; Kinning, E.; et al. Germline FH Mutations Presenting With Pheochromocytoma. J. Clin. Endocrinol. Metab. 2014, 99, E2046–E2050. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-C.; Chang, J.-H.; Jin, J. Regulation of nuclear factor-κB in autoimmunity. Trends Immunol. 2013, 34, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C. Non-canonical NF-κB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. Signaling to NF-kappaB. Genes & development. Genes Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef] [PubMed]
- Beinke, S.; Ley, S.C. Functions of NF-kappaB1 and NF-kappaB2 in immune cell biology. Biochem. J. 2004, 382, 393–409. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; West, A.P.; Ghosh, S. NF-κB and the immune response. Oncogene 2006, 25, 6758–6780. [Google Scholar] [CrossRef]
- Collignon, E.; Canale, A.; Al Wardi, C.; Bizet, M.; Calonne, E.; Dedeurwaerder, S.; Garaud, S.; Naveaux, C.; Barham, W.; Wilson, A.; et al. Immunity drives TET1 regulation in cancer through NF-κB. Sci. Adv. 2018, 4, eaap7309. [Google Scholar] [CrossRef]
- Arora, R.; Yates, C.; Gary, B.D.; McClellan, S.; Tan, M.; Xi, Y.; Reed, E.; Piazza, G.A.; Owen, L.B.; Dean-Colomb, W. Panepoxydone Targets NF-kB and FOXM1 to Inhibit Proliferation, Induce Apoptosis and Reverse Epithelial to Mesenchymal Transition in Breast Cancer. PLoS ONE 2014, 9, e98370. [Google Scholar] [CrossRef]
- Kaltschmidt, C.; Banz-Jansen, C.; Benhidjeb, T.; Beshay, M.; Förster, C.; Greiner, J.; Hamelmann, E.; Jorch, N.; Mertzlufft, F.; Pfitzenmaier, J.; et al. A Role for NF-κB in Organ Specific Cancer and Cancer Stem Cells. Cancers 2019, 11, 655. [Google Scholar] [CrossRef]
- Khongthong, P.; Roseweir, A.K.; Edwards, J. The NF-KB pathway and endocrine therapy resistance in breast cancer. Endocr.-Relat. Cancer 2019, 26, R369–R380. [Google Scholar] [CrossRef]
- Biswas, D.K.; Shi, Q.; Baily, S.; Strickland, I.; Ghosh, S.; Pardee, A.B.; Iglehart, J.D. NF-kappa B activation in human breast cancer specimens and its role in cell proliferation and apoptosis. Proc. Natl. Acad. Sci. USA 2004, 101, 10137–10142. [Google Scholar] [CrossRef] [PubMed]
- Bowie, A.; O’Neill, L.A.J. Vitamin C inhibits nfκb activation in endothelial cells. Biochem. Soc. Trans. 1997, 25, 131S. [Google Scholar] [CrossRef] [PubMed]
- Bowie, A.G.; O’Neill, L.A.J. Vitamin C inhibits NF-κB activation by TNF via the activation of p38 mitogen-activated protein kinase. J. Immunol. 2000, 165, 7180–7188. [Google Scholar] [CrossRef] [PubMed]
- Cárcamo, J.M.; Pedraza, A.; Bórquez-Ojeda, O.; Golde, D.W. Vitamin C Suppresses TNFα-Induced NFκB Activation by Inhibiting IκBα Phosphorylation. Biochemistry 2002, 41, 12995–13002. [Google Scholar] [CrossRef]
- Du, Y.-T.; Long, Y.; Tang, W.; Liu, X.-F.; Dai, F.; Zhou, B. Prooxidative inhibition against NF-κB-mediated inflammation by pharmacological vitamin C. Free. Radic. Biol. Med. 2022, 180, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.W.; Liu, H.C.; Yu, Y.L.; Hung, Y.T.; Wei, C.W.; Yiang, G.T. Combined treatment with vitamin C and methotrexate inhibits triple-negative breast cancer cell growth by increasing H2O2 accumulation and activating caspase-3 and p38 pathways. Oncol. Rep. 2017, 37, 2177–2184. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Latif, M.M.; Raouf, A.A.; Sabra, K.; Kelleher, D.; Reynolds, J.V. Vitamin C enhances chemosensitization of esophageal cancer cells in vitro. J. Chemother. 2005, 17, 539–549. [Google Scholar] [CrossRef]
- Murphy, S.H.; Suzuki, K.; Downes, M.; Welch, G.L.; De Jesus, P.; Miraglia, L.J.; Orth, A.P.; Chanda, S.K.; Evans, R.M.; Verma, I.M. Tumor suppressor protein (p)53, is a regulator of NF-κB repression by the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA 2011, 108, 17117–17122. [Google Scholar] [CrossRef]
- Kim, J.; Lee, S.D.; Chang, B.; Jin, D.H.; Jung, S.I.; Park, M.Y.; Han, Y.; Yang, Y.; Il Kim, K.; Lim, J.S.; et al. Enhanced antitumor activity of vitamin C via p53 in cancer cells. Free. Radic. Biol. Med. 2012, 53, 1607–1615. [Google Scholar] [CrossRef]
- Leekha, A.; Gurjar, B.S.; Tyagi, A.; Rizvi, M.A.; Verma, A.K. Vitamin C in synergism with cisplatin induces cell death in cervical cancer cells through altered redox cycling and p53 upregulation. J. Cancer Res. Clin. Oncol. 2016, 142, 2503–2514. [Google Scholar] [CrossRef]
- An, S.H.; Kang, J.H.; Kim, D.H.; Lee, M.S. Vitamin C increases the apoptosis via up-regulation p53 during cisplatin treatment in human colon cancer cells. BMB Rep. 2011, 44, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Hahm, E.; Jin, D.H.; Kang, J.S.; Kim, Y.I.; Hong, S.W.; Lee, S.K.; Kim, H.N.; Jung, D.J.; Kim, J.E.; Shin, D.H.; et al. The molecular mechanisms of vitamin C on cell cycle regulation in B16F10 murine melanoma. J. Cell. Biochem. 2007, 102, 1002–1010. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.G.; Khanna, N.; Singh, N. Vitamin C Augments Chemotherapeutic Response of Cervical Carcinoma HeLa Cells by Stabilizing P53. Biochem. Biophys. Res. Commun. 2001, 282, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Kiessling, M.K.; Klemke, C.D.; Kamiński, M.M.; Galani, I.E.; Krammer, P.H.; Gülow, K. Inhibition of Constitutively Activated Nuclear Factor-κB Induces Reactive Oxygen Species- and Iron-Dependent Cell Death in Cutaneous T-Cell Lymphoma. Cancer Res. 2009, 69, 2365–2374. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. Epigenetic Determinants of Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Mullally, A.; Hedvat, C.; Garcia-Manero, G.; Patel, J.; Wadleigh, M.; Malinge, S.; Yao, J.; Kilpivaara, O.; Bhat, R.; et al. Genetic characterization of TET1, TET2, and TET3 alterations in myeloid malignancies. Blood 2009, 114, 144–147. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef]
- Jin, S.G.; Jiang, Y.; Qiu, R.; Rauch, T.A.; Wang, Y.; Schackert, G.; Krex, D.; Lu, Q.; Pfeifer, G.P. 5-Hydroxymethylcytosine is strongly depleted in human cancers but its levels do not correlate with IDH1 mutations. Cancer Res. 2011, 71, 7360–7365. [Google Scholar] [CrossRef]
- Delhommeau, F.; Dupont, S.; Valle, V.D.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.-P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in Myeloid Cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F.; et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, N.; Bhagat, T.; Nieves, E.; Stenson, M.; Lawson, J.; Choudhary, G.S.; Habermann, T.; Nowakowski, G.; Singh, R.; Wu, X.; et al. Upregulation of TET activity with ascorbic acid induces epigenetic modulation of lymphoma cells. Blood Cancer J. 2017, 7, e587. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, N.; Bhagat, T.D.; Cheville, J.; Lohse, C.; Bhattacharyya, S.; Tischer, A.; Machha, V.; Gordon-Mitchell, S.; Choudhary, G.; Wong, L.-F.; et al. Ascorbic acid-induced TET activation mitigates adverse hydroxymethylcytosine loss in renal cell carcinoma. J. Clin. Investig. 2019, 129, 1612–1625. [Google Scholar] [CrossRef]
- Cimmino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.H.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.T.; Mitchell-Flack, M.; et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 2017, 170, 1079–1095.e1020. [Google Scholar] [CrossRef]
- Chen, L.; Song, H.; Luo, Z.; Cui, H.; Zheng, W.; Liu, Y.; Li, W.; Luo, F.; Liu, J. PHLPP2 is a novel biomarker and epigenetic target for the treatment of vitamin C in pancreatic cancer. Int. J. Oncol. 2020, 56, 1294–1303. [Google Scholar] [CrossRef]
- Mingay, M.; Chaturvedi, A.; Bilenky, M.; Cao, Q.; Jackson, L.; Hui, T.; Moksa, M.; Heravi-Moussavi, A.; Humphries, R.K.; Heuser, M.; et al. Vitamin C-induced epigenomic remodelling in IDH1 mutant acute myeloid leukaemia. Leukemia 2018, 32, 11–20. [Google Scholar] [CrossRef]
- Gerecke, C.; Schumacher, F.; Berndzen, A.; Homann, T.; Kleuser, B. Vitamin C in combination with inhibition of mutant IDH1 synergistically activates TET enzymes and epigenetically modulates gene silencing in colon cancer cells. Epigenetics 2020, 15, 307–322. [Google Scholar] [CrossRef]
- Peng, D.; Ge, G.; Gong, Y.; Zhan, Y.; He, S.; Guan, B.; Li, Y.; Xu, Z.; Hao, H.; He, Z.; et al. Vitamin C increases 5-hydroxymethylcytosine level and inhibits the growth of bladder cancer. Clin. Epigenet. 2018, 10, 94. [Google Scholar] [CrossRef]
- Gustafson, C.B.; Yang, C.; Dickson, K.M.; Shao, H.; Van Booven, D.; Harbour, J.W.; Liu, Z.-J.; Wang, G. Epigenetic reprogramming of melanoma cells by vitamin C treatment. Clin. Epigenet. 2015, 7, 51. [Google Scholar] [CrossRef]
- Gillberg, L.; Ørskov, A.D.; Nasif, A.; Ohtani, H.; Madaj, Z.; Hansen, J.W.; Rapin, N.; Mogensen, J.B.; Liu, M.; Dufva, I.H.; et al. Oral vitamin C supplementation to patients with myeloid cancer on azacitidine treatment: Normalization of plasma vitamin C induces epigenetic changes. Clin. Epigenet. 2019, 11, 143. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hong, J.; Han, H.; Park, J.; Kim, D.; Park, H.; Ko, M.; Koh, Y.; Shin, D.Y.; Yoon, S.S. Decreased vitamin C uptake mediated by SLC2A3 promotes leukaemia progression and impedes TET2 restoration. Br. J. Cancer 2020, 122, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- Momparler, R.L.; Côté, S.; Momparler, L.F. Enhancement of the Antileukemic Action of the Inhibitors of DNA and Histone Methylation: 5-Aza-2’-Deoxycytidine and 3-Deazaneplanocin-A by Vitamin C. Epigenomes 2021, 5, 7. [Google Scholar] [CrossRef]
- Zhang, X.; Li, S.; He, J.; Jin, Y.-J.; Zhang, R.; Dong, W.; Lin, M.; Yang, Y.; Tian, T.; Zhou, Y.; et al. TET2 suppresses VHL deficiency-driven clear cell renal cell carcinoma by inhibiting HIF signaling. Cancer Res. 2022. [Google Scholar] [CrossRef] [PubMed]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef]
- Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Luchtel, R.A.; Bhagat, T.; Pradhan, K.; Jacobs, W.R.; Levine, M.; Verma, A.; Shenoy, N. High-dose ascorbic acid synergizes with anti-PD1 in a lymphoma mouse model. Proc. Natl. Acad. Sci. USA 2020, 117, 1666–1677. [Google Scholar] [CrossRef]
- Peng, D.; He, A.; He, S.; Ge, G.; Wang, S.; Ci, W.; Li, X.; Xia, D.; Zhou, L. Ascorbic acid induced TET2 enzyme activation enhances cancer immunotherapy efficacy in renal cell carcinoma. Int. J. Biol. Sci. 2022, 18, 995–1007. [Google Scholar] [CrossRef]
- Xu, Y.-p.; Lv, L.; Liu, Y.; Smith, M.D.; Li, W.-C.; Tan, X.-m.; Cheng, M.; Li, Z.; Bovino, M.; Aubé, J.; et al. Tumor suppressor TET2 promotes cancer immunity and immunotherapy efficacy. J. Clin. Investig. 2019, 129, 4316–4331. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, C.; Shi, G.; Yue, D.; Shu, Y.; Hu, S.; Qi, Z.; Chen, Y.; Zhang, B.; Zhang, Y.; et al. High-dose VitC plus oncolytic adenoviruses enhance immunogenic tumor cell death and reprogram tumor immune microenvironment. Mol. Ther. 2022, 30, 644–661. [Google Scholar] [CrossRef]
- Ighodaro, O.M.; Akinloye, O.A. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): Their fundamental role in the entire antioxidant defence grid. Alex. J. Med. 2018, 54, 287–293. [Google Scholar] [CrossRef]
- Nicolussi, A.; D’Inzeo, S.; Capalbo, C.; Giannini, G.; Coppa, A. The role of peroxiredoxins in cancer. Mol. Clin. Oncol. 2017, 6, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.C.; Vousden, K.H. The role of ROS in tumour development and progression. Nat. Rev. Cancer 2022, 22, 280–297. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.G.; Bubici, C.; Zazzeroni, F.; Papa, S.; Jones, J.; Alvarez, K.; Jayawardena, S.; De Smaele, E.; Cong, R.; Beaumont, C.; et al. Ferritin Heavy Chain Upregulation by NF-κB Inhibits TNFα-Induced Apoptosis by Suppressing Reactive Oxygen Species. Cell 2004, 119, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Younus, H. Therapeutic potentials of superoxide dismutase. Int. J. Health Sci. 2018, 12, 88–93. [Google Scholar]
- Ekoue, D.N.; He, C.; Diamond, A.M.; Bonini, M.G. Manganese superoxide dismutase and glutathione peroxidase-1 contribute to the rise and fall of mitochondrial reactive oxygen species which drive oncogenesis. Biochim. Biophys. Acta (BBA) Bioenerg. 2017, 1858, 628–632. [Google Scholar] [CrossRef]
- Dhar, S.K.; St. Clair, D.K. Manganese superoxide dismutase regulation and cancer. Free. Radic. Biol. Med. 2012, 52, 2209–2222. [Google Scholar] [CrossRef]
- Becuwe, P.; Ennen, M.; Klotz, R.; Barbieux, C.; Grandemange, S. Manganese superoxide dismutase in breast cancer: From molecular mechanisms of gene regulation to biological and clinical significance. Free. Radic. Biol. Med. 2014, 77, 139–151. [Google Scholar] [CrossRef]
- Nandi, A.; Yan, L.-J.; Jana, C.K.; Das, N. Role of Catalase in Oxidative Stress- and Age-Associated Degenerative Diseases. Oxidative Med. Cell. Longev. 2019, 2019, 9613090. [Google Scholar] [CrossRef]
- Bauer, G. Tumor Cell-protective Catalase as a Novel Target for Rational Therapeutic Approaches Based on Specific Intercellular ROS Signaling. Anticancer. Res. 2012, 32, 2599–2624. [Google Scholar] [PubMed]
- Glorieux, C.; Calderon, P.B. Catalase, a remarkable enzyme: Targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem. 2017, 398, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione peroxidase-1 in health and disease: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta (BBA) Gen. Subj. 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Liu, J.; Hinkhouse, M.M.; Sun, W.; Weydert, C.J.; Ritchie, J.M.; Oberley, L.W.; Cullen, J.J. GoswamiRedox Regulation of Pancreatic Cancer Cell Growth: Role of Glutathione Peroxidase in the Suppression of the Malignant Phenotype. Hum. Gene Ther. 2004, 15, 239–250. [Google Scholar] [CrossRef]
- Jardim, B.V.; Moschetta, M.G.; Leonel, C.; Gelaleti, G.B.; Regiani, V.R.; Ferreira, L.C.; Lopes, J.R.; de Campos Zuccari, D.A.P. Glutathione and glutathione peroxidase expression in breast cancer: An immunohistochemical and molecular study. Oncol. Rep. 2013, 30, 1119–1128. [Google Scholar] [CrossRef]
- Meng, Q.; Shi, S.; Liang, C.; Liang, D.; Hua, J.; Zhang, B.; Xu, J.; Yu, X. Abrogation of glutathione peroxidase-1 drives EMT and chemoresistance in pancreatic cancer by activating ROS-mediated Akt/GSK3β/Snail signaling. Oncogene 2018, 37, 5843–5857. [Google Scholar] [CrossRef]
- Chang, X.-Z.; Li, D.-Q.; Hou, Y.-F.; Wu, J.; Lu, J.-S.; Di, G.-H.; Jin, W.; Ou, Z.-L.; Shen, Z.-Z.; Shao, Z.-M. Identification of the functional role of peroxiredoxin 6 in the progression of breast cancer. Breast Cancer Res. 2007, 9, 1–15. [Google Scholar] [CrossRef]
- Park, M.H.; Jo, M.; Kim, Y.; Lee, C.; Hong, J. Roles of peroxiredoxins in cancer, neurodegenerative diseases and inflammatory diseases. Pharmacol. Ther. 2016, 163, 1–23. [Google Scholar] [CrossRef]
- Lillig, C.H.; Holmgren, A. Thioredoxin and Related Molecules–From Biology to Health and Disease. Antioxid. Redox Signal. 2007, 9, 25–47. [Google Scholar] [CrossRef]
- Nordberg, J.; Arnér, E.S.J. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system1 1. This review is based on the licentiate thesis “Thioredoxin Reductase—Interactions with the Redox Active Compounds 1-chloro-2,4-dinitrobenzene and lipoic acid” by Jonas Nordberg, 2001, Karolinska Institute, Stockholm, ISBN 91-631-1064-4. Free. Radic. Biol. Med. 2001, 31, 1287–1312. [Google Scholar] [CrossRef]
- Cadenas, C.; Franckenstein, D.; Schmidt, M.; Gehrmann, M.; Hermes, M.; Geppert, B.; Schormann, W.; Maccoux, L.J.; Schug, M.; Schumann, A.; et al. Role of thioredoxin reductase 1 and thioredoxin interacting protein in prognosis of breast cancer. Breast Cancer Res. 2010, 12, R44. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free. Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef]
- Kim, S.J.; Miyoshi, Y.; Taguchi, T.; Tamaki, Y.; Nakamura, H.; Yodoi, J.; Kato, K.; Noguchi, S. High Thioredoxin Expression Is Associated with Resistance to Docetaxel in Primary Breast Cancer. Clin. Cancer Res. 2005, 11, 8425–8430. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L.; Generali, D. Inhibitors of tumor angiogenesis. Cancer Drug Des. Discov. 2014, 275–317. [Google Scholar]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione: New roles in redox signaling for an old antioxidant. Front. Pharmacol. 2014, 5, 196. [Google Scholar] [CrossRef] [PubMed]
- Balendiran, G.K.; Dabur, R.; Fraser, D. The role of glutathione in cancer. Cell Biochem. Funct. 2004, 22, 343–352. [Google Scholar] [CrossRef]
- Smith, P.F.; Alberts, D.W.; Rush, G.F. Role of glutathione reductase during menadione-induced NADPH oxidation in isolated rat hepatocytes. Biochem. Pharmacol. 1987, 36, 3879–3884. [Google Scholar] [CrossRef]
- Estrela, J.M.; Ortega, A.; Obrador, E. Glutathione in Cancer Biology and Therapy. Crit. Rev. Clin. Lab. Sci. 2006, 43, 143–181. [Google Scholar] [CrossRef]
- McIlwain, C.C.; Townsend, D.M.; Tew, K.D. Glutathione S-transferase polymorphisms: Cancer incidence and therapy. Oncogene 2006, 25, 1639–1648. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef] [PubMed]
- Fujitani, N.; Yoneda, A.; Takahashi, M.; Takasawa, A.; Aoyama, T.; Miyazaki, T. Retracted article: Silencing of Glutathione S-Transferase Pi Inhibits Cancer Cell Growth via Oxidative Stress Induced by Mitochondria Dysfunction. Sci. Rep. 2019, 9, 14764. [Google Scholar] [CrossRef] [PubMed]
- Shpyleva, S.I.; Tryndyak, V.P.; Kovalchuk, O.; Starlard-Davenport, A.; Chekhun, V.F.; Beland, F.A.; Pogribny, I.P. Role of ferritin alterations in human breast cancer cells. Breast Cancer Res. Treat. 2011, 126, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Nauman, G.; Gray, J.C.; Parkinson, R.; Levine, M.; Paller, C.J. Systematic Review of Intravenous Ascorbate in Cancer Clinical Trials. Antioxidants 2018, 7, 89. [Google Scholar] [CrossRef] [PubMed]
- Klimant, E.; Wright, H.; Rubin, D.; Seely, D.; Markman, M. Intravenous Vitamin C in the Supportive Care of Cancer Patients: A Review and Rational Approach. Curr. Oncol. 2018, 25, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Carr, A.C.; Cook, J. Intravenous Vitamin C for Cancer Therapy—Identifying the Current Gaps in Our Knowledge. Front. Physiol. 2018, 9, 1182. [Google Scholar] [CrossRef]
- Wang, F.; He, M.-M.; Wang, Z.-X.; Li, S.; Jin, Y.; Ren, C.; Shi, S.-M.; Bi, B.-T.; Chen, S.-Z.; Lv, Z.-D.; et al. Phase I study of high-dose ascorbic acid with mFOLFOX6 or FOLFIRI in patients with metastatic colorectal cancer or gastric cancer. BMC Cancer 2019, 19, 460. [Google Scholar] [CrossRef]
- Abdel-Latif, M.; Babar, M.; Kelleher, D.; Reynolds, J. A pilot study of the impact of Vitamin C supplementation with neoadjuvant chemoradiation on regulators of inflammation and carcinogenesis in esophageal cancer patients. J. Cancer Res. Ther. 2019, 15, 185–191. [Google Scholar] [CrossRef]
- Ou, J.; Zhu, X.; Zhang, H.; Du, Y.; Chen, P.; Wang, J.; Peng, X.; Bao, S.; Zhang, X.; Zhang, T.; et al. A Retrospective Study of Gemcitabine and Carboplatin With or Without Intravenous Vitamin C on Patients With Advanced Triple-Negative Breast Cancer. Integr. Cancer Ther. 2020, 19, 1534735419895591. [Google Scholar] [CrossRef]
- Qian, W.; Wang, L.; Li, P.; Hu, Y.; Wang, Q.; Yi, K.; Wu, M.; Xu, Y.; Song, J.; Chen, P.; et al. Efficiency and Tolerability of Induction and Consolidation Therapy with Arsenic Trioxide/Bortezomib/Ascorbic Acid/Dexamethasone (ABCD) Regimen Compared to Bortezomib/Dexamethasone (BD) Regimen in Newly Diagnosed Myeloma Patients. Cancer Manag. Res. 2020, 12, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, R.; Narui, R.; Morikawa, H.; Wada, H. Improved Chemotherapy Outcomes of Patients With Small-cell Lung Cancer Treated With Combined Alkalization Therapy and Intravenous Vitamin C. Cancer Diagn. Progn. 2021, 1, 157–163. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mussa, A.; Mohd Idris, R.A.; Ahmed, N.; Ahmad, S.; Murtadha, A.H.; Tengku Din, T.A.D.A.A.; Yean, C.Y.; Wan Abdul Rahman, W.F.; Mat Lazim, N.; Uskoković, V.; et al. High-Dose Vitamin C for Cancer Therapy. Pharmaceuticals 2022, 15, 711. https://doi.org/10.3390/ph15060711
Mussa A, Mohd Idris RA, Ahmed N, Ahmad S, Murtadha AH, Tengku Din TADAA, Yean CY, Wan Abdul Rahman WF, Mat Lazim N, Uskoković V, et al. High-Dose Vitamin C for Cancer Therapy. Pharmaceuticals. 2022; 15(6):711. https://doi.org/10.3390/ph15060711
Chicago/Turabian StyleMussa, Ali, Ros Akmal Mohd Idris, Naveed Ahmed, Suhana Ahmad, Ahmad Hafiz Murtadha, Tengku Ahmad Damitri Al Astani Tengku Din, Chan Yean Yean, Wan Faiziah Wan Abdul Rahman, Norhafiza Mat Lazim, Vuk Uskoković, and et al. 2022. "High-Dose Vitamin C for Cancer Therapy" Pharmaceuticals 15, no. 6: 711. https://doi.org/10.3390/ph15060711