

Gastrointestinal Permeation Enhancers for the Development of Oral Peptide Pharmaceuticals

Abstract
:1. Introduction
2. Approved Oral Peptide Drugs
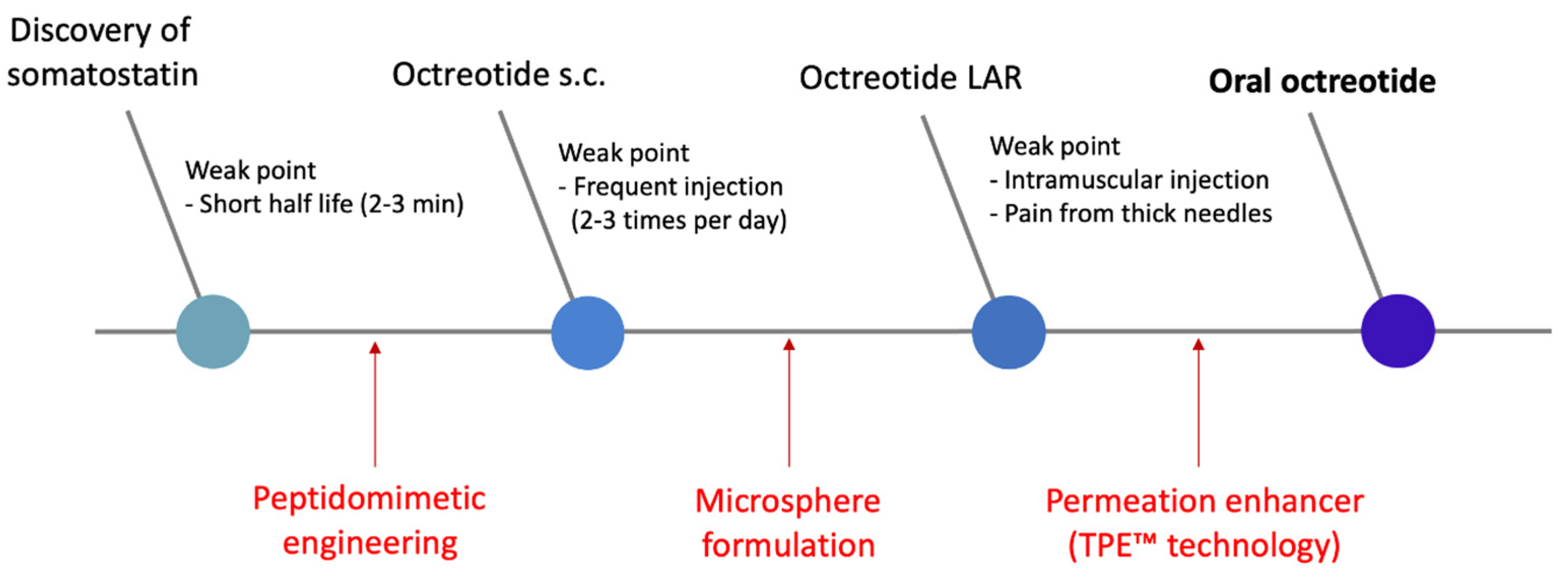
2.1. Mycapssa® (Oral Octreotide)
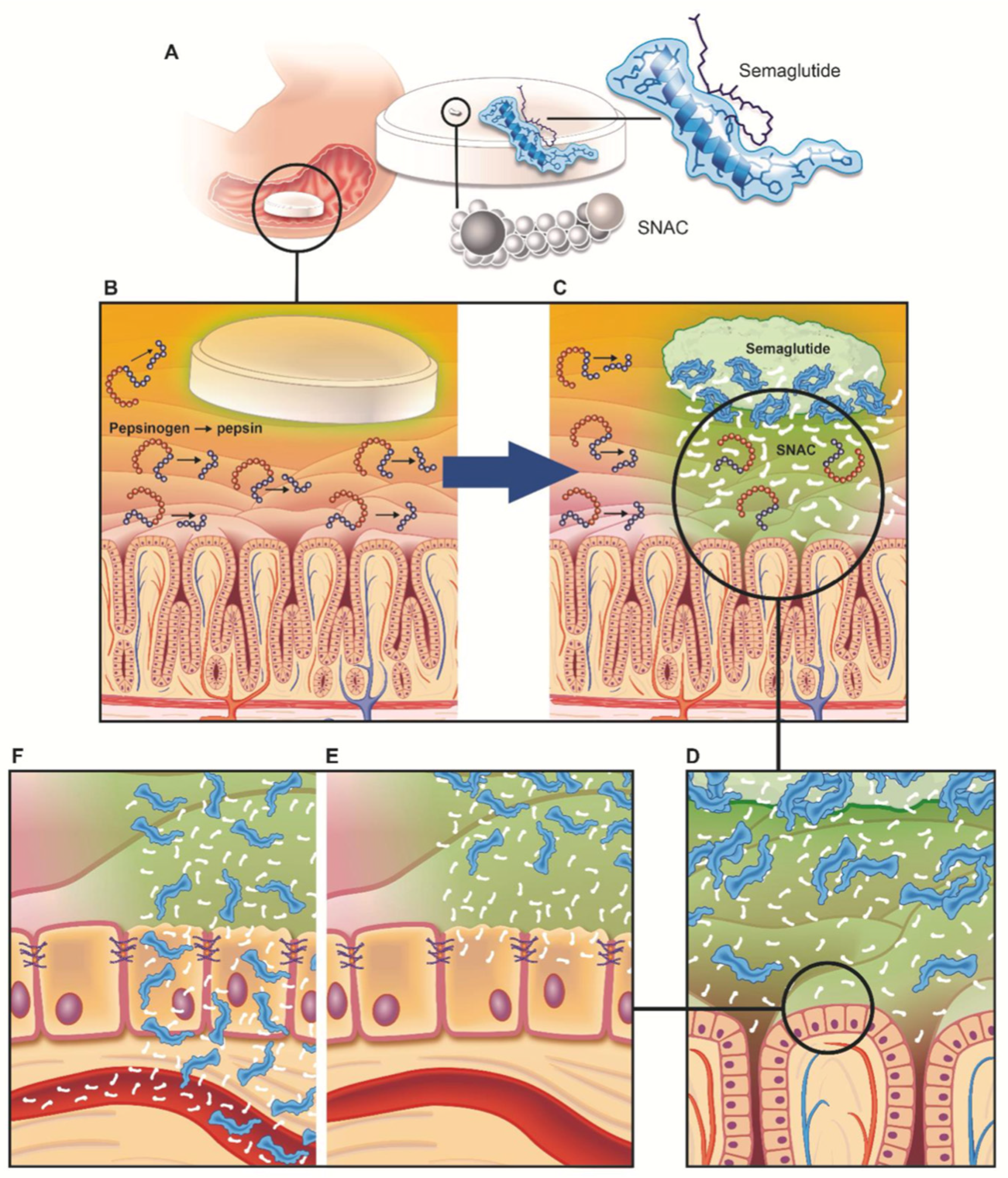
2.2. Rybelsus® (Oral Semaglutide)
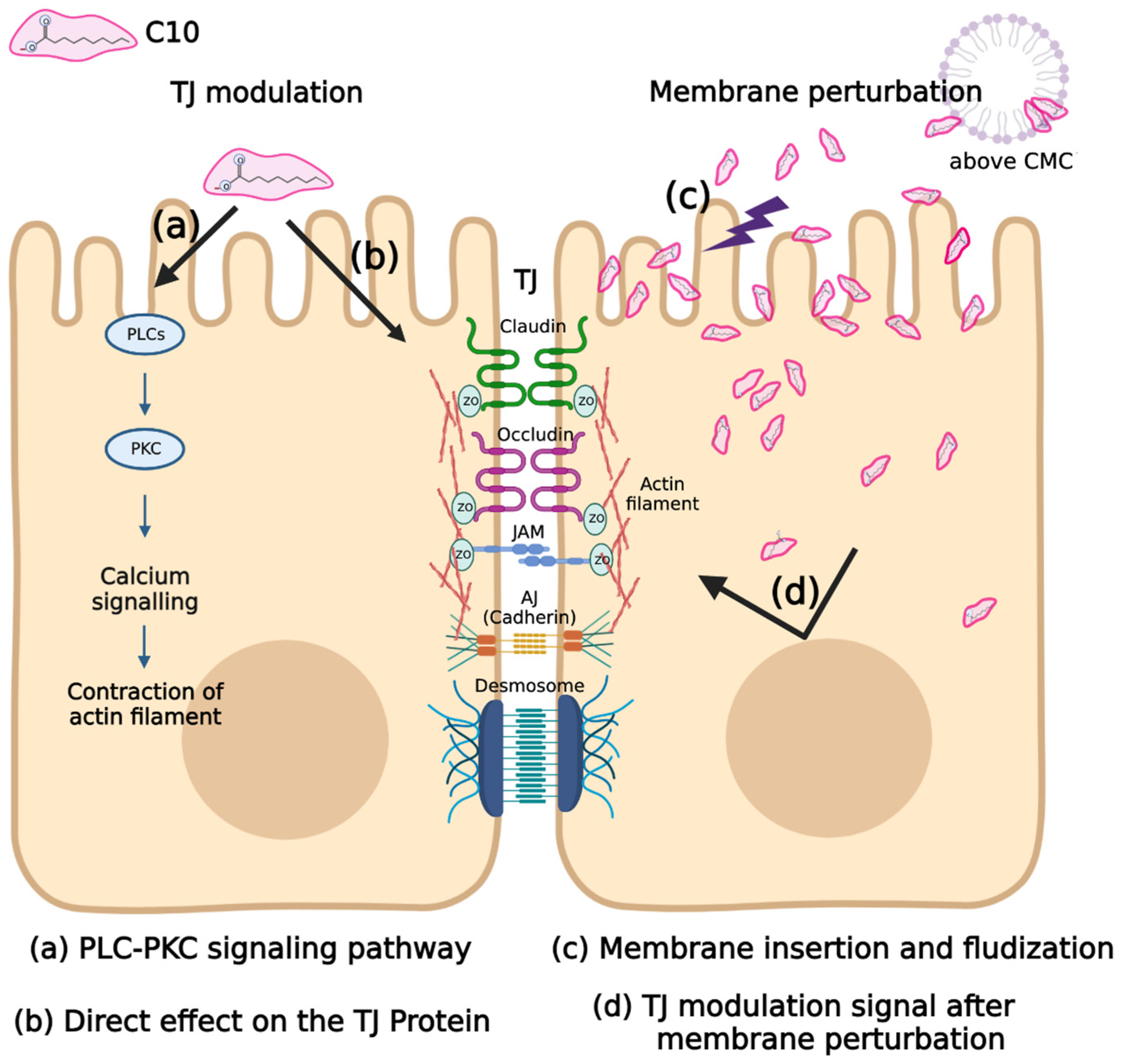
3. Permeation Enhancers–MCFAs (Medium Chain Fatty Acids)
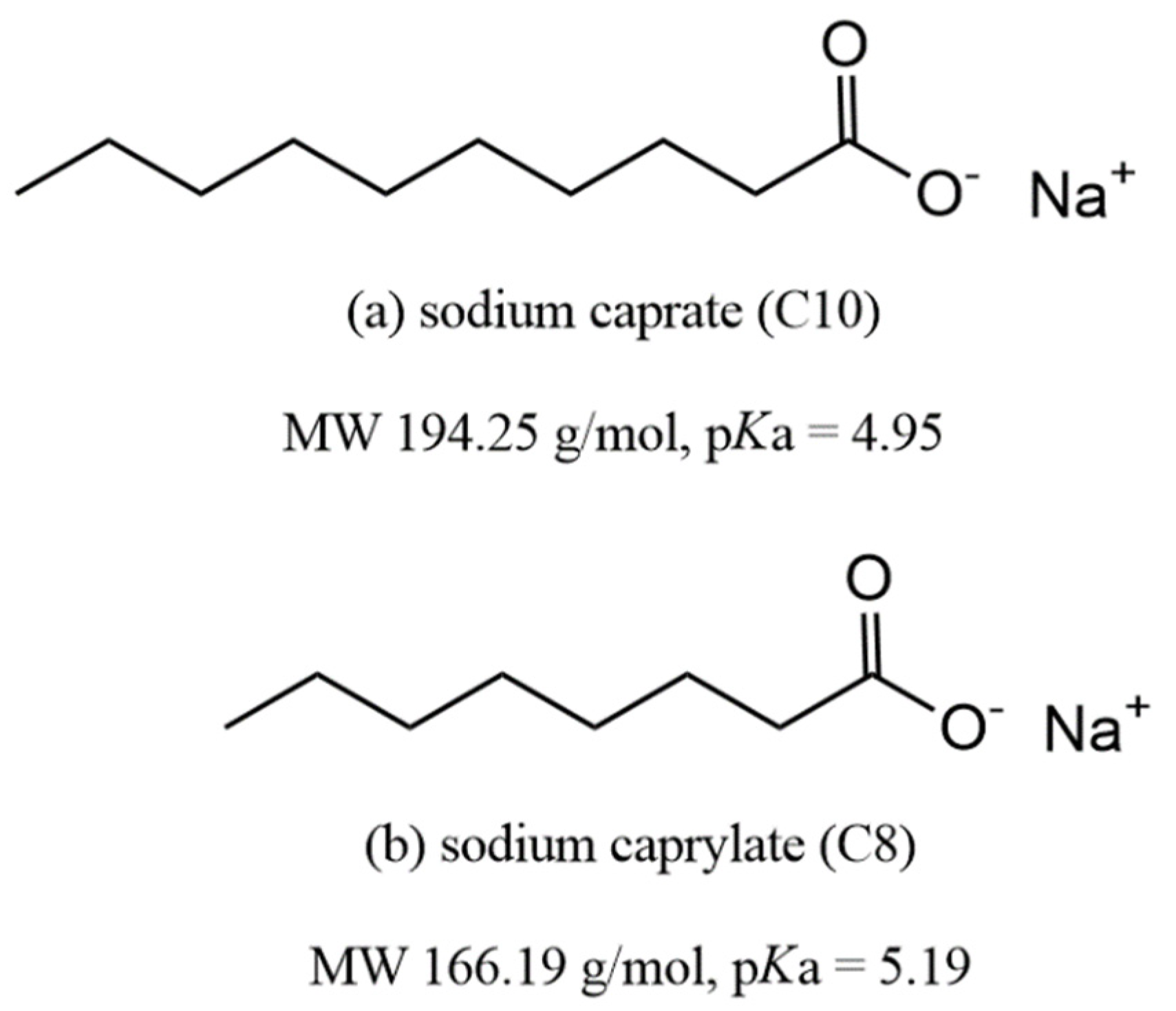
3.1. Sodium Caprylate (C8)
3.2. Sodium Caprate (C10)
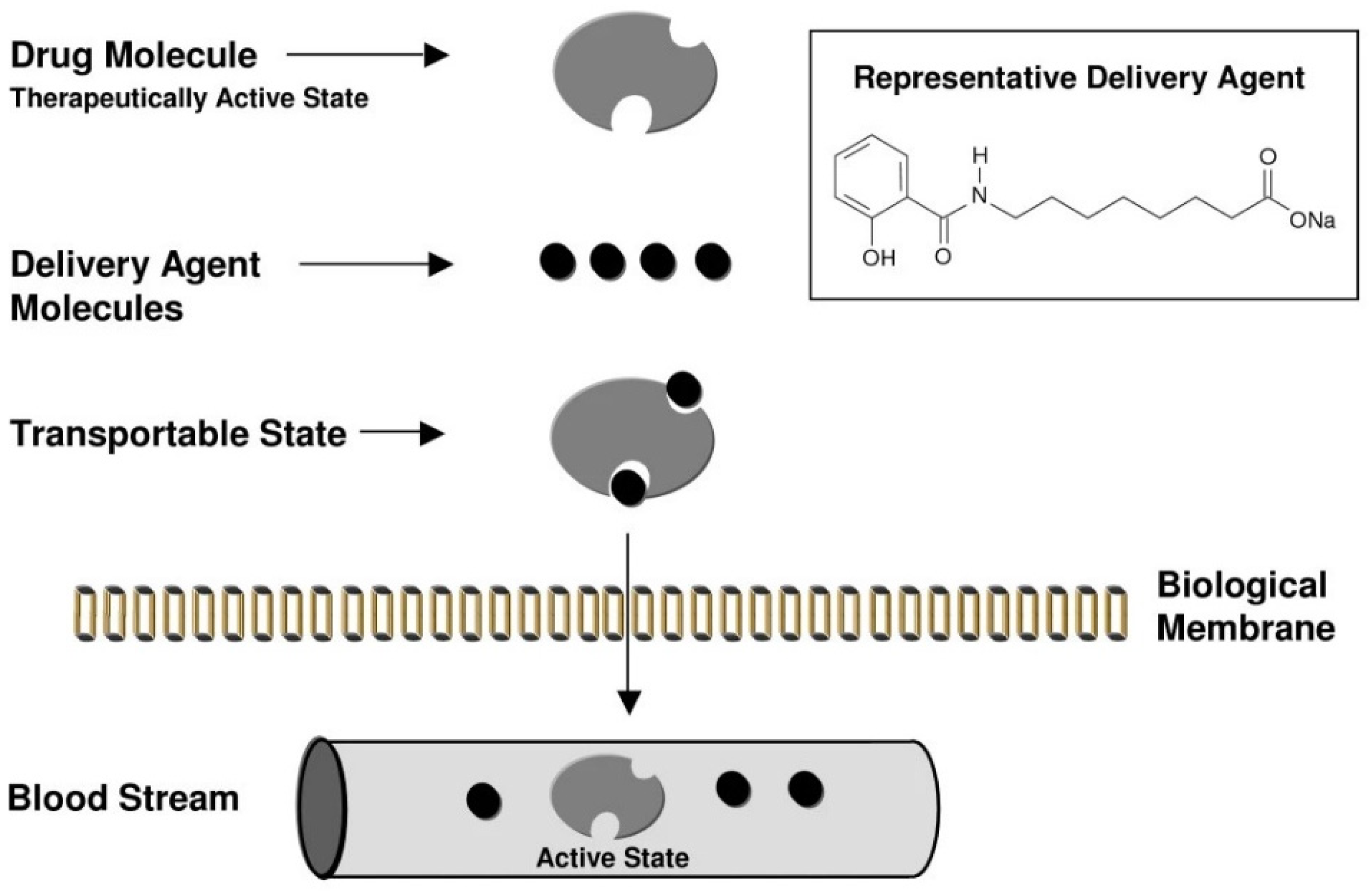
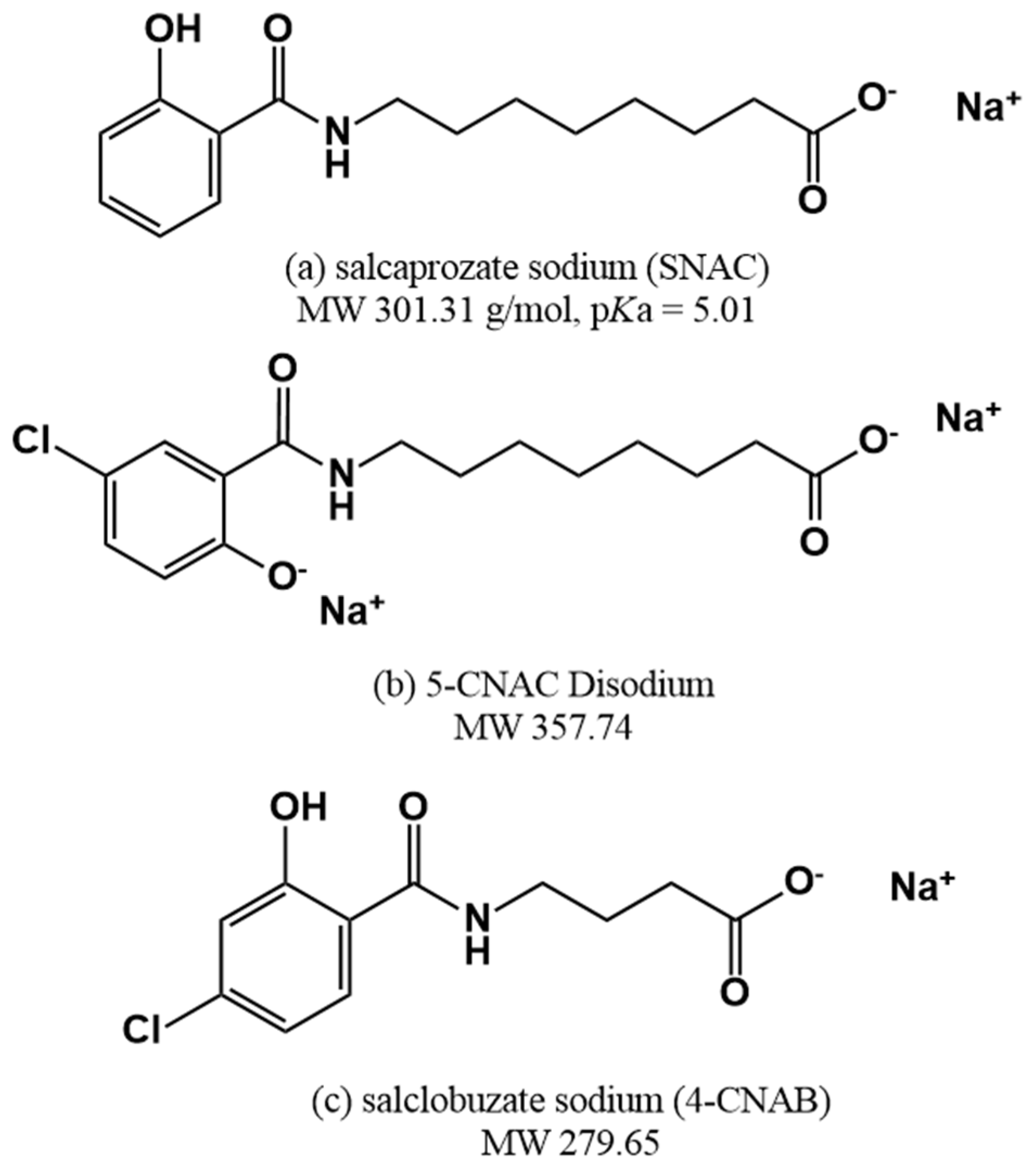
4. Permeation Enhancer: Eligen™ Technology-Based PEs
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lau, J.L.; Dunn, M.K. Therapeutic peptides: Historical perspectives, current development trends, and future directions. Bioorganic. Med. Chem. 2018, 26, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Al Shaer, D.; Al Musaimi, O.; Albericio, F.; de la Torre, B.G. 2019 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals 2020, 13, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Erak, M.; Bellmann-Sickert, K.; Els-Heindl, S.; Beck-Sickinger, A.G. Peptide chemistry toolbox—Transforming natural peptides into peptide therapeutics. Bioorganic Med. Chem. 2018, 26, 2759–2765. [Google Scholar] [CrossRef]
- Tsomaia, N. Peptide therapeutics: Targeting the undruggable space. Eur. J. Med. Chem. 2015, 94, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Trier, S.; Linderoth, L.; Bjerregaard, S.; Strauss, H.M.; Rahbek, U.L.; Andresen, T.L. Acylation of salmon calcitonin modulates in vitro intestinal peptide flux through membrane permeability enhancement. Eur. J. Pharm. Biopharm. 2015, 96, 329–337. [Google Scholar] [CrossRef] [Green Version]
- Na, D.H.; Youn, Y.S.; Park, E.J.; Lee, J.M.; Cho, O.R.; Lee, K.R.; Lee, S.D.; Yoo, S.D.; DeLuca, P.P.; Lee, K.C. Stability of PEGylated salmon calcitonin in nasal mucosa. J. Pharm. Sci. 2004, 93, 256–261. [Google Scholar] [CrossRef]
- Na, D.H.; Faraj, J.; Capan, Y.; Leung, K.P.; DeLuca, P.P. Stability of Antimicrobial Decapeptide (KSL) and Its Analogues for Delivery in the Oral Cavity. Pharm. Res. 2007, 24, 1544–1550. [Google Scholar] [CrossRef]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325. [Google Scholar] [CrossRef]
- Park, E.J.; Choi, J.; Lee, K.C.; Na, D.H. Emerging PEGylated non-biologic drugs. Expert Opin. Emerg. Drugs 2019, 24, 107–119. [Google Scholar] [CrossRef]
- Park, E.J.; Lim, S.M.; Lee, K.C.; Na, D.H. Exendins and exendin analogs for diabetic therapy: A patent review (2012-2015). Expert Opin. Ther. Pat. 2016, 26, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Sheahan, K.H.; Wahlberg, E.A.; Gilbert, M.P. An overview of GLP-1 agonists and recent cardiovascular outcomes trials. Postgrad. Med. J. 2019, 96, 156–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamberts, S.W.; van der Lely, A.-J.; de Herder, W.W.; Hofland, L.J. Octreotide. N. Engl. J. Med. 1996, 334, 246–254. [Google Scholar] [CrossRef] [Green Version]
- McKeage, K.; Cheer, S.; Wagstaff, A.J. Octreotide Long-Acting Release (LAR): A review of its use in the management of acromegaly. Drugs 2003, 63, 2473–2499. [Google Scholar] [CrossRef] [PubMed]
- Murty, S.B.; Na, D.H.; Thanoo, B.; DeLuca, P.P. Impurity formation studies with peptide-loaded polymeric microspheres: Part II. In vitro evaluation. Int. J. Pharm. 2005, 297, 62–72. [Google Scholar] [CrossRef]
- Na, D.H.; DeLuca, P.P. PEGylation of Octreotide: I. Separation of Positional Isomers and Stability Against Acylation by Poly(D,L-lactide-co-glycolide). Pharm. Res. 2005, 22, 736–742. [Google Scholar] [CrossRef]
- Ahn, J.H.; Park, E.J.; Lee, H.S.; Lee, K.C.; Na, D.H. Reversible Blocking of Amino Groups of Octreotide for the Inhibition of Formation of Acylated Peptide Impurities in Poly(Lactide-co-Glycolide) Delivery Systems. AAPS PharmSciTech 2011, 12, 1220–1226. [Google Scholar] [CrossRef] [Green Version]
- Gadelha, M.R.; Wildemberg, L.E.; Kasuki, L. The Future of Somatostatin Receptor Ligands in Acromegaly. J. Clin. Endocrinol. Metab. 2021, 107, 297–308. [Google Scholar] [CrossRef]
- Bornschein, J.; Drozdov, I.; Malfertheiner, P. Octreotide LAR: Safety and tolerability issues. Expert Opin. Drug Saf. 2009, 8, 755–768. [Google Scholar] [CrossRef]
- Geho, W.B.; Rosenberg, L.N.; Schwartz, S.L.; Lau, J.R.; Gana, T.J. A Single-blind, Placebo-controlled, Dose-ranging Trial of Oral Hepatic-directed Vesicle Insulin Add-on to Oral Antidiabetic Treatment in Patients With Type 2 Diabetes Mellitus. J. Diabetes Sci. Technol. 2014, 8, 551–559. [Google Scholar] [CrossRef]
- Rachmiel, M.; Barash, G.; Leshem, A.; Sagi, R.; Doenyas-Barak, K.; Koren, S. OR14-1 Pharmacodynamics, Safety, Tolerability, and Efficacy of Oral Insulin Formulation (Oshadi Icp) among Young Adults with Type 1 Diabetes: A Summary of Clinical Studies Phases I, Ib, and Ii. J. Endocr. Soc. 2019, 3. [Google Scholar] [CrossRef]
- Tan, X.; Liu, X.; Zhang, Y.; Zhang, H.; Lin, X.; Pu, C.; Gou, J.; He, H.; Yin, T.; Zhang, Y.; et al. Silica nanoparticles on the oral delivery of insulin. Expert Opin. Drug Deliv. 2018, 15, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Pangeni, R.; Kang, S.; Jha, S.K.; Subedi, L.; Park, J.W. Intestinal membrane transporter-mediated approaches to improve oral drug delivery. J. Pharm. Investig. 2021, 51, 137–158. [Google Scholar] [CrossRef]
- Leonaviciute, G.; Bernkop-Schnürch, A. Self-emulsifying drug delivery systems in oral (poly)peptide drug delivery. Expert Opin. Drug Deliv. 2015, 12, 1703–1716. [Google Scholar] [CrossRef] [PubMed]
- Noh, G.; Keum, T.; Bashyal, S.; Seo, J.-E.; Shrawani, L.; Kim, J.H.; Lee, S. Recent progress in hydrophobic ion-pairing and lipid-based drug delivery systems for enhanced oral delivery of biopharmaceuticals. J. Pharm. Investig. 2021, 52, 75–93. [Google Scholar] [CrossRef]
- Abramson, A.; Caffarel-Salvador, E.; Khang, M.; Dellal, D.; Silverstein, D.; Gao, Y.; Frederiksen, M.R.; Vegge, A.; Hubálek, F.; Water, J.J.; et al. An ingestible self-orienting system for oral delivery of macromolecules. Science 2019, 363, 611–615. [Google Scholar] [CrossRef] [Green Version]
- Abramson, A.; Caffarel-Salvador, E.; Soares, V.; Minahan, D.; Tian, R.Y.; Lu, X.; Dellal, D.; Gao, Y.; Kim, S.; Wainer, J.; et al. A luminal unfolding microneedle injector for oral delivery of macromolecules. Nat. Med. 2019, 25, 1512–1518. [Google Scholar] [CrossRef]
- Hashim, M.; Korupolu, R.; Syed, B.; Horlen, K.; Beraki, S.; Karamchedu, P.; Dhalla, A.K.; Ruffy, R.; Imran, M. Jejunal wall delivery of insulin via an ingestible capsule in anesthetized swine—A pharmacokinetic and pharmacodynamic study. Pharmacol. Res. Perspect. 2019, 7, e00522. [Google Scholar] [CrossRef] [Green Version]
- Fricker, G.; Fahr, A.; Beglinger, C.; Kissel, T.; Reiter, G.; Drewe, J. Permeation enhancement of octreotide by specific bile salts in rats and human subjects: In vitro, in vivo correlations. J. Cereb. Blood Flow Metab. 1996, 117, 217–223. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.-H.; Sinko, P.J. Oral delivery of salmon calcitonin. Adv. Drug Deliv. Rev. 2000, 42, 225–238. [Google Scholar] [CrossRef]
- Stern, W. DRUG DELIVERY—Oral Delivery of Peptides by Peptelligence Technology. Available online: https://drug-dev.com/oral-delivery-of-peptides-by-peptelligence-technology/ (accessed on 28 September 2022).
- Petersen, S.B.; Nielsen, L.G.; Rahbek, U.L.; Guldbrandt, M.; Brayden, D.J. Colonic absorption of salmon calcitonin using tetradecyl maltoside (TDM) as a permeation enhancer. Eur. J. Pharm. Sci. 2013, 48, 726–734. [Google Scholar] [CrossRef]
- Kondoh, M.; Masuyama, A.; Takahashi, A.; Asano, N.; Mizuguchi, H.; Koizumi, N.; Fujii, M.; Hayakawa, T.; Horiguchi, Y.; Watanbe, Y. A Novel Strategy for the Enhancement of Drug Absorption Using a Claudin Modulator. Mol. Pharmacol. 2004, 67, 749–756. [Google Scholar] [CrossRef] [Green Version]
- Gopalakrishnan, S.; Pandey, N.; Tamiz, A.P.; Vere, J.; Carrasco, R.; Somerville, R.; Tripathi, A.; Ginski, M.; Paterson, B.M.; Alkan, S.S. Mechanism of action of ZOT-derived peptide AT-1002, a tight junction regulator and absorption enhancer. Int. J. Pharm. 2009, 365, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Krug, S.M.; Hayaishi, T.; Iguchi, D.; Watari, A.; Takahashi, A.; Fromm, M.; Nagahama, M.; Takeda, H.; Okada, Y.; Sawasaki, T.; et al. Angubindin-1, a novel paracellular absorption enhancer acting at the tricellular tight junction. J. Control. Release 2017, 260, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Bocsik, A.; Gróf, I.; Kiss, L.; Ötvös, F.; Zsíros, O.; Daruka, L.; Fülöp, L.; Vastag, M.; Kittel, Á.; Imre, N.; et al. Dual Action of the PN159/KLAL/MAP Peptide: Increase of Drug Penetration across Caco-2 Intestinal Barrier Model by Modulation of Tight Junctions and Plasma Membrane Permeability. Pharmaceutics 2019, 11, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artursson, P.; Magnusson, C. Epithelial Transport of Drugs in Cell Culture. II: Effect of Extracellular Calcium Concentration on the Paracellular Transport of Drugs of Different Lipophilicities across Monolayers of Intestinal Epithelial (Caco-2) Cells. J. Pharm. Sci. 1990, 79, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Tomita, M.; Hayashi, M.; Awazu, S. Absorption-Enhancing Mechanism of EDTA, Caprate, and Decanoylcarnitine in Caco-2 Cells. J. Pharm. Sci. 1996, 85, 608–611. [Google Scholar] [CrossRef]
- Zhao, D.; Hirst, B.H. Comparison of Bile Salt Perturbation of Duodenal and Jejunal Isolated Brush-Border Membranes. Digestion 1990, 47, 200–207. [Google Scholar] [CrossRef]
- Bonengel, S.; Jelkmann, M.; Abdulkarim, M.; Gumbleton, M.; Reinstadler, V.; Oberacher, H.; Prüfert, F.; Bernkop-Schnürch, A. Impact of different hydrophobic ion pairs of octreotide on its oral bioavailability in pigs. J. Control. Release 2018, 273, 21–29. [Google Scholar] [CrossRef]
- Song, K.-H.; Chung, S.-J.; Shim, C.-K. Enhanced intestinal absorption of salmon calcitonin (sCT) from proliposomes containing bile salts. J. Control. Release 2005, 106, 298–308. [Google Scholar] [CrossRef]
- LeCluyse, E.; Appel, L.E.; Sutton, S.C. Relationship Between Drug Absorption Enhancing Activity and Membrane Perturbing Effects of Acylcarnitines. Pharm. Res. 1991, 8, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Sutton, S.C.; Forbes, A.E.; Cargill, R.; Hochman, J.H.; LeCluyse, E.L. Simultaneous in Vitro Measurement of Intestinal Tissue Permeability and Transepithelial Electrical Resistance (TEER) Using Sweetana–Grass Diffusion Cells. Pharm. Res. 1992, 09, 316–319. [Google Scholar] [CrossRef] [PubMed]
- LeCluyse, E.; Sutton, S.C.; Fix, J.A. In vitro effects of long-chain acylcarnitines on the permeability, transepithelial electrical resistance and morphology of rat colonic mucosa. J. Pharmacol. Exp. Ther. 1993, 265, 955–962. [Google Scholar] [PubMed]
- Petersen, S.B.; Nolan, G.; Maher, S.; Rahbek, U.L.; Guldbrandt, M.; Brayden, D.J. Evaluation of alkylmaltosides as intestinal permeation enhancers: Comparison between rat intestinal mucosal sheets and Caco-2 monolayers. Eur. J. Pharm. Sci. 2012, 47, 701–712. [Google Scholar] [CrossRef]
- Griesser, J.; Hetényi, G.; Moser, M.; Demarne, F.; Jannin, V.; Bernkop-Schnürch, A. Hydrophobic ion pairing: Key to highly payloaded self-emulsifying peptide drug delivery systems. Int. J. Pharm. 2017, 520, 267–274. [Google Scholar] [CrossRef]
- McCartney, F.; Rosa, M.; Brayden, D.J. Evaluation of Sucrose Laurate as an Intestinal Permeation Enhancer for Macromolecules: Ex Vivo and In Vivo Studies. Pharmaceutics 2019, 11, 565. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.; Ibsen, K.; Brown, T.; Chen, R.; Agatemor, C.; Mitragotri, S. Ionic liquids for oral insulin delivery. Proc. Natl. Acad. Sci. USA 2018, 115, 7296–7301. [Google Scholar] [CrossRef] [Green Version]
- Brayden, D.J.; Maher, S. Transient Permeation Enhancer® (TPE®) technology for oral delivery of octreotide: A technological evaluation. Expert Opin. Drug Deliv. 2021, 18, 1501–1512. [Google Scholar] [CrossRef]
- Biermasz, N.R. New medical therapies on the horizon: Oral octreotide. Pituitary 2017, 20, 149–153. [Google Scholar] [CrossRef] [Green Version]
- Tuvia, S.; Pelled, D.; Marom, K.; Salama, P.; Levin-Arama, M.; Karmeli, I.; Idelson, G.H.; Landau, I.; Mamluk, R. A Novel Suspension Formulation Enhances Intestinal Absorption of Macromolecules Via Transient and Reversible Transport Mechanisms. Pharm. Res. 2014, 31, 2010–2021. [Google Scholar] [CrossRef]
- Salama, P.; Mamluk., R.; Marom., K.; Weinstein., I.; Tzabari., M. Pharmaceuical Compositions and Related Methods of Delivery. US20100105627A1, 2009. [Google Scholar]
- Leone-Bay, A.; Santiago, N.; Achan, D.; Chaudhary, K.; DeMorin, F.; Falzarano, L.; Haas, S.; Kalbag, S.; Kaplan, D.; Leipold, H.; et al. N-Acylated.alpha.-Amino Acids as Novel Oral Delivery Agents for Proteins. J. Med. Chem. 1995, 38, 4263–4269. [Google Scholar] [CrossRef] [PubMed]
- Malkov, D.; Angelo, R.; Wang, H.-Z.; Flanders, E.; Tang, H.; Gomez-Orellana, I. Oral Delivery of Insulin with the eligen(®) Technology: Mechanistic Studies. Curr. Drug Deliv. 2005, 2, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.L.; McEntee, N.; Holland, J.; Patel, A. Development and approval of rybelsus (oral semaglutide): Ushering in a new era in peptide delivery. Drug Deliv. Transl. Res. 2021, 12, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-H.; Lee, S.; Youn, Y.S.; Na, D.H.; Chae, S.Y.; Byun, A.Y.; Lee, K.C. Synthesis, Characterization, and Pharmacokinetic Studies of PEGylated Glucagon-like Peptide-1. Bioconjugate Chem. 2005, 16, 377–382. [Google Scholar] [CrossRef]
- Son, S.; Lim, S.M.; Chae, S.Y.; Kim, K.; Park, E.J.; Lee, K.C.; Na, D.H. Mono-lithocholated exendin-4-loaded glycol chitosan nanoparticles with prolonged antidiabetic effects. Int. J. Pharm. 2015, 495, 81–86. [Google Scholar] [CrossRef]
- Lee, W.; Park, E.J.; Kwak, S.; Lee, K.C.; Na, D.H.; Bae, J.-S. Trimeric PEG-Conjugated Exendin-4 for the Treatment of Sepsis. Biomacromolecules 2016, 17, 1160–1169. [Google Scholar] [CrossRef]
- Husain, M.; Birkenfeld, A.L.; Donsmark, M.; Dungan, K.; Eliaschewitz, F.G.; Franco, D.R.; Jeppesen, O.K.; Lingvay, I.; Mosenzon, O.; Pedersen, S.D.; et al. Oral Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2019, 381, 841–851. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, L.B.; Lau, J. The Discovery and Development of Liraglutide and Semaglutide. Front. Endocrinol. 2019, 10, 155. [Google Scholar] [CrossRef] [Green Version]
- Buckley, S.T.; Baekdal, T.A.; Vegge, A.; Maarbjerg, S.J.; Pyke, C.; Ahnfelt-Ronne, J.; Madsen, K.G.; Scheele, S.G.; Alanentalo, T.; Kirk, R.K.; et al. Transcellular stomach absorption of aderivatized glucagon-like peptide-1 receptor agonist. Sci. Transl. Med. 2018, 10, eaar7047. [Google Scholar] [CrossRef]
- Kitao, K.; Nishimura, K. Adsuvant for Promoting Absorption of Pharmacologically Active Substances through the Rectum. US4338306A, 6 July 1982. [Google Scholar]
- Nishimura, K.; Nozaki, Y.; Yoshimi, A.; Nakamura, S.; Kitagawa, M.; Kakeya, N.; Kitao, K. Studies on the promoting effects of carboxylic acid derivatives on the rectal absorption of.BETA.-lactam antibiotics in rats. Chem. Pharm. Bull. 1985, 33, 282–291. [Google Scholar] [CrossRef]
- Lindmark, T.; Söderholm, J.D.; Olaison, G.; Alván, G.; Ocklind, G.; Artursson, P. Mechanism of Absorption Enhancement in Humans After Rectal Administration of Ampicillin in Suppositories Containing Sodium Caprate. Pharm. Res. 1997, 14, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Maher, S.; Geoghegan, C.; Brayden, D.J. Intestinal permeation enhancers to improve oral bioavailability of macromolecules: Reasons for low efficacy in humans. Expert Opin. Drug Deliv. 2020, 18, 273–300. [Google Scholar] [CrossRef] [PubMed]
- Hossain, S.; Berg, S.; Bergström, C.A.S.; Larsson, P. Aggregation Behavior of Medium Chain Fatty Acids Studied by Coarse-Grained Molecular Dynamics Simulation. AAPS PharmSciTech 2019, 20, 61. [Google Scholar] [CrossRef] [PubMed]
- Food Additive Status List. Available online: https://www.fda.gov/food/food-additives-petitions/food-additive-status-list#abb (accessed on 30 September 2022).
- Mortensen, A.; Aguilar, F.; Crebelli, R.; Di Domenico, A.; Dusemund, B.; Frutos, M.J.; Galtier, P.; Gott, D.; Gundert-Remy, U.; Leblanc, J.C.; et al. Re-evaluation of fatty acids (E 570) as a food additive. EFSA J. 2017, 15, e04785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, W.; Dong, X.; Shi, L.; Chu, X. Discrimination of Milk from Different Animal Species by a Foodomics Approach Based on High-Resolution Mass Spectrometry. J. Agric. Food Chem. 2020, 68, 6638–6645. [Google Scholar] [CrossRef]
- Sawada, T.; Ogawa, T.; Tomita, M.; Hayashi, M.; Awazu, S. Role of Paracellular Pathway in Nonelectrolyte Permeation Across Rat Colon Epithelium Enhanced by Sodium Caprate and Sodium Caprylate. Pharm. Res. 1991, 08, 1365–1371. [Google Scholar] [CrossRef]
- Mishima, M.; Wakita, Y.; Nakano, M. Studies on the promoting effects of medium chain fatty acid salts on the nasal absorption of insulin in rats. J. Pharmacobio-Dynamics 1987, 10, 624–631. [Google Scholar] [CrossRef] [Green Version]
- Hossain, S.; Joyce, P.; Parrow, A.; Jõemetsa, S.; Höök, F.; Larsson, P.; Bergström, C.A.S. Influence of Bile Composition on Membrane Incorporation of Transient Permeability Enhancers. Mol. Pharm. 2020, 17, 4226–4240. [Google Scholar] [CrossRef]
- Maher, S.; Mrsny, R.J.; Brayden, D.J. Intestinal permeation enhancers for oral peptide delivery. Adv. Drug Deliv. Rev. 2016, 106, 277–319. [Google Scholar] [CrossRef]
- Lindmark, T.; Nikkilä, T.; Artursson, P. Mechanisms of absorption enhancement by medium chain fatty acids in intestinal epithelial Caco-2 cell monolayers. J. Pharmacol. Exp. Ther. 1995, 275, 958–964. [Google Scholar]
- Kneiszl, R.; Hossain, S.; Larsson, P. In Silico-Based Experiments on Mechanistic Interactions between Several Intestinal Permeation Enhancers with a Lipid Bilayer Model. Mol. Pharm. 2021, 19, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Ates, M.; Kaynak, M.S.; Sahin, S. Effect of permeability enhancers on paracellular permeability of acyclovir. J. Pharm. Pharmacol. 2016, 68, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, S.; Im, H.T.; Sohn, Y.T.; Kim, K.S.; Kim, Y.-I.; Yong, C.S.; Kim, J.O.; Choi, H.-G.; Woo, J.S. Zanamivir Oral Delivery: Enhanced Plasma and Lung Bioavailability in Rats. Biomol. Ther. 2013, 21, 161–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, Y.-L.; Huang, J.-D. Effects of sodium deoxycholate and sodium caprate on the transport of epirubicin in human intestinal epithelial Caco-2 cell layers and everted gut sacs of rats. Biochem. Pharmacol. 2000, 59, 665–672. [Google Scholar] [CrossRef]
- Dos Santos, I.; Fawaz, F.; Lagueny, A.M.; Bonini, F. Improvement of norfloxacin oral bioavailability by EDTA and sodium caprate. Int. J. Pharm. 2003, 260, 1–4. [Google Scholar] [CrossRef]
- Raoof, A.A.; Chiu, P.; Ramtoola, Z.; Cumming, I.K.; Teng, C.; Weinbach, S.P.; Hardee, G.E.; Levin, A.A.; Geary, R. Oral bioavailability and multiple dose tolerability of an antisense oligonucleotide tablet formulated with sodium caprate. J. Pharm. Sci. 2004, 93, 1431–1439. [Google Scholar] [CrossRef]
- Raoof, A.A.; Ramtoola, Z.; McKenna, B.; Yu, R.Z.; Hardee, G.; Geary, R.S. Effect of sodium caprate on the intestinal absorption of two modified antisense oligonucleotides in pigs. Eur. J. Pharm. Sci. 2002, 17, 131–138. [Google Scholar] [CrossRef]
- Chao, A.C.; Nguyen, J.V.; Broughall, M.; Griffin, A.; Fix, J.A.; Daddona, P.E. In vitro and in vivo evaluation of effects of sodium caprate on enteral peptide absorption and on mucosal morphology. Int. J. Pharm. 1999, 191, 15–24. [Google Scholar] [CrossRef]
- Gleeson, J.; Frias, J.; Ryan, S.M.; Brayden, D.J. Sodium caprate enables the blood pressure-lowering effect of Ile-Pro-Pro and Leu-Lys-Pro in spontaneously hypertensive rats by indirectly overcoming PepT1 inhibition. Eur. J. Pharm. Biopharm. 2018, 128, 179–187. [Google Scholar] [CrossRef] [Green Version]
- Kamm, W.; Jonczyk, A.; Jung, T.; Luckenbach, G.; Raddatz, P.; Kissel, T. Evaluation of absorption enhancement for a potent cyclopeptidic ανβ3-antagonist in a human intestinal cell line (Caco-2). Eur. J. Pharm. Sci. 2000, 10, 205–214. [Google Scholar] [CrossRef]
- Lindmark, T.; Schipper, N.; Lazorová, L.; De Boer, A.G.; Artursson, P. Absorption Enhancement in Intestinal Epithelial Caco-2 Monolayers by Sodium Caprate: Assessment of Molecular Weight Dependence and Demonstration of Transport Routes. J. Drug Target. 1998, 5, 215–223. [Google Scholar] [CrossRef]
- Kim, I.-W.; Yoo, H.-J.; Song, I.-S.; Chung, Y.-B.; Moon, D.-C.; Chung, S.-J.; Shim, C.-K. Effect of excipients on the stability and transport of recombinant human epidermal growth factor (rhEGF) across Caco-2 cell monolayers. Arch. Pharmacal Res. 2003, 26, 330–337. [Google Scholar] [CrossRef]
- Yamamoto, A.; Okagawa, T.; Kotani, A.; Uchiyama, T.; Shimura, T.; Tabata, S.; Kondo, S.; Muranishi, S. Effects of Different Absorption Enhancers on the Permeation of Ebiratide, an ACTH Analogue, across Intestinal Membranes. J. Pharm. Pharmacol. 1997, 49, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, D.; Sjöblom, M.; Hedeland, M.; Lennernäs, H. The In Vivo Effect of Transcellular Permeation Enhancers on the Intestinal Permeability of Two Peptide Drugs Enalaprilat and Hexarelin. Pharmaceutics 2020, 12, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchiyama, T.; Sugiyama, T.; Quan, Y.-S.; Kotani, A.; Okada, N.; Fujita, T.; Muranishi, S.; Yamamoto, A. Enhanced Permeability of Insulin across the Rat Intestinal Membrane by Various Absorption Enhancers: Their Intestinal Mucosal Toxicity and Absorption-enhancing Mechanism of n-Lauryl-β-D-maltopyranoside. J. Pharm. Pharmacol. 1999, 51, 1241–1250. [Google Scholar] [CrossRef]
- Wallon, C.; Braaf, Y.; Wolving, M.; Olaison, G.; Söderholm, J.D. Endoscopic biopsies in Ussing chambers evaluated for studies of macromolecular permeability in the human colon. Scand. J. Gastroenterol. 2005, 40, 586–595. [Google Scholar] [CrossRef]
- Morishita, M.; Morishita, I.; Takayama, K.; Machida, Y.; Nagai, T. Site-Dependent Effect of Aprotinin, Sodium Caprate, Na2EDTA and Sodium Glycocholate on Intestinal Absorption of Insulin. Biol. Pharm. Bull. 1993, 16, 68–72. [Google Scholar] [CrossRef] [Green Version]
- Imai, T.; Sakai, M.; Ohtake, H.; Azuma, H.; Otagiri, M. Absorption-enhancing effect of glycyrrhizin induced in the presence of capric acid. Int. J. Pharm. 2005, 294, 11–21. [Google Scholar] [CrossRef]
- Muranushi, N.; Mack, E.; Kim, S.W. The Effects of Fatty Acids and Their Derivatives on the Intestinal Absorption of insulin in Rat. Drug Dev. Ind. Pharm. 1993, 19, 929–941. [Google Scholar] [CrossRef]
- Burcham, D.L.; Aungst, B.A.; Hussain, M.; Gorko, M.A.; Quon, C.Y.; Huang, S. The Effect of Absorption Enhancers on the Oral Absorption of the GP IIB/IIIA Receptor Antagonist, DMP 728, in Rats and Dogs. Pharm. Res. 1995, 12, 2065–2070. [Google Scholar] [CrossRef]
- Radwan, M.A.; Aboul-Enein, H.Y. The effect of oral absorption enhancers on the in vivo performance of insulin-loaded poly(ethylcyanoacrylate) nanospheres in diabetic rats. J. Microencapsul. 2002, 19, 225–235. [Google Scholar] [CrossRef]
- Watanabe, Y.; Mizufune, Y.; Kubomura, A.; Kiriyama, M.; Utoguchi, N.; Matsumoto, M. Studies of Drug Delivery Systems for a Therapeutic Agent Used in Osteoporosis. I. Pharmacodynamics (Hypocalcemic Effect) of Elcatonin in Rabbits Following Rectal Administration of Hollow-Type Suppositories Containing Elcatonin. Biol. Pharm. Bull. 1998, 21, 1187–1190. [Google Scholar] [CrossRef] [Green Version]
- Anderberg, E.K.; Lindmark, T.; Artursson, P. Sodium Caprate Elicits Dilatations in Human Intestinal Tight Junctions and Enhances Drug Absorption by the Paracellular Route. Pharm. Res. 1993, 10, 857–864. [Google Scholar] [CrossRef]
- Tomita, M.; Hayashi, M.; Awazu, S. Comparison of Absorption-Enhancing Effect between Sodium Caprate and Disodium Ethylenediaminetetraacetate in Caco-2 Cells. Biol. Pharm. Bull. 1994, 17, 753–755. [Google Scholar] [CrossRef] [Green Version]
- Soderholm, J.D.; Oman, H.; Blomquist, L.; Veen, J.; Lindmark, T.; Olaison, G. Reversible increase in tight junction permeability to macromolecules in rat ileal mucosa in vitro by sodium caprate, a constituent of milk fat. Dig. Dis. Sci. 1998, 43, 1547–1552. [Google Scholar] [CrossRef] [PubMed]
- Coyne, C.B.; Ribeiro, C.M.P.; Boucher, R.C.; Johnson, L.G. Acute Mechanism of Medium Chain Fatty Acid-Induced Enhancement of Airway Epithelial Permeability. J. Pharmacol. Exp. Ther. 2003, 305, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Sugibayashi, K.; Onuki, Y.; Takayama, K. Displacement of tight junction proteins from detergent-resistant membrane domains by treatment with sodium caprate. Eur. J. Pharm. Sci. 2009, 36, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Del Vecchio, G.; Tscheik, C.; Tenz, K.; Helms, H.C.; Winkler, L.; Blasig, R.; Blasig, I.E. Sodium Caprate Transiently Opens Claudin-5-Containing Barriers at Tight Junctions of Epithelial and Endothelial Cells. Mol. Pharm. 2012, 9, 2523–2533. [Google Scholar] [CrossRef]
- Krug, S.M.; Amasheh, M.; Dittmann, I.; Christoffel, I.; Fromm, M.; Amasheh, S. Sodium caprate as an enhancer of macromolecule permeation across tricellular tight junctions of intestinal cells. Biomaterials 2012, 34, 275–282. [Google Scholar] [CrossRef]
- Brayden, D.J.; Maher, S.; Bahar, B.; Walsh, E. Sodium caprate-induced increases in intestinal permeability and epithelial damage are prevented by misoprostol. Eur. J. Pharm. Biopharm. 2015, 94, 194–206. [Google Scholar] [CrossRef]
- Twarog, C.; Liu, K.; O’Brien, P.J.; Dawson, K.A.; Fattal, E.; Illel, B.; Brayden, D.J. A head-to-head Caco-2 assay comparison of the mechanisms of action of the intestinal permeation enhancers: SNAC and sodium caprate (C10). Eur. J. Pharm. Biopharm. 2020, 152, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.B.; Bailey, R.T., Jr. Misoprostol: A Prostaglandin E, Analog with Antisecretory and Cytoprotective Properties. DICP 1989, 23, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Maher, S.; Leonard, T.W.; Jacobsen, J.; Brayden, D.J. Safety and efficacy of sodium caprate in promoting oral drug absorption: From in vitro to the clinic. Adv. Drug Deliv. Rev. 2009, 61, 1427–1449. [Google Scholar] [CrossRef] [PubMed]
- Leonard, T.W.; Lynch, J.; McKenna, M.J.; Brayden, D. Promoting absorption of drugs in humans using medium-chain fatty acid-based solid dosage forms: GIPET™. Expert Opin. Drug Deliv. 2006, 3, 685–692. [Google Scholar] [CrossRef]
- Leonard, T.W.; Coughlan, D.C.; Cullen, A. Pharmaceutical Compositions of Selective Factor Xa Inhibitors for Oral Administration. WO2011120033A1, 29 September 2011. [Google Scholar]
- Walsh, E.G.; Adamczyk, B.E.; Chalasani, K.B.; Maher, S.; O’Toole, E.B.; Fox, J.S.; Leonard, T.W.; Brayden, D.J. Oral delivery of macromolecules: Rationale underpinning Gastrointestinal Permeation Enhancement Technology (GIPET®). Ther. Deliv. 2011, 2, 1595–1610. [Google Scholar] [CrossRef]
- Amory, J.K.; Leonard, T.W.; Page, S.T.; O’Toole, E.; McKenna, M.J.; Bremner, W.J. Oral administration of the GnRH antagonist acyline, in a GIPET®-enhanced tablet form, acutely suppresses serum testosterone in normal men: Single-dose pharmacokinetics and pharmacodynamics. Cancer Chemother. Pharmacol. 2009, 64, 641–645. [Google Scholar] [CrossRef] [Green Version]
- Twarog, C.; Fattah, S.; Heade, J.; Maher, S.; Fattal, E.; Brayden, D.J. Intestinal Permeation Enhancers for Oral Delivery of Macromolecules: A Comparison between Salcaprozate Sodium (SNAC) and Sodium Caprate (C10). Pharmaceutics 2019, 11, 78. [Google Scholar] [CrossRef] [Green Version]
- Ramaswamy, S.G.; Nayak, V.G.; Jha, S.K.; Hegde, V.; Waichale, V.S.; Melarkode, R.; Chirmule, N.; Rao, A.U.; Sengupta, N. Development and validation of an electrochemiluminescent ELISA for quantitation of oral insulin tregopil in diabetes mellitus serum. Bioanalysis 2017, 9, 975–986. [Google Scholar] [CrossRef]
- Hazra, P.; Adhikary, L.; Dave, N.; Khedkar, A.; Manjunath, H.S.; Anantharaman, R.; Iyer, H. Development of a process to manufacture PEGylated orally bioavailable insulin. Biotechnol. Prog. 2010, 26, 1695–1704. [Google Scholar] [CrossRef]
- Berg, S.; Krause, J.; Björkbom, A.; Walter, K.; Harun, S.; Granfeldt, A.; Janzén, D.; Nunes, S.F.; Antonsson, M.; Van Zuydam, N.; et al. In Vitro and In Vivo Evaluation of 3D Printed Capsules with Pressure Triggered Release Mechanism for Oral Peptide Delivery. J. Pharm. Sci. 2020, 110, 228–238. [Google Scholar] [CrossRef]
- Jørgensen, J.R.; Jepsen, M.L.; Nielsen, L.H.; Dufva, M.; Nielsen, H.M.; Rades, T.; Boisen, A.; Müllertz, A. Microcontainers for oral insulin delivery—In vitro studies of permeation enhancement. Eur. J. Pharm. Biopharm. 2019, 143, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Steiner., S.; Rosen., R. Delivery Systems for Pharmacological Agents Encapsulated with Proteinoids. US4925673A, 15 May 1990.
- Leone-Bay, A.; Ho, K.-K.; Agarwal, R.; Baughman, R.A.; Chaudhary, K.; DeMorin, F.; Genoble, L.; McInnes, C.; Lercara, C.; Milstein, S.; et al. 4-[4-[(2-Hydroxybenzoyl)amino]phenyl]butyric Acid as a Novel Oral Delivery Agent for Recombinant Human Growth Hormone. J. Med. Chem. 1996, 39, 2571–2578. [Google Scholar] [CrossRef]
- Milstein, S.J.; Leipold, H.; Sarubbi, D.; Leone-Bay, A.; Mlynek, G.M.; Robinson, J.R.; Kasimova, M.; Freire, E. Partially unfolded proteins efficiently penetrate cell membranes—implications for oral drug delivery. J. Control. Release 1998, 53, 259–267. [Google Scholar] [CrossRef]
- Arbit, E.; Goldberg, M.; Gomez-Orellana, I.; Majuru, S. Oral heparin: Status review. Thromb. J. 2006, 4, 6. [Google Scholar] [CrossRef] [Green Version]
- Kidron, M.; Dinh, S.; Menachem, Y.; Abbas, R.; Variano, B.; Goldberg, M.; Arbit, E.; Bar-On, H. A novel per-oral insulin formulation: Proof of concept study in non-diabetic subjects. Diabet. Med. 2004, 21, 354–357. [Google Scholar] [CrossRef] [PubMed]
- Mousa, S.A.; Zhang, F.; Aljada, A.; Chaturvedi, S.; Takieddin, M.; Zhang, H.; Chi, L.; Castelli, M.C.; Friedman, K.; Goldberg, M.M.; et al. Pharmacokinetics and Pharmacodynamics of Oral Heparin Solid Dosage Form in Healthy Human Subjects. J. Clin. Pharmacol. 2007, 47, 1508–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berkowitz, S.D.; Marder, V.J.; Kosutic, G.; Baughman, R.A. Oral heparin administration with a novel drug delivery agent (SNAC) in healthy volunteers and patients undergoing elective total hip arthroplasty. J. Thromb. Haemost. 2003, 1, 1914–1919. [Google Scholar] [CrossRef] [Green Version]
- Bittner, B.; McIntyre, C.; Tian, H.; Tang, K.; Shah, N.; Phuapradit, W.; Ahmed, H.; Chokshi, H.; Infeld, M.; Fotaki, N.; et al. Phase I clinical study to select a novel oral formulation for ibandronate containing the excipient sodium N-[8-(2-hydroxybenzoyl) amino] caprylate (SNAC). Die Pharm. 2012, 67, 233–241. [Google Scholar]
- Steinert, R.E.; Poller, B.; Castelli, M.C.; Drewe, J.; Beglinger, C. Oral administration of glucagon-like peptide 1 or peptide YY 3-36 affects food intake in healthy male subjects. Am. J. Clin. Nutr. 2010, 92, 810–817. [Google Scholar] [CrossRef] [Green Version]
- Karsdal, M.; Byrjalsen, I.; Alexandersen, P.; Bihlet, A.; Andersen, J.; Riis, B.; Bay-Jensen, A.; Christiansen, C. Treatment of symptomatic knee osteoarthritis with oral salmon calcitonin: Results from two phase 3 trials. Osteoarthr. Cartil. 2015, 23, 532–543. [Google Scholar] [CrossRef] [Green Version]
- Kapitza, C.; Zijlstra, E.; Heinemann, L.; Castelli, M.C.; Riley, G.; Heise, T. Oral Insulin: A Comparison With Subcutaneous Regular Human Insulin in Patients With Type 2 Diabetes. Diabetes Care 2010, 33, 1288–1290. [Google Scholar] [CrossRef] [Green Version]
- Fattah, S.; Ismaiel, M.; Murphy, B.; Rulikowska, A.; Frias, J.M.; Winter, D.C.; Brayden, D.J. Salcaprozate sodium (SNAC) enhances permeability of octreotide across isolated rat and human intestinal epithelial mucosae in Ussing chambers. Eur. J. Pharm. Sci. 2020, 154, 105509. [Google Scholar] [CrossRef]
- Salcaprozate Sodium. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Salcaprozate-sodium (accessed on 5 October 2022).
- Malkov, D.; Wang, H.; Dinh, S.; Gomez-Orellana, I. Pathway of oral absorption of heparin with sodium N-[8-(2-hydroxybenzoyl)amino] caprylate. Pharm. Res. 2002, 19, 1180–1184. [Google Scholar] [CrossRef]
- Alani, A.W.G.; Robinson, J.R. Mechanistic Understanding of Oral Drug Absorption Enhancement of Cromolyn Sodium by an Amino Acid Derivative. Pharm. Res. 2007, 25, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lu, Y.; Shi, S.; Zhang, Q.; Cao, X.; Sun, L.; An, D.; Zhang, X.; Kong, X.; Liu, J. Design and Development of a New Glucagon-Like Peptide-1 Receptor Agonist to Obtain High Oral Bioavailability. Pharm. Res. 2022, 39, 1891–1906. [Google Scholar] [CrossRef] [PubMed]
- Hess, S.; Rotshild, V.; Hoffman, A. Investigation of the enhancing mechanism of sodium N-[8-(2-hydroxybenzoyl)amino]caprylate effect on the intestinal permeability of polar molecules utilizing a voltage clamp method. Eur. J. Pharm. Sci. 2005, 25, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, W.; Shen, L.; Lang, L.; Huang, X.; Sheng, H.; Ning, G.; Wang, W. Safety, Pharmacokinetics, and Pharmacodynamics of Oral Insulin Administration in Healthy Subjects: A Randomized, Double-Blind, Phase 1 Trial. Clin. Pharmacol. Drug Dev. 2022, 11, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Binkley, N.; Bolognese, M.; Sidorowicz-Bialynicka, A.; Vally, T.; Trout, R.; Miller, C.; Buben, C.E.; Gilligan, J.P.; Krause, D.S. A phase 3 trial of the efficacy and safety of oral recombinant calcitonin: The oral calcitonin in postmenopausal osteoporosis (ORACAL) trial. J. Bone Miner. Res. 2012, 27, 1821–1829. [Google Scholar] [CrossRef]
- Bækdal, T.A.; Donsmark, M.; Hartoft-Nielsen, M.; Søndergaard, F.L.; Connor, A. Relationship Between Oral Semaglutide Tablet Erosion and Pharmacokinetics: A Pharmacoscintigraphic Study. Clin. Pharmacol. Drug Dev. 2021, 10, 453–462. [Google Scholar] [CrossRef]
- Aroda, V.R.; Rosenstock, J.; Terauchi, Y.; Altuntas, Y.; Lalic, N.M.; Villegas, E.C.M.; Jeppesen, O.K.; Christiansen, E.; Hertz, C.L.; Haluzík, M.; et al. PIONEER 1: Randomized Clinical Trial of the Efficacy and Safety of Oral Semaglutide Monotherapy in Comparison With Placebo in Patients With Type 2 Diabetes. Diabetes Care 2019, 42, 1724–1732. [Google Scholar] [CrossRef]
- Rodbard, H.W.; Rosenstock, J.; Canani, L.H.; Deerochanawong, C.; Gumprecht, J.; Lindberg, S.; Lingvay, I.; Søndergaard, A.L.; Treppendahl, M.B.; Montanya, E.; et al. Oral Semaglutide Versus Empagliflozin in Patients With Type 2 Diabetes Uncontrolled on Metformin: The PIONEER 2 Trial. Diabetes Care 2019, 42, 2272–2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granhall, C.; Søndergaard, F.L.; Thomsen, M.; Anderson, T.W. Pharmacokinetics, Safety and Tolerability of Oral Semaglutide in Subjects with Renal Impairment. Clin. Pharmacokinet. 2018, 57, 1571–1580. [Google Scholar] [CrossRef] [PubMed]
- Baekdal, T.A.; Thomsen, M.; Kupčová, V.; Hansen, C.W.; Msc, T.W.A. Pharmacokinetics, Safety, and Tolerability of Oral Semaglutide in Subjects With Hepatic Impairment. J. Clin. Pharmacol. 2018, 58, 1314–1323. [Google Scholar] [CrossRef]
- Overgaard, R.V.; Navarria, A.; Ingwersen, S.H.; Bækdal, T.A.; Kildemoes, R.J. Clinical Pharmacokinetics of Oral Semaglutide: Analyses of Data from Clinical Pharmacology Trials. Clin. Pharmacokinet. 2021, 60, 1335–1348. [Google Scholar] [CrossRef]
- Jordy, A.B.; Albayaty, M.; Breitschaft, A.; Anderson, T.W.; Christiansen, E.; Houshmand-Øregaard, A.; Manigandan, E.; Bækdal, T.A. Effect of Oral Semaglutide on the Pharmacokinetics of Levonorgestrel and Ethinylestradiol in Healthy Postmenopausal Women and Furosemide and Rosuvastatin in Healthy Subjects. Clin. Pharmacokinet. 2021, 60, 1171–1185. [Google Scholar] [CrossRef]
- Bækdal, T.A.; Borregaard, J.; Hansen, C.W.; Thomsen, M.; Anderson, T.W. Effect of Oral Semaglutide on the Pharmacokinetics of Lisinopril, Warfarin, Digoxin, and Metformin in Healthy Subjects. Clin. Pharmacokinet. 2019, 58, 1193–1203. [Google Scholar] [CrossRef] [Green Version]
- Hauge, C.; Breitschaft, A.; Hartoft-Nielsen, M.-L.; Jensen, S.; Bækdal, T.A. Effect of oral semaglutide on the pharmacokinetics of thyroxine after dosing of levothyroxine and the influence of co-administered tablets on the pharmacokinetics of oral semaglutide in healthy subjects: An open-label, one-sequence crossover, single-center, multiple-dose, two-part trial. Expert Opin. Drug Metab. Toxicol. 2021, 17, 1139–1148. [Google Scholar] [CrossRef]
- Riley, M.G.I.; Castelli, M.C.; Paehler, E.A. Subchronic Oral Toxicity of Salcaprozate Sodium (SNAC) in Sprague-Dawley and Wistar Rats. Int. J. Toxicol. 2009, 28, 278–293. [Google Scholar] [CrossRef] [PubMed]
- Riley, M.G.I.; York, R.G. Peri- and Postnatal Developmental Toxicity of Salcaprozate Sodium (SNAC) in Sprague-Dawley Rats. Int. J. Toxicol. 2009, 28, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Tuvia, S.; Atsmon, J.; Teichman, S.L.; Katz, S.; Salama, P.; Pelled, D.; Landau, I.; Karmeli, I.; Bidlingmaier, M.; Strasburger, C.J.; et al. Oral Octreotide Absorption in Human Subjects: Comparable Pharmacokinetics to Parenteral Octreotide and Effective Growth Hormone Suppression. J. Clin. Endocrinol. Metab. 2012, 97, 2362–2369. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.; Wong, J.; Gogoi, R.; Brown, T.; Mitragotri, S. Intestinal micropatches for oral insulin delivery. J. Drug Target. 2017, 25, 608–615. [Google Scholar] [CrossRef]
- Eiamtrakarn, S.; Itoh, Y.; Kishimoto, J.; Yoshikawa, Y.; Shibata, N.; Murakami, M.; Takada, K. Gastrointestinal mucoadhesive patch system (GI-MAPS) for oral administration of G-CSF, a model protein. Biomaterials 2002, 23, 145–152. [Google Scholar] [CrossRef]
- Jørgensen, J.R.; Yu, F.; Venkatasubramanian, R.; Nielsen, L.H.; Nielsen, H.M.; Boisen, A.; Rades, T.; Müllertz, A. In Vitro, Ex Vivo and In Vivo Evaluation of Microcontainers for Oral Delivery of Insulin. Pharmaceutics 2020, 12, 48. [Google Scholar] [CrossRef] [Green Version]
- Mazzoni, C.; Jacobsen, R.D.; Mortensen, J.; Jørgensen, J.R.; Vaut, L.; Jacobsen, J.; Gundlach, C.; Müllertz, A.; Nielsen, L.H.; Boisen, A. Polymeric Lids for Microcontainers for Oral Protein Delivery. Macromol. Biosci. 2019, 19, e1900004. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Yadav, V.; Smart, A.L.; Tajiri, S.; Basit, A.W. Toward Oral Delivery of Biopharmaceuticals: An Assessment of the Gastrointestinal Stability of 17 Peptide Drugs. Mol. Pharm. 2015, 12, 966–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almansour, K.; Taverner, A.; Turner, J.R.; Eggleston, I.M.; Mrsny, R.J. An intestinal paracellular pathway biased toward positively-charged macromolecules. J. Control. Release 2018, 288, 111–125. [Google Scholar] [CrossRef]
- Trier, S.; Linderoth, L.; Bjerregaard, S.; Andresen, T.L.; Rahbek, U.L. Acylation of Glucagon-Like Peptide-2: Interaction with Lipid Membranes and In Vitro Intestinal Permeability. PLoS ONE 2014, 9, e109939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofsäss, M.A.; Dressman, J.B. The Discriminatory Power of the BCS-Based Biowaiver: A Retrospective With Focus on Essential Medicines. J. Pharm. Sci. 2019, 108, 2824–2837. [Google Scholar] [CrossRef]
- Metry, M.; Polli, J.E. Evaluation of Excipient Risk in BCS Class I and III Biowaivers. AAPS J. 2022, 24, 20. [Google Scholar] [CrossRef] [PubMed]
- Benson, K.; Cramer, S.; Galla, H.-J. Impedance-based cell monitoring: Barrier properties and beyond. Fluids Barriers CNS 2013, 10, 5. [Google Scholar] [CrossRef]
- Capital Markets Day 2017. Available online: https://www.novonordisk.com/content/dam/nncorp/global/en/investors/irmaterial/cmd/2017/00_CMD%20Presentation%20combined.pdf (accessed on 5 October 2022).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Permeation Enhancer | Proposed Mechanism |
|---|---|
| MCFAs (C8, C10) | Paracellular
|
| Eligen™ technology-based PE (SNAC, 4-CNAB, 5-CNAC) | Transcellular
|
| EDTA | Paracellular
|
| Bile salt | Transcellular
|
| Acyl-carnitine | Transcellular
|
| Alkyl-maltoside | Combined transcellular, paracellular action
|
| Sodium docusate | Transcellular
|
| Sucrose laurate | Combined transcellular, paracellular action
|
| Choline geranate (CAGE, Ionic liquid) | Paracellular
|
| Peptide (MW) | Experimental Model | Dosing | Enhancement Ratio | Reference |
|---|---|---|---|---|
| In vitro assay | ||||
| D-decapeptide (~1.2 kDa) | Caco-2 cell monolayer | 20–25 mM | ~7 | [82] |
| Ile-Pro-Pro (325 Da) Leu-Lys-Pro (389 Da) | Caco-2 cell monolayer | 5 mM | ~2.5 (Ile-Pro-Pro) ~2 (Leu-Lys-Pro) | [83] |
| Cyclopeptide, (EMD121974) (589 Da) | Caco-2 cell monolayer | 10 mM | 10.6 | [84] |
| Vasopressin (1.2 kDa) | Caco-2 cell monolayer | 13 mM | 10 | [85] |
| Recombinant human epidermal growth factor (6 kDa) | Caco-2 cell monolayer | 1% (50 mM) | 10.6 | [86] |
| Ex vivo/in vivo assay | ||||
| D-decapeptide (~1.2 kDa) | Rat, ileal instillation | 0.5 mmol/kg | ~5 | [82] |
| Ebiratide (996 Da) | Rat, Ussing chamber (jejunum and colon) | 20 mM | 1.50 (jejunum) 3.84 (colon) | [87] |
| Enalaprilat (349 Da) | Rat, single-pass intestinal perfusion | 10 mg/mL | 9 | [88] |
| Hexarelin (887 Da) | Rat, single-pass intestinal perfusion | 5~20 mg/mL | Not enhanced | [88] |
| Insulin (5.8 kDa) | Rat, Ussing chamber (jejunum and colon) | 20 mM | 0.97 (jejunum) 2.50 (Colon) | [89] |
| Horseradish peroxidase (45 kDa) | Human, Ussing chamber (Colon) | 10 mM | ~2 | [90] |
| Insulin (5.8 kDa) | Rat, loop administration | 1% (50 mM) | Duodenum: not enhanced * Jejunum: enhanced * Ileum: 1.67 * Colon: 9 * | [91] |
| Salmon calcitonin (3.4 kDa) | Rat, colonal instillation | 0.1% (5 mM) | Enhanced * | [92] |
| Insulin (5.8 kDa) | Rat, rectal infusion | 50 mM | 24.31 * | [93] |
| DMP728 (657 Da) | Rat and dog, oral administration | Rat (8 mg/kg) Dog (2 mg/kg) | 2.70 (Rat) 1.36 (Dog) | [94] |
| Insulin (5.8 kDa) | Rat, oral administration | 0.5% (25 mM) | 3.79* | [95] |
| Elcatonin (3.4 kDa) | Rabbit, rectal suppository | 30 mg | 1.61* | [96] |
| Experimental Model | Proposed Mechanism of Action (Rationale) | Evidence | Reference |
|---|---|---|---|
| Caco-2 cell monolayer | TJ modulation | TEM
| [97] |
| Caco-2 cell monolayer | TJ modulation | TEM
| [74] |
| Caco-2 cell monolayer | TJ modulation (PLC activation and CaM-dependent contraction of actin filament) | Intracellular Ca2+ measurement (Fluorometric Ca2+ analyzer)
| [38] |
| Ex vivo Ussing chamber (Rat ileum) | TJ modulation | TEM
| [99] |
| Human airway epithelial cell | TJ modulation (Ca2+-independent mechanism and direct effect on the TJ protein) | Fluo-4 Ca2+ assay
| [100] |
| Membrane perturbation (Lipid raft disruption) TJ modulation (Displacement of specific TJ proteins) | Western-blot analysis
| [101] |
| TJ modulation (by reducing the membranous claudin-5 amount and the F-actin content) | Immunofluorescent Labeling and Confocal microscopy
| [102] |
| TJ modulation (by reversible removal of tricellulin from the tricellular TJ) | Two-path impedance spectroscopy
| [103] |
| Membrane perturbation | Quantitative real-time PCR and gene expression microarrays
| [104] |
| Caco-2 cell monolayer | Membrane perturbation (initial and fundamental mechanism) TJ modulation (by intracellular pathway arising from initial plasma membrane perturbation) | Immunofluorescence of TJ proteins
| [105] |
| Membrane perturbation (insertion of C10 into membrane and transmembrane perturbation) | CG-MD simulation
| [72] |
| Peptide (MW) | Model | Dosing | Enhancement Ratio | Reference |
|---|---|---|---|---|
| Insulin (5808 Da) | Caco-2 cell monolayer | 5 mM | ~10 | [54] |
| Semaglutide (4114 Da) | NCI-N87 cell monolayer | 80 mM | ~7 | [61] |
| Octreotide (1019 Da) | Ex vivo Ussing chamber of rat (colon, ileum, upper jejunum, duodenum, and stomach) and human (colon) | 20 mM 40 mM | Rat (20 mM): 1.4~3.4 Human: 1.5 (20 mM), 2.1 (40 mM) | [128] |
| SHR-2042 (GLP-1RA) (~4.5 kDa) | Rat, duodenal perfusion | 0.6 g/kg 1.2 g/kg | 7.8 (0.6 g/kg) 69 (1.2 g.kg) | [132] |
| Experimental Method and Model Drugs | Proposed Mechanism of Action | Evidence | Reference |
|---|---|---|---|
| Fluorescence microscopy, Heparin | The transcellular pathway that does not involve membrane permeabilization and does not appear to be endocytosis. | Fluorescence and confocal microscopy in the Caco-2 monolayer
| [130] |
| Voltage clamp method, 6-Carboxy-fluorescein (6-CF) | Transcellular pathway | Voltage clamp experiment
| [133] |
| Fluorescence microscopy, Insulin | Increased lipophilicity by non-covalent binding and the resulting transcellular pathway | Fluorescence and confocal microscopy in Caco-2 monolayer
| [54] |
| The standard shake-flask method and steady-state fluorescence emission anisotropy, Cromolyn sodium | Increased membrane fluidity, but not increased lipophilicity in cromolyn sodium | The standard shake-flask method
| [131] |
| Various in vitro, in vivo/ex vivo assays | Protection against enzymatic degradation via local buffering actions and semaglutide-specific transcellular absorption in the stomach | For transcellular mechanism
For semaglutide and SNAC specificity
| [61] |
| SNAC does not appear to exhibit a transcellular mechanism by membrane insertion and perturbation. | CG-MD simulation
| [72] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.C.; Park, E.J.; Na, D.H. Gastrointestinal Permeation Enhancers for the Development of Oral Peptide Pharmaceuticals. Pharmaceuticals 2022, 15, 1585. https://doi.org/10.3390/ph15121585
Kim JC, Park EJ, Na DH. Gastrointestinal Permeation Enhancers for the Development of Oral Peptide Pharmaceuticals. Pharmaceuticals. 2022; 15(12):1585. https://doi.org/10.3390/ph15121585
Chicago/Turabian StyleKim, Jae Cheon, Eun Ji Park, and Dong Hee Na. 2022. "Gastrointestinal Permeation Enhancers for the Development of Oral Peptide Pharmaceuticals" Pharmaceuticals 15, no. 12: 1585. https://doi.org/10.3390/ph15121585