
Targeted Treatment against Lipoprotein (a): The Coming Breakthrough in Lipid Lowering Therapy


Abstract
:1. Background
1.1. Atherosclerotic Cardiovascular Diseases
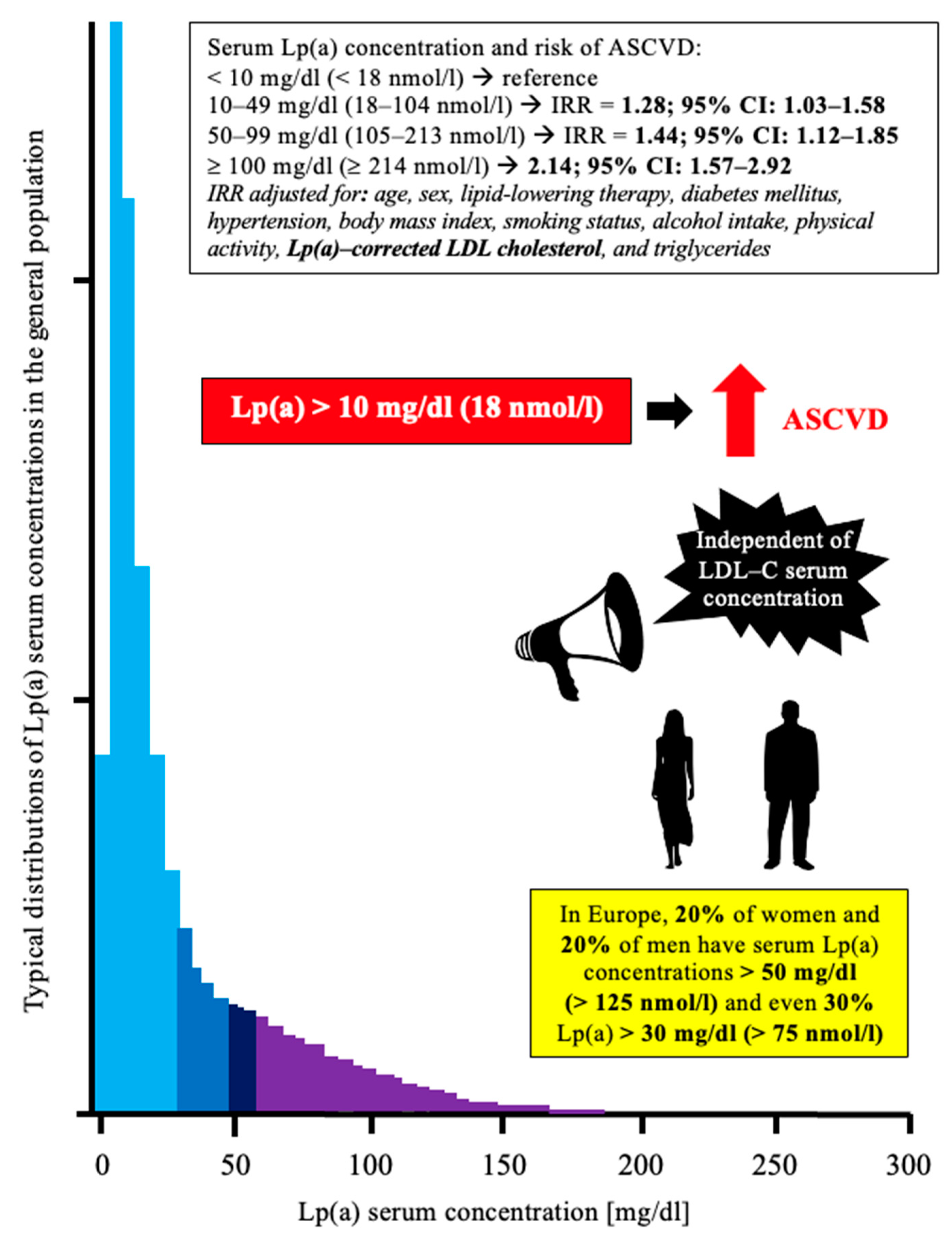
1.2. Lipoprotein (a)—Brief Overview for Clinicians
1.3. Lp(a) and ASCVD—Brief Overview of Pathophysiological Mechanisms
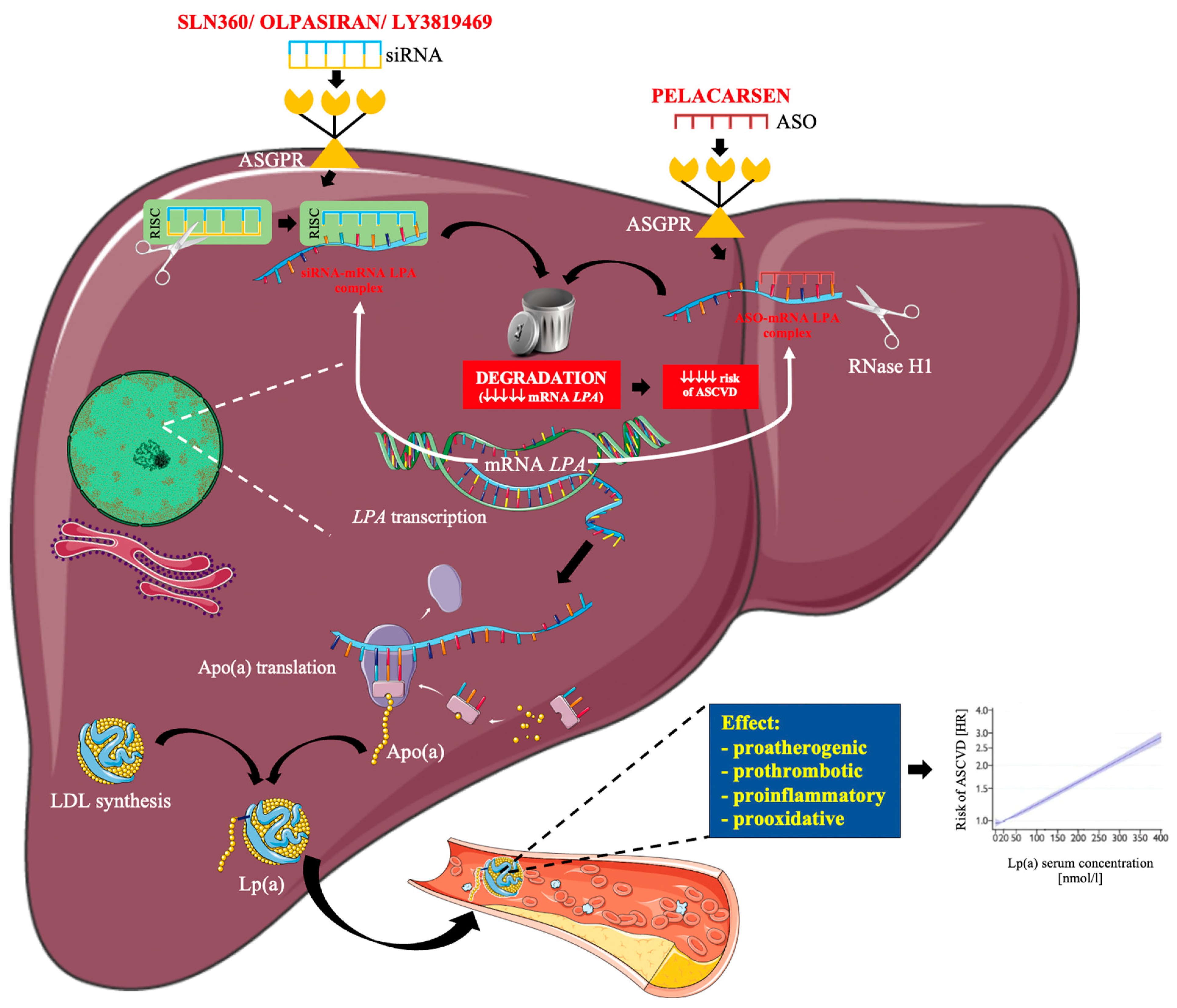
1.4. RNA-Based Therapy
2. Lp(a) Lowering Drugs under Clinical Development
2.1. Pelacarsen
2.2. Olpasiran
2.3. SLN360
2.4. LY3819469

{kind=link}
{kind=link}
| Drug | Clinical Phase/ Status | NCT Number/ Reference | Population/ Sample Size | Duration | Dose/ Treatment Arms | Key Results | Kay Safety Data |
|---|---|---|---|---|---|---|---|
| Pelacarsen | Phase 1/2a completed Viney et al., 2016 [51] | NCT02414594 | Healthy Lp(a) ≥ 75 nmol/L (30 mg/dL); n = 58 | 22 days | Single ascending dose: 10, 20, 40, 80, 120 mg; Multiple ascending doses: 10, 20, 40 mg in 6 doses each | Lp(a) reduction: Single dose −26% to −85%; Multiple doses −66% to −92% | No SAE |
| Phase 2 completed Tsimikas et al., 2020 [50] | NCT03070782 | Lp(a) ≥ 60 mg/dL and CVD; n= 286 | 6–12 months | 20 mg Q1W; 20 mg Q2W; 20, 40, 60 mg Q4W | Lp(a) reduction: −35% to −80% | Injection site reactions (27% drug vs. 6% placebo), urinary tract infection (13% drug vs. 6% placebo), myalgia (12% vs. 11%), headache (11% vs. 8%), influenza-like symptoms (7% vs. 6%) | |
| Phase 3 ongoing | NCT04023552 | Lp(a) ≥ 70 mg/dL and CVD; n = 8323 | 4–5 years | 80 mg Q4W | NA | NA | |
| Phase 1 completed | NCT05337878 | L(a) ≥ 15 nmol/L (8 mg/dL); healthy Japanese n = 29 | 204 days | Single ascending dose: 20, 40, 80 mg; Multiple ascending doses: 80 mg Q4W | NA | NA | |
| Phase 3 recruiting | NCT05305664 | Lp(a) > 60 mg/dL and CVD; n = 60 (target) | ~208 days | 80 mg Q4W vs. placebo | NA | NA | |
| Phase 1 recruiting | NCT05026996 | Subjects with mild hepatic impairment compared to matched healthy participants n = 16 | 60 days | Single dose | NA | NA | |
| Olpasiran | Phase 1 ongoing | NCT03626662 Koren et al., 2022 [58] | Lp(a) ≥ 70 nmol/L ≤ 199 nmol/L, n = 40; Lp(a) ≥ 200 nmol/L, n = 24 | 225 days | Single dose; 3 mg, 9 mg, 30 mg, 75 mg, 225 mg | Lp(a) reduction: −71 to −97% | Headache (10% drug vs. 25% placebo), upper respiratory tract infection (15% drug vs. 13% placebo), injection site reaction (one patient); no SAE |
| Phase 1 completed | NCT04987320 Sohn et al., 2022 [59] | Lp(a) ≥ 70 nmol/L (≥27 mg/dL); healthy Japanese and non-Japanese patients; n = 27 | 225 days | Single dose: 3 mg, 9 mg, 75 mg, 225 mg | Lp(a) reduction: –56% to –99% | Headache (one patient−16.7%), vitreous floaters (one patient 16.7%); no SAE | |
| Phase 2 ongoing | NCT04270760 O’Donghaou et al., 2022 [60,61] | L(a) > 150 nmol/L and ASCVD; n = 281 | 336 days | 10 mg Q12W, 75 mg Q12W, 225 mg Q12W, 225 mg Q24W, placebo | Lp(a) reduction: −70.5% to −100.5% | The most common AE: injection-site reaction (17% vs. 11% placebo) hypersensitivity reaction (6% vs. 2% placebo) | |
| Phase 3 not yet recruiting | NCT05581303 | Lp(a) ≥ 200 nmol/L and ASCVD n = 6000 (target) | 4 years | Q12W vs. placebo | NA | NA | |
| Phase 1 recruiting | NCT05481411 | Subjects with mild, moderate, or severe hepatic impairment compared to participants with normal hepatic function n = 24 (target) | 85 days | Single dose on day 1 | NA | NA | |
| Phase 1 recruiting | NCT05489614 | Subjects with normal renal function and participants with various degrees of renal impairment n = 32 (target) | 85 days | Single dose on day 1 | NA | NA | |
| SLN360 | Phase 1 completed | NCT04606602 Nissen et al., 2022 [65] | Lp(a) ≥ 150 nmol/L; n = 32 | 150 days | Single dose: 30; 100; 300 or 600 mg vs. placebo | Lp(a) reduction −46% to −98% | AEs were generally mild, most commonly low-grade injection site events (grades 1 and 2) and headache; no SAE |
| Phase 2 not yet recruiting | NCT05537571 | Lp(a) > 125 nmol/L and high risk of ASCVD event; n = 160 (target) | 240 days | Three different doses vs. placebo | NA | NA | |
| Phase 1 Recruiting | NCT04606602 | L(a) ≥ 125 nmol/L n = 88 (target) | 201 days | Single or multiple doses vs. placebo | NA | NA | |
| LY3819469 | Phase 1 ongoing | NCT04914546 | Part A: Healthy, high Lp(a) levels Part B Japanese participant; n = 66 (target) | part A: 53 weeks Part B: 29 weeks | NA | NA | NA |
3. Potential Risk Associated with Low Levels of Lp(a)
4. ASO versus siRNA in Lowering Lp(a)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global burden of cardiovascular diseases and risk factors, 1990–2019: Update from the GBD 2019 study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Surma, S.; Banach, M. Fibrinogen and atherosclerotic cardiovascular diseases—Review of the literature and clinical studies. Int. J. Mol. Sci. 2022, 23, 193. [Google Scholar] [CrossRef] [PubMed]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Atherosclerosis 2019, 290, 140–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visseren, F.L.J.; Mach, F.; Smulders, Y.M.; Carballo, D.; Koskinas, K.C.; Bäck, M.; Benetos, A.; Biffi, A.; Boavida, J.M.; Capodanno, D.; et al. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice. Eur. Heart J. 2021, 42, 3227–3337. [Google Scholar] [CrossRef] [PubMed]
- Banach, M.; Burchardt, P.; Chlebus, K.; Dobrowolski, P.; Dudek, D.; Dyrbuś, K.; Gąsior, M.; Jankowski, P.; Jóźwiak, J.; Kłosiewicz-Latoszek, L.; et al. PoLA/CFPiP/PCS/PSLD/PSD/PSH guidelines on diagnosis and therapy of lipid disorders in Poland 2021. Arch. Med. Sci. 2021, 17, 1447–1547. [Google Scholar] [CrossRef]
- Surma, S.; Banach, M. The role of atorvastatin and rosuvastatin and their combination with ezetimibe in the optimal therapy of lipid disorders. Świat. Med. Farm. 2022, 9, 64–79. [Google Scholar]
- Bianconi, V.; Banach, M.; Pirro, M. Why patients with familial hypercholesterolemia are at high cardiovascular risk? Beyond LDL-C levels. Trends Cardiovasc. Med. 2021, 31, 205–215. [Google Scholar] [CrossRef]
- Ference, B.A.; Graham, I.; Tokgozoglu, L.; Catapano, A.L. Impact of lipids on cardiovascular health: JACC Health Promotion Series. J. Am. Coll. Cardiol. 2018, 72, 1141–1156. [Google Scholar] [CrossRef]
- Rikhi, R.; Hammoud, A.; Ashburn, N.; Snavely, A.C.; Michos, E.D.; Chevli, P.; Tsai, M.Y.; Herrington, D.; Shapiro, M.D. Relationship of low-density lipoprotein-cholesterol and lipoprotein(a) to cardiovascular risk: The Multi-Ethnic Study of Atherosclerosis (MESA). Atherosclerosis 2022, 363, 102–108. [Google Scholar] [CrossRef]
- Reyes-Soffer, G.; Ginsberg, H.N.; Berglund, L.; Duell, P.B.; Heffron, S.P.; Kamstrup, P.R.; Lloyd-Jones, D.M.; Marcovina, S.M.; Yeang, C.; Koschinsky, M.L. Lipoprotein(a): A genetically determined, causal, and prevalent risk factor for atherosclerotic cardiovascular disease: A scientific statement from the American Heart Association. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 48–60. [Google Scholar] [CrossRef]
- Tsimikas, S. A test in context: Lipoprotein(a): Diagnosis, prognosis, controversies, and emerging therapies. J. Am. Coll. Cardiol. 2017, 69, 692–711. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Fazio, S.; Ferdinand, K.C.; Ginsberg, H.N.; Koschinsky, M.L.; Marcovina, S.M.; Moriarty, P.M.; Rader, D.J.; Remaley, A.T.; Reyes-Soffer, G.; et al. NHLBI Working Group Recommendations to Reduce Lipoprotein(a)-Mediated Risk of Cardiovascular Disease and Aortic Stenosis. J. Am. Coll. Cardiol. 2018, 71, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Madsen, C.M.; Kamstrup, P.R.; Langsted, A.; Varbo, A.; Nordestgaard, B.G. Lipoprotein(a)-lowering by 50 mg/dL (105 nmol/L) may be needed to reduce cardiovascular disease 20% in secondary prevention: A population-based study. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 255–266. [Google Scholar] [CrossRef]
- Cybulska, B.; Kłosiewicz-Latoszek, L.; Penson, P.E.; Banach, M. What do we know about the role of lipoprotein(a) in atherogenesis 57 years after its discovery? Prog. Cardiovasc. Dis. 2020, 63, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, F.; Mora, S.; Stroes, E.S.G.; Ference, B.A.; Arsenault, B.J.; Berglund, L.; Dweck, M.R.; Koschinsky, M.; Lambert, G.; Mach, F.; et al. Lipoprotein(a) in atherosclerotic cardiovascular disease and aortic stenosis: A European Atherosclerosis Society consensus statement. Eur. Heart J. 2022, 43, 3925–3946. [Google Scholar] [CrossRef]
- Afshar, M.; Rong, J.; Zhan, Y.; Chen, H.Y.; Engert, J.C.; Sniderman, A.D.; Larson, M.G.; Vasan, R.S.; Thanassoulis, G. Risks of incident cardiovascular disease associated with concomitant elevations in lipoprotein(a) and low-density lipoprotein cholesterol-the Framingham Heart Study. J. Am. Heart Assoc. 2020, 9, e014711. [Google Scholar] [CrossRef]
- Kaiser, Y.; Daghem, M.; Tzolos, E.; Meah, M.N.; Doris, M.K.; Moss, A.J.; Kwiecinski, J.; Kroon, J.; Nurmohamed, N.S.; van der Harst, P.; et al. Association of lipoprotein(a) with atherosclerotic plaque progression. J. Am. Coll. Cardiol. 2022, 79, 223–233. [Google Scholar] [CrossRef]
- Nurmohamed, N.S.; Kaiser, Y.; Schuitema, P.C.E.; Ibrahim, S.; Nierman, M.; Fischer, J.C.; Chamuleau, S.A.J.; Knaapen, P.; Stroes, E.S.G. Finding very high lipoprotein(a): The need for routine assessment. Eur. J. Prev. Cardiol. 2022, 29, 769–776. [Google Scholar] [CrossRef]
- Willeit, P.; Kiechl, S.; Kronenberg, F.; Witztum, J.L.; Santer, P.; Mayr, M.; Xu, Q.; Mayr, A.; Willeit, J.; Tsimikas, S. Discrimination and net reclassification of cardiovascular risk with lipoprotein(a): Prospective 15-year outcomes in the Bruneck Study. J. Am. Coll. Cardiol. 2014, 64, 851–860. [Google Scholar] [CrossRef]
- Kamstrup, P.R.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Extreme lipoprotein(a) levels and improved cardiovascular risk prediction. J. Am. Coll. Cardiol. 2013, 61, 1146–1156. [Google Scholar] [CrossRef] [Green Version]
- O’Donoghue, M.L.; Fazio, S.; Giugliano, R.P.; Stroes, E.S.G. Lipoprotein(a), PCSK9 inhibition, and cardiovascular risk. Insights from FOURIER trial. Circulation 2019, 139, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Bittner, V.A.; Szarek, M.; Aylward, P.E.; Bhatt, D.L.; Diaz, R.; Edelberg, J.M.; Fras, Z.; Goodman, S.G.; Halvorsen, S.; Hanotin, C.; et al. Effect of alirocumab on lipoprotein(a) and cardiovascular risk after acute coronary syndrome. J. Am. Coll. Cardiol. 2020, 75, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Szarek, M.; Bittner, V.A.; Aylward, P.; Baccara-Dinet, M.; Bhatt, D.L.; Diaz, R.; Fras, Z.; Goodman, S.G.; Halvorsen, S.; Harrington, R.A.; et al. Lipoprotein(a) lowering by alirocumab reduces the total burden of cardiovascular events independent of low-density lipoprotein cholesterol lowering: ODYSSEY OUTCOMES trial. Eur. Heart J. 2020, 41, 4245–4255. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Ference, B.A.; Staley, J.R.; Freitag, D.F.; Mason, A.M.; Nielsen, S.F.; Willeit, P.; Young, R.; Surendran, P.; Karthikeyan, S.; et al. Association of LPA Variants With Risk of Coronary Disease and the Implications for Lipoprotein(a)-Lowering Therapies: A Mendelian Randomization Analysis. JAMA Cardiol. 2018, 3, 619–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banach, M.; Penson, P.E. Statins and Lp(a): Do not make perfect the enemy of excellent. Eur. Heart J. 2020, 41, 190–191. [Google Scholar] [CrossRef]
- Awad, K.; Mikhailidis, D.P.; Katsiki, N.; Muntner, P.; Banach, M. Lipid and Blood Pressure Meta-Analysis Collaboration (LBPMC) Group. Effect of Ezetimibe Monotherapy on Plasma Lipoprotein(a) Concentrations in Patients with Primary Hypercholesterolemia: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Drugs 2018, 78, 453–462. [Google Scholar] [CrossRef]
- Momtazi-Borojeni, A.A.; Katsiki, N.; Pirro, M.; Banach, M.; Rasadi, K.A.; Sahebkar, A. Dietary natural products as emerging lipoprotein(a)-lowering agents. J. Cell. Physiol. 2019, 234, 12581–12594. [Google Scholar] [CrossRef]
- Schwartz, G.G.; Ballantyne, C.M. Existing and emerging strategies to lower Lipoprotein(a). Atherosclerosis 2022, 349, 110–122. [Google Scholar] [CrossRef]
- Di Fusco, S.A.; Arca, M.; Scicchitano, P.; Alonzo, A.; Perone, F.; Gulizia, M.M.; Gabrielli, D.; Oliva, F.; Imperoli, G.; Colivicchi, F. Lipoprotein(a): A risk factor for atherosclerosis and an emerging therapeutic target. Heart 2023, 109, 18–25. [Google Scholar] [CrossRef]
- Sahin, U.; Karikó, K.; Türeci, Ö. mRNA-based therapeutics--developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Witzigmann, D.; Thomson, S.B.; Chen, S.; Leavitt, B.R.; Cullis, P.R.; van der Meel, R. The current landscape of nucleic acid therapeutics. Nat. Nanotechnol. 2021, 16, 630–643. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T.; Witztum, J.L.; Bennett, C.F.; Baker, B.F. RNA-Targeted Therapeutics. Cell Metab. 2018, 27, 714–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crooke, S.T.; Liang, X.H.; Baker, B.F.; Crooke, R.M. Antisense technology: A review. J. Biol. Chem. 2021, 296, 100416. [Google Scholar] [CrossRef] [PubMed]
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 46–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crooke, S.T.; Liang, X.H.; Crooke, R.M.; Baker, B.F.; Geary, R.S. Antisense drug discovery and development technology considered in a pharmacological context. Biochem. Pharmacol. 2021, 189, 114196. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef]
- Hammond, S.M.; Bernstein, E.; Beach, D.; Hannon, G.J. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature 2000, 404, 293–296. [Google Scholar] [CrossRef]
- Meister, G.; Landthaler, M.; Patkaniowska, A.; Dorsett, Y.; Teng, G.; Tuschl, T. Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol. Cell 2004, 15, 185–197. [Google Scholar] [CrossRef]
- Burnett, J.C.; Rossi, J.J. RNA-Based therapeutics: Current progress and future prospects. Chem. Biol. 2012, 19, 60–71. [Google Scholar] [CrossRef] [Green Version]
- Chi, X.; Gatti, P.; Papoian, T. Safety of antisense oligonucleotide and siRNA-based therapeutics. Drug Discov. Today 2017, 22, 823–833. [Google Scholar] [CrossRef]
- Judge, D.P.; Kristen, A.V.; Grogan, M.; Maurer, M.S.; Falk, R.H.; Hanna, M.; Gillmore, J.; Garg, P.; Vaishnaw, A.K.; Harrop, J.; et al. Phase 3 Multicenter Study of Revusiran in Patients with Hereditary Transthyretin-Mediated (hATTR) Amyloidosis with Cardiomyopathy (ENDEAVOUR). Cardiovasc. Drugs Ther. 2020, 34, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.A.; Scheinberg, M.; Waddington-Cruz, M.; Heitner, S.B.; Karam, C.; Drachman, B.; Khella, S.; Whelan, C.; Obici, L. Inotersen for the treatment of adults with polyneuropathy caused by hereditary transthyretin-mediated amyloidosis. Expert Rev. Clin. Pharmacol. 2019, 12, 701–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlegel, M.K.; Janas, M.M.; Jiang, Y.; Barry, J.D.; Davis, W.; Agarwal, S.; Berman, D.; Brown, C.R.; Castoreno, A.; LeBlanc, S.; et al. From bench to bedside: Improving the clinical safety of GalNAc-siRNA conjugates using seed-pairing destabilization. Nucleic Acids Res. 2022, 50, 6656–6670. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Corey, D.R. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018, 46, 1584–1600. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Kato, Y.; Hayasi, E.; Tabata, T.; Suzuki, M.; Takahara, Y.; Sugiyama, Y. A novel hepatic-targeting system for therapeutic cytokines that delivers to the hepatic asialoglycoprotein receptor, but avoids receptor-mediated endocytosis. Pharm. Res. 2002, 19, 1736–1744. [Google Scholar] [CrossRef] [PubMed]
- Springer, A.D.; Dowdy, S.F. GalNAc-siRNA conjugates: Leading the way for delivery of RNAi therapeutics. Nucleic Acid Ther. 2018, 28, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Nair, J.K.; Willoughby, J.L.; Chan, A.; Charisse, K.; Alam, M.R.; Wang, Q.; Hoekstra, M.; Kandasamy, P.; Kel’in, A.V.; Milstein, S.; et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014, 136, 16958–16961. [Google Scholar] [CrossRef] [Green Version]
- Gareri, C.; Polimeni, A.; Giordano, S.; Tammè, L.; Curcio, A.; Indolfi, C. Antisense Oligonucleotides and Small Interfering RNA for the Treatment of Dyslipidemias. J. Clin. Med. 2022, 11, 3884. [Google Scholar] [CrossRef]
- Bennett, C.F. Therapeutic antisense oligonucleotides are coming of age. Annu. Rev. Med. 2019, 70, 307–321. [Google Scholar] [CrossRef]
- Tsimikas, S.; Viney, N.J.; Hughes, S.G.; Singleton, W.; Graham, M.J.; Baker, B.F.; Burkey, J.L.; Yang, Q.; Marcovina, S.M.; Geary, R.S.; et al. Antisense therapy targeting apolipoprotein(a): A randomised, double-blind, placebo-controlled phase 1 study. Lancet 2015, 386, 1472–1483. [Google Scholar] [CrossRef]
- Viney, N.J.; van Capelleveen, J.C.; Geary, R.S.; Xia, S.; Tami, J.A.; Yu, R.Z.; Marcovina, S.M.; Hughes, S.G.; Graham, M.J.; Crooke, R.M.; et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): Two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet 2016, 388, 2239–2253. [Google Scholar] [CrossRef] [PubMed]
- Prakash, T.P.; Graham, M.J.; Yu, J.; Carty, R.; Low, A.; Chappell, A.; Schmidt, K.; Zhao, C.; Aghajan, M.; Murray, H.F.; et al. Targeted delivery of antisenseoligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res. 2014, 42, 8796–8807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeang, C.; Karwatowska-Prokopczuk, E.; Su, F.; Dinh, B.; Xia, S.; Witztum, J.L.; Tsimikas, S. Effect of pelacarsen on lipoprotein(a) cholesterol and corrected low-density lipoprotein cholesterol. J. Am. Coll. Cardiol. 2022, 79, 1035–1046. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Karwatowska-Prokopczuk, E.; Gouni-Berthold, I.; Tardif, J.C.; Baum, S.J.; Steinhagen-Thiessen, E.; Shapiro, M.D.; Stroes, E.S.; Moriarty, P.M.; Nordestgaard, B.G.; et al. Lipoprotein(a) reduction in persons with cardiovascular disease. N. Engl. J. Med. 2020, 382, 244–255. [Google Scholar] [CrossRef]
- Karwatowska-Prokopczuk, E.; Clouet-Foraison, N.; Xia, S.; Viney, N.J.; Witztum, J.L.; Marcovina, S.M.; Tsimikas, S. Prevalence and influence of LPA gene variants and isoform size on the Lp(a)-lowering effect of pelacarsen. Atherosclerosis 2021, 324, 102–108. [Google Scholar] [CrossRef]
- Swerdlow, D.I.; Rider, D.A.; Yavari, A.; Wikström Lindholm, M.; Campion, G.V.; Nissen, S.E. Treatment and prevention of lipoprotein(a)-mediated cardiovascular disease: The emerging potential of RNA interference therapeutics. Cardiovasc. Res. 2022, 118, 1218–1231. [Google Scholar] [CrossRef]
- Subhan, M.A.; Torchilin, V.P. siRNA based drug design, quality, delivery and clinical translation. Nanomedicine 2020, 29, 102239. [Google Scholar] [CrossRef]
- Koren, M.J.; Moriarty, P.M.; Baum, S.J.; Neutel, J.; Hernandez-Illas, M.; Weintraub, H.S.; Florio, M.; Kassahun, H.; Melquist, S.; Varrieur, T.; et al. Preclinical development and phase 1 trial of a novel siRNA targeting lipoprotein(a). Nat. Med. 2022, 28, 96–103. [Google Scholar] [CrossRef]
- Sohn, W.; Winkle, P.; Neutel, J.; Wu, Y.; Jabari, F.; Terrio, C.; Varrieur, T.; Wang, J.; Hellawell, J. Pharmacokinetics, Pharmacodynamics, and Tolerability of Olpasiran in Healthy Japanese and Non-Japanese Participants: Results from a Phase I, Single-dose, Open-label Study. Clin. Ther. 2022, 44, 1237–1247. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; López, J.A.G.; Knusel, B.; Gencer, B.; Wang, H.; Wu, Y.; Kassahun, H.; Sabatine, M.S. Study design and rationale for the Olpasiran trials of Cardiovascular Events And lipoproteiN(a) reduction-DOSE finding study (OCEAN(a)-DOSE). Am. Heart J. 2022, 251, 61–69. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; Rosenson, R.S.; Gencer, B.; López, J.A.G.; Lepor, N.E.; Baum, S.J.; Stout, E.; Gaudet, D.; Knusel, B.; Kuder, J.F.; et al. OCEAN(a)-DOSE Trial Investigators. Small Interfering RNA to Reduce Lipoprotein(a) in Cardiovascular Disease. N. Engl. J. Med. 2022, 387, 1855–1864. [Google Scholar] [CrossRef] [PubMed]
- Nurmohamed, N.S.; Kraaijenhof, J.M.; Stroes, E.S.G. Lp(a): A new pathway to target? Curr. Atheroscler. Rep. 2022, 24, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Rider, D.A.; Eisermann, M.; Löffler, K.; Aleku, M.; Swerdlow, D.I.; Dames, S.; Hauptmann, J.; Morrison, E.; Lindholm, M.W.; Schubert, S.; et al. Pre-clinical assessment of SLN360, a novel siRNA targeting LPA, developed to address elevated lipoprotein (a) in cardiovascular disease. Atherosclerosis 2022, 349, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Rider, D.; Chivers, S.; Aretz, J.; Eisermann, M.; Löffler, K.; Hauptmann, J.; Morrison, E.; Campion, G. Preclinical Toxicological Assessment of A Novel siRNA, SLN360, Targeting Elevated Lipoprotein (a) in Cardiovascular Disease. Toxicol. Sci. 2022, 189, 237–249. [Google Scholar] [CrossRef]
- Nissen, S.E.; Wolski, K.; Balog, C.; Swerdlow, D.I.; Scrimgeour, A.C.; Rambaran, C.; Wilson, R.J.; Boyce, M.; Ray, K.K.; Cho, L.; et al. Single ascending dose study of a short interfering RNA targeting lipoprotein(a) production in individuals with elevated plasma lipoprotein(a) levels. JAMA 2022, 327, 1679–1687. [Google Scholar] [CrossRef]
- Sheridan, C. RNA drugs lower lipoprotein(a) and genetically driven cholesterol. Nat. Biotechnol. 2022, 40, 983–985. [Google Scholar] [CrossRef]
- Gudbjartsson, D.F.; Thorgeirsson, G.; Sulem, P.; Helgadottir, A.; Gylfason, A.; Saemundsdottir, J.; Bjornsson, E.; Norddahl, G.L.; Jonasdottir, A.; Jonasdottir, A.; et al. Lipoprotein(a) concentration and risks of cardiovascular disease and diabetes. J. Am. Coll. Cardiol. 2019, 74, 2982–2994. [Google Scholar] [CrossRef]
- Mora, S.; Kamstrup, P.R.; Rifai, N.; Nordestgaard, B.G.; Buring, J.E.; Ridker, P.M. Lipoprotein(a) and risk of type 2 diabetes. Clin. Chem. 2010, 56, 1252–1260. [Google Scholar] [CrossRef] [Green Version]
- Kamstrup, P.R.; Nordestgaard, B.G. Lipoprotein(a) concentrations, isoform size, and risk of type 2 diabetes: A Mendelian randomisation study. Lancet Diabetes Endocrinol. 2013, 1, 220–227. [Google Scholar] [CrossRef]
- Langsted, A.; Nordestgaard, B.G.; Kamstrup, P.R. Low lipoprotein(a) levels and risk of disease in a large, contemporary, general population study. Eur. Heart J. 2021, 42, 1147–1156. [Google Scholar] [CrossRef]
- Tsimikas, S. In search of a physiological function of lipoprotein(a): Causality of elevated Lp(a) levels and reduced incidence of type 2 diabetes. J. Lipid Res. 2018, 59, 741–744. [Google Scholar] [CrossRef] [PubMed]
- Farukhi, Z.M.; Mora, S. Lifelong low Lp(a) levels: Genetics give a green light? Eur. Heart J. 2021, 42, 1157–1159. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Haycock, P.C.; Gurdasani, D.; Pomilla, C.; Boekholdt, S.M.; Tsimikas, S.; Khaw, K.T.; Wareham, N.J.; Sandhu, M.S.; Forouhi, N.G. The association between circulating lipoprotein(a) and type 2 diabetes: Is it causal? Diabetes 2014, 63, 332–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banach, M.; López-Sendon, J.L.; Averna, M.; Cariou, B.; Loy, M.; Manvelian, G.; Batsu, I.; Poulouin, Y.; Gaudet, D. Treatment adherence and effect of concurrent statin intensity on the efficacy and safety of alirocumab in a real-life setting: Results from ODYSSEY APPRISE. Arch. Med. Sci. 2021, 18, 285–292. [Google Scholar] [CrossRef]
- Solnica, B.; Sygitowicz, G.; Sitkiewicz, D.; Cybulska, B.; Jóźwiak, J.; Odrowąż-Sypniewska, G.; Banach, M. 2020 Guidelines of the Polish Society of Laboratory Diagnostics (PSLD) and the Polish Lipid Association (PoLA) on laboratory diagnostics of lipid metabolism disorders. Arch. Med. Sci. 2020, 16, 237–252. [Google Scholar] [CrossRef]
- Blom, D.J.; Marais, A.D.; Moodley, R.; van der Merwe, N.; van Tonder, A.; Raal, F.J. RNA-based therapy in the management of lipid disorders: A review. Lipids Health Dis. 2022, 21, 41. [Google Scholar] [CrossRef]


| Author, Year, [Ref] | Characteristics of the Study Group | Key Results |
|---|---|---|
| Lp(a) and ASCVD Risk | ||
| Rikhi R. et al., 2022, [9] | 4585 subjects from Multi-Ethnic Study of Atherosclerosis (MESA), follow-up: 13.4 years | Elevated Lp(a), regardless of baseline LDL-C, significant increased ASCVD risk |
| Afshar M. et al., 2020, [16] | 2606 subjects from Framingham Offspring | Regardless of LDL-C, were associated with an increased risk of ASCVD |
| Kaiser Y. et al., 2022, [17] | 191 patients with ASCVD | Increased Lp(a) are associated with accelerated progression of low-attenuation plaque in patients with advanced multivessel CAD despite receiving guideline- based preventative therapies |
| Nurmohamed N. et al., 2022, [18] | 12,437 subjects with ASCVD | Patients with Lp(a) > 99th percentile had an OR of 2.64 for ASCVD [95% CI: 1.45–4.89] and 3.39 for MI (95% CI: 1.56–7.94). Importantly, the addition of Lp(a) to ASCVD risk algorithms led to 31% and 63% being reclassified into a higher risk category for SCORE and Second Manifestations of ARTerial disease (SMART), respectively |
| Willeit P. et al., 2014 [19] | 826 subjects | Increased Lp(a) predict 15-year ASCVD outcomes and improves ASCVD risk prediction, particularly in intermediate-risk groups |
| Kampstrup P. et al., 2013, [20] | 8720 subjects | Extreme Lp(a) level substantially improved MI and CAD risk prediction |
| Lowering of Lp(a) and the risk of ASCVD | ||
| O’Donoghue M. et al., 2019, [21] | 25,069 patients with stable CAD treated with evolocumab; follow-up: 2.2 years | Significant correlation between a 15% reduction in the risk of major coronary events (95% Cl: 2–25%; p = 0.0199) and a reduction in serum Lp(a) concentration by 25 nmol/L after adjustment for LDL-C |
| Bittner V. et al., 2020, [22] and Szarek M. et al., 2020, [23] | ODYSSEY OUTCOMES study in 18,924 ACS patients; follow-up: 2.8 years | A 5 mg/dL reduction in Lp(a) was associated with a significant 2.5% reduction in cardiovascular events |
| Burgess S. et al., 2018, [24] | 62,240 patients CAD compared with a control group of 127,000 subjects | Each 10 mg/dL reduction in Lp(a) was associated with a 5.8% reduction in the risk of CAD (OR = 0.94; 95% Cl: 0.93–0.95) |
| Lipid-Lowering Drug | Effect on Lp(a) Serum Concentration |
|---|---|
| Niacin | Reduction; 30% |
| Statins | Possible increase; 6–10% |
| Ezetimibe | Possible reduction; 0–7% |
| Bempedoic acid | No effect |
| Fibrates | Minimal, possible increase in setting of HTG |
| Bile acid sequestrants | No effect |
| PCSK9 inhibitors | Reduction; 20–30% |
| Inclisiran | Reduction; 15–26% |
| Mipomersen | Reduction; 25% |
| CETP inhibitors | Reduction; 25% |
| ASO based drugs | Reduction; 70–90% |
| siRNA based drugs | Reduction 70–98% |
| Lipoprotein apheresis | Reduction; 20–30% |
| Lipoprotein (a) apheresis | Reduction; 70–80% |
| Recommendations | Class | Level |
|---|---|---|
| Lp(a) concentration should be measured at least once in every adult individual’s life. | IIa | C |
| Measurement of Lp(a) should be considered in all patients with premature onset of cardiovascular disease, the lack of expected statin therapy effect, and in those with a borderline risk between moderate and high, for better risk stratification. | IIa | C |
| Measurement of Lp(a) may be considered in patients with very high cardiovascular risk and atherosclerotic cardiovascular disease, in patients with familial hypercholesterolaemia, and in pregnant women as a prevention of pre-eclampsia or miscarriage, in recurrent pregnancy loss, or intrauterine growth restriction. | IIb | C |
| Drug | Type | Chemical Modification | Delivery System | Targeting Gene | Company |
|---|---|---|---|---|---|
| Pelacarsen (TQJ230; formerly IONIS-APO(a)-LRX, AKCEA-APO(a)-LRX, ISIS 681257) | ASO | 2′-O-MOE | GalNAc | LPA | Novartis Pharmaceuticals |
| Olpasiran (AMG-890, ARO-LPA) | siRNA | 2′-O-Me | GalNAc | LPA | Amgen, Arrowhead Pharmaceuticals |
| SLN360 | siRNA | 2′-O-Me | GalNAc | LPA | Silence Therapeutics plc |
| LY3819469 | siRNA | 2′-O-Me; 2′-F | GalNAc | LPA | Eli Lilly and Company |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sosnowska, B.; Surma, S.; Banach, M. Targeted Treatment against Lipoprotein (a): The Coming Breakthrough in Lipid Lowering Therapy. Pharmaceuticals 2022, 15, 1573. https://doi.org/10.3390/ph15121573
Sosnowska B, Surma S, Banach M. Targeted Treatment against Lipoprotein (a): The Coming Breakthrough in Lipid Lowering Therapy. Pharmaceuticals. 2022; 15(12):1573. https://doi.org/10.3390/ph15121573
Chicago/Turabian StyleSosnowska, Bożena, Stanisław Surma, and Maciej Banach. 2022. "Targeted Treatment against Lipoprotein (a): The Coming Breakthrough in Lipid Lowering Therapy" Pharmaceuticals 15, no. 12: 1573. https://doi.org/10.3390/ph15121573