
Reactivity of Covalent Fragments and Their Role in Fragment Based Drug Discovery
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. The Role of Covalent Inhibitors and Warheads in Drug Design
2. Predicting Warhead Reactivity
3. FBDD and the Role of Covalents within
4. Covalent FBDD Case Studies
4.1. GTPases
4.2. SARS-Cov-2
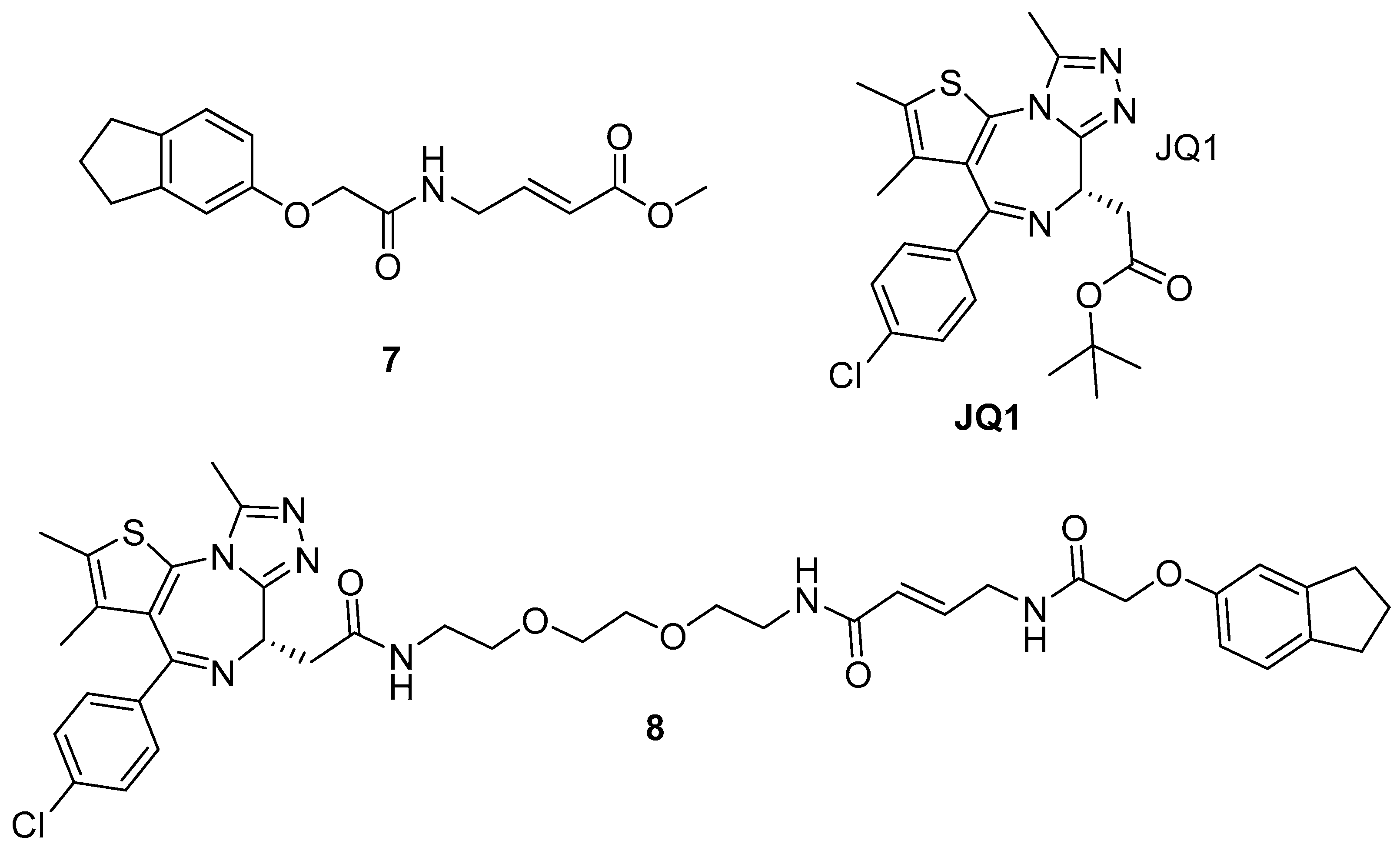
4.3. BRD4-BET2
4.4. Enzymes Involved in Ubiquitination/Deubiquitination
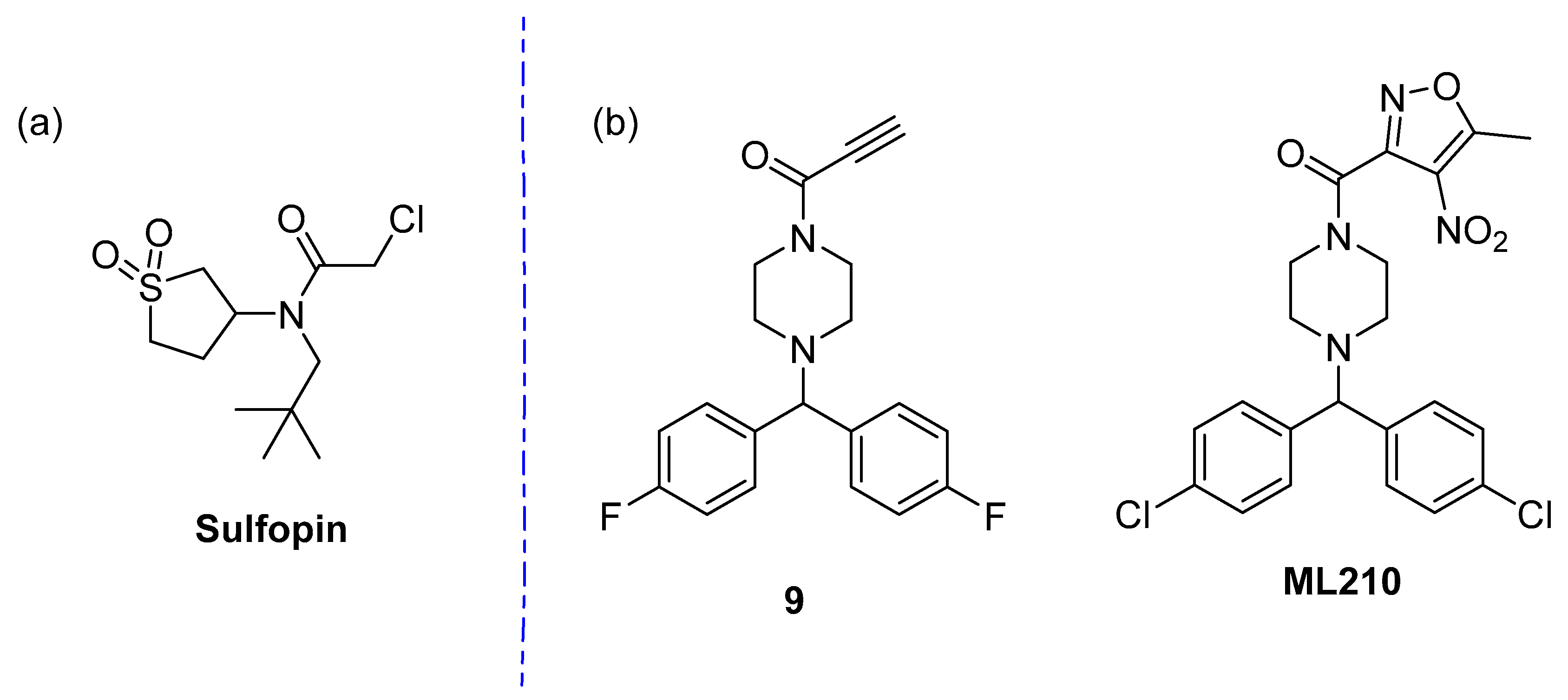
4.5. Pin1
4.6. GPX4
4.7. LP-Pla2
4.8. Tau
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Singh, J.; Petter, R.C.; Baillie, T.A.; Whitty, A. The resurgence of covalent drugs. Nat. Rev. Drug. Discov. 2011, 10, 307–317. [Google Scholar] [CrossRef]
- Zhao, Z.; Bourne, P.E. Progress with covalent small-molecule kinase inhibitors. Drug. Discov. Today 2018, 23, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Baillie, T.A. Targeted Covalent Inhibitors for Drug Design. Angew. Chem. Int. Ed. 2016, 55, 13408–13421. [Google Scholar] [CrossRef] [PubMed]
- Chaikuad, A.; Koch, P.; Laufer, S.A.; Knapp, S. The Cysteinome of Protein Kinases as a Target in Drug Development. Angew. Chem. Int. Ed. 2018, 57, 4372–4385. [Google Scholar] [CrossRef]
- Dalton, S.E.; Campos, S. Covalent Small Molecules as Enabling Platforms for Drug Discovery. Chem. Biochem. 2020, 21, 1080–1100. [Google Scholar] [CrossRef] [PubMed]
- Sutanto, F.; Konstantinidou, M.; Dömling, A. Covalent inhibitors: A rational approach to drug discovery. RSC Med. Chem. 2020, 11, 876–884. [Google Scholar] [CrossRef]
- Boike, L.; Henning, N.J.; Nomura, D.K. Advances in covalent drug discovery. Nat. Rev. Drug. Discov. 2022, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Bauer, R.A. Covalent inhibitors in drug discovery: From accidental discoveries to avoided liabilities and designed therapies. Drug. Discov. Today 2015, 20, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
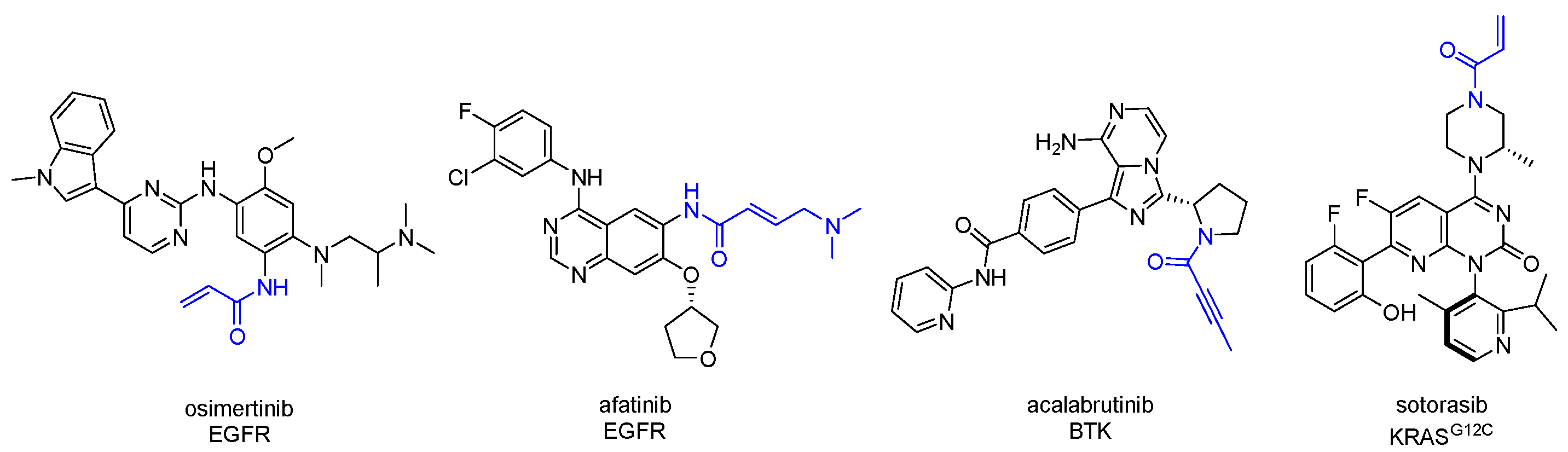
- Butterworth, S.; Cross, D.A.E.; Finlay, M.R.V.; Ward, R.A.; Waring, M.J. The structure-guided discovery of osimertinib: The first U.S. FDA approved mutant selective inhibitor of EGFR T790M. Med. Chem. Comm. 2017, 8, 820–822. [Google Scholar] [CrossRef]
- Barf, T.; Covey, T.; Izumi, R.; van de Kar, B.; Gulrajani, M.; van Lith, B.; van Hoek, M.; de Zwart, E.; Mittag, D.; Demont, D.; et al. Acalabrutinib (ACP-196): A Covalent Bruton Tyrosine Kinase Inhibitor with a Differentiated Selectivity and In Vivo Potency Profile. J. Pharm. Exp. Ther. 2017, 363, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Smaill, J.B.; Patterson, A.V.; Ding, K. Discovery of Cysteine-targeting Covalent Protein Kinase Inhibitors. J. Med. Chem. 2022, 65, 58–83. [Google Scholar] [CrossRef]
- Liu, Q.; Sabnis, Y.; Zhao, Z.; Zhang, T.; Buhrlage, S.J.; Jones, L.H.; Gray, N.S. Developing Irreversible Inhibitors of the Protein Kinase Cysteinome. Chem. Biol. 2013, 20, 146–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef]
- Ábrányi-Balogh, P.; Keserű, G.M. Chapter 2—Warheads for designing covalent inhibitors and chemical probes. In Advances in Chemical Proteomics; Yao, X., Ed.; Elsevier: Amsterdam, The Netherlands, 2022; pp. 47–73. [Google Scholar]
- Barf, T.; Kaptein, A. Irreversible Protein Kinase Inhibitors: Balancing the Benefits and Risks. J. Med. Chem. 2012, 55, 6243–6262. [Google Scholar] [CrossRef]
- Lonsdale, R.; Ward, R.A. Structure-based design of targeted covalent inhibitors. Chem. Soc. Rev. 2018, 47, 3816–3830. [Google Scholar] [CrossRef]
- Tan, L.; Wang, J.; Tanizaki, J.; Huang, Z.; Aref, A.R.; Rusan, M.; Zhu, S.-J.; Zhang, Y.; Ercan, D.; Liao, R.G.; et al. Development of covalent inhibitors that can overcome resistance to first-generation FGFR kinase inhibitors. Proc. Natl. Acad. Sci. USA 2014, 111, E4869–E4877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]
- Nakayama, S.; Atsumi, R.; Takakusa, H.; Kobayashi, Y.; Kurihara, A.; Nagai, Y.; Nakai, D.; Okazaki, O. A zone classification system for risk assessment of idiosyncratic drug toxicity using daily dose and covalent binding. Drug Metab. Dispos. 2009, 37, 1970–1977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baillie, T.A. Approaches to mitigate the risk of serious adverse reactions in covalent drug design. Expert Opin. Drug. Discov. 2021, 16, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Xue, G.; Chen, J.; Liu, L.; Zhou, D.; Zuo, Y.; Fu, T.; Pan, Z. Protein degradation through covalent inhibitor-based PROTACs. Chem. Comm. 2020, 56, 1521–1524. [Google Scholar] [CrossRef]
- Lebraud, H.; Wright, D.J.; Johnson, C.N.; Heightman, T.D. Protein Degradation by In-Cell Self-Assembly of Proteolysis Targeting Chimeras. ACS Cent. Sci. 2016, 2, 927–934. [Google Scholar] [CrossRef] [Green Version]
- Bond, M.J.; Chu, L.; Nalawansha, D.A.; Li, K.; Crews, C.M. Targeted Degradation of Oncogenic KRASG12C by VHL-Recruiting PROTACs. ACS Cent. Sci. 2020, 6, 1367–1375. [Google Scholar] [CrossRef]
- Ward, C.C.; Kleinman, J.I.; Brittain, S.M.; Lee, P.S.; Chung, C.Y.S.; Kim, K.; Petri, Y.; Thomas, J.R.; Tallarico, J.A.; McKenna, J.M.; et al. Covalent Ligand Screening Uncovers a RNF4 E3 Ligase Recruiter for Targeted Protein Degradation Applications. ACS Chem. Biol. 2019, 14, 2430–2440. [Google Scholar] [CrossRef] [PubMed]
- Gabizon, R.; London, N. The rise of covalent proteolysis targeting chimeras. Curr. Opin. Chem. Biol. 2021, 62, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Kiely-Collins, H.; Winter, G.E.; Bernardes, G.J.L. The role of reversible and irreversible covalent chemistry in targeted protein degradation. Cell. Chem. Biol. 2021, 28, 952–968. [Google Scholar] [CrossRef]
- Grimster, N.P. Covalent PROTACs: The best of both worlds? RSC Med. Chem. 2021, 12, 1452–1458. [Google Scholar] [CrossRef]
- Tinworth, C.P.; Lithgow, H.; Dittus, L.; Bassi, Z.I.; Hughes, S.E.; Muelbaier, M.; Dai, H.; Smith, I.E.D.; Kerr, W.J.; Burley, G.A.; et al. PROTAC-Mediated Degradation of Bruton’s Tyrosine Kinase Is Inhibited by Covalent Binding. ACS Chem. Biol. 2019, 14, 342–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabizon, R.; Shraga, A.; Gehrtz, P.; Livnah, E.; Shorer, Y.; Gurwicz, N.; Avram, L.; Unger, T.; Aharoni, H.; Albeck, S.; et al. Efficient Targeted Degradation via Reversible and Irreversible Covalent PROTACs. J. Am. Chem. Soc. 2020, 142, 11734–11742. [Google Scholar] [CrossRef]
- Guo, W.-H.; Qi, X.; Yu, X.; Liu, Y.; Chung, C.-I.; Bai, F.; Lin, X.; Lu, D.; Wang, L.; Chen, J.; et al. Enhancing intracellular accumulation and target engagement of PROTACs with reversible covalent chemistry. Nat. Comm. 2020, 11, 4268. [Google Scholar] [CrossRef] [PubMed]
- Tong, B.; Luo, M.; Xie, Y.; Spradlin, J.N.; Tallarico, J.A.; McKenna, J.M.; Schirle, M.; Maimone, T.J.; Nomura, D.K. Bardoxolone conjugation enables targeted protein degradation of BRD4. Sci. Rep. 2020, 10, 15543. [Google Scholar] [CrossRef]
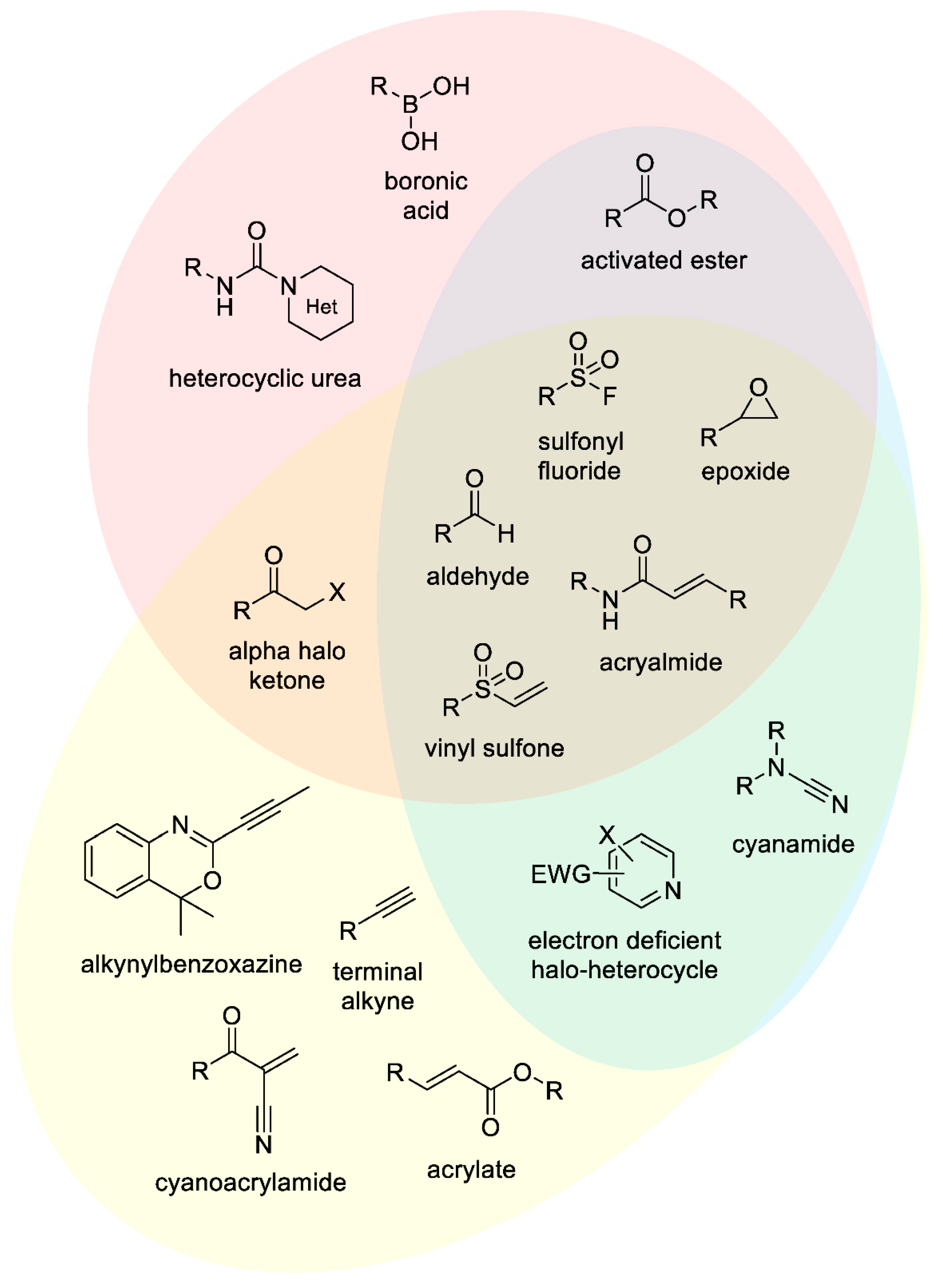
- Gehringer, M.; Laufer, S.A. Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2019, 62, 5673–5724. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhang, P.; Xu, F.; Hu, S.; Liu, J.; Tan, Y.; Tu, Z.; Sun, H.; Zhang, Z.-M.; He, Q.-Y.; et al. Ynamide Electrophile for the Profiling of Ligandable Carboxyl Residues in Live Cells and the Development of New Covalent Inhibitors. J. Med. Chem. 2022, 65, 10408–10418. [Google Scholar] [CrossRef]
- Zhang, Z.; Morstein, J.; Ecker, A.K.; Guiley, K.Z.; Shokat, K.M. Chemoselective Covalent Modification of K-Ras(G12R) with a Small Molecule Electrophile. J. Am. Chem. Soc. 2022, 144, 15916–15921. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.-C.; May, V.K.; Bedford, G.C.; Tuley, A.A.; Fast, W. Discovery of 4,4′-Dipyridylsulfide Analogs as “Switchable Electrophiles” for Covalent Inhibition. ACS Chem. Biol. 2021, 16, 264–269. [Google Scholar] [CrossRef] [PubMed]
- McAulay, K.; Hoyt, E.A.; Thomas, M.; Schimpl, M.; Bodnarchuk, M.S.; Lewis, H.J.; Barratt, D.; Bhavsar, D.; Robinson, D.M.; Deery, M.J.; et al. Alkynyl Benzoxazines and Dihydroquinazolines as Cysteine Targeting Covalent Warheads and Their Application in Identification of Selective Irreversible Kinase Inhibitors. J. Am. Chem. Soc. 2020, 142, 10358–10372. [Google Scholar] [CrossRef]
- Keeley, A.; Ábrányi-Balogh, P.; Keserű, G.M. Design and characterization of a heterocyclic electrophilic fragment library for the discovery of cysteine-targeted covalent inhibitors. MedChemComm 2019, 10, 263–267. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Guo, D.; Yan, Z.; Zhao, Y. Allenamide as a bioisostere of acrylamide in the design and synthesis of targeted covalent inhibitors. MedChemComm 2018, 9, 244–253. [Google Scholar] [CrossRef]
- Casimiro-Garcia, A.; Trujillo, J.I.; Vajdos, F.; Juba, B.; Banker, M.E.; Aulabaugh, A.; Balbo, P.; Bauman, J.; Chrencik, J.; Coe, J.W.; et al. Identification of Cyanamide-Based Janus Kinase 3 (JAK3) Covalent Inhibitors. J. Med. Chem. 2018, 61, 10665–10699. [Google Scholar] [CrossRef] [PubMed]
- Al-Khawaldeh, I.; Al Yasiri, M.J.; Aldred, G.G.; Basmadjian, C.; Bordoni, C.; Harnor, S.J.; Heptinstall, A.B.; Hobson, S.J.; Jennings, C.E.; Khalifa, S.; et al. An Alkynylpyrimidine-Based Covalent Inhibitor That Targets a Unique Cysteine in NF-κB-Inducing Kinase. J. Med. Chem. 2021, 64, 10001–10018. [Google Scholar] [CrossRef]
- Reddi, R.N.; Resnick, E.; Rogel, A.; Rao, B.V.; Gabizon, R.; Goldenberg, K.; Gurwicz, N.; Zaidman, D.; Plotnikov, A.; Barr, H.; et al. Tunable Methacrylamides for Covalent Ligand Directed Release Chemistry. J. Am. Chem. Soc. 2021, 143, 4979–4992. [Google Scholar] [CrossRef] [PubMed]
- Mons, E.; Jansen, I.D.C.; Loboda, J.; van Doodewaerd, B.R.; Hermans, J.; Verdoes, M.; van Boeckel, C.A.A.; van Veelen, P.A.; Turk, B.; Turk, D.; et al. The Alkyne Moiety as a Latent Electrophile in Irreversible Covalent Small Molecule Inhibitors of Cathepsin, K. J. Am. Chem. Soc. 2019, 141, 3507–3514. [Google Scholar] [CrossRef] [Green Version]
- Cossar, P.J.; Wolter, M.; van Dijck, L.; Valenti, D.; Levy, L.M.; Ottmann, C.; Brunsveld, L. Reversible Covalent Imine-Tethering for Selective Stabilization of 14-3-3 Hub Protein Interactions. J. Am. Chem. Soc. 2021, 143, 8454–8464. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.M.; Paavilainen, V.O.; Krishnan, S.; Serafimova, I.M.; Taunton, J. Electrophilic Fragment-Based Design of Reversible Covalent Kinase Inhibitors. J. Am. Chem. Soc. 2013, 135, 5298–5301. [Google Scholar] [CrossRef] [Green Version]
- Reja, R.M.; Wang, W.; Lyu, Y.; Haeffner, F.; Gao, J. Lysine-Targeting Reversible Covalent Inhibitors with Long Residence Time. J. Am. Chem. Soc. 2022, 144, 1152–1157. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.; Miller, R.M.; Tian, B.; Mullins, R.D.; Jacobson, M.P.; Taunton, J. Design of Reversible, Cysteine-Targeted Michael Acceptors Guided by Kinetic and Computational Analysis. J. Am. Chem. Soc. 2014, 136, 12624–12630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanier, M.; Cole, D.C.; Istratiy, Y.; Klein, M.G.; Schwartz, P.A.; Tjhen, R.; Jennings, A.; Hixon, M.S. Repurposing Suzuki Coupling Reagents as a Directed Fragment Library Targeting Serine Hydrolases and Related Enzymes. J. Med. Chem. 2017, 60, 5209–5215. [Google Scholar] [CrossRef] [Green Version]
- Zheng, M.; Chen, F.-J.; Li, K.; Reja, R.M.; Haeffner, F.; Gao, J. Lysine-Targeted Reversible Covalent Ligand Discovery for Proteins via Phage Display. J. Am. Chem. Soc. 2022, 144, 15885–15893. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, J.M.; McFarland, J.M.; Paavilainen, V.O.; Bisconte, A.; Tam, D.; Phan, V.T.; Romanov, S.; Finkle, D.; Shu, J.; Patel, V.; et al. Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat. Chem. Biol. 2015, 11, 525–531. [Google Scholar] [CrossRef] [Green Version]
- Du, H.; Gao, J.; Weng, G.; Ding, J.; Chai, X.; Pang, J.; Kang, Y.; Li, D.; Cao, D.; Hou, T. CovalentInDB: A comprehensive database facilitating the discovery of covalent inhibitors. Nucleic Acids Res. 2021, 49, D1122–D1129. [Google Scholar] [CrossRef]
- Péczka, N.; Orgován, Z.; Ábrányi-Balogh, P.; Keserű, G.M. Electrophilic warheads in covalent drug discovery: An overview. Expert Opin. Drug. Discov. 2022, 17, 413–422. [Google Scholar] [CrossRef]
- Wang, X.; Lin, Z.; Bustin, K.A.; McKnight, N.R.; Parsons, W.H.; Matthews, M.L. Discovery of Potent and Selective Inhibitors against Protein-Derived Electrophilic Cofactors. J. Am. Chem. Soc. 2022, 144, 5377–5388. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Kerry, P.S.; Nanson, J.D.; Bosanac, T.; Sasaki, Y.; Krauss, R.; Saikot, F.K.; Adams, S.E.; Mosaiab, T.; Masic, V.; et al. Structural basis of SARM1 activation, substrate recognition, and inhibition by small molecules. Mol. Cell 2022, 82, 1643–1659. [Google Scholar] [CrossRef]
- Mao, Y.; Soni, K.; Sangani, C.; Yao, Y. An Overview of Privileged Scaffold: Quinolines and Isoquinolines in Medicinal Chemistry as Anticancer Agents. Curr. Top. Med. Chem. 2020, 20, 2599–2633. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, E.J. Chapter 1 Privileged Scaffolds in Medicinal Chemistry: An Introduction. In Privileged Scaffolds in Medicinal Chemistry: Design, Synthesis, Evaluation; The Royal Society of Chemistry: London, UK, 2016; pp. 1–15. [Google Scholar]
- Montaño, J.L.; Wang, B.J.; Volk, R.F.; Warrington, S.E.; Garda, V.G.; Hofmann, K.L.; Chen, L.C.; Zaro, B.W. Improved Electrophile Design for Exquisite Covalent Molecule Selectivity. ACS Chem. Biol. 2022, 17, 1440–1449. [Google Scholar] [CrossRef]
- Jöst, C.; Nitsche, C.; Scholz, T.; Roux, L.; Klein, C.D. Promiscuity and Selectivity in Covalent Enzyme Inhibition: A Systematic Study of Electrophilic Fragments. J. Med. Chem. 2014, 57, 7590–7599. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.S.; MacKenzie, C.J.; Fletcher, D.; Gilbert, I.H. Characterising covalent warhead reactivity. Bioorg. Med. Chem. 2019, 27, 2066–2074. [Google Scholar] [CrossRef]
- Awoonor-Williams, E.; Rowley, C.N. How Reactive are Druggable Cysteines in Protein Kinases? J. Chem. Inf. Model. 2018, 58, 1935–1946. [Google Scholar] [CrossRef] [Green Version]
- Awoonor-Williams, E.; Kennedy, J.; Rowley, C.N. Chapter Six—Measuring and predicting warhead and residue reactivity. In Annual Reports in Medicinal Chemistry; Ward, R.A., Grimster, N.P., Eds.; Academic Press: Cambridge, MA, USA, 2021; pp. 203–227. [Google Scholar]
- Flanagan, M.E.; Abramite, J.A.; Anderson, D.P.; Aulabaugh, A.; Dahal, U.P.; Gilbert, A.M.; Li, C.; Montgomery, J.; Oppenheimer, S.R.; Ryder, T.; et al. Chemical and Computational Methods for the Characterization of Covalent Reactive Groups for the Prospective Design of Irreversible Inhibitors. J. Med. Chem. 2014, 57, 10072–10079. [Google Scholar] [CrossRef]
- Ábrányi-Balogh, P.; Petri, L.; Imre, T.; Szijj, P.; Scarpino, A.; Hrast, M.; Mitrović, A.; Fonovič, U.P.; Németh, K.; Barreteau, H.; et al. A road map for prioritizing warheads for cysteine targeting covalent inhibitors. Eur. J. Med. Chem. 2018, 160, 94–107. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Hatcher, J.M.; Teng, M.; Gray, N.S.; Kostic, M. Recent Advances in Selective and Irreversible Covalent Ligand Development and Validation. Cell. Chem. Biol. 2019, 26, 1486–1500. [Google Scholar] [CrossRef] [PubMed]
- Böhme, A.; Thaens, D.; Paschke, A.; Schüürmann, G. Kinetic Glutathione Chemoassay to Quantify Thiol Reactivity of Organic Electrophiles—Application to α,β-Unsaturated Ketones, Acrylates, and Propiolates. Chem. Res. Toxicol. 2009, 22, 742–750. [Google Scholar] [CrossRef]
- Cee, V.J.; Volak, L.P.; Chen, Y.; Bartberger, M.D.; Tegley, C.; Arvedson, T.; McCarter, J.; Tasker, A.S.; Fotsch, C. Systematic Study of the Glutathione (GSH) Reactivity of N-Arylacrylamides: 1. Effects of Aryl Substitution. J. Med. Chem. 2015, 58, 9171–9178. [Google Scholar] [CrossRef] [PubMed]
- Birkholz, A.; Kopecky, D.J.; Volak, L.P.; Bartberger, M.D.; Chen, Y.; Tegley, C.M.; Arvedson, T.; McCarter, J.D.; Fotsch, C.; Cee, V.J. Systematic Study of the Glutathione Reactivity of N-Phenylacrylamides: 2. Effects of Acrylamide Substitution. J. Med. Chem. 2020, 63, 11602–11614. [Google Scholar] [CrossRef]
- Dahal, U.P.; Gilbert, A.M.; Obach, R.S.; Flanagan, M.E.; Chen, J.M.; Garcia-Irizarry, C.; Starr, J.T.; Schuff, B.; Uccello, D.P.; Young, J.A. Intrinsic reactivity profile of electrophilic moieties to guide covalent drug design: N-α-acetyl-l-lysine as an amine nucleophile. MedChemComm 2016, 7, 864–872. [Google Scholar] [CrossRef]
- Mayer, R.J.; Ofial, A.R. Nucleophilicity of Glutathione: A Link to Michael Acceptor Reactivities. Angew. Chem. Int. Ed. 2019, 58, 17704–17708. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, P.A.; Kuzmic, P.; Solowiej, J.; Bergqvist, S.; Bolanos, B.; Almaden, C.; Nagata, A.; Ryan, K.; Feng, J.; Dalvie, D.; et al. Covalent EGFR inhibitor analysis reveals importance of reversible interactions to potency and mechanisms of drug resistance. Proc. Natl. Acad. Sci. USA 2014, 111, 173–178. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Jiang, W.; Chatterjee, P.; Luo, Y. Ranking Reversible Covalent Drugs: From Free Energy Perturbation to Fragment Docking. J. Chem. Inf. Model. 2019, 59, 2093–2102. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.; Botello-Smith, W.M.; Zhang, H.; Qian, L.; Alsamarah, A.; Kent, D.; Lacroix, J.J.; Baudry, M.; Luo, Y. Can Relative Binding Free Energy Predict Selectivity of Reversible Covalent Inhibitors? J. Am. Chem. Soc. 2017, 139, 17945–17952. [Google Scholar] [CrossRef] [Green Version]
- Resnick, E.; Bradley, A.; Gan, J.; Douangamath, A.; Krojer, T.; Sethi, R.; Geurink, P.P.; Aimon, A.; Amitai, G.; Bellini, D.; et al. Rapid Covalent-Probe Discovery by Electrophile-Fragment Screening. J. Am. Chem. Soc. 2019, 141, 8951–8968. [Google Scholar] [CrossRef] [Green Version]
- Craven, G.B.; Affron, D.P.; Allen, C.E.; Matthies, S.; Greener, J.G.; Morgan, R.M.L.; Tate, E.W.; Armstrong, A.; Mann, D.J. High-Throughput Kinetic Analysis for Target-Directed Covalent Ligand Discovery. Angew. Chem. Int. Ed. 2018, 57, 5257–5261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Craven, G.B.; Affron, D.P.; Kösel, T.; Wong, T.L.M.; Jukes, Z.H.; Liu, C.-T.; Morgan, R.M.L.; Armstrong, A.; Mann, D.J. Multiparameter Kinetic Analysis for Covalent Fragment Optimization by Using Quantitative Irreversible Tethering (qIT). Chembiochem 2020, 21, 3417–3422. [Google Scholar] [CrossRef]
- Schultz, T.W.; Yarbrough, J.W.; Johnson, E.L. Structure–activity relationships for reactivity of carbonyl-containing compounds with glutathione. SAR QSAR Environ. Res. 2005, 16, 313–322. [Google Scholar] [CrossRef]
- Mayr, H.; Ofial, A.R. A quantitative approach to polar organic reactivity. SAR QSAR Environ. Res. 2015, 26, 619–646. [Google Scholar] [CrossRef]
- Palazzesi, F.; Hermann, M.R.; Grundl, M.A.; Pautsch, A.; Seeliger, D.; Tautermann, C.S.; Weber, A. BIreactive: A Machine-Learning Model to Estimate Covalent Warhead Reactivity. J. Chem. Inf. Model. 2020, 60, 2915–2923. [Google Scholar] [CrossRef]
- Krenske, E.H.; Petter, R.C.; Houk, K.N. Kinetics and Thermodynamics of Reversible Thiol Additions to Mono- and Diactivated Michael Acceptors: Implications for the Design of Drugs That Bind Covalently to Cysteines. J. Org. Chem. 2016, 81, 11726–11733. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, R.; Burgess, J.; Colclough, N.; Davies, N.L.; Lenz, E.M.; Orton, A.L.; Ward, R.A. Expanding the Armory: Predicting and Tuning Covalent Warhead Reactivity. J. Chem. Inf. Model. 2017, 57, 3124–3137. [Google Scholar] [CrossRef]
- Voice, A.; Tresadern, G.; van Vlijmen, H.; Mulholland, A. Limitations of Ligand-Only Approaches for Predicting the Reactivity of Covalent Inhibitors. J. Chem. Inf. Model. 2019, 59, 4220–4227. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.M.; Rowley, C.N. Automated computational screening of the thiol reactivity of substituted alkenes. J. Comput. Aided Mol. Des. 2015, 29, 725–735. [Google Scholar] [CrossRef]
- Palazzesi, F.; Grundl, M.A.; Pautsch, A.; Weber, A.; Tautermann, C.S. A Fast Ab Initio Predictor Tool for Covalent Reactivity Estimation of Acrylamides. J. Chem. Inf. Model. 2019, 59, 3565–3571. [Google Scholar] [CrossRef] [PubMed]
- Hermann, M.R.; Pautsch, A.; Grundl, M.A.; Weber, A.; Tautermann, C.S. Covalent inhibitor reactivity prediction by the electrophilicity index—In and out of scope. J. Comput. Aided Mol. Des. 2021, 35, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Sure, R.; El Mahdali, M.; Plajer, A.; Deglmann, P. Towards a converged strategy for including microsolvation in reaction mechanism calculations. J. Comput. Aided Mol. Des. 2021, 35, 473–492. [Google Scholar] [CrossRef]
- Tavakoli, M.; Mood, A.; Van Vranken, D.; Baldi, P. Quantum Mechanics and Machine Learning Synergies: Graph Attention Neural Networks to Predict Chemical Reactivity. J. Chem. Inf. Model. 2022, 62, 2121–2132. [Google Scholar] [CrossRef] [PubMed]
- Palazzesi, F.; Pozzan, A. Deep Learning Applied to Ligand-Based De Novo Drug Design. In Artificial Intelligence in Drug Design; Heifetz, A., Ed.; Springer: New York, NY, USA, 2022; pp. 273–299. [Google Scholar]
- Zhang, Y.; Zhang, D.; Tian, H.; Jiao, Y.; Shi, Z.; Ran, T.; Liu, H.; Lu, S.; Xu, A.; Qiao, X.; et al. Identification of Covalent Binding Sites Targeting Cysteines Based on Computational Approaches. Mol. Pharm. 2016, 13, 3106–3118. [Google Scholar] [CrossRef]
- Awoonor-Williams, E.; Walsh, A.G.; Rowley, C.N. Modeling covalent-modifier drugs. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 1664–1675. [Google Scholar] [CrossRef] [PubMed]
- Schirmeister, T.; Kesselring, J.; Jung, S.; Schneider, T.H.; Weickert, A.; Becker, J.; Lee, W.; Bamberger, D.; Wich, P.R.; Distler, U.; et al. Quantum Chemical-Based Protocol for the Rational Design of Covalent Inhibitors. J. Am. Chem. Soc. 2016, 138, 8332–8335. [Google Scholar] [CrossRef] [PubMed]
- Arafet, K.; Serrano-Aparicio, N.; Lodola, A.; Mulholland, A.J.; González, F.V.; Świderek, K.; Moliner, V. Mechanism of inhibition of SARS-CoV-2 Mpro by N3 peptidyl Michael acceptor explained by QM/MM simulations and design of new derivatives with tunable chemical reactivity. Chem. Sci. 2021, 12, 1433–1444. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, A.M.; Oliveira, A.R.S.; da Costa, C.H.S.; Kenny, P.W.; Montanari, C.A.; Varela, J.d.J.G.; Lameira, J. Assessment of Reversibility for Covalent Cysteine Protease Inhibitors Using Quantum Mechanics/Molecular Mechanics Free Energy Surfaces. J. Chem. Inf. Model. 2022, 62, 4083–4094. [Google Scholar] [CrossRef]
- Abe, Y.; Shoji, M.; Nishiya, Y.; Aiba, H.; Kishimoto, T.; Kitaura, K. The reaction mechanism of sarcosine oxidase elucidated using FMO and QM/MM methods. Phys. Chem. Chem. Phys. 2017, 19, 9811–9822. [Google Scholar] [CrossRef]
- Tautermann, C.S. Current and Future Challenges in Modern Drug Discovery. In Quantum Mechanics in Drug Discovery; Heifetz, A., Ed.; Springer: New York, NY, USA, 2020; pp. 1–17. [Google Scholar]
- Mihalovits, L.M.; Ferenczy, G.G.; Keserű, G.M. The role of quantum chemistry in covalent inhibitor design. Int. J. Quantum Chem. 2022, 122, e26768. [Google Scholar] [CrossRef]
- Mihalovits, L.M.; Ferenczy, G.G.; Keserű, G.M. Affinity and Selectivity Assessment of Covalent Inhibitors by Free Energy Calculations. J. Chem. Inf. Model. 2020, 60, 6579–6594. [Google Scholar] [CrossRef]
- Sotriffer, C. Docking of Covalent Ligands: Challenges and Approaches. Mol. Inform. 2018, 37, 1800062. [Google Scholar] [CrossRef] [PubMed]
- Kumalo, H.M.; Bhakat, S.; Soliman, M.E.S. Theory and Applications of Covalent Docking in Drug Discovery: Merits and Pitfalls. Molecules 2015, 20, 1984–2000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianco, G.; Goodsell, D.S.; Forli, S. Selective and Effective: Current Progress in Computational Structure-Based Drug Discovery of Targeted Covalent Inhibitors. Trends Pharmacol. Sci. 2020, 41, 1038–1049. [Google Scholar] [CrossRef] [PubMed]
- Borsari, C.; Keles, E.; McPhail, J.A.; Schaefer, A.; Sriramaratnam, R.; Goch, W.; Schaefer, T.; De Pascale, M.; Bal, W.; Gstaiger, M.; et al. Covalent Proximity Scanning of a Distal Cysteine to Target PI3Kα. J. Am. Chem. Soc. 2022, 144, 6326–6342. [Google Scholar] [CrossRef]
- Chowdhury, S.R.; Kennedy, S.; Zhu, K.; Mishra, R.; Chuong, P.; Nguyen, A.U.; Kathman, S.G.; Statsyuk, A.V. Discovery of covalent enzyme inhibitors using virtual docking of covalent fragments. Bioorg. Med. Chem. Lett. 2019, 29, 36–39. [Google Scholar] [CrossRef]
- Wen, C.; Yan, X.; Gu, Q.; Du, J.; Wu, D.; Lu, Y.; Zhou, H.; Xu, J. Systematic Studies on the Protocol and Criteria for Selecting a Covalent Docking Tool. Molecules 2019, 24, 2183. [Google Scholar] [CrossRef] [Green Version]
- Zaidman, D.; Gehrtz, P.; Filep, M.; Fearon, D.; Gabizon, R.; Douangamath, A.; Prilusky, J.; Duberstein, S.; Cohen, G.; Owen, C.D.; et al. An automatic pipeline for the design of irreversible derivatives identifies a potent SARS-CoV-2 M(pro) inhibitor. Cell Chem. Biol. 2021, 28, 1795–1806. [Google Scholar] [CrossRef]
- Scarpino, A.; Ferenczy, G.G.; Keserű, G.M. Covalent Docking in Drug Discovery: Scope and Limitations. Curr. Pharm. Des. 2020, 26, 5684–5699. [Google Scholar] [CrossRef] [PubMed]
- Mortenson, D.E.; Brighty, G.J.; Plate, L.; Bare, G.; Chen, W.; Li, S.; Wang, H.; Cravatt, B.F.; Forli, S.; Powers, E.T.; et al. “Inverse Drug Discovery” Strategy to Identify Proteins That Are Targeted by Latent Electrophiles As Exemplified by Aryl Fluorosulfates. J. Am. Chem. Soc. 2018, 140, 200–210. [Google Scholar] [CrossRef]
- Zheng, Q.; Woehl, J.L.; Kitamura, S.; Santos-Martins, D.; Smedley, C.J.; Li, G.; Forli, S.; Moses, J.E.; Wolan, D.W.; Sharpless, K.B. SuFEx-enabled, agnostic discovery of covalent inhibitors of human neutrophil elastase. Proc. Natl. Acad. Sci. USA 2019, 116, 18808–18814. [Google Scholar] [CrossRef]
- Vinogradova, E.V.; Zhang, X.; Remillard, D.; Lazar, D.C.; Suciu, R.M.; Wang, Y.; Bianco, G.; Yamashita, Y.; Crowley, V.M.; Schafroth, M.A.; et al. An Activity-Guided Map of Electrophile-Cysteine Interactions in Primary Human T Cells. Cell 2020, 182, 1009–1026. [Google Scholar] [CrossRef]
- Bon, M.; Bilsland, A.; Bower, J.; McAulay, K. Fragment-based drug discovery-the importance of high-quality molecule libraries. Mol. Oncol. 2022, 16, 3761–3777. [Google Scholar] [CrossRef]
- Wan, X.; Yang, T.; Cuesta, A.; Pang, X.; Balius, T.E.; Irwin, J.J.; Shoichet, B.K.; Taunton, J. Discovery of Lysine-Targeted eIF4E Inhibitors through Covalent Docking. J. Am. Chem. Soc. 2020, 142, 4960–4964. [Google Scholar] [CrossRef]
- Hatmal, M.m.M.; Abuyaman, O.; Taha, M. Docking-generated multiple ligand poses for bootstrapping bioactivity classifying Machine Learning: Repurposing covalent inhibitors for COVID-19-related TMPRSS2 as case study. Comput. Struct. Biotechnol. 2021, 19, 4790–4824. [Google Scholar] [CrossRef] [PubMed]
- Erlanson, D.A.; Fesik, S.W.; Hubbard, R.E.; Jahnke, W.; Jhoti, H. Twenty years on: The impact of fragments on drug discovery. Nat. Rev. Drug. Discov. 2016, 15, 605–619. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Zhao, T.; Kang, D.; Zhang, J.; Song, Y.; Namasivayam, V.; Kongsted, J.; Pannecouque, C.; De Clercq, E.; Poongavanam, V.; et al. Overview of Recent Strategic Advances in Medicinal Chemistry. J. Med. Chem. 2019, 62, 9375–9414. [Google Scholar] [CrossRef]
- Jahnke, W.; Erlanson, D.A.; de Esch, I.J.P.; Johnson, C.N.; Mortenson, P.N.; Ochi, Y.; Urushima, T. Fragment-to-Lead Medicinal Chemistry Publications in 2019. J. Med. Chem. 2020, 63, 15494–15507. [Google Scholar] [CrossRef]
- De Esch, I.J.P.; Erlanson, D.A.; Jahnke, W.; Johnson, C.N.; Walsh, L. Fragment-to-Lead Medicinal Chemistry Publications in 2020. J. Med. Chem. 2022, 65, 84–99. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L.; Groom, C.R.; Alex, A. Ligand efficiency: A useful metric for lead selection. Drug. Discov. Today 2004, 9, 430–431. [Google Scholar] [CrossRef]
- Kuntz, I.D.; Chen, K.; Sharp, K.A.; Kollman, P.A. The maximal affinity of ligands. Proc. Natl. Acad. Sci. USA 1999, 96, 9997–10002. [Google Scholar] [CrossRef]
- Murray, C.W.; Rees, D.C. The rise of fragment-based drug discovery. Nat. Chem. 2009, 1, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Carbery, A.; Skyner, R.; von Delft, F.; Deane, C.M. Fragment Libraries Designed to Be Functionally Diverse Recover Protein Binding Information More Efficiently Than Standard Structurally Diverse Libraries. J. Med. Chem. 2022, 65, 11404–11413. [Google Scholar] [CrossRef]
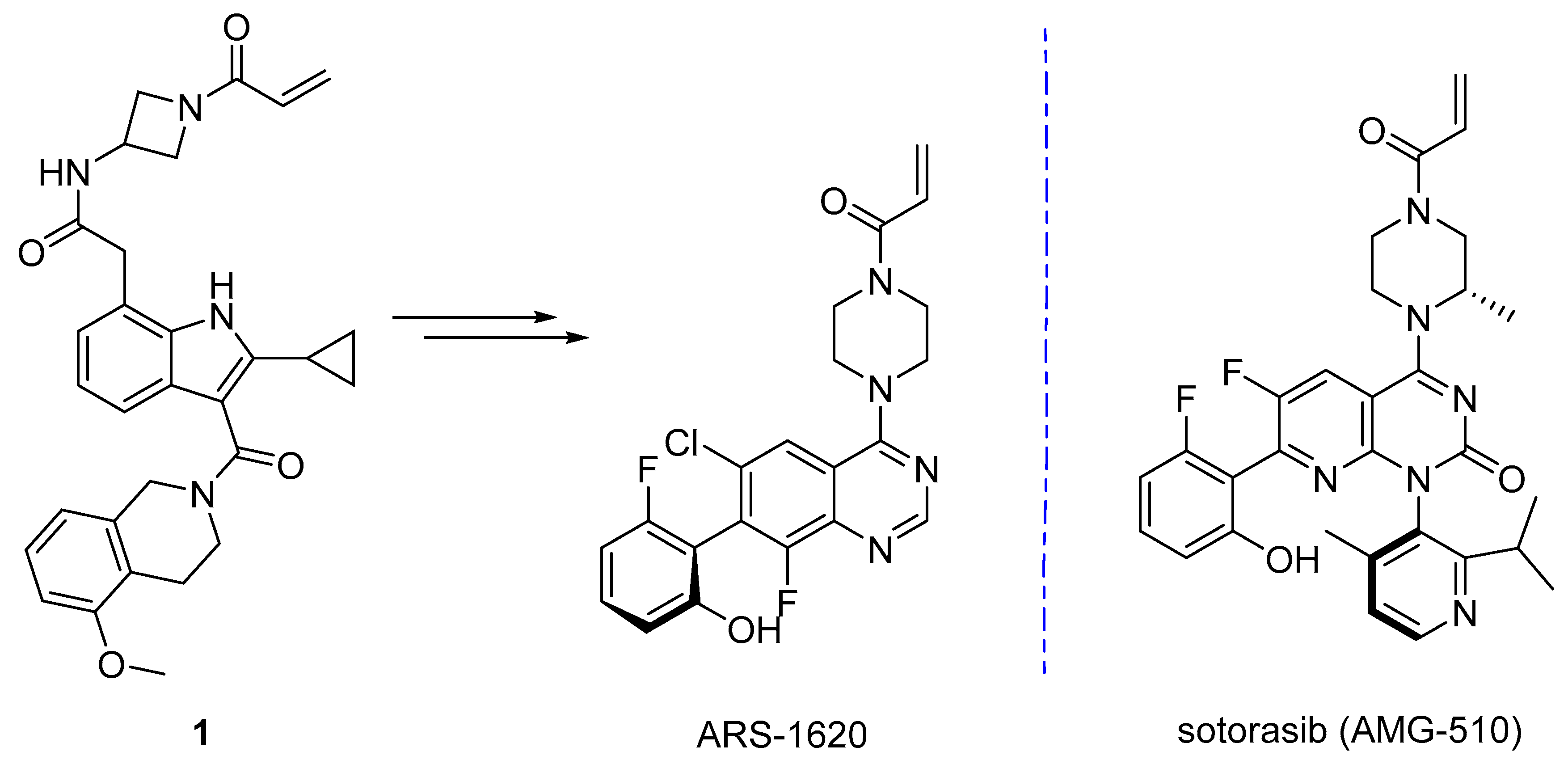
- Lanman, B.A.; Allen, J.R.; Allen, J.G.; Amegadzie, A.K.; Ashton, K.S.; Booker, S.K.; Chen, J.J.; Chen, N.; Frohn, M.J.; Goodman, G.; et al. Discovery of a Covalent Inhibitor of KRAS(G12C) (AMG 510) for the Treatment of Solid Tumors. J. Med. Chem. 2020, 63, 52–65. [Google Scholar] [CrossRef] [Green Version]
- Congreve, M.; Carr, R.; Murray, C.; Jhoti, H. A ’rule of three’ for fragment-based lead discovery? Drug. Discov. Today 2003, 8, 876–877. [Google Scholar] [CrossRef]
- Jhoti, H.; Williams, G.; Rees, D.C.; Murray, C.W. The ’rule of three’ for fragment-based drug discovery: Where are we now? Nat. Rev. Drug. Discov. 2013, 12, 644. [Google Scholar] [CrossRef] [Green Version]
- Köster, H.; Craan, T.; Brass, S.; Herhaus, C.; Zentgraf, M.; Neumann, L.; Heine, A.; Klebe, G. A Small Nonrule of 3 Compatible Fragment Library Provides High Hit Rate of Endothiapepsin Crystal Structures with Various Fragment Chemotypes. J. Med. Chem. 2011, 54, 7784–7796. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, P.; Hartman, A.M.; Hirsch, A.K.H.; Empting, M. Concepts and Core Principles of Fragment-Based Drug Design. Molecules 2019, 24, 4309. [Google Scholar] [CrossRef] [Green Version]
- Edfeldt, F.N.; Folmer, R.H.; Breeze, A.L. Fragment screening to predict druggability (ligandability) and lead discovery success. Drug. Discov. Today 2011, 16, 284–287. [Google Scholar] [CrossRef]
- Lagoutte, R.; Patouret, R.; Winssinger, N. Covalent inhibitors: An opportunity for rational target selectivity. Curr. Opin. Chem. Biol. 2017, 39, 54–63. [Google Scholar] [CrossRef] [Green Version]
- Olp, M.D.; Sprague, D.J.; Goetz, C.J.; Kathman, S.G.; Wynia-Smith, S.L.; Shishodia, S.; Summers, S.B.; Xu, Z.; Statsyuk, A.V.; Smith, B.C. Covalent-Fragment Screening of BRD4 Identifies a Ligandable Site Orthogonal to the Acetyl-Lysine Binding Sites. ACS Chem. Biol. 2020, 15, 1036–1049. [Google Scholar] [CrossRef] [PubMed]
- Darby, J.F.; Atobe, M.; Firth, J.D.; Bond, P.; Davies, G.J.; O’Brien, P.; Hubbard, R.E. Increase of enzyme activity through specific covalent modification with fragments. Chem. Sci. 2017, 8, 7772–7779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browne, C.M.; Jiang, B.; Ficarro, S.B.; Doctor, Z.M.; Johnson, J.L.; Card, J.D.; Sivakumaren, S.C.; Alexander, W.M.; Yaron, T.M.; Murphy, C.J.; et al. A Chemoproteomic Strategy for Direct and Proteome-Wide Covalent Inhibitor Target-Site Identification. J. Am. Chem. Soc. 2019, 141, 191–203. [Google Scholar] [CrossRef]
- Backus, K.M.; Correia, B.E.; Lum, K.M.; Forli, S.; Horning, B.D.; González-Páez, G.E.; Chatterjee, S.; Lanning, B.R.; Teijaro, J.R.; Olson, A.J.; et al. Proteome-wide covalent ligand discovery in native biological systems. Nature 2016, 534, 570–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowley, V.M.; Thielert, M.; Cravatt, B.F. Functionalized Scout Fragments for Site-Specific Covalent Ligand Discovery and Optimization. ACS Cent. Sci. 2021, 7, 613–623. [Google Scholar] [CrossRef]
- Kuljanin, M.; Mitchell, D.C.; Schweppe, D.K.; Gikandi, A.S.; Nusinow, D.P.; Bulloch, N.J.; Vinogradova, E.V.; Wilson, D.L.; Kool, E.T.; Mancias, J.D.; et al. Reimagining high-throughput profiling of reactive cysteines for cell-based screening of large electrophile libraries. Nat. Biotechnol. 2021, 39, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Jia, G.; Guo, J.; Liu, Y.; Wang, C. Quantitative Chemoproteomic Profiling with Data-Independent Acquisition-Based Mass Spectrometry. J. Am. Chem. Soc. 2022, 144, 901–911. [Google Scholar] [CrossRef]
- Hacker, S.M.; Backus, K.M.; Lazear, M.R.; Forli, S.; Correia, B.E.; Cravatt, B.F. Global profiling of lysine reactivity and ligandability in the human proteome. Nat. Chem. 2017, 9, 1181–1190. [Google Scholar] [CrossRef]
- Yan, T.; Desai, H.S.; Boatner, L.M.; Yen, S.L.; Cao, J.; Palafox, M.F.; Jami-Alahmadi, Y.; Backus, K.M. SP3-FAIMS Chemoproteomics for High-Coverage Profiling of the Human Cysteinome. Chembiochem 2021, 22, 1841–1851. [Google Scholar] [CrossRef] [PubMed]
- Abbasov, M.E.; Kavanagh, M.E.; Ichu, T.-A.; Lazear, M.R.; Tao, Y.; Crowley, V.M.; am Ende, C.W.; Hacker, S.M.; Ho, J.; Dix, M.M.; et al. A proteome-wide atlas of lysine-reactive chemistry. Nat. Chem. 2021, 13, 1081–1092. [Google Scholar] [CrossRef]
- Litwin, K.; Crowley, V.M.; Suciu, R.M.; Boger, D.L.; Cravatt, B.F. Chemical proteomic identification of functional cysteines with atypical electrophile reactivities. Tetrahedron Lett. 2021, 67, 152861. [Google Scholar] [CrossRef]
- Tolmachova, K.A.; Moroz, Y.S.; Konovets, A.; Platonov, M.O.; Vasylchenko, O.V.; Borysko, P.; Zozulya, S.; Gryniukova, A.; Bogolubsky, A.V.; Pipko, S.; et al. (Chlorosulfonyl)benzenesulfonyl Fluorides—Versatile Building Blocks for Combinatorial Chemistry: Design, Synthesis and Evaluation of a Covalent Inhibitor Library. ACS Comb. Sci. 2018, 20, 672–680. [Google Scholar] [CrossRef]
- Liu, R.; Yue, Z.; Tsai, C.-C.; Shen, J. Assessing Lysine and Cysteine Reactivities for Designing Targeted Covalent Kinase Inhibitors. J. Am. Chem. Soc. 2019, 141, 6553–6560. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Liu, Q.; Bliven, S.; Xie, L.; Bourne, P.E. Determining Cysteines Available for Covalent Inhibition Across the Human Kinome. J. Med. Chem. 2017, 60, 2879–2889. [Google Scholar] [CrossRef] [Green Version]
- McGregor, L.M.; Jenkins, M.L.; Kerwin, C.; Burke, J.E.; Shokat, K.M. Expanding the Scope of Electrophiles Capable of Targeting K-Ras Oncogenes. Biochemistry 2017, 56, 3178–3183. [Google Scholar] [CrossRef] [Green Version]
- Shraga, A.; Resnick, E.; Gabizon, R.; London, N. Chapter Eight—Covalent fragment screening. In Annual Reports in Medicinal Chemistry; Ward, R.A., Grimster, N.P., Eds.; Academic Press: Cambridge, MA, USA, 2021; pp. 243–265. [Google Scholar]
- Lu, W.; Kostic, M.; Zhang, T.; Che, J.; Patricelli, M.P.; Jones, L.H.; Chouchani, E.T.; Gray, N.S. Fragment-based covalent ligand discovery. RSC Chem. Biol. 2021, 2, 354–367. [Google Scholar] [CrossRef]
- Parker, C.G.; Galmozzi, A.; Wang, Y.; Correia, B.E.; Sasaki, K.; Joslyn, C.M.; Kim, A.S.; Cavallaro, C.L.; Lawrence, R.M.; Johnson, S.R.; et al. Ligand and Target Discovery by Fragment-Based Screening in Human Cells. Cell 2017, 168, 527–541.e29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.; Murray, J.B.; Luo, H.; Cheng, X.; Chen, Q.; Song, C.; Duan, C.; Tan, P.; Zhang, L.; Liu, J.; et al. PAC-FragmentDEL—Photoactivated covalent capture of DNA-encoded fragments for hit discovery. RSC Med. Chem. 2022. [Google Scholar] [CrossRef]
- Mullard, A. Fragment-based screening sees the light. Nat. Rev. Drug. Discov. 2020, 19, 742–743. [Google Scholar] [CrossRef]
- Erlanson, D.A.; Hansen, S.K. Making drugs on proteins: Site-directed ligand discovery for fragment-based lead assembly. Curr. Opin. Chem. Biol. 2004, 8, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Erlanson, D.A.; Wells, J.A.; Braisted, A.C. Tethering: Fragment-Based Drug Discovery. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 199–223. [Google Scholar] [CrossRef]
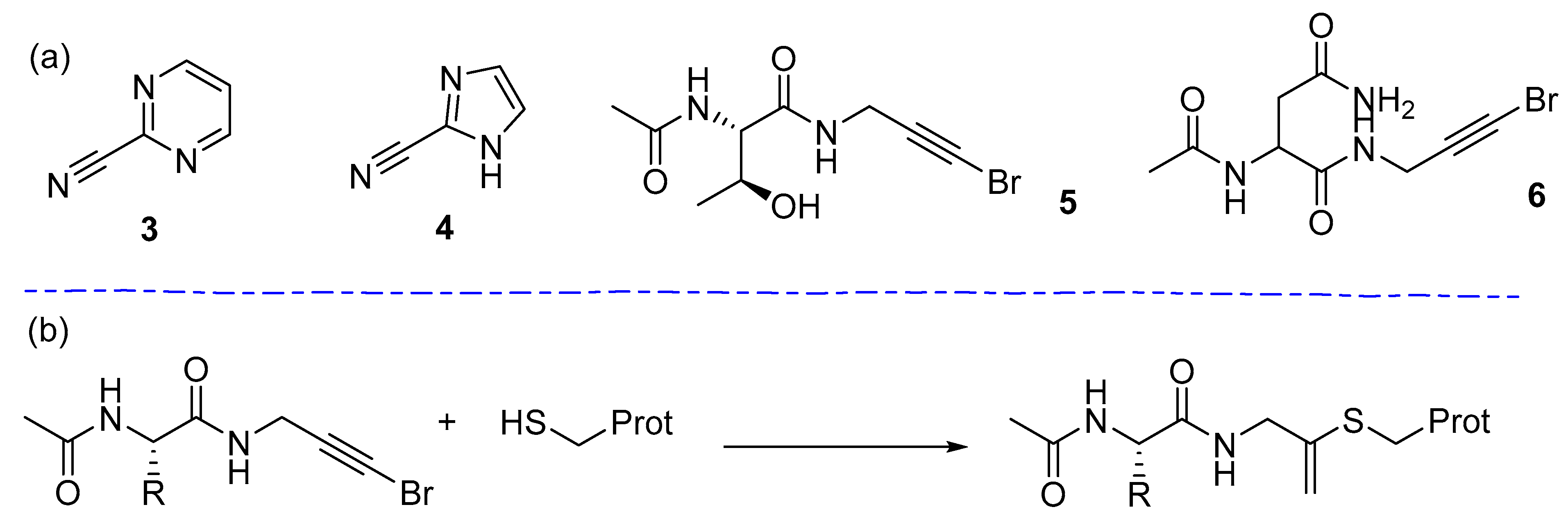
- Kathman, S.G.; Statsyuk, A.V. Covalent tethering of fragments for covalent probe discovery. MedChemComm 2016, 7, 576–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [Green Version]
- Shin, Y.; Jeong, J.W.; Wurz, R.P.; Achanta, P.; Arvedson, T.; Bartberger, M.D.; Campuzano, I.D.G.; Fucini, R.; Hansen, S.K.; Ingersoll, J.; et al. Discovery of N-(1-Acryloylazetidin-3-yl)-2-(1H-indol-1-yl)acetamides as Covalent Inhibitors of KRASG12C. ACS Med. Chem. Lett. 2019, 10, 1302–1308. [Google Scholar] [CrossRef]
- Dalvit, C. NMR methods in fragment screening: Theory and a comparison with other biophysical techniques. Drug. Discov. Today 2009, 14, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Phan, J.; Friberg, A.R.; Camper, D.V.; Olejniczak, E.T.; Fesik, S.W. A method for the second-site screening of K-Ras in the presence of a covalently attached first-site ligand. J. Biomol. NMR 2014, 60, 11–14. [Google Scholar] [CrossRef] [Green Version]
- Keeley, A.; Petri, L.; Ábrányi-Balogh, P.; Keserű, G.M. Covalent fragment libraries in drug discovery. Drug. Discov. Today 2020, 25, 983–996. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Akahane, K.; McNally, R.; Reyskens, K.M.S.E.; Ficarro, S.B.; Liu, S.; Herter-Sprie, G.S.; Koyama, S.; Pattison, M.J.; Labella, K.; et al. Development of Selective Covalent Janus Kinase 3 Inhibitors. J. Med. Chem. 2015, 58, 6589–6606. [Google Scholar] [CrossRef] [Green Version]
- London, N.; Miller, R.M.; Krishnan, S.; Uchida, K.; Irwin, J.J.; Eidam, O.; Gibold, L.; Cimermančič, P.; Bonnet, R.; Shoichet, B.K.; et al. Covalent docking of large libraries for the discovery of chemical probes. Nat. Chem. Biol. 2014, 10, 1066–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffer, L.; Saez-Ayala, M.; Horvath, D.; Varnek, A.; Morelli, X.; Roche, P. CovaDOTS: In Silico Chemistry-Driven Tool to Design Covalent Inhibitors Using a Linking Strategy. J. Chem. Inf. Model. 2019, 59, 1472–1485. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Wen, W.; Rao, L.; Huang, Y.; Lei, M.; Liu, K.; Hu, S.; Song, R.; Ren, Y.; Wan, J. Cov_FB3D: A De Novo Covalent Drug Design Protocol Integrating the BA-SAMP Strategy and Machine-Learning-Based Synthetic Tractability Evaluation. J. Chem. Inf. Model. 2020, 60, 4388–4402. [Google Scholar] [CrossRef] [PubMed]
- Yoshimori, A.; Miljković, F.; Bajorath, J. Approach for the Design of Covalent Protein Kinase Inhibitors via Focused Deep Generative Modeling. Molecules 2022, 27, 570. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal Transduct. Target Ther. 2021, 6, 386. [Google Scholar] [CrossRef]
- Janes, M.R.; Zhang, J.; Li, L.-S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, E.C.; Drezner, N.; Li, X.; Mishra-Kalyani, P.S.; Liu, Y.; Zhao, H.; Bi, Y.; Liu, J.; Rahman, A.; Wearne, E.; et al. FDA Approval Summary: Sotorasib for KRAS G12C-Mutated Metastatic NSCLC. Clin. Cancer Res. 2022, 28, 1482–1486. [Google Scholar] [CrossRef]
- Kwan, A.K.; Piazza, G.A.; Keeton, A.B.; Leite, C.A. The path to the clinic: A comprehensive review on direct KRASG12C inhibitors. J. Exp. Clin. Cancer Res. 2022, 41, 27. [Google Scholar] [CrossRef]
- Bum-Erdene, K.; Ghozayel, M.K.; Xu, D.; Meroueh, S.O. Covalent Fragment Screening Identifies Rgl2 RalGEF Cysteine for Targeted Covalent Inhibition of Ral GTPase Activation. ChemMedChem 2022, 17, e202100750. [Google Scholar] [CrossRef] [PubMed]
- Jamshidiha, M.; Lanyon-Hogg, T.; Sutherell, C.L.; Craven, G.B.; Tersa, M.; De Vita, E.; Brustur, D.; Pérez-Dorado, I.; Hassan, S.; Petracca, R.; et al. Identification of the first structurally validated covalent ligands of the small GTPase RAB27A. RSC Med. Chem. 2022, 13, 150–155. [Google Scholar] [CrossRef]
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Clyde, A.; Galanie, S.; Kneller, D.W.; Ma, H.; Babuji, Y.; Blaiszik, B.; Brace, A.; Brettin, T.; Chard, K.; Chard, R.; et al. High-Throughput Virtual Screening and Validation of a SARS-CoV-2 Main Protease Noncovalent Inhibitor. J. Chem. Inf. Model. 2022, 62, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Rossetti, G.G.; Ossorio, M.A.; Rempel, S.; Kratzel, A.; Dionellis, V.S.; Barriot, S.; Tropia, L.; Gorgulla, C.; Arthanari, H.; Thiel, V.; et al. Non-covalent SARS-CoV-2 Mpro inhibitors developed from in silico screen hits. Sci. Rep. 2022, 12, 2505. [Google Scholar] [CrossRef]
- Huff, S.; Kummetha, I.R.; Tiwari, S.K.; Huante, M.B.; Clark, A.E.; Wang, S.; Bray, W.; Smith, D.; Carlin, A.F.; Endsley, M.; et al. Discovery and Mechanism of SARS-CoV-2 Main Protease Inhibitors. J. Med. Chem. 2022, 65, 2866–2879. [Google Scholar] [CrossRef]
- Ullrich, S.; Nitsche, C. The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett. 2020, 30, 127377. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, N.; Sacco, M.D.; Ma, C.; Hu, Y.; Townsend, J.A.; Meng, X.; Zhang, F.; Zhang, X.; Ba, M.; Szeto, T.; et al. Expedited Approach toward the Rational Design of Noncovalent SARS-CoV-2 Main Protease Inhibitors. J. Med. Chem. 2022, 65, 2848–2865. [Google Scholar] [CrossRef]
- Hoffman, R.L.; Kania, R.S.; Brothers, M.A.; Davies, J.F.; Ferre, R.A.; Gajiwala, K.S.; He, M.; Hogan, R.J.; Kozminski, K.; Li, L.Y.; et al. Discovery of Ketone-Based Covalent Inhibitors of Coronavirus 3CL Proteases for the Potential Therapeutic Treatment of COVID-19. J. Med. Chem. 2020, 63, 12725–12747. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Yao, S.; Zhao, W.; Zhang, Y.; Liu, J.; Shao, Q.; Wang, Q.; Li, M.; Xie, H.; Shang, W. Identification of pyrogallol as a warhead in design of covalent inhibitors for the SARS-CoV-2 3CL protease. Nat. Comm. 2021, 12, 3623. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Vankadara, S.; Dawson, M.D.; Fong, J.Y.; Oh, Q.Y.; Ang, Q.A.; Liu, B.; Chang, H.Y.; Koh, J.; Koh, X.; Tan, Q.W.; et al. A Warhead Substitution Study on the Coronavirus Main Protease Inhibitor Nirmatrelvir. ACS Med. Chem. Lett. 2022, 13, 1345–1350. [Google Scholar] [CrossRef]
- Konno, S.; Kobayashi, K.; Senda, M.; Funai, Y.; Seki, Y.; Tamai, I.; Schäkel, L.; Sakata, K.; Pillaiyar, T.; Taguchi, A.; et al. 3CL Protease Inhibitors with an Electrophilic Arylketone Moiety as Anti-SARS-CoV-2 Agents. J. Med. Chem. 2022, 65, 2926–2939. [Google Scholar] [CrossRef]
- Hirose, Y.; Shindo, N.; Mori, M.; Onitsuka, S.; Isogai, H.; Hamada, R.; Hiramoto, T.; Ochi, J.; Takahashi, D.; Ueda, T.; et al. Discovery of Chlorofluoroacetamide-Based Covalent Inhibitors for Severe Acute Respiratory Syndrome Coronavirus 2 3CL Protease. J. Med. Chem. 2022, 65, 13852–13865. [Google Scholar] [CrossRef] [PubMed]
- La Monica, G.; Bono, A.; Lauria, A.; Martorana, A. Targeting SARS-CoV-2 Main Protease for Treatment of COVID-19: Covalent Inhibitors Structure–Activity Relationship Insights and Evolution Perspectives. J. Med. Chem. 2022, 65, 12500–12534. [Google Scholar] [CrossRef]
- Douangamath, A.; Fearon, D.; Gehrtz, P.; Krojer, T.; Lukacik, P.; Owen, C.D.; Resnick, E.; Strain-Damerell, C.; Aimon, A.; Ábrányi-Balogh, P.; et al. Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease. Nat. Comm. 2020, 11, 5047. [Google Scholar] [CrossRef]
- Miura, C.; Shindo, N.; Okamoto, K.; Kuwata, K.; Ojida, A. Fragment-Based Discovery of Irreversible Covalent Inhibitors of Cysteine Proteases Using Chlorofluoroacetamide Library. Chem. Pharm. Bull. 2020, 68, 1074–1081. [Google Scholar] [CrossRef] [PubMed]
- Kathman, S.G.; Xu, Z.; Statsyuk, A.V. A Fragment-Based Method to Discover Irreversible Covalent Inhibitors of Cysteine Proteases. J. Med. Chem. 2014, 57, 4969–4974. [Google Scholar] [CrossRef]
- Schulz, R.; Atef, A.; Becker, D.; Gottschalk, F.; Tauber, C.; Wagner, S.; Arkona, C.; Abdel-Hafez, A.A.; Farag, H.H.; Rademann, J.; et al. Phenylthiomethyl Ketone-Based Fragments Show Selective and Irreversible Inhibition of Enteroviral 3C Proteases. J. Med. Chem. 2018, 61, 1218–1230. [Google Scholar] [CrossRef] [PubMed]
- McShan, D.; Kathman, S.; Lowe, B.; Xu, Z.; Zhan, J.; Statsyuk, A.; Ogungbe, I.V. Identification of non-peptidic cysteine reactive fragments as inhibitors of cysteine protease rhodesain. Bioorg. Med. Chem. Lett. 2015, 25, 4509–4512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wood, D.J.; Lopez-Fernandez, J.D.; Knight, L.E.; Al-Khawaldeh, I.; Gai, C.; Lin, S.; Martin, M.P.; Miller, D.C.; Cano, C.; Endicott, J.A.; et al. FragLites—Minimal, Halogenated Fragments Displaying Pharmacophore Doublets. An Efficient Approach to Druggability Assessment and Hit Generation. J. Med. Chem. 2019, 62, 3741–3752. [Google Scholar] [CrossRef]
- Shorstova, T.; Foulkes, W.D.; Witcher, M. Achieving clinical success with BET inhibitors as anti-cancer agents. Br. J. Cancer 2021, 124, 1478–1490. [Google Scholar] [CrossRef]
- Lewin, J.; Soria, J.-C.; Stathis, A.; Delord, J.-P.; Peters, S.; Awada, A.; Aftimos, P.G.; Bekradda, M.; Rezai, K.; Zeng, Z.; et al. Phase Ib Trial with Birabresib, a Small-Molecule Inhibitor of Bromodomain and Extraterminal Proteins, in Patients With Selected Advanced Solid Tumors. J. Clin. Oncol. 2018, 36, 3007–3014. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.-P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Müller, S.; Pawson, T.; et al. Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef] [Green Version]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Crews, C.M. Electrophilic Screening Platforms for Identifying Novel Covalent Ligands for E3 Ligases. Biochemistry 2021, 60, 2367–2370. [Google Scholar] [CrossRef] [PubMed]
- Kathman, S.G.; Span, I.; Smith, A.T.; Xu, Z.; Zhan, J.; Rosenzweig, A.C.; Statsyuk, A.V. A Small Molecule That Switches a Ubiquitin Ligase From a Processive to a Distributive Enzymatic Mechanism. J. Am. Chem. Soc. 2015, 137, 12442–12445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, H.; Isabella Tsai, Y.-C.; Fantom, K.; Chung, C.-W.; Kümper, S.; Martino, L.; Thomas, D.A.; Eberl, H.C.; Muelbaier, M.; House, D.; et al. Fragment-Based Covalent Ligand Screening Enables Rapid Discovery of Inhibitors for the RBR E3 Ubiquitin Ligase HOIP. J. Am. Chem. Soc. 2019, 141, 2703–2712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubiella, C.; Pinch, B.J.; Koikawa, K.; Zaidman, D.; Poon, E.; Manz, T.D.; Nabet, B.; He, S.; Resnick, E.; Rogel, A.; et al. Sulfopin is a covalent inhibitor of Pin1 that blocks Myc-driven tumors in vivo. Nat. Chem. Biol. 2021, 17, 954–963. [Google Scholar] [CrossRef]
- Cordon, M.B.; Jacobsen, K.M.; Nielsen, C.S.; Hjerrild, P.; Poulsen, T.B. Forward Chemical Genetic Screen for Oxygen-Dependent Cytotoxins Uncovers New Covalent Fragments that Target GPX4. Chembiochem 2022, 23, e202100253. [Google Scholar] [CrossRef]
- Eaton, J.K.; Furst, L.; Ruberto, R.A.; Moosmayer, D.; Hilpmann, A.; Ryan, M.J.; Zimmermann, K.; Cai, L.L.; Niehues, M.; Badock, V.; et al. Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat. Chem. Biol. 2020, 16, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Karaj, E.; Sindi, S.H.; Kuganesan, N.; Perera, L.; Taylor, W.; Tillekeratne, L.M.V. Tunable Cysteine-Targeting Electrophilic Heteroaromatic Warheads Induce Ferroptosis. J. Med. Chem. 2022, 65, 11788–11817. [Google Scholar] [CrossRef]
- Huang, F.; Hu, H.; Wang, K.; Peng, C.; Xu, W.; Zhang, Y.; Gao, J.; Liu, Y.; Zhou, H.; Huang, R.; et al. Identification of Highly Selective Lipoprotein-Associated Phospholipase A2 (Lp-PLA2) Inhibitors by a Covalent Fragment-Based Approach. J. Med. Chem. 2020, 63, 7052–7065. [Google Scholar] [CrossRef] [PubMed]
- Petri, L.; Ábrányi-Balogh, P.; Vagrys, D.; Imre, T.; Varró, N.; Mándity, I.; Rácz, A.; Wittner, L.; Tóth, K.; Tóth, E.Z.; et al. A covalent strategy to target intrinsically disordered proteins: Discovery of novel tau aggregation inhibitors. Eur. J. Med. Chem. 2022, 231, 114163. [Google Scholar] [CrossRef]
- Petri, L.; Ábrányi-Balogh, P.; Tímea, I.; Pálfy, G.; Perczel, A.; Knez, D.; Hrast, M.; Gobec, M.; Sosič, I.; Nyíri, K.; et al. Assessment of Tractable Cysteines for Covalent Targeting by Screening Covalent Fragments. Chembiochem 2021, 22, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Petri, L.; Egyed, A.; Bajusz, D.; Imre, T.; Hetényi, A.; Martinek, T.; Ábrányi-Balogh, P.; Keserű, G.M. An electrophilic warhead library for mapping the reactivity and accessibility of tractable cysteines in protein kinases. Eur. J. Med. Chem. 2020, 207, 112836. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McAulay, K.; Bilsland, A.; Bon, M. Reactivity of Covalent Fragments and Their Role in Fragment Based Drug Discovery. Pharmaceuticals 2022, 15, 1366. https://doi.org/10.3390/ph15111366
McAulay K, Bilsland A, Bon M. Reactivity of Covalent Fragments and Their Role in Fragment Based Drug Discovery. Pharmaceuticals. 2022; 15(11):1366. https://doi.org/10.3390/ph15111366
Chicago/Turabian StyleMcAulay, Kirsten, Alan Bilsland, and Marta Bon. 2022. "Reactivity of Covalent Fragments and Their Role in Fragment Based Drug Discovery" Pharmaceuticals 15, no. 11: 1366. https://doi.org/10.3390/ph15111366