
Investigation of the Mechanisms of Tramadol-Induced Seizures in Overdose in the Rat
 , , , and
, , , and
Abstract
:1. Introduction
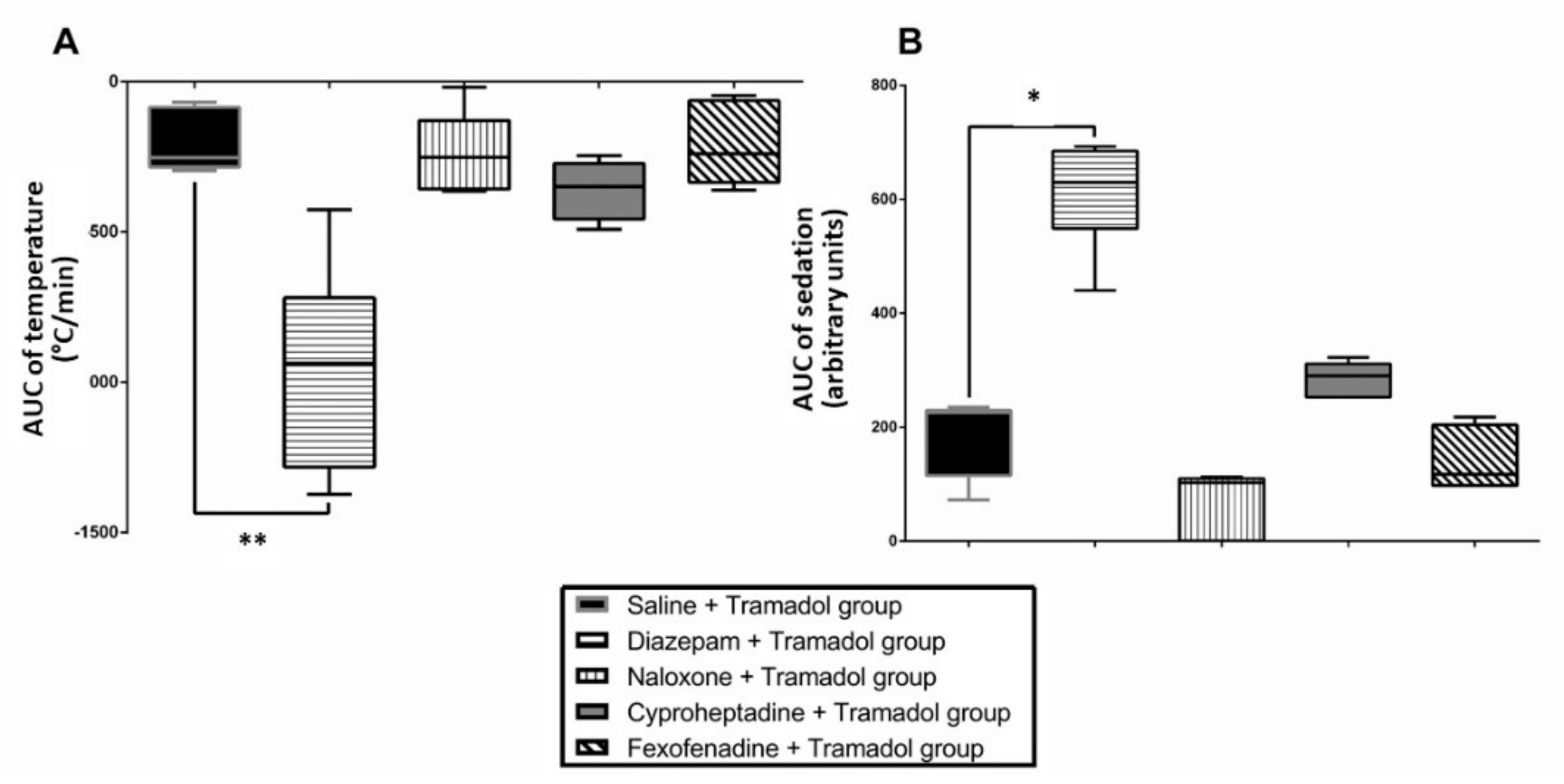
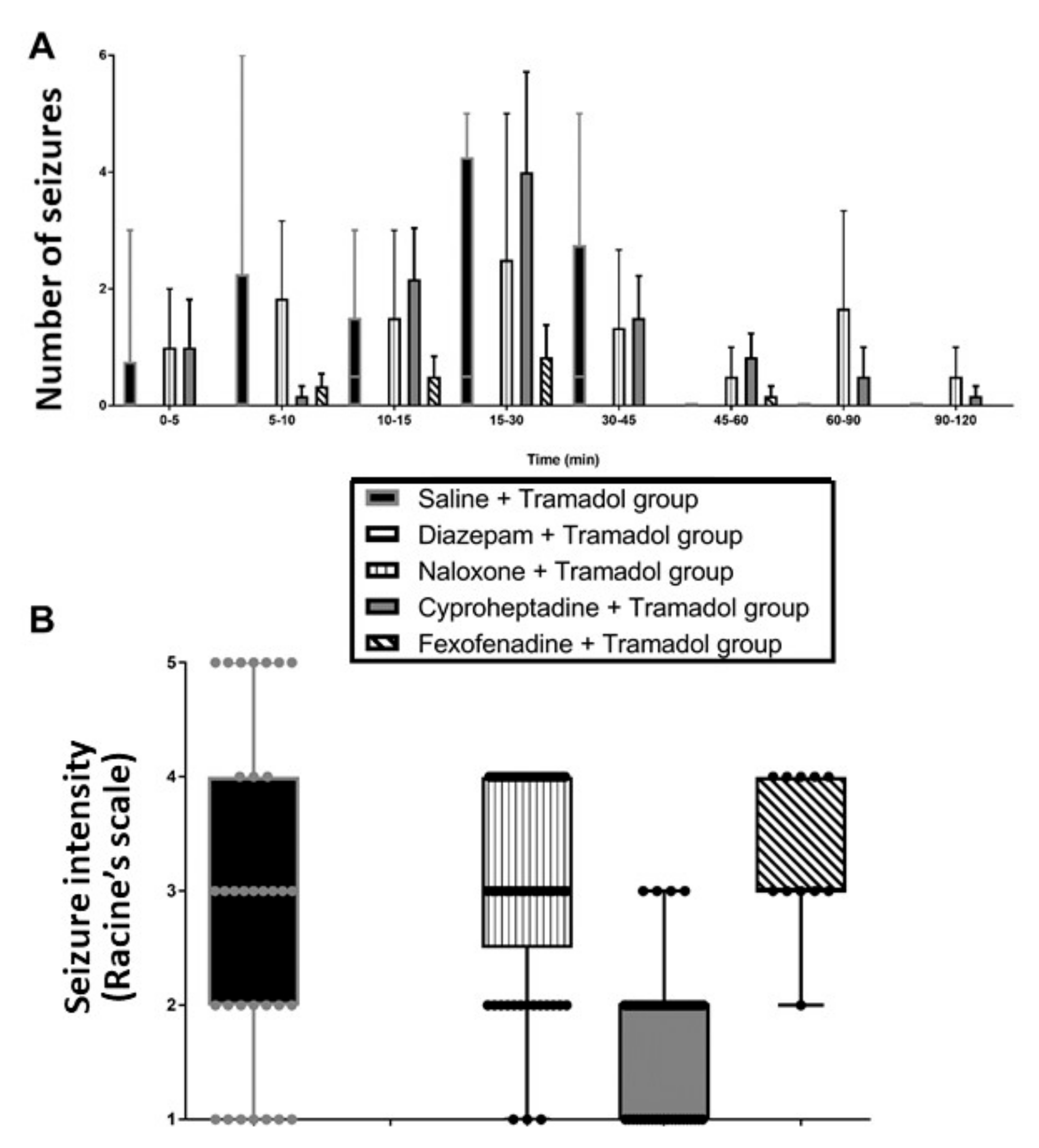
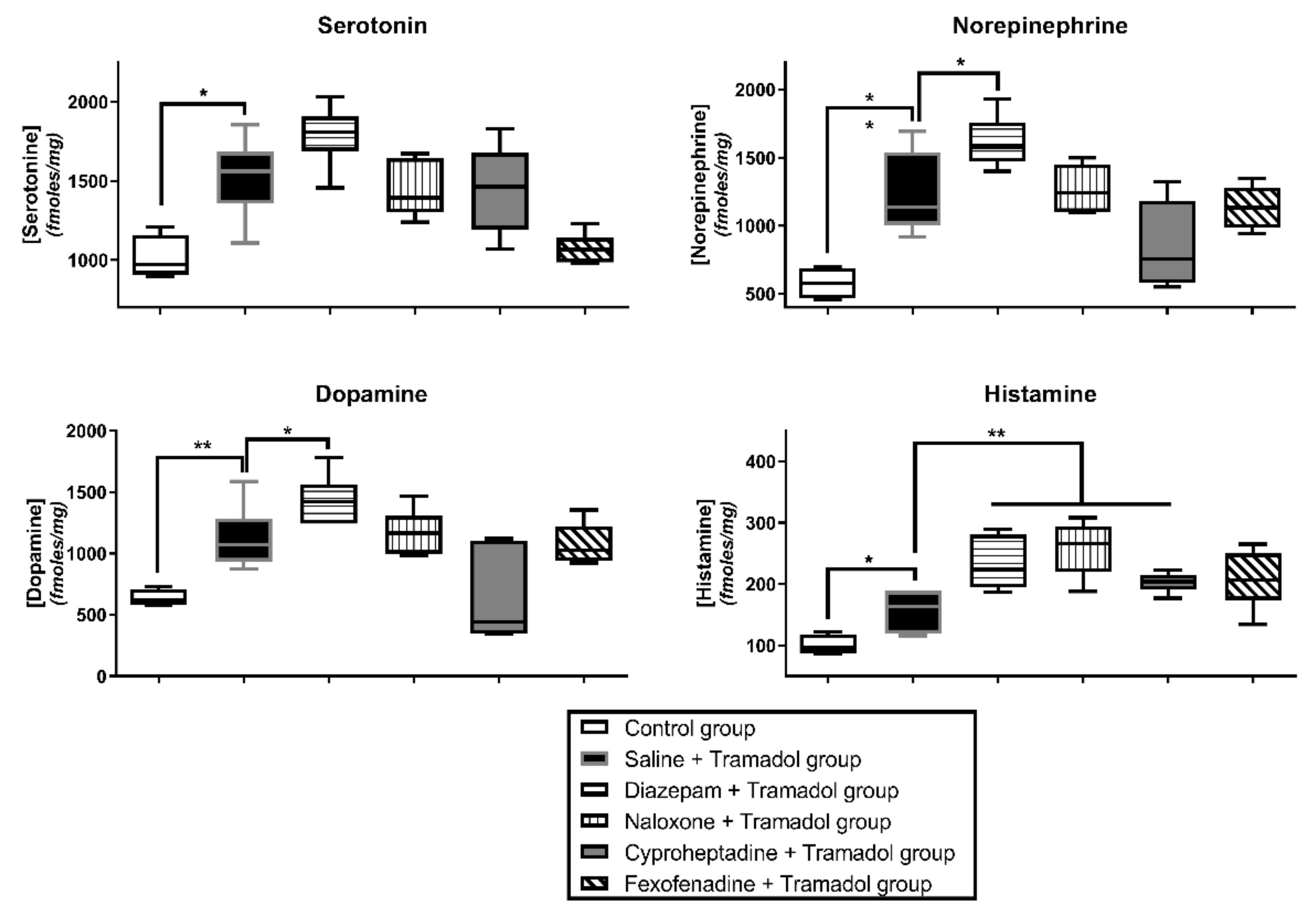
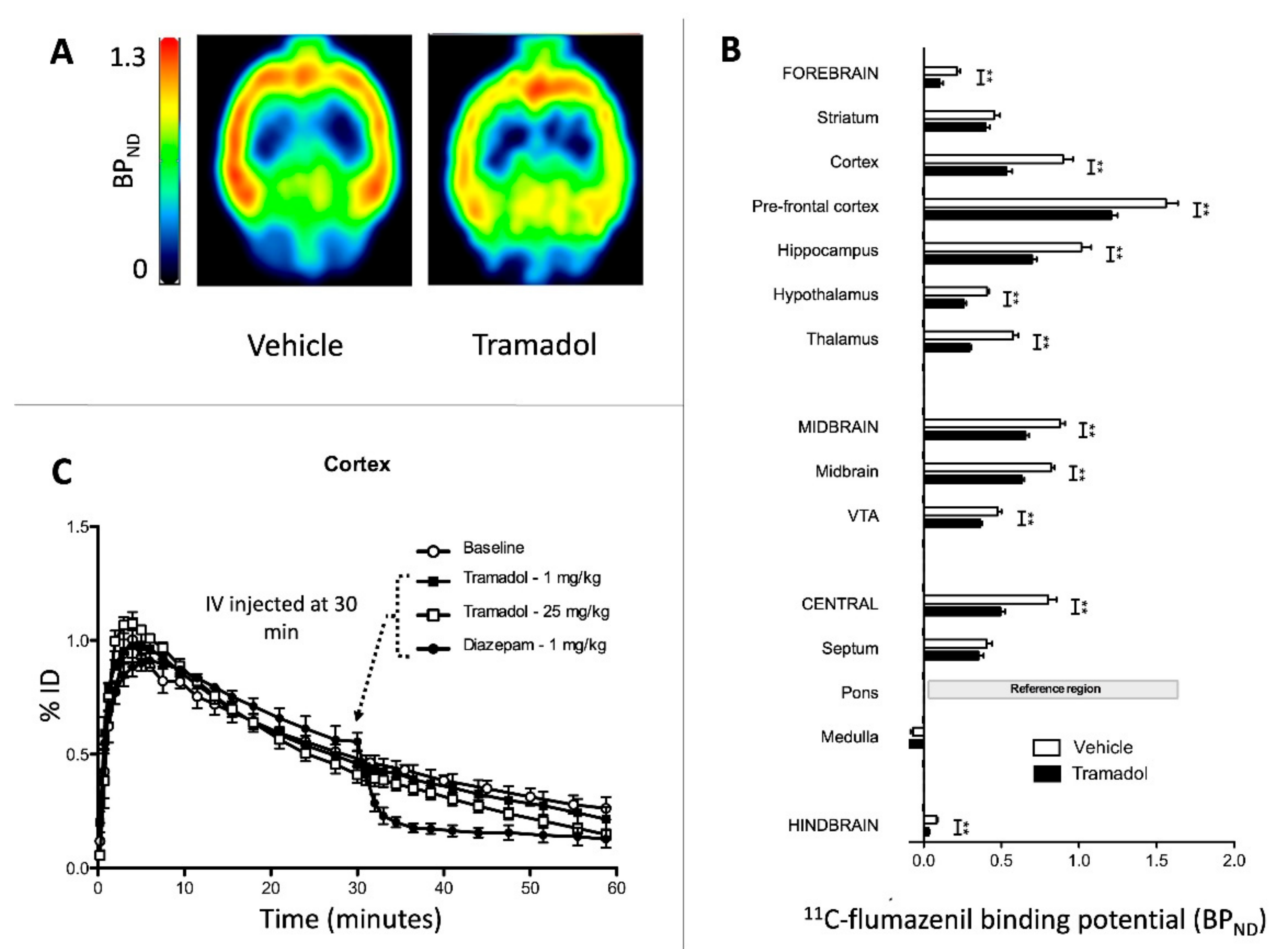
2. Results
2.1. Study 1-Effects of Pretreatments on Tramadol-Induced Sedation, Seizures, Effects on Temperature and Brain Monoamines
2.2. Study 2-Interactions of Tramadol with GABAA Receptors
3. Discussion
4. Materials and Methods
4.1. Animals and Drugs
4.2. Jugular Catheterization
4.3. Clinical Parameters
4.4. Measurement of Monoamine Concentrations in the Brain
4.5. 11C-Flumazenil Positron Emission Tomography (PET) Imaging
4.6. Study Design
4.6.1. Study 1-Effects of Pretreatments on Tramadol-Induced Sedation, Seizures, Temperature and Brain Monoamines
4.6.2. Study 2-Interactions of Tramadol with GABAA Receptors
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dart, R.C.; Surratt, H.L.; Cicero, T.J.; Parrino, M.W.; Severtson, S.G.; Bucher-Bartelson, B.; Green, J.L. Trends in opioid analgesic abuse and mortality in the United States. N. Engl. J. Med. 2015, 372, 241–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawton, K.; Bergen, H.; Simkin, S.; Wells, C.; Kapur, N.; Gunnell, D. Six-year followup of impact of co-proxamol withdrawal in England and Wales on prescribing and deaths: Time-series study. PLoS Med. 2012, 9, e1001213. [Google Scholar] [CrossRef] [Green Version]
- Grond, S.; Sablotzki, A. Clinical pharmacology of tramadol. Clin. Pharmacokinet. 2004, 43, 879–923. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, K.A.; Alsop, J.A.; Albertson, T.E. Tramadol exposures reported to statewide poison control system. Ann. Pharmacother. 2005, 39, 1039–1044. [Google Scholar] [CrossRef]
- Shadnia, S.; Soltaninejad, K.; Heydari, K.; Sasanian, G.; Abdollahi, M. Tramadol intoxication: A review of 114 cases. Hum. Exp. Toxicol. 2008, 27, 201–205. [Google Scholar] [CrossRef]
- Tsutaoka, B.T.; Ho, R.Y.; Fung, S.M.; Kearney, T.E. Comparative Toxicity of Tapentadol and Tramadol Utilizing Data Reported to the National Poison Data System. Ann. Pharmacother. 2015, 49, 1311–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jovanović-Cupić, V.; Martinović, Z.; Nesić, N. Seizures associated with intoxication and abuse of tramadol. Clin. Toxicol. 2006, 44, 143–146. [Google Scholar] [CrossRef]
- Talaie, H.; Panahandeh, R.; Fayaznouri, M.; Asadi, Z.; Abdollahi, M. Dose-independent occurrence of seizure with tramadol. J. Med. Toxicol. 2009, 5, 63–67. [Google Scholar] [CrossRef] [Green Version]
- Taghaddosinejad, F.; Mehrpour, O.; Afshari, R.; Seghatoleslami, A.; Abdollahi, M.; Dart, R.C. Factors related to seizure in tramadol poisoning and its blood concentration. J. Med. Toxicol. 2011, 7, 183–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, B.P.; Carpenter, J.E.; Dunkley, C.A.; Moran, T.P.; Alfaifi, M.; Alsukaiti, W.S.; Kazzi, Z. Seizures in tramadol overdoses reported in the ToxIC registry: Predisposing factors and the role of naloxone. Clin. Toxicol. 2019, 57, 692–696. [Google Scholar] [CrossRef]
- Nakhaee, S.; Amirabadizadeh, A.; Brent, J.; Miri-Moghaddam, E.; Foadoddini, M.; Farrokhfall, K.; Hosseini, M.; Abdollahi, M.; Mehrpour, O. Tramadol and the occurrence of seizures: A systematic review and meta-analysis. Crit. Rev. Toxicol. 2019, 49, 710–723. [Google Scholar] [CrossRef] [PubMed]
- Ryan, N.M.; Isbister, G.K. Tramadol overdose causes seizures and respiratory depression but serotonin toxicity appears unlikely. Clin. Toxicol. 2015, 53, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Tashakori, A.; Afshari, R. Tramadol overdose as a cause of serotonin syndrome: A case series. Clin. Toxicol. 2010, 48, 337–341. [Google Scholar] [CrossRef]
- Boyer, E.W.; Shannon, M. The serotonin syndrome. N. Engl. J. Med. 2005, 352, 1112–1120. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, Y.; Funao, T.; Suehiro, K.; Takahashi, R.; Mori, T.; Nishikawa, K. Brain serotonin content regulates the manifestation of tramadol-induced seizures in rats: Disparity between tramadol-induced seizure and serotonin syndrome. Anesthesiology 2015, 122, 178–189. [Google Scholar] [CrossRef]
- Lagard, C.; Chevillard, L.; Malissin, I.; Risède, P.; Callebert, J.; Labat, L.; Launay, J.M.; Laplanche, J.L.; Mégarbane, B. Mechanisms of tramadol-related neurotoxicity in the rat: Does diazepam/tramadol combination play a worsening role in overdose? Toxicol. Appl. Pharmacol. 2016, 310, 108–119. [Google Scholar] [CrossRef]
- Rehni, A.K.; Singh, I.; Kumar, M. Tramadol-induced seizurogenic effect: A possible role of opioid-dependent gamma-amino butyric acid inhibitory pathway. Basic Clin. Pharmacol. Toxicol. 2008, 103, 262–266. [Google Scholar] [CrossRef]
- Rehni, A.K.; Singh, T.G.; Singh, N.; Arora, S. Tramadol-induced seizurogenic effect: A possible role of opioid-dependent histamine H1 receptor activation-linked mechanism. Naunyn Schmiedebergs Arch. Pharmacol. 2010, 381, 11–19. [Google Scholar] [CrossRef]
- Bameri, B.; Shaki, F.; Ahangar, N.; Ataee, R.; Samadi, M.; Mohammadi, H. Evidence for the involvement of the dopaminergic system in seizure and oxidative damage induced by tramadol. Int. J. Toxicol. 2018, 37, 164–170. [Google Scholar] [CrossRef]
- Parente, A.; Vállez García, D.; Shoji, A.; Lopes Alves, I.; Maas, B.; Zijlma, R.; Dierckx, R.A.; Buchpiguel, C.A.; de Vries, E.F.; Doorduin, J. Contribution of neuroinflammation to changes in [11C]flumazenil binding in the rat brain: Evaluation of the inflamed pons as reference tissue. Nucl. Med. Biol. 2017, 49, 50–56. [Google Scholar] [CrossRef]
- Beaune, S.; Callebert, J.; Baud, F.J.; Risède, P.; Juvin, P.; Mégarbane, B. Mechanisms of high-dose citalopram-induced death in a rat model. Toxicology 2012, 302, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Saboory, E.; Derchansky, M.; Ismaili, M.; Jahromi, S.S.; Brull, R.; Carlen, P.L.; El Beheiry, H. Mechanisms of morphine enhancement of spontaneous seizure activity. Anesth. Analg. 2007, 105, 1729–1735. [Google Scholar] [CrossRef] [PubMed]
- Manocha, A.; Sharma, K.K.; Mediratta, P.K. On the mechanism of anticonvulsant effect of tramadol in mice. Pharmacol. Biochem. Behav. 2005, 82, 74–81. [Google Scholar] [CrossRef]
- Raffa, R.B.; Friderichs, E.; Reimann, W.; Shank, R.P.; Codd, E.E.; Vaught, J.L. Opioid and nonopioid components independently contribute to the mechanism of action of tramadol, an ‘atypical’ opioid analgesic. J. Pharmacol. Exp. Ther. 1992, 260, 275–285. [Google Scholar] [PubMed]
- Gilbert, P.E.; Martin, W.R. Antagonism of the convulsant effects of heroin, d-propoxyphene, meperidine, normeperidine and thebaine by naloxone in mice. J. Pharmacol. Exp. Ther. 1975, 192, 538–541. [Google Scholar] [PubMed]
- Yang, L.; Li, F.; Ge, W.; Mi, C.; Mi, C.; Wang, R.; Sun, R. Protective effects of naloxone in two-hit seizure model. Epilepsia 2010, 51, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Omrani, A.; Ghadami, M.R.; Fathi, N.; Tahmasian, M.; Fathollahi, Y.; Touhidi, A. Naloxone improves impairment of spatial performance induced by pentylenetetrazol kindling in rats. Neuroscience 2007, 145, 824–831. [Google Scholar] [CrossRef]
- Raffa, R.B.; Stone, D.J. Unexceptional seizure potential of tramadol or its enantiomers or metabolites in mice. J. Pharmacol. Exp. Ther. 2008, 325, 500–506. [Google Scholar] [CrossRef] [Green Version]
- Yokoyama, H.; Onodera, K.; Iinuma, K.; Watanabe, T. 2-Thiazolylethylamine, a selective histamine H1 agonist, decreases seizure susceptibility in mice. Pharmacol. Biochem. Behav. 1994, 47, 503–507. [Google Scholar] [CrossRef]
- Soria-Jasso, L.; Arias-Montano, J. Histamine H1 receptor activation stimulates [3H] GABA release from human astrocytoma U373 MG cells. Eur. J. Pharmacol. 1996, 318, 185–192. [Google Scholar] [CrossRef]
- Sangalli, B.C. Role of the central histaminergic neuronal system in the CNS toxicity of the first generation H1-antagonists. Prog. Neurobiol. 1997, 52, 145–157. [Google Scholar] [CrossRef]
- Kamei, C.; Ohuchi, M.; Sugimoto, Y.; Okuma, C. Mechanism responsible for epileptogenic activity by first-generation H1-antagonists in rats. Brain Res. 2000, 887, 183–186. [Google Scholar] [CrossRef]
- Fujii, Y.; Tanaka, T.; Harada, C.; Hirai, T.; Kamei, C. Epileptogenic activity induced by histamine H1 antagonists in amygdala-kindled rats. Brain Res. 2003, 991, 258–261. [Google Scholar] [CrossRef]
- Samadi, M.; Shaki, F.; Bameri, B.; Fallah, M.; Ahangar, N.; Mohammadi, H. Caffeine attenuates seizure and brain mitochondrial disruption induced by Tramadol: The role of adenosinergic pathway. Drug Chem. Toxicol. 2021, 44, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Eizadi-Mood, N.; Ozcan, D.; Sabzghabaee, A.M.; Mirmoghtadaee, P.; Hedaiaty, M. Does naloxone prevent seizure in tramadol-intoxicated patients? Int. J. Prev. Med. 2014, 5, 302–307. [Google Scholar]
- Shadnia, S.; Brent, J.; Mousavi-Fatemi, K.; Hafezi, P.; Soltaninejad, K. Recurrent seizures in tramadol intoxication: Implications for therapy based on 100 patients. Basic Clin. Pharmacol. Toxicol. 2012, 111, 133–136. [Google Scholar] [CrossRef]
- Giorgi, F.S.; Ferrucci, M.; Lazzeri, G.; Pizzanelli, C.; Lenzi, P.; Alessandrl, M.G.; Murri, L.; Fornai, F. A damage to locus coeruleus neurons converts sporadic seizures into self-sustaining limbic status epilepticus. Eur. J. Neurosci. 2003, 17, 2593–2601. [Google Scholar] [CrossRef]
- Ahern, T.H.; Javors, M.A.; Eagles, D.A.; Martillotti, J.; Mitchell, H.A.; Liles, L.C.; Weinshenker, D. The effects of chronic norepinephrine transporter inactivation on seizure susceptibility in mice. Neuropsychopharmacology 2006, 31, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Jahan, K.; Pillai, K.K.; Vohora, D. DSP-4 induced depletion of brain noradrenaline and increased 6-hertz psychomotor seizure susceptibility in mice is prevented by sodium valproate. Brain Res. Bull. 2018, 142, 263–269. [Google Scholar] [CrossRef]
- Biagioni, F.; Celli, R.; Puglisi-Allegra, S.; Nicoletti, F.; Giorgi, F.S.; Fornai, F. Noradrenaline and seizures: A perspective on the role of adrenergic receptors in limbic seizures. Curr. Neuropharmacol. 2022. [Google Scholar] [CrossRef]
- Hara, K.; Minami, K.; Sata, T. The effects of tramadol and its metabolite on glycine, gamma-aminobutyric acidA, and N-methyl-D-aspartate receptors expressed in Xenopus oocytes. Anesth. Analg. 2005, 100, 1400–1405. [Google Scholar] [CrossRef]
- Safavynia, S.A.; Keating, G.; Speigel, I.; Fidler, J.A.; Kreuzer, M.; Rye, D.B.; Jenkins, A.; García, P.S. Effects of γ-Aminobutyric Acid Type A Receptor Modulation by Flumazenil on Emergence from General Anesthesia. Anesthesiology 2016, 125, 147–158. [Google Scholar] [CrossRef] [Green Version]
- Liefaard, L.C.; Ploeger, B.A.; Molthoff, C.F.; de Jong, H.W.; Dijkstra, J.; van der Weerd, L.; Lammertsma, A.A.; Danhof, M.; Voskuyl, R.A. Changes in GABAA receptor properties in amygdala kindled animals: In vivo studies using [11C]flumazenil and positron emission tomography. Epilepsia 2009, 50, 88–98. [Google Scholar] [CrossRef]
- Syvänen, S.; Labots, M.; Tagawa, Y.; Eriksson, J.; Windhorst, A.D.; Lammertsma, A.A.; de Lange, E.C.; Voskuyl, R.A. Altered GABAA receptor density and unaltered blood-brain barrier transport in a kainate model of epilepsy: An in vivo study using 11C-flumazenil and PET. J. Nucl. Med. 2012, 53, 1974–1983. [Google Scholar] [CrossRef] [Green Version]
- Naylor, D.E.; Liu, H.; Wasterlain, C.G. Trafficking of GABA(A) receptors, loss of inhibition, and a mechanism for pharmacoresistance in status epilepticus. J. Neurosci. 2005, 25, 7724–7733. [Google Scholar] [CrossRef] [Green Version]
- Goodkin, H.P.; Joshi, S.; Mtchedlishvili, Z.; Brar, J.; Kapur, J. Subunit-specific trafficking of GABA(A) receptors during status epilepticus. J. Neurosci. 2008, 28, 2527–2538. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, H.; Hirabayashi, Y.; Shimizu, R.; Saitoh, K.; Mitsuhata, H. Sevoflurane is equivalent to isoflurane for attenuating bupivacaine-induced arrhythmias and seizures in rats. Anesth. Analg. 1996, 83, 570–573. [Google Scholar] [CrossRef]
- Bar-Klein, G.; Klee, R.; Brandt, C.; Bankstahl, M.; Bascuñana, P.; Töllner, K.; Dalipaj, H.; Bankstahl, J.P.; Friedman, A.; Löscher, W. Isoflurane prevents acquired epilepsy in rat models of temporal lobe epilepsy. Ann. Neurol. 2016, 80, 896–908. [Google Scholar] [CrossRef]
- Chua, H.C.; Chebib, M. GABA(A) Receptors and the diversity in their structure and pharmacology. Adv. Pharmacol. 2017, 79, 1–34. [Google Scholar]
- Gholami, M.; Saboory, E.; Roshan-Milani, S. Proconvulsant effects of tramadol and morphine on pentylenetetrazol-induced seizures in adult rats using different routes of administration. Epilepsy Behav. 2014, 36, 90–96. [Google Scholar] [CrossRef]
- Olsen, R.W. GABA(A) receptor: Positive and negative allosteric modulators. Neuropharmacology 2018, 136, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Ashton, D.; Wauquier, A. Effects of some anti-epileptic, neuroleptic and gabaminergic drugs on convulsions induced by D.,L-allylglycine. Pharmacol. Biochem. Behav. 1979, 11, 221–226. [Google Scholar] [CrossRef]
- Valian, N.; Sorayya, M.; Asadi, S.; Sherafati, F.; Ershad, A.; Savaheli, S.; Ahmadiani, A. Preconditioning by ultra-low dose of tramadol reduces the severity of tramadol-induced seizure: Contribution of glutamate receptors. Biomed. Pharmacother. 2021, 133, 111031. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.D.; Warner, D.S.; Todd, M.M.; Baker, M.T.; Jensen, N.F. The influence of inhalational anesthetics on in vivo and in vitro benzodiazepine receptor binding in the rat cerebral cortex. Anesthesiology 1991, 74, 97–104. [Google Scholar] [CrossRef]
- Mukaida, K.; Shichino, T.; Koyanagi, S.; Himukashi, S.; Fukuda, K. Activity of the serotonergic system during isoflurane anesthesia. Anesth. Analg. 2007, 104, 836–839. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, Y.; Hashimoto, Y.; Hirota, K.; Matsuki, A. Inhibited hypothalamic histamine metabolism during isoflurane and sevoflurane anesthesia in rats. Acta Anaesthesiol. Scand. 1998, 42, 858–863. [Google Scholar] [CrossRef]
- Votaw, J.R.; Byas-Smith, M.G.; Voll, R.; Halkar, R.; Goodman, M.M. Isoflurane alters the amount of dopamine transporter expressed on the plasma membrane in humans. Anesthesiology 2004, 101, 1128–1135. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S. Effect of isoflurane anesthesia on norepinephrine metabolism in rat brain. Masui 1992, 41, 522–531. [Google Scholar]
- Larsen, P.; Ulin, J.; Dahlstrom, K. A New Method for Production of 11C-Labelled Methyl Iodide from 11C-Methane. J. Label. Compd. Radiopharm. 1995, 37, 73–75. [Google Scholar]
- Jewett, D.M. A simple synthesis of [11C]methyl triflate. Int. J. Rad. Appl. Instrum. A 1992, 43, 1383–1385. [Google Scholar] [CrossRef] [Green Version]
- Pirnay, S.O.; Mégarbane, B.; Borron, S.W.; Risède, P.; Monier, C.; Ricordel, I.; Baud, F.J. Effects of various combinations of benzodiazepines with buprenorphine on arterial blood gases in rats. Basic Clin. Pharmacol. Toxicol. 2008, 103, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Racine, R.J. Modification of seizure activity by electrical stimulation. Electroencephalogr. Clin. Neurophysiol. 1972, 32, 269–299. [Google Scholar] [CrossRef]
- Jang, H.S.; Jang, I.S.; Lee, M.G. The effects of tramadol on electroencephalographic spectral parameters and analgesia in rats. Korean J. Physiol. Pharmacol. 2008, 14, 191–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kema, I.P.; Schellings, A.M.; Hoppenbrouwers, C.J.; Rutgers, H.M.; De Vries, E.G.; Muskiet, F.A. High performance liquid chromatographic profiling of tryptophan and related indoles in body fluids and tissues of carcinoid patients. Clin. Chim. Acta 1993, 221, 143–158. [Google Scholar] [CrossRef]
- Haimart, M.; Launay, J.M.; Zürcher, G.; Cauet, N.; Dreux, C.; Da Prada, M. Simultaneous determination of histamine and N alpha-methylhistamine in biological samples by an improved enzymatic single isotope assay. Agents Actions 1985, 16, 71–75. [Google Scholar] [CrossRef]
- Bao, Q.; Newport, D.; Chen, M.; Stout, D.B.; Chatziioannou, A.F. Performance evaluation of the inveon dedicated PET preclinical tomograph based on the NEMA NU-4 standards. J. Nucl. Med. 2009, 50, 401–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiffer, W.K.; Mirrione, M.M.; Biegon, A.; Alexoff, D.L.; Patel, V.; Dewey, S.L. Serial microPET measures of the metabolic reaction to a microdialysis probe implant. J. Neurosci. Methods 2006, 155, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Alves, I.L.; Vállez García, D.; Parente, A.; Doorduin, J.; Dierckx, R.; Marques da Silva, A.M.; Koole, M.; Willemsen, A.; Boellaard, R. Pharmacokinetic modeling of [11C]flumazenil kinetics in the rat brain. EJNMMI Res. 2017, 7, 17. [Google Scholar] [CrossRef] [Green Version]
- Derlet, R.W.; Albertson, T.E. Diazepam in the prevention of seizures and death in cocaine-intoxicated rats. Ann. Emerg. Med. 1989, 18, 542–546. [Google Scholar] [CrossRef]
- Haglind, E. Effects of continuous naloxone infusion in intestinal ischemia shoch in the rat. Circ. Shock 1992, 32, 269–299. [Google Scholar]
- Ma, Z.; Zhang, G.; Jenney, C.; Krishnamoorthy, S.; Tao, R. Characterization of serotonin-toxicity syndrome (toxidrome) elicited by 5-hydroxy-l-tryptophan in clorgyline-pretreated rats. Eur. J. Pharmacol. 2008, 588, 198–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anoush, M.; Mohammad Khani, M.R. Evaluating the Anti-nociceptive and Anti-inflammatory Effects of Ketotifen and Fexofenadine in Rats. Adv. Pharm. Bull. 2015, 5, 217–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pottier, G.; Marie, S.; Goutal, S.; Auvity, S.; Peyronneau, M.A.; Stute, S.; Boisgard, R.; Dollé, F.; Buvat, I.; Caillé, F.; et al. Imaging the Impact of the P-Glycoprotein (ABCB1) Function on the Brain Kinetics of Metoclopramide. J. Nucl. Med. 2016, 57, 309–314. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Saline + Tramadol Group | Diazepam + Tramadol Group | Naloxone + Tramadol Group | Cyproheptadine + Tramadol Group | Fexofenadine + Tramadol Group | Control Group | |
|---|---|---|---|---|---|---|---|
| Time (min) | |||||||
| T-30 | ø | ø | ø | ø | 15 mg/kg fexofenadine i.p. | ø | |
| T-15 | 4% tween i.p. | 1.77 mg/kg diazepam i.p. | 4% tween i.p. | 10 mg/kg cyproheptadine i.p. | ø | 4% tween i.p. | |
| T-10 | Saline i.v. | Saline i.v. | 2 mg/kg i.v. bolus followed by 4 mg/kg/h infusion | Saline i.v. | Saline i.v. | Saline i.v. | |
| T0 | 75 mg/kg tramadol i.p. | 75 mg/kg tramadol i.p. | 75 mg/kg tramadol i.p. | 75 mg/kg tramadol i.p. | 75 mg/kg tramadol i.p. | Saline i.p. | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lagard, C.; Vodovar, D.; Chevillard, L.; Callebert, J.; Caillé, F.; Pottier, G.; Liang, H.; Risède, P.; Tournier, N.; Mégarbane, B. Investigation of the Mechanisms of Tramadol-Induced Seizures in Overdose in the Rat. Pharmaceuticals 2022, 15, 1254. https://doi.org/10.3390/ph15101254
Lagard C, Vodovar D, Chevillard L, Callebert J, Caillé F, Pottier G, Liang H, Risède P, Tournier N, Mégarbane B. Investigation of the Mechanisms of Tramadol-Induced Seizures in Overdose in the Rat. Pharmaceuticals. 2022; 15(10):1254. https://doi.org/10.3390/ph15101254
Chicago/Turabian StyleLagard, Camille, Dominique Vodovar, Lucie Chevillard, Jacques Callebert, Fabien Caillé, Géraldine Pottier, Hao Liang, Patricia Risède, Nicolas Tournier, and Bruno Mégarbane. 2022. "Investigation of the Mechanisms of Tramadol-Induced Seizures in Overdose in the Rat" Pharmaceuticals 15, no. 10: 1254. https://doi.org/10.3390/ph15101254