
Pharmacological Properties of Trichostatin A, Focusing on the Anticancer Potential: A Comprehensive Review

 ,
,  , ,
, ,  , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Research Methodology
3. Antioxidant Properties of TSA
4. Antidiabetic Activity of TSA
5. Anti-Inflammatory Activity of TSA
6. Anticancer Activity of TSA
6.1. Direct Anticancer Mechanisms of TSA
6.1.1. Anticancer Action of TSA against Brain Cancer Cells
6.1.2. Anticancer Action of TSA on Neuroblastoma (NB)
6.1.3. Anticancer Activity of TSA on Human Tongue Squamous Cell Carcinoma
6.1.4. Anticancer Activity of TSA on Nasopharyngeal Carcinoma
6.1.5. Anticancer Activity of TSA on Lung Cancer Cells
6.1.6. Gastric Cancer, Colorectal Cancer, and Esophageal Cancer
6.1.7. Anticancer Effect of TSA on Hepatocellular Carcinoma
6.1.8. Anticancer Effect of TSA on Pancreatic Adenocarcinoma
6.1.9. Anticancer Effect of TSA on Leukemia
6.1.10. Anticancer Effect of TSA on Osteosarcoma/Excessive Bone Resorption
6.1.11. Anticancer Action of TSA on Musculoskeletal Sarcomas (Rhabdomyosarcoma)
6.1.12. Anticancer Action of TSA on Mast Cell Tumor (MCT) and Breast Cancer Cells
6.1.13. Anticancer Action of TSA on Endometriosis/Cervical Cancer Cells
6.1.14. Anticancer Action of TSA on Ovarian Cancer Cells
6.1.15. Anticancer Action of TSA on Urinary Bladder Cancer Cells
6.1.16. Anticancer Action of TSA on Prostate Cancer/Spermatogenesis
6.2. Anticancer Activity of TSA through Sensitization
6.3. Effect of Other Molecules on Enhancing the Anticancer Activity of TSA
6.4. Anticancer Effect of TSA in Combination with Chemotherapy
6.5. TSA Targets Epigenetic Modifications in Cancer
7. Conclusions and Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| BDNF | Brain Derived Neurotrophic Factor |
| CDK | Cyclin Dependent Kinase |
| CLL | Lymphocytic Leukemia |
| CytoD | Cytochalasin D |
| EBV | Epstein-Barr Virus |
| GPNMB | Glycoprotein non-metastatic melanoma protein B |
| hBM-MSCs | Human bone marrow-mesenchymal stem cells |
| HIF | Hypoxia-Inducible Factor |
| LatB | Latrunculin B |
| LPS | Lipopolysaccharide |
| MCT | Mast Cell Tumor |
| MDA | Malondialdehyde |
| NPBMNC | Normal Peripheral Blood Mononuclear Cells |
| NPC | Nasopharyngeal Carcinoma |
| OPN | Osteopontin |
| PARP | PolyADP-Ribose Polymerase |
| PCNA | Proliferating Cell Nuclear Antigen |
| RMS | Rhabdomyosarcoma |
| ROS | Reactive Oxygen Species |
| RSC96 | Cultured Rat Schwann Cells |
| SOD | Superoxide Dismutase |
| STZ | Streptozotocin |
| T-AOC | Total Antioxidant Capacity |
| TRAIL | TNF-Related Apoptosis-inducing Ligand |
| TSA | Trichostatin A |
| UPR | Unfolded Protein response |
| UVB | Following Ultraviolet-B |
| VEGF | Vascular endothelial growth factor |
| WAF1 | Wild-type P53-activated Fragment 1 |
References
- Sharifi-Rad, J.; Dey, A.; Koirala, N.; Shaheen, S.; El Omari, N.; Salehi, B.; Goloshvili, T.; Cirone Silva, N.C.; Bouyahya, A.; Vitalini, S. Cinnamomum Species: Bridging Phytochemistry Knowledge, Pharmacological Properties and Toxicological Safety for Health Benefits. Front. Pharmacol. 2021, 12, 600139. [Google Scholar] [CrossRef] [PubMed]
- Bouyahya, A.; Chamkhi, I.; Benali, T.; Guaouguaou, F.-E.; Balahbib, A.; El Omari, N.; Taha, D.; Belmehdi, O.; Ghokhan, Z.; El Menyiy, N. Traditional Use, Phytochemistry, Toxicology, and Pharmacology of Origanum Majorana L. J. Ethnopharmacol. 2021, 265, 113318. [Google Scholar] [CrossRef] [PubMed]
- El Omari, N.; Bakrim, S.; Bakha, M.; Lorenzo, J.M.; Rebezov, M.; Shariati, M.A.; Aboulaghras, S.; Balahbib, A.; Khayrullin, M.; Bouyahya, A. Natural Bioactive Compounds Targeting Epigenetic Pathways in Cancer: A Review on Alkaloids, Terpenoids, Quinones, and Isothiocyanates. Nutrients 2021, 13, 3714. [Google Scholar] [CrossRef] [PubMed]
- Bouyahya, A.; Guaouguaou, F.-E.; El Omari, N.; El Menyiy, N.; Balahbib, A.; El-Shazly, M.; Bakri, Y. Anti-Inflammatory and Analgesic Properties of Moroccan Medicinal Plants: Phytochemistry, in Vitro and in Vivo Investigations, Mechanism Insights, Clinical Evidences and Perspectives. J. Pharm. Anal. 2021, 12, 35–57. [Google Scholar] [CrossRef]
- El Omari, N.; Bakha, M.; Imtara, H.; Guaouguaoua, F.E.; Balahbib, A.; Zengin, G.; Bouyahya, A. Anticancer mechanisms of phytochemical compounds: Focusing on epigenetic targets. Environ. Sci. Pollut. Res. 2021, 28, 47869–47903. [Google Scholar] [CrossRef]
- Alqahtani, A.S.; Ullah, R.; Shahat, A.A. Bioactive Constituents and Toxicological Evaluation of Selected Antidiabetic Medicinal Plants of Saudi Arabia. Evid. Based Complement. Altern. Med. 2022, 2022, 7123521. [Google Scholar] [CrossRef]
- Morgan, E.W.; Perdew, G.H.; Patterson, A.D. Multi-Omics Strategies for Investigating the Microbiome in Toxicology Research. Toxicol. Sci. 2022, 187, 189–213. [Google Scholar] [CrossRef]
- El Menyiy, N.; Mrabti, H.N.; El Omari, N.; Bakili, A.E.; Bakrim, S.; Mekkaoui, M.; Balahbib, A.; Amiri-Ardekani, E.; Ullah, R.; Alqahtani, A.S. Medicinal Uses, Phytochemistry, Pharmacology, and Toxicology of Mentha Spicata. Evid. Based Complement. Altern. Med. 2022, 2022, 7990508. [Google Scholar] [CrossRef]
- Tsuji, N.; Kobayashi, M.; Nagashima, K.; Wakisaka, Y.; Koizumi, K. A New Antifungal Antibiotic, Trichostatin. J. Antibiot. 1976, 29, 1–6. [Google Scholar] [CrossRef]
- Singh, S.B.; Genilloud, O.; Peláez, F. 2.05-Terrestrial Microorganisms–Filamentous Bacteria. In Comprehensive Natural Products II: Chemistry and Biology; Liu, H.-W., Mander, L., Eds.; Elsevier: Oxford, UK, 2010; pp. 109–140. ISBN 978-0-08-045382-8. [Google Scholar]
- Guo, Y.; Li, Z.; Shi, C.; Li, J.; Yao, M.; Chen, X. Trichostatin A Attenuates Oxidative Stress-Mediated Myocardial Injury through the FoxO3a Signaling Pathway. Int. J. Mol. Med. 2017, 40, 999–1008. [Google Scholar] [CrossRef]
- Jeong, S.-G.; Cho, G.-W. Trichostatin A Modulates Intracellular Reactive Oxygen Species through SOD2 and FOXO1 in Human Bone Marrow-Mesenchymal Stem Cells. Cell Biochem. Funct. 2015, 33, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Rong, X.; Yang, J.; Lu, Y. Evaluation of the Antioxidant Effects of Different Histone Deacetylase Inhibitors (HDACis) on Human Lens Epithelial Cells (HLECs) after UVB Exposure. BMC Ophthalmol. 2019, 19, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Qu, M.; Wang, Y.; Duan, H.; Chen, P.; Wang, Y.; Shi, W.; Danielson, P.; Zhou, Q. Trichostatin A Inhibits Transforming Growth Factor-β-Induced Reactive Oxygen Species Accumulation and Myofibroblast Differentiation via Enhanced NF-E2-Related Factor 2-Antioxidant Response Element Signaling. Mol. Pharmacol. 2013, 83, 671–680. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Zhang, X.; Jia, K.; Zhang, C.; Zhu, L.; Cheng, M.; Li, F.; Zhao, S.; Hao, J. Trichostatin A Increases BDNF Protein Expression by Improving XBP-1s/ATF6/GRP78 Axis in Schwann Cells of Diabetic Peripheral Neuropathy. Biomed. Pharm. 2021, 133, 111062. [Google Scholar] [CrossRef] [PubMed]
- Noh, H.; Oh, E.Y.; Seo, J.Y.; Yu, M.R.; Kim, Y.O.; Ha, H.; Lee, H.B. Histone Deacetylase-2 Is a Key Regulator of Diabetes- and Transforming Growth Factor-Beta1-Induced Renal Injury. Am. J. Physiol. Renal Physiol. 2009, 297, F729–F739. [Google Scholar] [CrossRef] [Green Version]
- Tiernan, A.R.; Champion, J.A.; Sambanis, A. Trichostatin A Affects the Secretion Pathways of Beta and Intestinal Endocrine Cells. Exp. Cell Res. 2015, 330, 212–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.-H.; Oh, S.-W.; Kang, M.-S.; Kwon, H.J.; Oh, G.-T.; Kim, D.-Y. Trichostatin A Attenuates Airway Inflammation in Mouse Asthma Model. Clin. Exp. Allergy 2005, 35, 89–96. [Google Scholar] [CrossRef]
- Han, S.-B.; Lee, J.K. Anti-Inflammatory Effect of Trichostatin-A on Murine Bone Marrow-Derived Macrophages. Arch. Pharm. Res. 2009, 32, 613–624. [Google Scholar] [CrossRef]
- Ling, T.; Xie, J. Trichostatin A Exerts Anti-Inflammation Functions in LPS-Induced Acute Lung Injury Model through Inhibiting TNF-α and Upregulating MicorRNA-146a Expression. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 3935–3942. [Google Scholar]
- Sato, T.; Kotake, D.; Hiratsuka, M.; Hirasawa, N. Enhancement of Inflammatory Protein Expression and Nuclear Factor Κb (NF-Κb) Activity by Trichostatin A (TSA) in OP9 Preadipocytes. PLoS ONE 2013, 8, e59702. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Yang, F.; Li, X.; Wang, L.; Chu, X.; Zhang, H.; Gong, Z. Trichostatin A Inhibits Inflammation in Phorbol Myristate Acetate-Induced Macrophages by Regulating the Acetylation of Histone and/or Non-Histone Proteins. Mol. Med. Rep. 2016, 13, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.-G. The Histone Deacetylase Inhibitor, Trichostatin A, Induces G2/M Phase Arrest and Apoptosis in YD-10B Oral Squamous Carcinoma Cells. Oncol. Rep. 2011, 27, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Alao, J.P.; Stavropoulou, A.V.; Lam, E.W.; Coombes, R.C. Role of Glycogen Synthase Kinase 3 Beta (GSK3β) in Mediating the Cytotoxic Effects of the Histone Deacetylase Inhibitor Trichostatin A (TSA) in MCF-7 Breast Cancer Cells. Mol. Cancer 2006, 5, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y. Induction of Apoptosis by Trichostatin A, a Histone Deacetylase Inhibitor, Is Associated with Inhibition of Cyclooxygenase-2 Activity in Human Non-Small Cell Lung Cancer Cells. Int. J. Oncol. 2005, 27, 473–479. [Google Scholar] [CrossRef]
- Deng, Z.; Liu, X.; Jin, J.; Xu, H.; Gao, Q.; Wang, Y.; Zhao, J. Histone Deacetylase Inhibitor Trichostatin a Promotes the Apoptosis of Osteosarcoma Cells through P53 Signaling Pathway Activation. Int. J. Biol. Sci. 2016, 12, 1298–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Höring, E.; Podlech, O.; Silkenstedt, B.; Rota, I.A.; Adamopoulou, E.; Naumann, U. The Histone Deacetylase Inhibitor Trichostatin A Promotes Apoptosis and Antitumor Immunity in Glioblastoma Cells. Anticancer Res. 2013, 33, 1351–1360. [Google Scholar]
- Hwang, J.W.; Kim, Y.M.; Hong, S.H.; Choi, B.T.; Lee, W.H.; Choi, Y.H. Modulacon of Cell Cycle Control by Histone Deacetylase Inhibitor Trichostatin A in A549 Human Non-Small Cell Lung Cancer Cells. J. Life Sci. 2005, 15, 726–733. [Google Scholar]
- Rhodes, L.V.; Nitschke, A.M.; Segar, H.C.; Martin, E.C.; Driver, J.L.; Elliott, S.; Nam, S.Y.; Li, M.; Nephew, K.P.; Burow, M.E. The Histone Deacetylase Inhibitor Trichostatin A Alters MicroRNA Expression Profiles in Apoptosis-Resistant Breast Cancer Cells. Oncol. Rep. 2012, 27, 10–16. [Google Scholar]
- Emonds, E. Molecular Determinants of the Antitumor Effects of Trichostatin A in Pancreatic Cancer Cells. World J. Gastroenterol. 2010, 16, 1970. [Google Scholar] [CrossRef]
- Hong, Z.; Han, Z.; Xiao, M.; Yang, Y.; Xia, X.; Zhou, J. Microarray Study of Mechanism of Trichostatin a Inducing Apoptosis of Molt-4 Cells. J. Huazhong Univ. Sci. Technol. Med. Sci. 2009, 29, 445–450. [Google Scholar] [CrossRef]
- Li, G.-C.; Zhang, X.; Pan, T.-J.; Chen, Z.; Ye, Z.-Q. Histone Deacetylase Inhibitor Trichostatin A Inhibits the Growth of Bladder Cancer Cells through Induction of P21WAF1 and G1 Cell Cycle Arrest. Int. J. Urol. 2006, 13, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Wu, Y.; Wang, X.; Liu, X.; Zhong, K.; Huang, Y.; Chen, Y.; Gao, H. In Vitro and in Vivo Characterization of the Antibacterial Activity and Membrane Damage Mechanism of Quinic Acid against Staphylococcus Aureus. J. Food Saf. 2018, 38, e12416. [Google Scholar] [CrossRef]
- Bai, Y.; Chen, Y.; Chen, X.; Jiang, J.; Wang, X.; Wang, L.; Wang, J.; Zhang, J.; Gao, L. Trichostatin A Activates FOXO1 and Induces Autophagy in Osteosarcoma. Arch. Med. Sci. 2019, 15, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Sun, X.; Zhang, Q.; Chen, X.; Zhao, T.; Lu, L.; Zhang, J.; Hong, Y. Histone Deacetylase Inhibitor Trichostatin A and Autophagy Inhibitor Chloroquine Synergistically Exert Anti-Tumor Activity in H-Ras Transformed Breast Epithelial Cells. Mol. Med. Rep. 2018, 17, 4345–4350. [Google Scholar] [CrossRef]
- Eriksson, I.; Joosten, M.; Roberg, K.; Öllinger, K. The Histone Deacetylase Inhibitor Trichostatin A Reduces Lysosomal PH and Enhances Cisplatin-Induced Apoptosis. Exp. Cell Res. 2013, 319, 12–20. [Google Scholar] [CrossRef]
- Karagiannis, T.C.; Harikrishnan, K.; El-Osta, A. The Histone Deacetylase Inhibitor, Trichostatin A, Enhances Radiation Sensitivity and Accumulation of GammaH2A.X. Cancer Biol. Ther. 2005, 4, 787–793. [Google Scholar] [CrossRef] [Green Version]
- Karagiannis, T.C.; Smith, A.J.; El’Osta, A. Radio-and Chemo-Sensitization of Human Erythroleukemic K562 Cells by the Histone Deacetylase Inhibitor Trichostatin A. Hell. J. Nucl. Med. 2004, 7, 184–191. [Google Scholar]
- Lambert, I.H.; Nielsen, D.; Stürup, S. Impact of the Histone Deacetylase Inhibitor Trichostatin A on Active Uptake, Volume-Sensitive Release of Taurine, and Cell Fate in Human Ovarian Cancer Cells. Am. J. Physiol. Cell Physiol. 2020, 318, C581–C597. [Google Scholar] [CrossRef]
- Piacentini, P.; Donadelli, M.; Costanzo, C.; Moore, P.S.; Palmieri, M.; Scarpa, A. Trichostatin A Enhances the Response of Chemotherapeutic Agents in Inhibiting Pancreatic Cancer Cell Proliferation. Virchows Arch. 2006, 448, 797–804. [Google Scholar] [CrossRef]
- Yoon, C.Y.; Park, M.J.; Lee, J.S.; Lee, S.C.; Oh, J.J.; Park, H.; Chung, C.W.; Abdullajanov, M.M.; Jeong, S.J.; Hong, S.K.; et al. The Histone Deacetylase Inhibitor Trichostatin A Synergistically Resensitizes a Cisplatin Resistant Human Bladder Cancer Cell Line. J. Urol. 2011, 185, 1102–1111. [Google Scholar] [CrossRef]
- Donadelli, M.; Costanzo, C.; Beghelli, S.; Scupoli, M.T.; Dandrea, M.; Bonora, A.; Piacentini, P.; Budillon, A.; Caraglia, M.; Scarpa, A.; et al. Synergistic Inhibition of Pancreatic Adenocarcinoma Cell Growth by Trichostatin A and Gemcitabine. Biochim. Biophys. Acta Mol. Cell Res. 2007, 1773, 1095–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray-Zmijewski, F.; Lane, D.P.; Bourdon, J.C. P53/P63/P73 Isoforms: An Orchestra of Isoforms to Harmonise Cell Differentiation and Response to Stress. Cell Death Differ. 2006, 13, 962–972. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Kukita, A.; Kukita, T.; Shobuike, T.; Nakamura, T.; Kohashi, O. Two Histone Deacetylase Inhibitors, Trichostatin A and Sodium Butyrate, Suppress Differentiation into Osteoclasts but Not into Macrophages. Blood 2003, 101, 3451–3459. [Google Scholar] [CrossRef]
- Touma, S.E.; Goldberg, J.S.; Moench, P.; Guo, X.; Tickoo, S.K.; Gudas, L.J.; Nanus, D.M. Retinoic Acid and the Histone Deacetylase Inhibitor Trichostatin A Inhibit the Proliferation of Human Renal Cell Carcinoma in a Xenograft Tumor Model. Clin. Cancer Res. 2005, 11, 3558–3566. [Google Scholar] [CrossRef] [Green Version]
- Januchowski, R.; Jagodzinski, P.P. Trichostatin A Down-Regulates ZAP-70, LAT and SLP-76 Content in Jurkat T Cells. Int. Immunopharmacol. 2007, 7, 198–204. [Google Scholar] [CrossRef]
- Sanaei, F.; Amin, M.M.; Alavijeh, Z.P.; Esfahani, R.A.; Sadeghi, M.; Bandarrig, N.S.; Fatehizadeh, A.; Taheri, E.; Rezakazemi, M. Health Risk Assessment of Potentially Toxic Elements Intake via Food Crops Consumption: Monte Carlo Simulation-Based Probabilistic and Heavy Metal Pollution Index. Environ. Sci. Pollut. Res. 2021, 28, 1479–1490. [Google Scholar] [CrossRef] [PubMed]
- Sanaei, M.; Kavoosi, F. Effect of 5-Aza-2′-Deoxycytidine in Comparison to Valproic Acid and Trichostatin a on Histone Deacetylase 1, Dna Methyltransferase 1, and Cip/Kip Family (P21, P27, and P57) Genes Expression, Cell Growth Inhibition, and Apoptosis Induction in Colon Cancer Sw480 Cell Line. Adv. Biomed. Res. 2019, 8, 52. [Google Scholar] [CrossRef]
- Vincent, A.; Ducourouble, M.; Van Seuningen, I. Epigenetic Regulation of the Human Mucin Gene MUC4 in Epithelial Cancer Cell Lines Involves Both DNA Methylation and Histone Modifications Mediated by DNA Methyltransferases and Histone Deacetylases. FASEB j. 2008, 22, 3035–3045. [Google Scholar] [CrossRef]
- Geng, Y.; Liu, J.; Xie, Y.; Jiang, H.; Zuo, K.; Li, T.; Liu, Z. Trichostatin A Promotes GLI1 Degradation and P21 Expression in Multiple Myeloma Cells. Cancer Manag. Res. 2018, 10, 2905–2914. [Google Scholar] [CrossRef] [Green Version]
- Alao, J.P.; Lam, E.W.-F.; Ali, S.; Buluwela, L.; Bordogna, W.; Lockey, P.; Varshochi, R.; Stavropoulou, A.V.; Coombes, R.C.; Vigushin, D.M. Histone Deacetylase Inhibitor Trichostatin A Represses Estrogen Receptor α-Dependent Transcription and Promotes Proteasomal Degradation of Cyclin D1 in Human Breast Carcinoma Cell Lines. Clin. Cancer Res. 2004, 10, 8094–8104. [Google Scholar] [CrossRef] [Green Version]
- Sassi, F.d.A.; Caesar, L.; Jaeger, M.; Nör, C.; Abujamra, A.L.; Schwartsmann, G.; de Farias, C.B.; Brunetto, A.L.; Lopez, P.L.d.C.; Roesler, R. Inhibitory Activities of Trichostatin A in U87 Glioblastoma Cells and Tumorsphere-Derived Cells. J. Mol. Neurosci. 2014, 54, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Aziee, S.; Haiyuni, M.Y.; Al-Jamal, H.A.N.; Shafini, M.Y.; Wahab, R.A.; Shamsuddin, S.; Johan, M.F. Apoptotic Induction in CCRF-CEM and HL-60 Human Leukemic Cell Lines by 5-Azacitidine and Trichostatin A. J. Biomed. Clin. Sci. 2018, 3, 54–61. [Google Scholar]
- Balaguer, T.M.; Gómez-Martínez, A.; García-Morales, P.; Lacueva, J.; Calpena, R.; Reverte, L.R.; Riquelme, N.L.; Martinez-Lacaci, I.; Ferragut, J.A.; Saceda, M. Dual Regulation of P-Glycoprotein Expression by Trichostatin A in Cancer Cell Lines. BMC Mol. Biol. 2012, 13, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buishand, F.O.; Cardin, E.; Hu, Y.; Ried, T. Trichostatin A Preferentially Reverses the Upregulation of Gene-Expression Levels Induced by Gain of Chromosome 7 in Colorectal Cancer Cell Lines. Genes Chromosomes Cancer 2018, 57, 35–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cecconi, D.; Donadelli, M.; Rinalducci, S.; Zolla, L.; Scupoli, M.T.; Scarpa, A.; Palmieri, M.; Righetti, P.G. Proteomic Analysis of Pancreatic Endocrine Tumor Cell Lines Treated with the Histone Deacetylase Inhibitor Trichostatin A. Proteomics 2007, 7, 1644–1653. [Google Scholar] [CrossRef]
- Chen, C.; Chen, C.; Chen, J.; Zhou, L.; Xu, H.; Jin, W.; Wu, J.; Gao, S. Histone Deacetylases Inhibitor Trichostatin A Increases the Expression of Dleu2/MiR-15a/16-1 via HDAC3 in Non-Small Cell Lung Cancer. Mol. Cell Biochem. 2013, 383, 137–148. [Google Scholar] [CrossRef]
- Chen, Z.; Yang, Y.; Liu, B.; Wang, B.; Sun, M.; Zhang, L.; Chen, B.; You, H.; Zhou, M. Promotion of Metastasis-Associated Gene Expression in Survived PANC-1 Cells Following Trichostatin A Treatment. Anti-Cancer Agents Med. Chem. 2015, 15, 1317–1325. [Google Scholar] [CrossRef]
- Cheng, D.-D.; Yang, Q.-C.; Zhang, Z.-C.; Yang, C.-X.; Liu, Y.-W. Antitumor Activity of Histone Deacetylase Inhibitor Trichostatin A in Osteosarcoma Cells. Asian Pac. J. Cancer Prev. 2012, 13, 1395–1399. [Google Scholar] [CrossRef] [Green Version]
- Chiba, T.; Yokosuka, O.; Fukai, K.; Kojima, H.; Tada, M.; Arai, M.; Imazeki, F.; Saisho, H. Cell Growth Inhibition and Gene Expression Induced by the Histone Deacetylase Inhibitor, Trichostatin A, on Human Hepatoma Cells. Oncology 2004, 66, 481–491. [Google Scholar] [CrossRef]
- Chodkowska, A.; Bieńkowska, A.; S\lyk, Ż.; Giebu\ltowicz, J.; Ma\lecki, M. Anticancer Activity of Topical Ointments with Histone Deacetylase Inhibitor, Trichostatin A. Adv. Clin. Exp. Med. 2020, 29, 1039–1049. [Google Scholar] [CrossRef]
- de Oliveira Santos, J.; Zuma, A.A.; de Luna Vitorino, F.N.; da Cunha, J.P.C.; de Souza, W.; Motta, M.C.M. Trichostatin A Induces Trypanosoma Cruzi Histone and Tubulin Acetylation: Effects on Cell Division and Microtubule Cytoskeleton Remodelling. Parasitology 2019, 146, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Diao, J.-S.; Xia, W.-S.; Yi, C.-G.; Wang, Y.-M.; Li, B.; Xia, W.; Liu, B.; Guo, S.-Z.; Sun, X.-D. Trichostatin A Inhibits Collagen Synthesis and Induces Apoptosis in Keloid Fibroblasts. Arch. Dermatol. Res. 2011, 303, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Drzewiecka, H.; Jagodzinski, P.P. Trichostatin A Reduced Phospholipase C Gamma-1 Transcript and Protein Contents in MCF-7 Breast Cancer Cells. Biomed. Pharmacother. 2012, 66, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Han, M.H.; Park, C.; Kwon, T.K.; Kim, G.-Y.; Kim, W.-J.; Hong, S.H.; Yoo, Y.H.; Choi, Y.H. The Histone Deacetylase Inhibitor Trichostatin A Sensitizes Human Renal Carcinoma Cells to TRAIL-Induced Apoptosis through Down-Regulation of c-FLIPL. Biomol. Ther. 2015, 23, 31–38. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Liu, H.; Chen, Y. Effects of Trichostatin A on HDAC8 Expression, Proliferation and Cell Cycle of Molt-4 Cells. J. Huazhong Univ. Sci. Technol. 2006, 26, 531–533. [Google Scholar] [CrossRef]
- Hong, S.; Chang, S.-Y.; Yeom, D.-H.; Kang, J.-H.; Hong, K.-J. Differential Regulation of Thrombospondin-1 Expression and Antiangiogenesis of ECV304 Cells by Trichostatin A and Helixor A. Anti-Cancer Drugs 2007, 18, 1005–1014. [Google Scholar] [CrossRef]
- Hrgovic, I.; Doll, M.; Kleemann, J.; Wang, X.-F.; Zoeller, N.; Pinter, A.; Kippenberger, S.; Kaufmann, R.; Meissner, M. The Histone Deacetylase Inhibitor Trichostatin a Decreases Lymphangiogenesis by Inducing Apoptosis and Cell Cycle Arrest via P21-Dependent Pathways. BMC Cancer 2016, 16, 763. [Google Scholar] [CrossRef] [Green Version]
- Hsu, Y.-F.; Sheu, J.-R.; Hsiao, G.; Lin, C.-H.; Chang, T.-H.; Chiu, P.-T.; Wang, C.-Y.; Hsu, M.-J. P53 in Trichostatin A Induced C6 Glioma Cell Death. Biochim. Biophys. Acta Gen. Subj. 2011, 1810, 504–513. [Google Scholar] [CrossRef]
- Zhang, X.F.; Yan, Q.; Shen, W.; Gurunathan, S. Trichostatin A enhances the apoptotic potential of palladium nanoparticles in human cervical cancer cells. Int. J. Mol. Sci. 2016, 17, 1354. [Google Scholar] [CrossRef]
- Huang, K.; Liu, Y.; Gu, C.; Liu, D.; Zhao, B. Trichostatin A Augments Esophageal Squamous Cell Carcinoma Cells Migration by Inducing Acetylation of RelA at K310 Leading Epithelia–Mesenchymal Transition. Anti-Cancer Drugs 2020, 31, 567–574. [Google Scholar] [CrossRef]
- Huang, X.-Y.; Xiao, G.-T.; Huang, T.-X.; Chen, Z.-X.; Gao, W.-Y.; Zheng, B.-Y.; Wang, X. Trichostatin A Alleviates HBx-Induced HCC Metastasis in Metabolic Stress through Up-Regulating SIRT3 Expression; In Review. 2021. Available online: https://assets.researchsquare.com/files/rs-420738/v1/5af26051-9733-43fd-810f-6817798dfef0.pdf?c=1631882363 (accessed on 25 August 2022).
- Kang, J.-H.; Kim, S.-A.; Chang, S.-Y.; Hong, S.; Hong, K.-J. Inhibition of Trichostatin A-Induced Antiangiogenesis by Small-Interfering RNA for Thrombospondin-1. Exp. Mol. Med. 2007, 39, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Kashiwagi, Y.; Horie, K.; Kanno, C.; Inomata, M.; Imamura, T.; Kato, M.; Yamamoto, T.; Yamashita, H. Trichostatin A–Induced TGF-β Type II Receptor Expression in Retinoblastoma Cell Lines. Invest. Ophthalmol. Vis. Sci. 2010, 51, 679. [Google Scholar] [CrossRef] [PubMed]
- Katsura, T.; Iwai, S.; Ota, Y.; Shimizu, H.; Ikuta, K.; Yura, Y. The Effects of Trichostatin A on the Oncolytic Ability of Herpes Simplex Virus for Oral Squamous Cell Carcinoma Cells. Cancer Gene Ther. 2009, 16, 237–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.B.; Yoshida, M.; Horinouchi, S. Selective Induction of Cyclin-Dependent Kinase Inhibitors and Their Roles in Cell Cycle Arrest Caused by Trichostatin A, an Inhibitor of Histone Deacetylase. Ann. N. Y. Acad Sci. 1999, 886, 200–203. [Google Scholar] [CrossRef] [PubMed]
- Yoo, Y.C.; Lee, H.Y.; Kwak, S.T.; Lee, K.B. Regulatory Effect of Chondroitin Sulfates Derived Form Human Placenta on Mitogen-Induced Activation of Murine Splenocytes. In Proceedings of the PSK Conference, Japon, Tokai, 1 April 2000; p. 1441. [Google Scholar]
- Kim, H.-N.; Ha, H.; Lee, J.-H.; Jung, K.; Yang, D.; Woo, K.M.; Lee, Z.H. Trichostatin A Inhibits Osteoclastogenesis and Bone Resorption by Suppressing the Induction of C-Fos by RANKL. Eur. J. Pharmacol. 2009, 623, 22–29. [Google Scholar] [CrossRef]
- Li, H.; Wu, X. Histone Deacetylase Inhibitor, Trichostatin A, Activates P21WAF1/CIP1 Expression through Downregulation of c-Myc and Release of the Repression of c-Myc from the Promoter in Human Cervical Cancer Cells. Biochem. Biophys. Res. Commun. 2004, 324, 860–867. [Google Scholar] [CrossRef]
- Li, C.; Tao, Y.; Li, C.; Liu, B.; Liu, J.; Wang, G.; Liu, H. PU.1-Bim Axis Is Involved in Trichostatin A-Induced Apoptosis in Murine pro-B Lymphoma FL5.12 Cells. Acta Biochim. Biophys. Sin. 2016, 48, 850–855. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-W.; Wang, S.-A.; Hsu, T.-Y.; Chen, T.-A.; Chang, W.-C.; Hung, J.-J. Inhibition of LPS-Induced C/EBPδ by Trichostatin a Has a Positive Effect on LPS-Induced Cyclooxygenase 2 Expression in RAW264.7 Cells. J. Cell. Biochem. 2010, 110, 1430–1438. [Google Scholar] [CrossRef]
- Liu, Y.; He, G.; Wang, Y.; Guan, X.; Pang, X.; Zhang, B. MCM-2 Is a Therapeutic Target of Trichostatin A in Colon Cancer Cells. Toxicol. Lett. 2013, 221, 23–30. [Google Scholar] [CrossRef]
- Liu, J.; Li, Y.; Dong, F.; Li, L.; Masuda, T.; Allen, T.D.; Lobe, C.G. Trichostatin A Suppresses Lung Adenocarcinoma Development in Grg1 Overexpressing Transgenic Mice. Biochem. Biophys. Res. Commun. 2015, 463, 1230–1236. [Google Scholar] [CrossRef]
- Liu, J.-H.; Cao, Y.-M.; Rong, Z.-P.; Ding, J.; Pan, X. Trichostatin A Induces Autophagy in Cervical Cancer Cells by Regulating the PRMT5-STC1-TRPV6-JNK Pathway. Pharmacology 2021, 106, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Łuczak, M.W.; Jagodziński, P.P. Trichostatin A Down-Regulates CYP19 Transcript and Protein Levels in MCF-7 Breast Cancer Cells. Biomed. Pharmacother. 2009, 63, 262–266. [Google Scholar] [CrossRef]
- Ma, J.; Guo, X.; Zhang, S.; Liu, H.; Lu, J.; Dong, Z.; Liu, K.; Ming, L. Trichostatin A, a Histone Deacetylase Inhibitor, Suppresses Proliferation and Promotes Apoptosis of Esophageal Squamous Cell Lines. Mol. Med. Rep. 2015, 11, 4525–4531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzio, E.A.; Soliman, K.F.A. Whole-Transcriptomic Profile of SK-MEL-3 Melanoma Cells Treated with the Histone Deacetylase Inhibitor: Trichostatin A. Cancer Genom. Proteom. 2018, 15, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Meng, C.; Dai, D.; Guo, K. Comparative Evaluation of the Effects of 5-Aza-2′-Deoxycytidine and Trichostatin A on Reactivation of HMLH1 in COC1/DDP Ovarian Cancer Cell Line. Chin. J. Cancer Res. 2009, 21, 102–108. [Google Scholar] [CrossRef]
- Meng, F.; Sun, G.; Zhong, M.; Yu, Y.; Brewer, M.A. Anticancer Efficacy of Cisplatin and Trichostatin A or 5-Aza-2′-Deoxycytidine on Ovarian Cancer. Br. J. Cancer 2013, 108, 579–586. [Google Scholar] [CrossRef]
- Miyanaga, A.; Gemma, A.; Noro, R.; Kataoka, K.; Matsuda, K.; Nara, M.; Okano, T.; Seike, M.; Yoshimura, A.; Kawakami, A.; et al. Antitumor Activity of Histone Deacetylase Inhibitors in Non-Small Cell Lung Cancer Cells: Development of a Molecular Predictive Model. Mol. Cancer Ther. 2008, 7, 1923–1930. [Google Scholar] [CrossRef] [Green Version]
- Moore, P.S.; Barbi, S.; Donadelli, M.; Costanzo, C.; Bassi, C.; Palmieri, M.; Scarpa, A. Gene Expression Profiling after Treatment with the Histone Deacetylase Inhibitor Trichostatin A Reveals Altered Expression of Both Pro- and Anti-Apoptotic Genes in Pancreatic Adenocarcinoma Cells. Biochim. Biophys. Acta Mol. Cell Res. 2004, 1693, 167–176. [Google Scholar] [CrossRef] [Green Version]
- Moreira, J.M.A.; Scheipers, P.; Sørensen, P. The Histone Deacetylase Inhibitor Trichostatin A Modulates CD4+ T Cell Responses. BMC Cancer 2003, 3, 30. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, N.K.; Weisberg, E.; Gilchrist, D.; Bueno, R.; Sugarbaker, D.J.; Jaklitsch, M.T. Effectiveness of Trichostatin A as a Potential Candidate for Anticancer Therapy in Non–Small-Cell Lung Cancer. Ann. Thorac. Surg. 2006, 81, 1034–1042. [Google Scholar] [CrossRef]
- Nagamine, M.K.; Sanches, D.S.; Pinello, K.C.; Torres, L.N.; Mennecier, G.; Latorre, A.O.; Fukumasu, H.; Dagli, M.L.Z. In Vitro Inhibitory Effect of Trichostatin A on Canine Grade 3 Mast Cell Tumor. Vet. Res. Commun. 2011, 35, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Nagaraja, S.S.; Krishnamoorthy, V.; Raviraj, R.; Paramasivam, A.; Nagarajan, D. Effect of Trichostatin A on Radiation Induced Epithelial-Mesenchymal Transition in A549 Cells. Biochem. Biophys. Res. Commun. 2017, 493, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- Noh, E.J.; Lim, D.-S.; Jeong, G.; Lee, J.-S. An HDAC Inhibitor, Trichostatin A, Induces a Delay at G2/M Transition, Slippage of Spindle Checkpoint, and Cell Death in a Transcription-Dependent Manner. Biochem. Biophys. Res. Commun. 2009, 378, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Noh, H.; Park, J.; Shim, M.; Lee, Y. Trichostatin A Enhances Estrogen Receptor-Alpha Repression in MCF-7 Breast Cancer Cells under Hypoxia. Biochem. Biophys. Res. Commun. 2016, 470, 748–752. [Google Scholar] [CrossRef] [PubMed]
- Olaharski, A.J.; Ji, Z.; Woo, J.-Y.; Lim, S.; Hubbard, A.E.; Zhang, L.; Smith, M.T. The Histone Deacetylase Inhibitor Trichostatin A Has Genotoxic Effects in Human Lymphoblasts In Vitro. Toxicol. Sci. 2006, 93, 341–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, X.; He, G.; Luo, C.; Wang, Y.; Zhang, B. Knockdown of Rad9A Enhanced DNA Damage Induced by Trichostatin A in Esophageal Cancer Cells. Tumor. Biol. 2016, 37, 963–970. [Google Scholar] [CrossRef]
- Papeleu, P.; Loyer, P.; Vanhaecke, T.; Elaut, G.; Geerts, A.; Guguen-Guillouzo, C.; Rogiers, V. Trichostatin A Induces Differential Cell Cycle Arrests but Does Not Induce Apoptosis in Primary Cultures of Mitogen-Stimulated Rat Hepatocytes. J. Hepatol. 2003, 39, 374–382. [Google Scholar] [CrossRef]
- Park, S.-J.; Kim, M.-J.; Kim, H.-B.; Sohn, H.-Y.; Bae, J.-H.; Kang, C.-D.; Kim, S.-H. Trichostatin A Sensitizes Human Ovarian Cancer Cells to TRAIL-Induced Apoptosis by down-Regulation of c-FLIPL via Inhibition of EGFR Pathway. Biochem. Pharmacol. 2009, 77, 1328–1336. [Google Scholar] [CrossRef]
- Peiffer, L.; Poll-Wolbeck, S.J.; Flamme, H.; Gehrke, I.; Hallek, M.; Kreuzer, K.-A. Trichostatin A Effectively Induces Apoptosis in Chronic Lymphocytic Leukemia Cells via Inhibition of Wnt Signaling and Histone Deacetylation. J. Cancer Res. Clin. Oncol. 2014, 140, 1283–1293. [Google Scholar] [CrossRef]
- Platta, C.S.; Greenblatt, D.Y.; Kunnimalaiyaan, M.; Chen, H. The HDAC Inhibitor Trichostatin A Inhibits Growth of Small Cell Lung Cancer Cells. J. Surg. Res. 2007, 142, 219–226. [Google Scholar] [CrossRef]
- Ruan, W.-M.; Li, Y.-L.; Nie, G.; Zhou, W.-X.; Zou, X.-M. Differential Expression of Glycoprotein Non-Metastatic Melanoma Protein B (GPNMB) Involved in Trichostatin A-Induced Apoptosis in Gastric Cancer. Int. J. Clin. Exp. Med. 2014, 7, 4857–4866. [Google Scholar] [PubMed]
- Salvi, V.; Bosisio, D.; Mitola, S.; Andreoli, L.; Tincani, A.; Sozzani, S. Trichostatin A Blocks Type I Interferon Production by Activated Plasmacytoid Dendritic Cells. Immunobiology 2010, 215, 756–761. [Google Scholar] [CrossRef] [PubMed]
- Sanaei, M.; Kavoosi, F.; Arabloo, M. Effect of Curcumin in Comparison with Trichostatin A on the Reactivation of Estrogen Receptor Alpha Gene Expression, Cell Growth Inhibition and Apoptosis Induction in Hepatocellular Carcinoma Hepa 1-6 Cell LLine. Asian Pac. J. Cancer Prev. 2020, 21, 1045–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, J.; Cho, N.; Kim, H.; Tsurumi, T.; Jang, Y.; Lee, W.; Lee, S. Cell Cycle Arrest and Lytic Induction of EBV-Transformed B Lymphoblastoid Cells by a Histone Deacetylase Inhibitor, Trichostatin A. Oncol. Rep. 2008, 19, 93–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Kumar, S.; Kundu, G.C. Transcriptional Regulation of Human Osteopontin Promoter by Histone Deacetylase Inhibitor, Trichostatin A in Cervical Cancer Cells. Mol. Cancer 2010, 9, 178. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Bhat, A.A.; Krishnan, M.; Singh, A.B.; Dhawan, P. Trichostatin-A Modulates Claudin-1 MRNA Stability through the Modulation of Hu Antigen R and Tristetraprolin in Colon Cancer Cells. Carcinogenesis 2013, 34, 2610–2621. [Google Scholar] [CrossRef]
- Shen, Z.; Liao, X.; Shao, Z.; Feng, M.; Yuan, J.; Wang, S.; Gan, S.; Ha, Y.; He, Z.; Jie, W. Short-Term Stimulation with Histone Deacetylase Inhibitor Trichostatin a Induces Epithelial-Mesenchymal Transition in Nasopharyngeal Carcinoma Cells without Increasing Cell Invasion Ability. BMC Cancer 2019, 19, 262. [Google Scholar] [CrossRef]
- Shindo, Y.; Arai, W.; Konno, T.; Kohno, T.; Kodera, Y.; Chiba, H.; Miyajima, M.; Sakuma, Y.; Watanabe, A.; Kojima, T. Effects of Histone Deacetylase Inhibitors Tricostatin A and Quisinostat on Tight Junction Proteins of Human Lung Adenocarcinoma A549 Cells and Normal Lung Epithelial Cells. Histochem. Cell Biol. 2021, 155, 637–653. [Google Scholar] [CrossRef]
- Song, W.-Y.; Yang, Q.-L.; Zhao, W.-L.; Jin, H.-X.; Yao, G.-D.; Peng, Z.-F.; Shi, S.-L.; Yang, H.-Y.; Zhang, X.-Y.; Sun, Y.-P. The Effects of Anticancer Drugs TSA and GSK on Spermatogenesis in Male Mice. Am. J. Transl. Res. 2016, 8, 221. [Google Scholar]
- Strait, K.A.; Dabbas, B.; Hammond, E.H.; Warnick, C.T.; Ilstrup, S.J.; Ford, C.D. Cell Cycle Blockade and Differentiation of Ovarian Cancer Cells by the Histone Deacetylase Inhibitor Trichostatin A Are Associated with Changes in P21, Rb, and Id Proteins 1 Supported by Grants from Feature Films for Families Cancer Research Fund (CEO, Forrest S. Baker III) and The Deseret Foundation. 1. Mol. Cancer Ther. 2002, 1, 1181–1190. [Google Scholar]
- Subramanian, C.; Jarzembowski, J.A.; Opipari, A.W.; Castle, V.P.; Kwok, R.P.S. CREB-Binding Protein Is a Mediator of Neuroblastoma Cell Death Induced By the Histone Deacetylase Inhibitor Trichostatin A. Neoplasia 2007, 9, 495–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Liu, X.; Chen, Y.; Liu, F. Anticancer Activities of Trichostatin A on Malignant Lymphoid Cells. J. Huazhong Univ. Sci. Technol. 2006, 26, 538–541. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Han, Y.; Liu, J.; Fang, Y.; Tian, Y.; Zhou, J.; Ma, D.; Wu, P. Trichostatin A Targets the Mitochondrial Respiratory Chain, Increasing Mitochondrial Reactive Oxygen Species Production to Trigger Apoptosis in Human Breast Cancer Cells. PLoS ONE 2014, 9, e91610. [Google Scholar] [CrossRef] [PubMed]
- Tarnowski, M.; Tkacz, M.; Kopytko, P.; Bujak, J.; Piotrowska, K.; Pawlik, A. Trichostatin A Inhibits Rhabdomyosarcoma Proliferation and Induces Differentiation through MyomiR Reactivation. Folia Biol. 2019, 65, 43–52. [Google Scholar]
- Tavakoli-Yaraki, M.; Karami-Tehrani, F.; Salimi, V.; Sirati-Sabet, M. Induction of Apoptosis by Trichostatin A in Human Breast Cancer Cell Lines: Involvement of 15-Lox-1. Tumor. Biol. 2013, 34, 241–249. [Google Scholar] [CrossRef]
- Toki, S.; Goleniewska, K.; Reiss, S.; Zhou, W.; Newcomb, D.C.; Bloodworth, M.H.; Stier, M.T.; Boyd, K.L.; Polosukhin, V.V.; Subramaniam, S.; et al. The Histone Deacetylase Inhibitor Trichostatin A Suppresses Murine Innate Allergic Inflammation by Blocking Group 2 Innate Lymphoid Cell (ILC2) Activation. Thorax 2016, 71, 633–645. [Google Scholar] [CrossRef] [Green Version]
- Urbinati, G.; Marsaud, V.; Nicolas, V.; Vergnaud-Gauduchon, J.; Renoir, J.-M. Liposomal Trichostatin A: Therapeutic Potential in Hormone-Dependent and -Independent Breast Cancer Xenograft Models. Horm. Mol. Biol. Clin. Investig. 2011, 6, 215–225. [Google Scholar] [CrossRef]
- Vigushin, D.M.; Ali, S.; Pace, P.E.; Mirsaidi, N.; Ito, K.; Adcock, I.; Coombes, R.C. Trichostatin A Is a Histone Deacetylase Inhibitor with Potent Antitumor Activity against Breast Cancer in Vivo. Clin. Cancer Res. 2001, 7, 971–976. [Google Scholar]
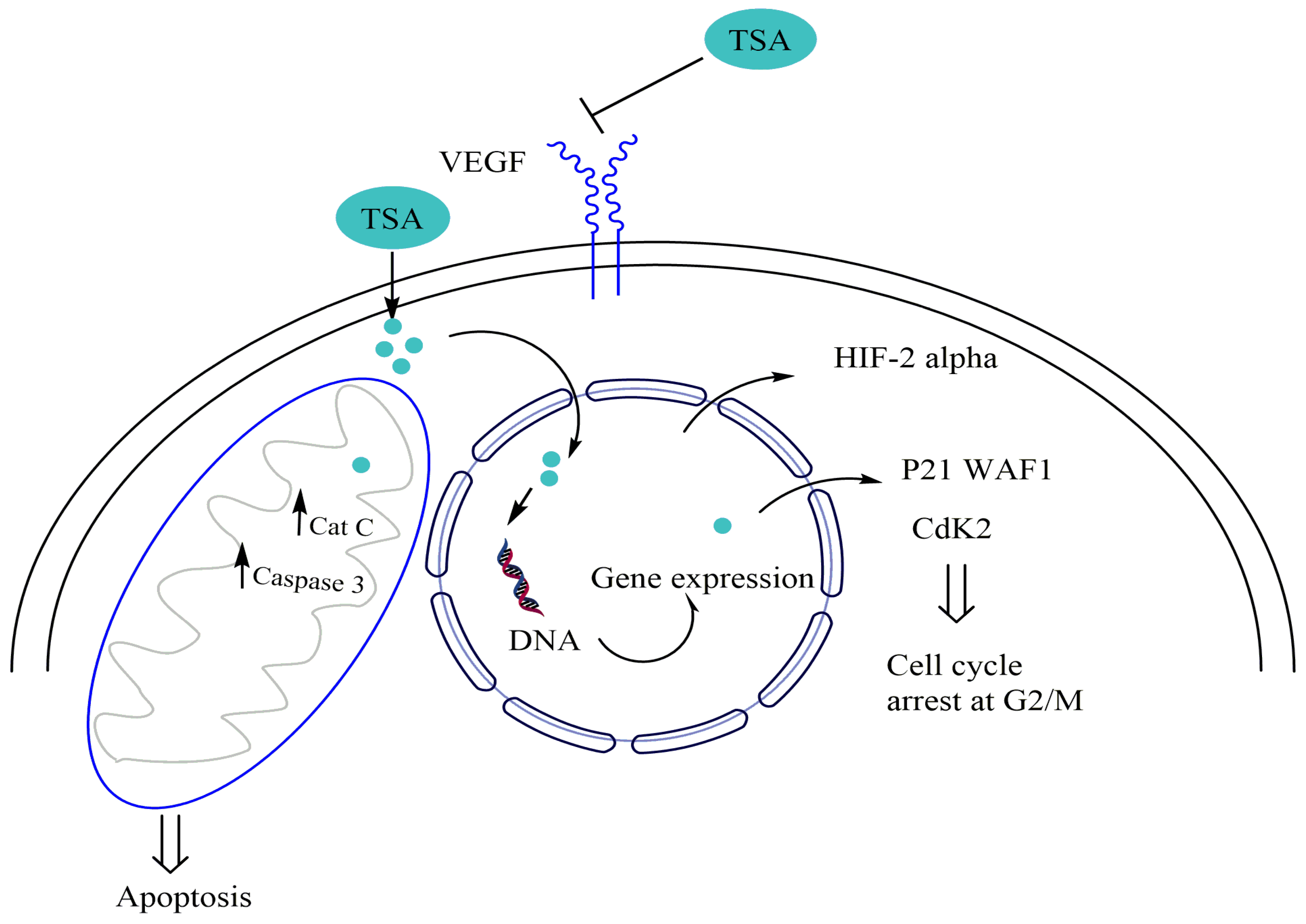
- Kang, F.-W.; Que, L.; Wu, M.; Wang, Z.-L.; Sun, J. Effects of Trichostatin A on HIF-1α and VEGF Expression in Human Tongue Squamous Cell Carcinoma Cells in Vitro. Oncol. Rep. 2012, 28, 193–199. [Google Scholar]
- Wang, F.; Qi, Y.; Li, X.; He, W.; Fan, Q.-X.; Zong, H. HDAC Inhibitor Trichostatin A Suppresses Esophageal Squamous Cell Carcinoma Metastasis through HADC2 Reduced MMP-2/9. Clin. Investig. Med. 2013, E87–E94. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Xu, J.; Wang, H.; Wu, L.; Yuan, W.; Du, J.; Cai, S. Trichostatin A, a Histone Deacetylase Inhibitor, Reverses Epithelial–Mesenchymal Transition in Colorectal Cancer SW480 and Prostate Cancer PC3 Cells. Biochem. Biophys. Res. Commun. 2015, 456, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-C.; Wang, S.-T.; Liu, H.-T.; Wang, X.-Y.; Wu, S.-C.; Chen, L.-C.; Liu, Y.-W. Trichostatin A Induces Bladder Cancer Cell Death via Intrinsic Apoptosis at the Early Phase and Sp1-Survivin Downregulation at the Late Phase of Treatment. Oncol. Rep. 2017, 38, 1587–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, H.J.; Choi, Y.H. G1 Phase Arrest of the Cell Cycle by Histone Deacetylase Inhibitor Trichostatin A in U937 Human Leukemic Cells. J. Cancer Prev. 2006, 11, 114–122. [Google Scholar]
- Wu, Y.; Guo, S.-W. Inhibition of Proliferation of Endometrial Stromal Cells by Trichostatin A, RU486, CDB-2914, N-Acetylcysteine, and ICI 182780. Gynecol. Obs. Invest. 2006, 62, 193–205. [Google Scholar] [CrossRef]
- Xingang, L.; Weikai, C.; Junxia, G.; Guohui, C.; Yan, C. Regulation of Histone Acetylation and Apoptosis by Trichostatin in HL-60 Cells. J. Huazhong Univ. Sci. Technol. Med. Sci. 2004, 24, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Wang, Y.; Mei, Q.; Chen, J.; Du, J.; Wei, Y.; Xu, Y. Trichostatin A Inhibits Proliferation, Induces Apoptosis and Cell Cycle Arrest in HeLa Cells. Chin. J. Cancer Res. 2006, 18, 188–192. [Google Scholar] [CrossRef]
- Yang, D.-H.; Lee, J.-W.; Lee, J.; Moon, E.-Y. Dynamic Rearrangement of F-Actin Is Required to Maintain the Antitumor Effect of Trichostatin A. PLoS ONE 2014, 9, e97352. [Google Scholar] [CrossRef] [Green Version]
- Yi, T.; Baek, J.-H.; Kim, H.-J.; Choi, M.-H.; Seo, S.-B.; Ryoo, H.-M.; Kim, G.-S.; Woo, K.M. Trichostatin A-Mediated Upregulation of P21WAF1 Contributes to Osteoclast Apoptosis. Exp. Mol. Med. 2007, 39, 213–221. [Google Scholar] [CrossRef] [Green Version]
- You, B.R.; Park, W.H. Trichostatin A Induces Apoptotic Cell Death of HeLa Cells in a Bcl-2 and Oxidative Stress-Dependent Manner. Int. J. Oncol. 2013, 42, 359–366. [Google Scholar] [CrossRef] [Green Version]
- ZHANG, X.; YU, X.; ZHAO, M.; YI, X.; DU, Z.; XU, Y. Trichostatin A Induces Mitotic Catastrophe of Prostate Cancer DU145 Cells. Chin. J. Cancer Biother. 2007. [Google Scholar]
- Zhang, S.; Cai, X.; Huang, F.; Zhong, W.; Yu, Z. Effect of Trichostatin A on Viability and Microrna Expression in Human Pancreatic Cancer Cell Line Bxpc-3. Exp. Oncol. 2008, 30, 265–268. [Google Scholar] [PubMed]
- Zhang, C.Z.Y.; Zhang, H.T.; Chen, G.G.; Lai, P.B.S. Trichostatin A Sensitizes HBx-Expressing Liver Cancer Cells to Etoposide Treatment. Apoptosis 2011, 16, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.-C.; Jiang, S.-J.; Zhang, S.; Ma, X.-B. Histone Deacetylase Inhibitor Trichostatin A Enhances Antitumor Effects of Docetaxel or Erlotinib in A549 Cell Line. Asian Pac. J. Cancer Prev. 2012, 13, 3471–3476. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Jiang, S.-J.; Shang, B.; Jiang, H.-J. Effects of Histone Deacetylase Inhibitor Trichostatin A Combined with Cisplatin on Apoptosis of A549 Cell Line: TSA Combined with Cisplatin. Thorac. Cancer 2015, 6, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Shao, C.; Chen, Z.; Li, Y.; Jing, X.; Huang, Q. Low Dose of Trichostatin a Improves Radiation Resistance by Activating Akt/Nrf2-Dependent Antioxidation Pathway in Cancer Cells. Radiat. Res. 2021, 195, 366–377. [Google Scholar] [CrossRef]
- ZHU, Y.; PAN, M.; WEI, Q.; CAO, X. A Study on Transcription Regulation Induced by Trichostatin A during Cytotoxicity on MCF-7 Cells. Acta Univ. Med. Nanjing Nat. Sci. 2007, 27, 546. [Google Scholar]
- Zohre, S.; Kazem, N.-K.; Abolfazl, A.; Mohammad, R.-Y.; Aliakbar, M.; Effat, A.; Zahra, D.; Hassan, D.; Nosratollah, Z. Trichostatin A-Induced Apoptosis Is Mediated by Krüppel-like Factor 4 in Ovarian and Lung Cancer. Asian Pac. J. Cancer Prev. 2014, 15, 6581–6586. [Google Scholar] [CrossRef] [Green Version]
- Bajbouj, K.; Mawrin, C.; Hartig, R.; Schulze-Luehrmann, J.; Wilisch-Neumann, A.; Roessner, A.; Schneider-Stock, R. P53-Dependent Antiproliferative and pro-Apoptotic Effects of Trichostatin A (TSA) in Glioblastoma Cells. J. Neuro. Oncol. 2012, 107, 503–516. [Google Scholar] [CrossRef]
- Wetzel, M.; Premkumar, D.R.; Arnold, B.; Pollack, I.F. Effect of Trichostatin A, a Histone Deacetylase Inhibitor, on Glioma Proliferation in Vitro by Inducing Cell Cycle Arrest and Apoptosis. J. Neurosurg. Pediatr. 2005, 103, 549–556. [Google Scholar] [CrossRef]
- Foltz, G.; Yoon, J.-G.; Lee, H.; Ma, L.; Tian, Q.; Hood, L.; Madan, A. Epigenetic Regulation of Wnt Pathway Antagonists in Human Glioblastoma Multiforme. Genes Cancer 2010, 1, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Kang, J. Quercetin and Trichostatin A Cooperatively Kill Human Leukemia Cells. Die Pharm. Int. J. Pharm. Sci. 2005, 60, 856–860. [Google Scholar]
- Bouyahya, A.; Mechchate, H.; Oumeslakht, L.; Zeouk, I.; Aboulaghras, S.; Balahbib, A.; El Omari, N. The role of epigenetic modifications in human cancers and the use of natural compounds as epidrugs: Mechanistic pathways and pharmacodynamic actions. Biomolecules 2022, 12, 367. [Google Scholar] [CrossRef] [PubMed]
- Park, I.-H.; Kang, J.-H.; Shin, J.-M.; Lee, H.-M. Trichostatin A Inhibits Epithelial Mesenchymal Transition Induced by TGF-Β1 in Airway Epithelium. PLoS ONE 2016, 11, e0162058. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Liu, Y.; Qi, B.; Gu, C.; Huo, S.; Zhao, B. Trichostatin A Promotes Esophageal Squamous Cell Carcinoma Cell Migration and EMT through BRD4/ERK1/2-dependent Pathway. Cancer Med. 2021, 10, 5235–5245. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; He, G.; Zhang, K.; Guan, X.; Wang, Y.; Zhang, B. Trichostatin A Induces P53-dependent Endoplasmic Reticulum Stress in Human Colon Cancer Cells. Oncol. Lett. 2018, 17, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Han, R.-F.; Li, K.; Yang, Z.-S.; Chen, Z.-G.; Yang, W.-C. Trichostatin A Induces Mesenchymal-like Morphological Change and Gene Expression but Inhibits Migration and Colony Formation in Human Cancer Cells. Mol. Med. Rep. 2014, 10, 3211–3216. [Google Scholar] [CrossRef] [Green Version]
- Pei, Y.; Robertson, E.S. The Crosstalk of Epigenetics and Metabolism in Herpesvirus Infection. Viruses 2020, 12, 1377. [Google Scholar] [CrossRef]
- Park, H.; Lee, Y.J.; Kim, T.H.; Lee, J.; Yoon, S.; Choi, W.S.; Myung, C.-S.; Kim, H.S. Effects of Trichostatin A, a Histone Deacetylase Inhibitor, on the Regulation of Apoptosis in H-Ras-Transformed Breast Epithelial Cells. Int. J. Mol. Med. 2008, 22, 605–611. [Google Scholar]
- Saito, Y.; Jones, P.M. Epigenetic Activation of Tumor Suppressor MicroRNAs in Human Cancer Cells. Cell Cycle 2006, 5, 2220–2222. [Google Scholar] [CrossRef]
- Wang, X.; Chen, S.; Shen, T.; Lu, H.; Xiao, D.; Zhao, M.; Yao, Y.; Li, X.; Zhang, G.; Zhou, X.; et al. Trichostatin A Reverses Epithelial-mesenchymal Transition and Attenuates Invasion and Migration in MCF-7 Breast Cancer Cells. Exp. Ther. Med. 2020, 19, 1687–1694. [Google Scholar] [CrossRef] [Green Version]
- Xiong, J.; Xu, X.; Zhou, X.; Liu, J.; Gong, Z.; Wu, P.; Li, W. USP22 Transcriptional Activity Is Negatively Regulated by the Histone Deacetylase Inhibitor Trichostatin A. Mol. Med. Rep. 2014, 10, 3343–3347. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xia, Y.; Sun, J. Breast and Gut Microbiome in Health and Cancer. Genes Dis. 2021, 8, 581–589. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.Y.; Yee, Z.Y.; Mai, C.W.; Fang, C.-M.; Abdullah, S.; Ngai, S.C. Zebularine and Trichostatin A Sensitized Human Breast Adenocarcinoma Cells towards Tumor Necrosis Factor-Related Apoptosis Inducing Ligand (TRAIL)-Induced Apoptosis. Heliyon 2019, 5, e02468. [Google Scholar] [CrossRef]
- Tan, L.; Kwok, R.P.; Shukla, A.; Kshirsagar, M.; Zhao, L.; Opipari, A.W.; Liu, J.R. Trichostatin A Restores Apaf-1 Function in Chemoresistant Ovarian Cancer Cells. Cancer 2011, 117, 784–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Hu, C.; Gu, Q.; Li, Y.; Song, M. Trichostatin A Sensitizes Cisplatin-Resistant A549 Cells to Apoptosis by up-Regulating Death-Associated Protein Kinase. Acta Pharm. Sin. 2010, 31, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.-C.; Hsu, F.-S.; Kuo, K.-L.; Liu, S.-H.; Shun, C.-T.; Shi, C.-S.; Chang, H.-C.; Tsai, Y.-C.; Lin, M.-C.; Wu, J.-T.; et al. Trichostatin A, a Histone Deacetylase Inhibitor, Induces Synergistic Cytotoxicity with Chemotherapy via Suppression of Raf/MEK/ERK Pathway in Urothelial Carcinoma. J. Mol. Med. 2018, 96, 1307–1318. [Google Scholar] [CrossRef]
- Hajji, N.; Wallenborg, K.; Vlachos, P.; Nyman, U.; Hermanson, O.; Joseph, B. Combinatorial Action of the HDAC Inhibitor Trichostatin A and Etoposide Induces Caspase-Mediated AIF-Dependent Apoptotic Cell Death in Non-Small Cell Lung Carcinoma Cells. Oncogene 2008, 27, 3134–3144. [Google Scholar] [CrossRef] [Green Version]
- Jasek, E.; Lis, G.J.; Jasińska, M.; Jurkowska, H.; Litwin, J.A. Effect of Histone Deacetylase Inhibitors Trichostatin A and Valproic Acid on Etoposide-Induced Apoptosis in Leukemia Cells. Anticancer Res. 2012, 32, 2791–2799. [Google Scholar]
- Fandy, T.E.; Srivastava, R.K. Trichostatin A Sensitizes TRAIL-Resistant Myeloma Cells by Downregulation of the Antiapoptotic Bcl-2 Proteins. Cancer Chemother. Pharm. 2006, 58, 471–477. [Google Scholar] [CrossRef]
- Zhang, X.; Yashiro, M.; Ren, J.; Hirakawa, K. Histone Deacetylase Inhibitor, Trichostatin A, Increases the Chemosensitivity of Anticancer Drugs in Gastric Cancer Cell Lines. Oncol. Rep. 2006, 16, 563–568. [Google Scholar] [CrossRef]
- Jang, E.R.; Lim, S.-J.; Lee, E.S.; Jeong, G.; Kim, T.-Y.; Bang, Y.-J.; Lee, J.-S. The Histone Deacetylase Inhibitor Trichostatin A Sensitizes Estrogen Receptor α-Negative Breast Cancer Cells to Tamoxifen. Oncogene 2004, 23, 1724–1736. [Google Scholar] [CrossRef] [Green Version]
- Sato, H.; Kashiba, T.; Uzu, M.; Fujiwara, T.; Shibata, Y.; Suzuki, R.; Yamaura, K.; Hisaka, A. Combined Treatment of Trichostatin A Enhances Cytotoxic Effects of Sunitinib on Renal Cell Carcinoma Cells. Am. Assoc. Cancer Res. 2015, 75, 5375. [Google Scholar] [CrossRef]
- Sato, H.; Uzu, M.; Kashiba, T.; Fujiwara, T.; Hatakeyama, H.; Ueno, K.; Hisaka, A. Trichostatin A Modulates Cellular Metabolism in Renal Cell Carcinoma to Enhance Sunitinib Sensitivity. Eur. J. Pharmacol. 2019, 847, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Donia, T.; Khedr, S.; Salim, E.I.; Hessien, M. Trichostatin A Sensitizes Hepatoma Cells to Taxol More than 5-Aza-DC and Dexamethasone. Drug Metab. Pers. Ther. 2021, 36, 299–309. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Shin, J.H.; Chie, E.K.; Wu, H.-G.; Kim, J.S.; Kim, I.H.; Ha, S.W.; Park, C.I.; Kang, W.-S. Trichostatin A, a Histone Deacetylase Inhibitor, Potentiated Cytotoxic Effect of Ionizing Radiation in Human Head and Neck Cancer Cell Lines. Radiat. Oncol. J. 2004, 22, 138–141. [Google Scholar]
- Kim, I.A.; Kim, J.H.; Shin, J.H.; Kim, I.H.; Kim, J.S.; Wu, H.-G.; Chie, E.K.; Kim, Y.H.; Kim, B.-K.; Hong, S.; et al. A Histone Deacetylase Inhibitor, Trichostatin A, Enhances Radiosensitivity by Abrogating G2/M Arrest in Human Carcinoma Cells. Cancer Res. Treat. 2005, 37, 122. [Google Scholar] [CrossRef]
- Igaz, N.; Kovács, D.; Rázga, Z.; Kónya, Z.; Boros, I.M.; Kiricsi, M. Modulating Chromatin Structure and DNA Accessibility by Deacetylase Inhibition Enhances the Anti-Cancer Activity of Silver Nanoparticles. Colloids Surf. B Biointerfaces 2016, 146, 670–677. [Google Scholar] [CrossRef]
- Shin, S.; Kim, M.; Lee, S.-J.; Park, K.-S.; Lee, C.H. Trichostatin A Sensitizes Hepatocellular Carcinoma Cells to Enhanced NK Cell-Mediated Killing by Regulating Immune-Related Genes. Cancer Genom. Proteom. 2017, 14, 349–362. [Google Scholar]
- Roh, M.S.; Kim, C.W.; Park, B.S.; Kim, G.C.; Jeong, J.H.; Kwon, H.C.; Suh, D.J.; Cho, K.H.; Yee, S.-B.; Yoo, Y.H. Mechanism of Histone Deacetylase Inhibitor Trichostatin A Induced Apoptosis in Human Osteosarcoma Cells. Apoptosis 2004, 9, 583–589. [Google Scholar] [CrossRef]
- Lin, L.; Wei, Y.; Zhu, W.; Wang, C.; Su, R.; Feng, H.; Yang, H. Prevalence, Risk Factors and Associated Adverse Pregnancy Outcomes of Anaemia in Chinese Pregnant Women: A Multicentre Retrospective Study. BMC Pregnancy Childbirth 2018, 18, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Zhang, T.; Teng, Z.; Zhang, R.; Wang, J.-B.; Mei, Q.-B. Sensitization to γ-Irradiation-Induced Cell Cycle Arrest and Apoptosis by the Histone Deacetylase Inhibitor Trichostatin A in Non-Small Cell Lung Cancer (NSCLC) Cells. Cancer Biol. Ther. 2009, 8, 823–831. [Google Scholar] [CrossRef] [Green Version]
- Chan, S.-T.; Yang, N.-C.; Huang, C.-S.; Liao, J.-W.; Yeh, S.-L. Quercetin Enhances the Antitumor Activity of Trichostatin A through Upregulation of P53 Protein Expression In Vitro and In Vivo. PLoS ONE 2013, 8, e54255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, S.-T.; Chuang, C.-H.; Lin, Y.-C.; Liao, J.-W.; Lii, C.-K.; Yeh, S.-L. Quercetin Enhances the Antitumor Effect of Trichostatin A and Suppresses Muscle Wasting in Tumor-Bearing Mice. Food Funct. 2018, 9, 871–879. [Google Scholar] [CrossRef]
- Lee, C.S.; Jang, E.-R.; Kim, Y.J.; Myung, S.C.; Kim, W. Casein Kinase 2 Inhibition Differentially Modulates Apoptotic Effect of Trichostatin A against Epithelial Ovarian Carcinoma Cell Lines. Mol. Cell. Biochem. 2010, 338, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Yang, J.C.; Kim, Y.J.; Jang, E.-R.; Kim, W.; Myung, S.C. 18β-Glycyrrhetinic Acid Potentiates Apoptotic Effect of Trichostatin A on Human Epithelial Ovarian Carcinoma Cell Lines. Eur. J. Pharmacol. 2010, 649, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.-C.; Yang, Y.-C.; Huang, P.-R.; Wen, Y.-D.; Yeh, S.-L. Genistein Enhances the Effect of Trichostatin A on Inhibition of A549 Cell Growth by Increasing Expression of TNF Receptor-1. Toxicol. Appl. Pharmacol. 2012, 262, 247–254. [Google Scholar] [CrossRef]
- Chen, Z.; Clark, S.; Birkeland, M.; Sung, C.-M.; Lago, A.; Liu, R.; Kirkpatrick, R.; Johanson, K.; Winkler, J.D.; Hu, E. Induction and Superinduction of Growth Arrest and DNA Damage Gene 45 (GADD45) α and β Messenger RNAs by Histone Deacetylase Inhibitors Trichostatin A (TSA) and Butyrate in SW620 Human Colon Carcinoma Cells. Cancer Lett. 2002, 188, 127–140. [Google Scholar] [CrossRef]
- Wang, B.X.; Yin, B.L.; He, B.; Chen, C.; Zhao, M.; Zhang, W.X.; Xia, Z.K.; Pan, Y.Z.; Tang, J.Q.; Zhou, X.M.; et al. Overexpression of DNA Damage-Induced 45 α Gene Contributes to Esophageal Squamous Cell Cancer by Promoter Hypomethylation. J. Exp. Clin. Cancer Res. 2012, 31, 11–13. [Google Scholar] [CrossRef] [Green Version]
- Min, K.N.; Cho, M.J.; Kim, D.-K.; Sheen, Y.Y. Estrogen Receptor Enhances the Antiproliferative Effects of Trichostatin A and HC-Toxin in Human Breast Cancer Cells. Arch. Pharmacal Res. 2004, 27, 554–561. [Google Scholar] [CrossRef]
- Kang, J.; Zhang, D.; Chen, J.; Liu, Q.; Lin, C. Antioxidants and Trichostatin A Synergistically Protect against in Vitro Cytotoxicity of Ni2+ in Human Hepatoma Cells. Toxicology 2005, 19, 173–182. [Google Scholar] [CrossRef]
- Kang, J.; Chen, J.; Zhang, D.; Da, W.; Ou, Y. Synergistic Killing of Human Leukemia Cells by Antioxidants and Trichostatin A. Cancer Chemother. Pharm. 2004, 54, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Grigorov, B. Reactive Oxygen Species and Their Relation to Carcinogenesis. Trakia J. Sci. 2012, 10, 83–92. [Google Scholar]
- Maxhimer, J.B.; Reddy, R.M.; Zuo, J.; Cole, G.W.; Schrump, D.S.; Nguyen, D.M. Induction of Apoptosis of Lung and Esophageal Cancer Cells Treated with the Combination of Histone Deacetylase Inhibitor (Trichostatin A) and Protein Kinase C Inhibitor (Calphostin C). J. Thorac. Cardiovasc. Surg. 2005, 129, 53–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, H.G.; Yoon, C.Y.; Yu, J.H.; Park, M.J.; Lee, J.E.; Jeong, S.J.; Hong, S.K.; Byun, S.-S.; Lee, S.E. Induction of Caspase Mediated Apoptosis and Down-Regulation of Nuclear Factor-ΚB and Akt Signaling Are Involved in the Synergistic Antitumor Effect of Gemcitabine and the Histone Deacetylase Inhibitor Trichostatin A in Human Bladder Cancer Cells. J. Urol. 2011, 186, 2084–2093. [Google Scholar] [CrossRef]
- Hamner, J.B.; Sims, T.L.; Cutshaw, A.; Dickson, P.V.; Rosati, S.; McGee, M.; Ng, C.Y.; Davidoff, A.M. The Efficacy of Combination Therapy Using Adeno-Associated Virus—Interferon β and Trichostatin A in Vitro and in a Murine Model of Neuroblastoma. J. Pediatr. Surg. 2008, 43, 177–183. [Google Scholar] [CrossRef]
- Hřebačková, J.; Poljakova, J.; Eckschlager, T.; Hraběta, J.; Prochazka, P.; Smutnỳ, S.; Stiborova, M. Histone Deacetylase Inhibitors Valproate and Trichostatin A Are Toxic to Neuroblastoma Cells and Modulate Cytochrome P450 1A1, 1B1 and 3A4 Expression in These Cells. Interdiscip. Toxicol. 2009, 2, 205. [Google Scholar] [CrossRef] [Green Version]
- Jin, X.; Fang, Y.; Hu, Y.; Chen, J.; Liu, W.; Chen, G.; Gong, M.; Wu, P.; Zhu, T.; Wang, S.; et al. Synergistic Activity of the Histone Deacetylase Inhibitor Trichostatin A and the Proteasome Inhibitor PS-341 against Taxane-Resistant Ovarian Cancer Cell Lines. Oncol. Lett. 2017, 13, 4619–4626. [Google Scholar] [CrossRef] [Green Version]
- Montagut, C.; Settleman, J. Targeting the RAF–MEK–ERK pathway in cancer therapy. Cancer Lett. 2009, 283, 125–134. [Google Scholar] [CrossRef]
- Yan, G.; Graham, K.; Lanza-Jacoby, S. Curcumin Enhances the Anticancer Effects of Trichostatin a in Breast Cancer Cells. Mol. Carcinog. 2013, 52, 404–411. [Google Scholar] [CrossRef]
- Piao, J.; Chen, L.; Quan, T.; Li, L.; Quan, C.; Piao, Y.; Jin, T.; Lin, Z. Superior Efficacy of Co-Treatment with the Dual PI3K/MTOR Inhibitor BEZ235 and Histone Deacetylase Inhibitor Trichostatin A against NSCLC. Oncotarget 2016, 7, 60169–60180. [Google Scholar] [CrossRef]
- Chen, L.; Jin, T.; Zhu, K.; Piao, Y.; Quan, T.; Quan, C.; Lin, Z. PI3K/MTOR Dual Inhibitor BEZ235 and Histone Deacetylase Inhibitor Trichostatin A Synergistically Exert Anti-Tumor Activity in Breast Cancer. Oncotarget 2017, 8, 11937–11949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, J.; Demirjian, A.; Sui, J.; Marasco, W.; Callery, M.P. Histone Deacetylase Inhibitor Trichostatin A and Proteasome Inhibitor PS-341 Synergistically Induce Apoptosis in Pancreatic Cancer Cells. Biochem. Biophys. Res. Commun. 2006, 348, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.-Y.; Kim, S.-R.; Hwang, J.-W.; Bae, M.-K.; Wee, H.-J.; Choi, Y.-H.; Oh, S.-O.; Kim, B.-S.; Yoon, S.; Bae, S.-K. Combined Treatment of Trichostatin A and Heat Shock Increases Apoptosis in STAT3 Dependent Astrocytoma Cells. Cancer Prev. Res. 2006, 11, 205–210. [Google Scholar]
- Baek, S.-Y.; Kim, S.-R.; Bae, M.-K.; Hwang, J.-W.; Kim, J.-S.; Choi, Y.H.; Wee, H.-J.; Kim, B.-S.; Kim, J.-B.; Yoon, S.; et al. Trichostatin A Increases the Thermosensitivity of Human Glioblastoma A172 Cells. Neurosci. Lett. 2006, 396, 230–234. [Google Scholar] [CrossRef]
- Chen, J.; Bai, H.; Wang, C.; Kang, J. Trichostatin A Improves the Anticancer Activity of Low Concentrations of Curcumin in Human Leukemia Cells. Cell Death 2006, 61, 710–716. [Google Scholar]
- Piotrowska, H.; Jagodzinski, P.P. Trichostatin A, Sodium Butyrate, and 5-Aza-2′-Deoxycytidine Alter the Expression of Glucocorticoid Receptor α and β Isoforms in Hut-78 T- and Raji B-Lymphoma Cell Lines. Biomed. Pharmacother. 2007, 61, 451–454. [Google Scholar] [CrossRef]
- Jiang, S.-J.; Zhang, S.; Mu, X.-Y.; Li, W.; Wang, Y. Effects of Trichostatin A and Paclitaxel on Apoptosis and Microtubule Stabilization in Endometrial Carcinoma Cells: An in Vitro Research. Zhonghua Yi Xue Za Zhi 2008, 88, 2427–2431. [Google Scholar]
- Liu, T.-C.; Castelo-Branco, P.; Rabkin, S.D.; Martuza, R.L. Trichostatin A and Oncolytic HSV Combination Therapy Shows Enhanced Antitumoral and Antiangiogenic Effects. Mol. Ther. 2008, 16, 1041–1047. [Google Scholar] [CrossRef]
- Liu, Z.; Marquez, M.; Nilsson, S.; Holmberg, A. Incubation with Somatostatin, 5-Aza Decitabine and Trichostatin up-Regulates Somatostatin Receptor Expression in Prostate Cancer Cells. Oncol. Rep. 2008, 20, 151–154. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.-C.; Du, Y.-C.; Chuu, J.-J.; Hwang, S.-L.; Hsieh, P.-C.; Hung, C.-S.; Chang, F.-R.; Wu, Y.-C. Active Extracts of Wild Fruiting Bodies of Antrodia Camphorata (EEAC) Induce Leukemia HL 60 Cells Apoptosis Partially through Histone Hypoacetylation and Synergistically Promote Anticancer Effect of Trichostatin A. Arch. Toxicol. 2009, 83, 121–129. [Google Scholar] [CrossRef]
- Hrabeta, J.; Poljakova, J.; Frei, E.; Stiborova, M.; Eckschlager, T. Inhibitors of Histone Deacetylase, Valproic Acid and Trichostatin A, Increase Cytotoxicity of Anticancer Drug Ellipticine to Neuroblastoma Cells. In Proceedings of the Cancer Research, Philadelphia, PA, USA, 9–13 December 2009. [Google Scholar]
- Cecconi, D.; Donadelli, M.; Dalla Pozza, E.; Rinalducci, S.; Zolla, L.; Scupoli, M.T.; Righetti, P.G.; Scarpa, A.; Palmieri, M. Synergistic Effect of Trichostatin A and 5-Aza-2′-Deoxycytidine on Growth Inhibition of Pancreatic Endocrine Tumour Cell Lines: A Proteomic Study. Proteomics 2009, 9, 1952–1966. [Google Scholar] [CrossRef] [PubMed]
- Pouliot, K. The Histone Deacetylase Inhibitors, Trichostatin A and Apicidin, Enhance the Radiosensitivity of Ovarian Carcinoma Cells in Vitro. In Proceedings of the Cancer Research, Philadelphia, PA, USA, 9–13 December 2009. [Google Scholar]
- Shiau, R.-J.; Chen, K.-Y.; Wen, Y.-D.; Chuang, C.-H.; Yeh, S.-L. Genistein and β-Carotene Enhance the Growth-Inhibitory Effect of Trichostatin A in A549 Cells. Eur. J. Nutr. 2010, 49, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Poljakova, J.; Hrebackova, J.; Dvorakova, M.; Moserova, M.; Eckschlager, T.; Hrabeta, J.; Göttlicherova, M.; Kopejtkova, B.; Frei, E.; Kizek, R.; et al. Anticancer Agent Ellipticine Combined with Histone Deacetylase Inhibitors, Valproic Acid and Trichostatin A, Is an Effective DNA Damage Strategy in Human. Neuroblastoma 2011, 32 (Suppl. S1), 101–116. [Google Scholar]
- Jiang, L.; Lian, M.; Wang, H.; Fang, J.; Wang, Q. Inhibitory Effects of 5-Aza-2′-Deoxycytidine and Trichostatin A in Combination with P53-Expressing Adenovirus on Human Laryngocarcinoma Cells. Chin. J. Cancer Res. 2012, 24, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.; Li, H.; Ma, Y.; Tang, B.; Tian, J.; Akers, W.; Achilefu, S.; Gu, Y. The Enhanced Antiproliferative Response to Combined Treatment of Trichostatin A with Raloxifene in MCF-7 Breast Cancer Cells and Its Relevance to Estrogen Receptor β Expression. Mol. Cell. Biochem. 2012, 366, 111–122. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, Q.; Jiang, S. Effect of Trichostatin A and Paclitaxel on the Proliferation and Apoptosis of Lung Adenocarcinoma Cells. Chin. Med. J. 2013, 126, 129–134. [Google Scholar]
- Tran, H.T.T.; Kim, H.N.; Lee, I.-K.; Nguyen-Pham, T.-N.; Ahn, J.-S.; Kim, Y.-K.; Lee, J.-J.; Park, K.-S.; Kook, H.; Kim, H.-J. Improved Therapeutic Effect against Leukemia by a Combination of the Histone Methyltransferase Inhibitor Chaetocin and the Histone Deacetylase Inhibitor Trichostatin A. J. Korean Med. Sci. 2013, 28, 237. [Google Scholar] [CrossRef] [Green Version]
- Duo, J.; Ma, Y.; Wang, G.; Han, X.; Zhang, C. Metformin Synergistically Enhances Antitumor Activity of Histone Deacetylase Inhibitor Trichostatin A Against Osteosarcoma Cell Line. DNA Cell Biol. 2013, 32, 156–164. [Google Scholar] [CrossRef]
- Kiliccioglu, I.; Konac, E.; Varol, N.; Gurocak, S.; Yucel Bilen, C. Apoptotic Effects of Proteasome and Histone Deacetylase Inhibitors in Prostate Cancer Cell Lines. Genet. Mol. Res. 2014, 13, 3721–3731. [Google Scholar] [CrossRef]
- Asgar, M.A.; Senawong, G.; Sripa, B.; Senawong, T. Synergistic Anticancer Effects of Cisplatin and Histone Deacetylase Inhibitors (SAHA and TSA) on Cholangiocarcinoma Cell Lines. Int. J. Oncol. 2016, 48, 409–420. [Google Scholar] [CrossRef] [Green Version]
- Du, R.; Liu, Z.; Hou, X.; Fu, G.; An, N.; Wang, L. Trichostatin A Potentiates Genistein-Induced Apoptosis and Reverses EMT in HEp2 Cells. Mol. Med. Rep. 2016, 13, 5045–5052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Jayaram, P.; Kabekkodu, S.P.; Satyamoorthy, K. Targeted drug delivery in cervical cancer: Current perspectives. Eur. J. Pharmacol. 2022, 917, 174751. [Google Scholar] [CrossRef]
- Mazzio, E.A.; Soliman, K.F.A. HTP Nutraceutical Screening for Histone Deacetylase Inhibitors and Effects of HDACis on Tumor-Suppressing MiRNAs by Trichostatin A and Grapeseed (Vitis Vinifera) in HeLa Cells. Cancer Genom. Proteom. 2017, 14, 17–34. [Google Scholar] [CrossRef] [Green Version]
- Gilardini Montani, M.S.; Granato, M.; Santoni, C.; Del Porto, P.; Merendino, N.; D’Orazi, G.; Faggioni, A.; Cirone, M. Histone Deacetylase Inhibitors VPA and TSA Induce Apoptosis and Autophagy in Pancreatic Cancer Cells. Cell. Oncol. 2017, 40, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Yang, Y.; Xu, J.; Pan, Y.; Zhang, W.; Xing, Y.; Ni, H.; Sun, Y.; Hou, Y.; Li, N. Tamarix Hohenackeri Bunge Exerts Anti-Inflammatory Effects on Lipopolysaccharide-Activated Microglia in Vitro. Phytomedicine 2018, 40, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Zhou, X.; He, Y.; Liu, H.; Wang, Y.; Chen, Y.; Li, M.; He, Y.; Li, G.; Li, Y. Effect of Trichostatin A on Burkitt’s Lymphoma Cells: Inhibition of EPS8 Activity through Phospho-Erk1/2 Pathway. Biochem. Biophys. Res. Commun. 2018, 497, 990–996. [Google Scholar] [CrossRef]
- Wu, N.; Zhu, Y.; Xu, X.; Zhu, Y.; Song, Y.; Pang, L.; Chen, Z. The Anti-Tumor Effects of Dual PI3K/MTOR Inhibitor BEZ235 and Histone Deacetylase Inhibitor Trichostatin A on Inducing Autophagy in Esophageal Squamous Cell Carcinoma. J. Cancer 2018, 9, 987–997. [Google Scholar] [CrossRef] [Green Version]
- Chuang, C.-H.; Chan, S.-T.; Chen, C.-H.; Yeh, S.-L. Quercetin Enhances the Antitumor Activity of Trichostatin A through Up-Regulation of P300 Protein Expression in P53 Null Cancer Cells. Chem. Biol. Interact. 2019, 306, 54–61. [Google Scholar] [CrossRef]
- Hsu, F.-S.; Wu, J.-T.; Lin, J.-Y.; Yang, S.-P.; Kuo, K.-L.; Lin, W.-C.; Shi, C.-S.; Chow, P.-M.; Liao, S.-M.; Pan, C.-I.; et al. Histone Deacetylase Inhibitor, Trichostatin A, Synergistically Enhances Paclitaxel-Induced Cytotoxicity in Urothelial Carcinoma Cells by Suppressing the ERK Pathway. Int. J. Mol. Sci. 2019, 20, 1162. [Google Scholar] [CrossRef] [Green Version]
- Ren, C.; Gao, C.; Li, X.; Xiong, J.; Shen, H.; Wang, L.; Zhu, D.; Wu, P.; Ding, W.; Wang, H. The Antitumor Efficiency of Zinc Finger Nuclease Combined with Cisplatin and Trichostatin A in Cervical Cancer Cells. Anti-Cancer Agents Med. Chem. 2020, 20, 2125–2135. [Google Scholar] [CrossRef]
- Wong, S.H.M.; Fang, C.-M.; Loh, H.-S.; Ngai, S.C. Trichostatin A and Zebularine along with E-Cadherin Re-Expression Enhance Tumour Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL)-Mediated Cell Cycle Arrest in Human Breast Adenocarcinoma Cells. Asia Pac. J. Mol. Biol. Biotechnol. 2021, 29, 26–41. [Google Scholar] [CrossRef]
- Ou, J.-N.; Torrisani, J.; Unterberger, A.; Provençal, N.; Shikimi, K.; Karimi, M.; Ekström, T.J.; Szyf, M. Histone Deacetylase Inhibitor Trichostatin A Induces Global and Gene-Specific DNA Demethylation in Human Cancer Cell Lines. Biochem. Pharmacol. 2007, 73, 1297–1307. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Meeran, S.M.; Patel, S.N.; Chen, H.; Hardy, T.M.; Tollefsbol, T.O. Epigenetic Reactivation of Estrogen Receptor-α (ERα) by Genistein Enhances Hormonal Therapy Sensitivity in ERα-Negative Breast Cancer. Mol. Cancer 2013, 12, 9. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; Sun, G.; Zhong, M.; Yu, Y.; Brewer, M.A. Inhibition of DNA Methyltransferases, Histone Deacetylases and Lysine-Specific Demethylase-1 Suppresses the Tumorigenicity of the Ovarian Cancer Ascites Cell Line SKOV3. Int. J. Oncol. 2013, 43, 495–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, E.; Bandle, R.; Clair, T.; Roberts, D.D.; Stracke, M.L. Trichostatin A and 5-Aza-2′-Deoxycytidine Switch S1P from an Inhibitor to a Stimulator of Motility through Epigenetic Regulation of S1P Receptors. Cancer Lett. 2007, 250, 53–62. [Google Scholar] [CrossRef]
- Choi, J.-H.; Min, N.Y.; Park, J.; Kim, J.H.; Park, S.H.; Ko, Y.J.; Kang, Y.; Moon, Y.J.; Rhee, S.; Ham, S.W.; et al. TSA-Induced DNMT1 down-Regulation Represses HTERT Expression via Recruiting CTCF into Demethylated Core Promoter Region of HTERT in HCT116. Biochem. Biophys. Res. Commun. 2010, 391, 449–454. [Google Scholar] [CrossRef] [PubMed]
- Sanaei, M.; Kavoosi, F. Effect of Curcumin and Trichostatin A on the Expression of DNA Methyltransfrase 1 in Hepatocellular Carcinoma Cell Line Hepa 1-6. Iran. J. Pediatr. Hematol. Oncol. 2018, 8, 10. [Google Scholar]
- Januchowski, R.; Dąbrowski, M.; Ofori, H.; Jagodzinski, P.P. Trichostatin A Down-Regulate DNA Methyltransferase 1 in Jurkat T Cells. Cancer Lett. 2007, 246, 313–317. [Google Scholar] [CrossRef]
- Wang, H.; Li, Q.; Chen, H. Genistein Affects Histone Modifications on Dickkopf-Related Protein 1 (DKK1) Gene in SW480 Human Colon Cancer Cell Line. PLoS ONE 2012, 7, e40955. [Google Scholar] [CrossRef]
- Wu, D.-S.; Shen, J.-Z.; Yu, A.-F.; Fu, H.-Y.; Zhou, H.-R.; Shen, S.-F. Epigallocatechin-3-Gallate and Trichostatin A Synergistically Inhibit Human Lymphoma Cell Proliferation through Epigenetic Modification of P16INK4a. Oncol. Rep. 2013, 30, 2969–2975. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Approach | Key Results | Ref |
|---|---|---|
| TGF-β-induced myofibroblast differentiation of corneal fibroblasts Immunofluorescence staining Reverse transcription quantitative-polymerase chain reaction Western blot analysis | Elevated intracellular GSH level and cellular total antioxidant capacity Decreased cellular ROS and H2O2 accumulation. Induced Nrf2 nuclear translocation Upregulated the expression of Nrf2-ARE downstream antioxidant genes | [14] |
| Human bone marrow-mesenchymal stem cells MTT assay Immunoblot analysis | Increased SOD2 Decreased intracellular ROS Suppressed H2O2-induced ROS generation Modulates FOXO1 | [12] |
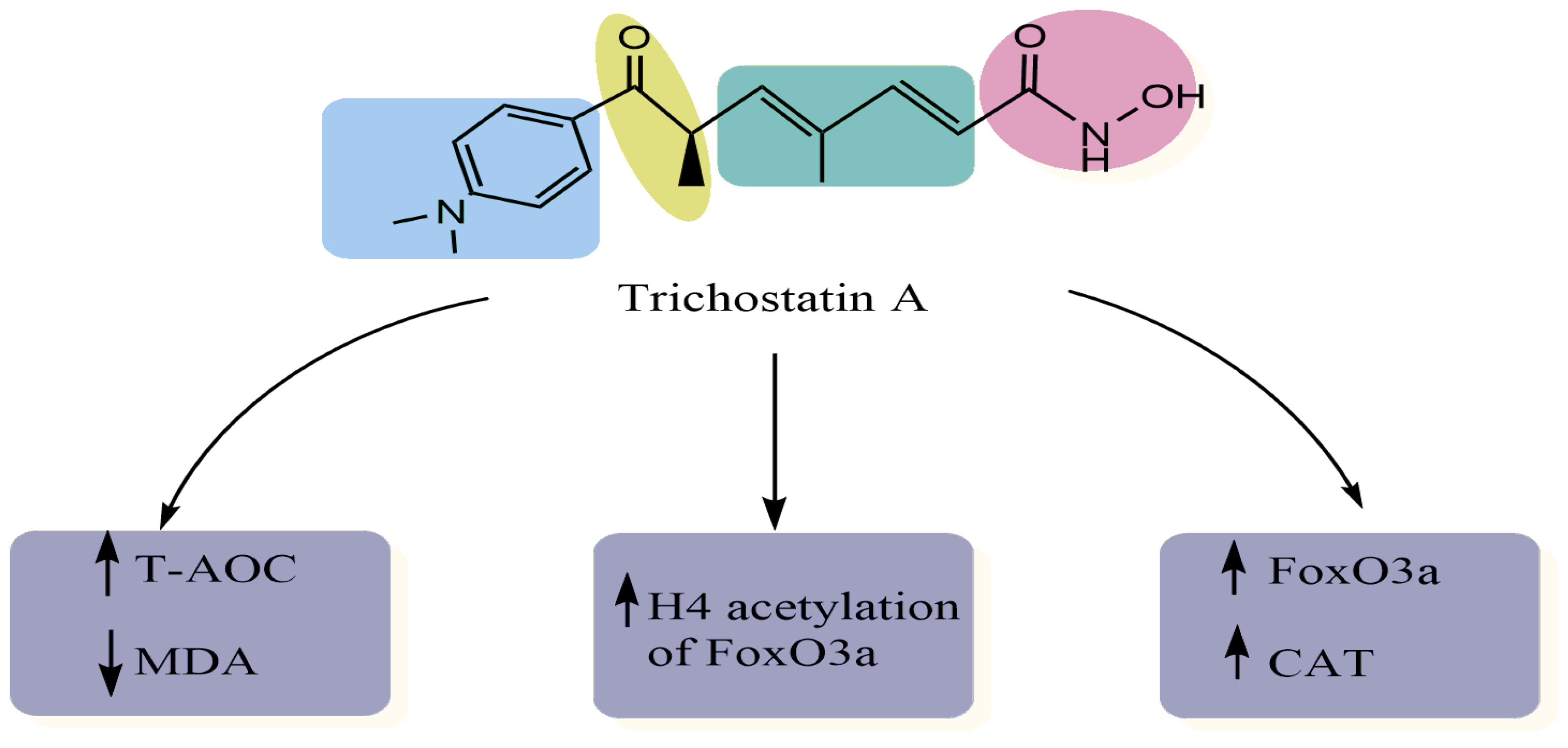
| H9c2 rat myocardial cell line Western blot analysis Chromatin immunoprecipitation assay | Decreased the levels of MDA Decreased the H2O2-induced levels of ROS Increased the expression of FoxO3a, SOD2 and CAT, and increased H4 acetylation of the FoxO3a promoter region | [11] |
| Human lens epithelial cells (HLECs) after UVB exposure Cell viability Western blot assay Enzyme-linked immunosorbent assay Real-time PCR | Suppressed BAX and caspase-3 expression Suppressed the expression of FOXO3A and MT2 Increased SOD levels Decreased MDA levels Decreased ROS levels Increased total antioxidant capacity | [13] |
| Experimental Approach | Key Results | Ref. |
|---|---|---|
|
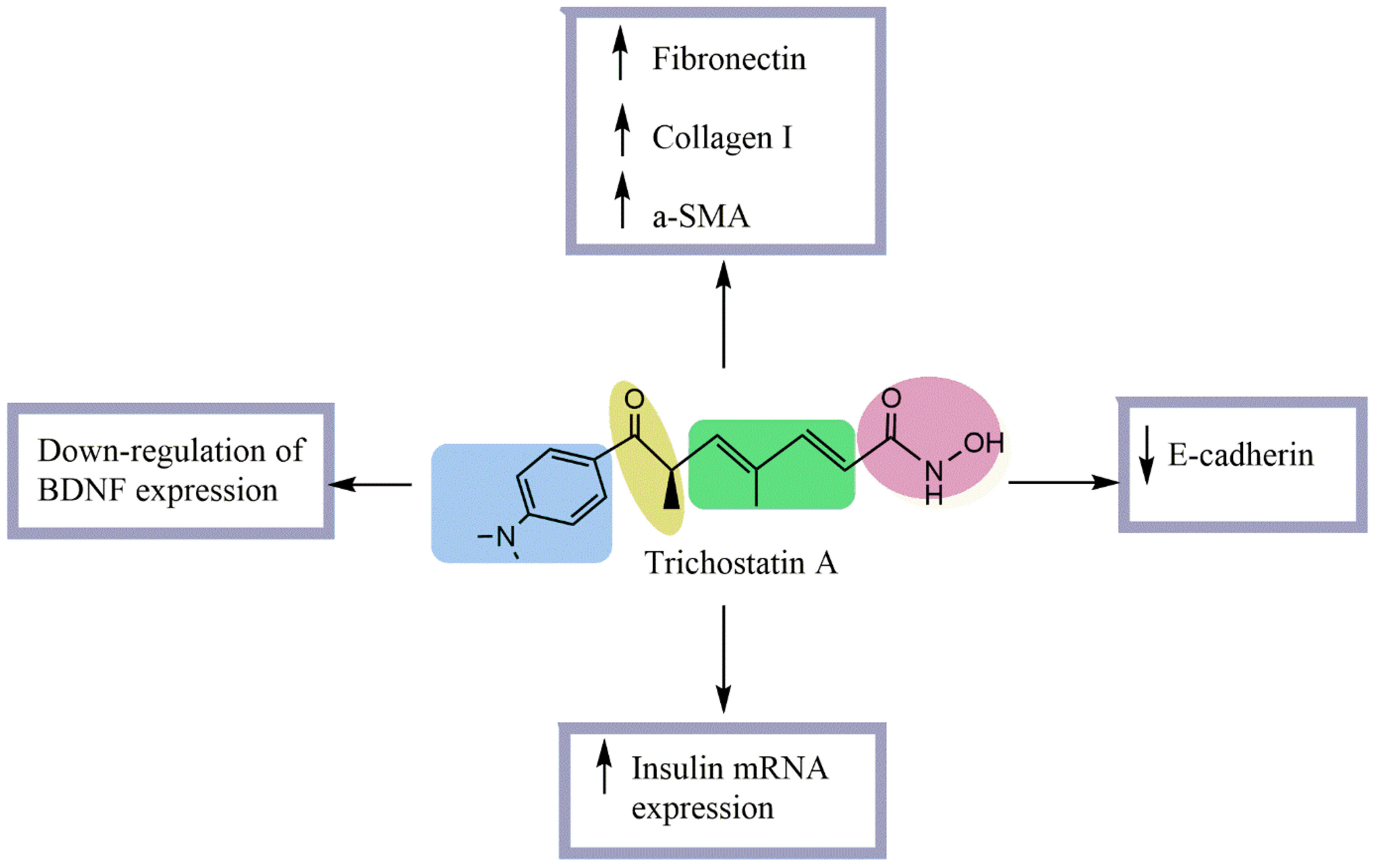
Streptozotocin (STZ)-induced diabetic rats Normal rat kidney tubular epithelial cells (NRK52-E) stimulated with TGF-β1 | No effect on blood glucose or kidney/body weight ratio. Significantly decreased urinary protein/creatinine excretion Significantly increased glomerular and tubular fibronectin and collagen I and tubular α-SMA expression. Significantly decreased tubular E-cadherin expression. Increased E-cadherin expression at both mRNA and protein levels. Prevented ECM upregulation and EMT in NRK52-E cells. Prevented TGF-β1-induced downregulation of E-cadherin and upregulation of collagen I. | [16] |
| β-cell line βTC-tet, L-cell line GLUTag, or recombinant insulin-secreting L-cell lines Real-time PCR, ELISA, and radioimmunoassay | Significantly promoted insulin mRNA secretion in TSA-treated βTC-tet cells. Significantly promoted GLP-1 mRNA secretion in TSA-treated GLUTag cells. Significantly promoted insulin mRNA secretion in TSA-treated GLUTag-INS and EINS cells. Decreased mRNA levels of insulin and GLP-1 in β- and L-cells Caused a 2.5-fold increase in stored insulin and a 2-fold increase in glucose-stimulated insulin secretion in βTC-tet cells. Increased stored and MH-stimulated GLP-1 in GLUTag cells. Significantly increased EINS proinsulin and insulin secretion | [17] |
|
Streptozotocin (STZ)-induced diabetic rats Rat Schwann cells |
Enhanced the action potential amplitude of sciatic nerves. Increased BDNF expression Increased GRP78 expression regulated BDNF protein level Improved XBP-1s/ ATF6/GRP78 axis. Improved the binding of GRP78 and BDNF Improved the differentiation of SH-SY5Y cells | [15] |
| Experimental Approach | Key Results | Ref |
|---|---|---|
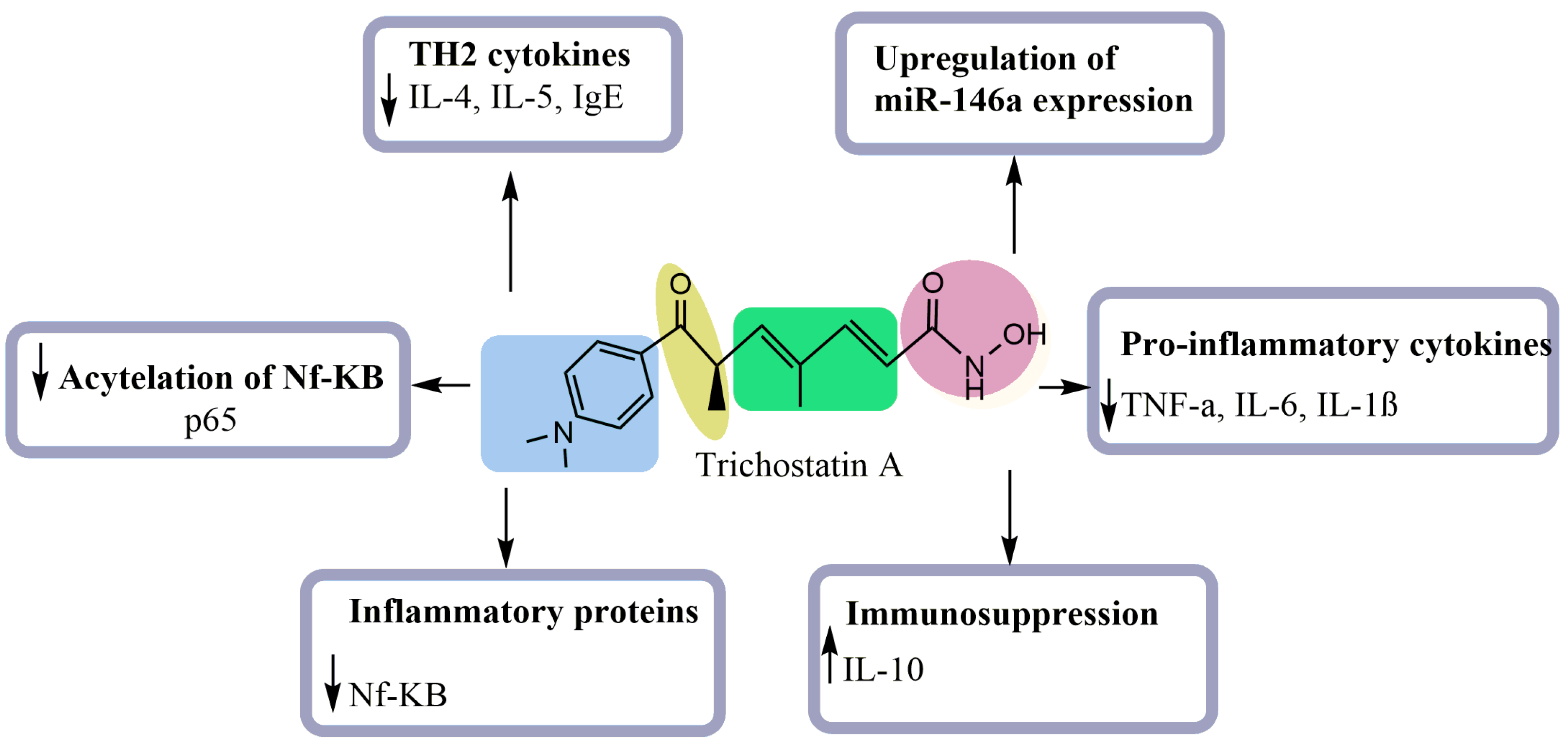
| Allergen-induced airway inflammation in a mouse asthma mode | Decreased inflammatory cells Reduced IL-4, IL-5, and IgE levels Reduced Th2 cytokines expression Decreased infiltration of CD4+ | [18] |
| Lipopolysaccharide-(LPS)-stimulated macrophages | Inhibited the production of nitric oxide (NO) Reduced the mRNA and protein levels of the proinflammatory cytokines (TNF-α, IL-6, and IL-1β) Increased the level of the immunosuppressive cytokine IL-10 Decreased the cell surface markers of the maturity of the macrophages | [19] |
| Lipopolysaccharide (LPS)-induced production of IL-6 in OP9 Preadipocytes | Enhanced palmitic acid-induced IL-6 production Enhanced the expression of inflammatory genes. Increased the level of NF-kB p65 acetylation | [21] |
| Phorbol myristate acetate-induced macrophages | Reduced TNF-α, IFN-γ, IL-10 and IL-18 levels Suppressed the expression of class I HDACs Inhibited apoptosis of macrophages Reduced the viability of PMA-induced macrophages Suppressed the expression of proinflammatory genes Enhanced the acetylation of NF-κB p65 Promoted histone acetylation | [22] |
| LPS-induced acute lung injury model in vitro | Enhanced LPS-stimulated NR8383 cells Decreased the levels of TNF-α Upregulated the micorRNA-146a expression | [20] |
| Origin | Used Model | Experimental Approach | Key Results | References |
|---|---|---|---|---|
| Purchased | RPMI8226 and MM.1S cells | Immunofluorescence Immunoprecipitation Western blot analysis qPCR | Induced cytotoxic effect in multiple myeloma cell lines Induced cell apoptosis Inhibited hedgehog signaling pathway | [50] |
| Purchased | YD-10B oral squamous carcinoma cells | MTT assay Cell cycle analysis Western blot analysis DAPI staining | Inhibited cell proliferation Arrested cell cycle progression at the G2/M phase Induced mitochondrial membrane destruction Induced cyto-c release and proteolytic activation of caspases-3 and -7 | [23] |
| Purchased | MDA-MB-231 and MCF-7 human breast carcinoma and SK-UT-1B uterine cancer cell lines | Flow cytometry analysis RT-PCP | Induced cyclin D1 downregulation through both ERα-dependent and ERα-independent mechanisms | [51] |
| Purchased | MCF-7 cells | Cell proliferation assay Immunoblotting Flow cytometry analysis | Induced Akt dephosphorylation in a PP1-dependent manner, resulting in the activation of GSK3β in MCF-7 cells TSA-induced cytotoxicity was attenuated by the selective inhibition of GSK3β resulting in increased proliferation | [24] |
| Not reported | U87 glioblastoma cells | RT-PCP | Reduced proliferation and colony sizes resulting in G2/M arrest Inhibited tumorsphere formation | [52] |
| Not reported | Gastric cancer cells (MKN-45 and SGC-7901 cells) | MTT and BrdU immunofluorescence assays Soft agar assay Flow cytometry analysis Western blot analysis | Suppressed cell proliferation Induced apoptosis by regulating the PI3K/AKT signaling pathway in gastric cancer cells Induced cell cycle arrest at the G1 phase and apoptosis | [15] |
| Not reported | Two leukemic cell lines (CCRF-CEM and HL-60) | Flow cytometry analysis | The IC50 value of CCRF-CEM was 2.65 ± 0.3 μM The IC50 value of HL-60 was 2.35 ± 0.2 μM CCRF-CEM cells were reduced to 56.5%, 45.3%, and 40.2% on the first, third, and sixth days HL-60 cells were reduced to 55.6%, 45.2%, and 36.3% on the first, second, and fourth days | [53] |
| Purchased | Human osteosarcoma cells | Confocal microscopy Western blot analysis Flow cytometry analysis | Promoted osteosarcoma cell death Induced autophagy in U2OS cells Inhibited mTOR signaling pathway and enhanced FOXO1 transcriptional activity | [34] |
| Not reported | Pancreatic and colon carcinoma cell lines | Western blot analysis Real-time RT- PCR | Increased MDR1 mRNA levels Downregulated the upstream promoter responsible for the active P-glycoprotein expression | [54] |
| Purchased | Human colon adenocarcinoma cell lines DLD-1 and SW480 | Viability assays Western blot analysis Gene expression microarrays | Reduced cell viability Reversed the upregulation of gene expression levels induced by gain of chromosome 7 | [55] |
| Purchased | Human pancreatic endocrine tumor cell lines (CM, BON, and QGP-1) | Cell proliferation assay Cell cycle analysis 2-D gel electrophoresis | Inhibited cell growth by arresting the cell cycle in the G2/M phase and inducing apoptosis | [56] |
| Purchased | Lung cancer cells | mRNA extraction and qRT-PCR Colony formation assay Flow cytometry analysis Cell cycle analysis Western blot analysis | Inhibited proliferation, reduced colony formation, and induced cell cycle arrest and apoptosis Reduced the expression of Bcl-2 through the upregulation of miR-15a/16-1 | [57] |
| Not reported | Human pancreatic cancer cell lines (PANC-1, SW1990, and MIATACA-2 cells) | MTT assay Hoechst 33258 staining Flow cytometry analysis RT-PCR and western blot analyses | Decreased cell viability in a dose-dependent manner in PANC-1 cells Increased apoptosis of PANC-1 cells Increased the expression levels of Bax and caspase-3 Downregulated the expression level of Bcl-2 | [58] |
| Purchased | Osteosarcoma MG-63 cells | MTT assay TUNEL assay Annexin V staining Flow cytometry analysis | Inhibited cell proliferation Induced apoptosis of MG-63 cells Arrested the cell cycle in G1/G2 phase Inhibited the invasiveness of MG-63 cells | [59] |
| Purchased | Five human hepatoma cell lines | MTT assay TUNEL assay Semi-Quantitative RT-PCR Chromatin Immunoprecipitation (ChIP) assay | Inhibited cell growth Induced apoptosis Inhibited the gene expression profile in hepatoma cell lines | [60] |
| Not reported | Mouse model with L1 neoplastic tumors | Measurement of tumor size and mice body weight Preparation of four formulations for the in vivo study | Reduced neoplastic tumor growth using the semi-solid formulation applied to the skin Impaired the skin barrier function of neoplastic tumors | [61] |
| Purchased | A549 cells | DNA fragmentation assay Flow cytometry analysis RNA extraction and RT-PCR Western blot analysis | Inhibited the cell viability Induced the apoptosis of A549 cells Induced the proteolytic activation of caspases-3 and -9 Induced a concomitant degradation of poly(ADP-ribose)-polymerase protein Decreased the levels of COX-2 mPvNA | |
| Purchased | HCT116 human colon cancer cell lines | MTT assay Reporter assay RNA extraction and RT-qPCR Western blot analysis ChIP assay | Induced the endoplasmic reticulum (ER) stress in wild-type (WT) HCT116 cells Induced apoptosis and cell viability depending on p53 | |
| Purchased | Trypanosoma cruzi | Flow cytometry analysis Transmission electron microscopy LC-MS/MS | Reduced protozoa proliferation and viability Altered the dynamics of the microtubule cytoskeleton Altered the segregation of kDNA, generating polynuclear cells with atypical morphology | [62] |
| Purchased | Human osteosarcoma MG63 cell line Human osteoblastic cell line hFOB 1.19 | MTT assay Flow cytometry analysis Western blot analysis | Inhibited the growth of MG63 cells Promoted apoptosis through activation of p53 signaling pathway | [26] |
| Not reported | Keloid fibroblasts | MTT viability assay Hoechst staining Flow cytometry analysis RNA extraction and real time RT-PCR Western blot analysis | Inhibited the collagen synthesis and induced apoptosis in keloid fibroblasts | [63] |
| Purchased | MCF-7 cells | RQ-PCR analysis Western blot analysis | Reduced the phospholipase C gamma-1 (PLCγ1) transcript and protein levels in MCF-7 cells | [64] |
| Purchased | Human pancreatic carcinoma cell lines (BxPC-3, AsPC-1, and CAPAN-1) | Real-time PCR Immunoblotting | Inhibited the incorporation of BrdU into BxPC-3 cells. Inhibited the phosphorylation of ERK 1/2 and AKT in BxPC-3 cells. Induced an activation of the MAP kinase p38 in all three cell lines especially in BxPC-3 cells Increased the mRNA levels of bax in BxPC-3 cells only Increased cell cycle inhibitor protein p21Waf1 levels in BxPC-3 and AsPC-1 cells | [30] |
| Purchased | MCF10A and MCF10A-ras cells | RT-PCR Western blot analysis | Activated apoptosis in MCF10A-ras cells only Activated the FOXO1 via P21 upregulation Induced autophagy in MCF10A and MCF10A-ras cells by blocking the mammalian target of rapamycin signaling pathway | [35] |
| Purchased | BGC-823 human gastric cancer cell line, MCF-7 cells, and KYSE-510 human esophageal squamous cell carcinoma (ESCC) | Immunocytochemistry assay RNA isolation and qPCR Western blot analysis Colony forming assay | Induced mesenchymal-like morphological changes in human cancer cells Increased the expression levels of mesenchymal markers and E-cadherin Reduced cancer cell mobility Reduced cancer cell colony formation | |
| Purchased | Human renal cell carcinoma (RCC) caki cell line | Flow cytometry analysis Western blot analysis Measurement of mitochondrial membrane potential Determination of caspase activity | Increased TRAIL-induced apoptotic cell death in Caki cells Elevated TRAIL-induced activation of caspases in Caki cells Enhanced the downregulation of Bcl-2 and truncation of Bid in TRAIL-treated Caki cells | [65] |
| Purchased | Molt-4 cell line | MTT assay Flow cytometry analysis Immunocytochemistry Western blot analysis | Inhibited the proliferation of Molt-4 cells (IC50 = 254.32 μg/L after 24 h of exposure) Decreased the percentage of G0/G1 cells and arrested cells in G2/M phase | [66] |
| Purchased | Human endothelial cell line (ECV304 cells) | MTT assay Northern blot analysis Western blot analysis Wounded cell migration assay | Increased thrombospondin-1 expression, which reduced ECV 304 cell migration Inhibited tube formation regardless of the presence of exogenous vascular endothelial growth factor | [67] |
| Purchased | Human leukemia cell line Molt-4 | MTT assay Annexin-V-FITC staining RT-PCR Western blot analysis | Induced Molt-4 apoptosis Upregulated 310 genes and downregulated 313 genes | [31] |
| Purchased | Human malignant glioma LNT-229 and LN-308 cell lines NMRI nude mice | Viability and cell growth assays PCR analysis Caspase activity assay Athymic CD1-deficient NMRI nude mice | Induced the upregulation of natural killer group-2 member-D (NKG2D) ligands and immunogenicity in glioblastoma (GBM) cells Suppressed tumor growth of GBM xenografts (in vivo) | [27] |
| Purchased | Human dermal lymphatic endothelial cells | BrdU assay Flow cytometry analysis Western blot analysis Semi-quantitative RT-PCR | Decreased lymphangiogenesis by inducing apoptosis and cell cycle arrest via p21-dependent pathways | [68] |
| Not reported | C6 glioma cell line | Immunoblot analysis MTT assay Flow cytometry analysis ChIP assay | Decreased cell viability Induced C6 cell apoptosis Induced the p38MAPK and AMPK activation in C6 cells | [69] |
| Not reported | Human cervical carcinoma cell (Hela cells) | MTT assay RT-PCR | Inhibited cell viability Induced cell apoptosis Promoted the expression of apoptosis-related genes | [70] |
| Not reported | Two human ESCC cell lines, KYSE-150 and KYSE-450 | Western blot analysis Transwell migration assay | Promoted cell migration by RelA K310ac-slug-EMT pathway | [71] |
| Not reported | Hepatocellular carcinoma (HCC) cell line Huh7 | qRT-PCR Western blot and immunoprecipitation | Alleviated the specific subset of HCC, the hepatitis B virus X protein (HBx)-induced HCC in metabolic stress, through promoting sirtuin 3 (SIRT3) transcription | [72] |
| Not reported | A549 cells | Flow cytometry analysis | Induced the growth inhibition and morphological changes Inhibited cyclins and cyclin-dependent kinases (CDKs) expression Induced tumor suppressor p53 and Cdk inhibitors such as p21 and p27 | [28] |
| Purchased | Oral squamous cell carcinoma (OSCC) lines HSC-3 and Ca9.22 | Trypan blue staining MTT assay Western blot analysis | Decreased OSCC cell viability and proliferation Enhanced the expression levels of Bim protein Damaged mitochondrial membrane potential and increased cytosolic apoptosis-inducing factor (AIF) in Ca9.22 cells | |
| Purchased | Jurkat leukemia T cell clone E6-1 cells | RQ-PCR Western blot analysis | Induced ZAP-70, LAT, and SLP-76 transcript and protein downregulation in Jurkat leukemia T cells Reduced the half-life of ZAP-70, LAT, and SLP-76 mRNAs | [46] |
| Purchased | Keloid fibroblasts | MTT assay RNA extraction and RT-qPCR Flow cytometry analysis Western blot analysis | Inhibited cell proliferation in a time- and dose-dependent manner Induced alterations in the expression of numerous miRNA sequences Downregulated the expression of miR-30a-5p | |
| Not reported | HeLa and bovine aortic endothelial (BAE) cells | Western blot analysis Northern blot analysis MTT assay | Increased thrombospondin-1 (TSP-1) expression at both the mRNA and protein levels through transcriptional activation | [73] |
| Purchased | Four retinoblastoma cell lines | RT-PCR Western blot analysis ChIP assay Luciferase activity assay | Induced the expression of TβR-II mRNA Activated the TβR-II promoter Inhibited cell growth | [74] |
| Purchased | Human oral SCC cell line SAS, Ca9-22, and HSC | MTT assay Flow cytometry analysis Western blot analysis RT-PCR Confocal laser microscopic analysis | Enhanced the replication of the HSV-1 mutant through the activation of NF-κB Inhibited cell growth by inducing cell cycle arrest at G1 | [75] |
| Not reported | HeLa cells | RT-PCR Western blot analysis | Upregulated the expression of p21WAF1 and p16INK4A in various cell lines Downregulated the expression of cyclin A Upregulated the expression of gelsolin and fibronectin | [76] |
| Not reported | MDA-MB-231 human breast cancer cell | MTT assay | Decreased cell viability (IC50 = 100 ng/mL) Induced apoptosis Induced poly (ADP-ribose) polymerase-1 (PARP-1) cleavage and caspase-3 activation Upregulated the expression of CDK inhibitor p21(WAF1/CIP1) protein Downregulated the expression of Bcl-2 | [77] |
| Purchased | Bone marrow cells and calvarial osteoblasts collected from the tibias and femurs of ICR mice | TRAP staining RT-PCR Western blot analysis In vivo experiment | Inhibited osteoclastogenesis and bone resorption by suppressing the induction of c-Fos by RANKL | [78] |
| Not reported | HeLa cells | RT-PCR Western blot analysis ChIP assay | Activated p21WAF1/CIP1 expression through the downregulation of c-myc and the release of the repression of c-myc from the promoter | [79] |
| Purchased | Human bladder cancer cell line, BIU-87 | MTT assay Flow cytometry analysis RT-PCR DNA fragmentation analysis | Inhibited bladder cancer cell proliferation Induced cell cycle arrest at the G1 phase Increased apoptotic cell death Increased p21WAF1 mRNA expression | [32] |
| Purchased | Murine pro-B lymphoma FL5.12 cells | MTT assay DNA fragmentation assay Flow cytometry analysis Western blot analysis RT-PCR | Inhibited cellular proliferation Induced apoptosis Induced DNA fragmentation Increased the protein levels of cleaved caspase-3 and PARP Induced apoptotic protein Bim Inhibited PU.1 | [80] |
| Not reported | RAW264.7 cells | RT-PCR Western blot analysis ChIP assay | Inhibited LPS-induced C/EBPδ, resulting in a positive effect on LPS-induced cox-2 expression in RAW264.7 cells | [81] |
| Not reported | Human colon cancer cell lines HCT116, HT29, SW480 | Annexin-V staining qRT-PCR Western blot analysis | Altered the expression of cell cycle-associated genes in HCT116 cells Downregulated the gene expression of minichromosome maintenance protein-2 (MCM-2) Increased phosphorylated JNK, which was involved in the downregulation of MCM-2 | [82] |
| Not reported | ZAP-Grg1 transgenic mouse line (in vivo) A549 cells Human umbilical vein endothelial cells (HUVECs) | Western blot analysis qRT-PCR MTT assay Electric cell-substrate impedance sensing (ECIS) analysis | Inhibited lung tumorigenesis in Grg1 transgenic mice Reduced the expression of ErbB1 and ErbB2 Reduced the expression of VEGF and VEGFR2 | [83] |
| Purchased | Human ESCC cell lines KYSE-150 and EC9706 | Transwell migration assay qRT-PCR Western blot analysis | Promoted esophageal squamous cell carcinoma cell migration and EMT through BRD4/ERK1/2-dependent pathway | |
| Purchased | HeLa and Caski cervical cancer cell lines | MTT assay Flow cytometry analysis qRT-PCR Western blot analysis | Suppressed cervical cancer cell proliferation and induced apoptosis and autophagy through the regulation of the PRMT5/STC1/TRPV6/JNK axis | [84] |
| Purchased | MCF-7 cells | Trypan blue staining qRT-PCR Western blot analysis | Reduced CYP19 transcript and protein contents in MCF-7 cells Lowered CYP19 transcript stability and significantly decreased the transcript’s half-life | [85] |
| Purchased | EC9706 cells | Annexin V-FITC/PI staining Western blot analysis MTT assay Flow cytometry analysis | Suppressed ESCC cell growth by inhibiting the activation of the PI3K/Akt and ERK1/2 pathways | [86] |
| Purchased | SK-MEL-3 melanoma cells | Fluorescence microscopy Flow cytometry analysis | Downregulated critical components of the MAPK/MEK/BRAF oncogenic pathway, initiating a mitotic arrest | [87] |
| Purchased | Human ovarian cancer cell lines, COC1 and its DDP-resistant subline, COC1/DDP | RT-PCR Western blot analysis MSP assay ChIP assay | No effect on the reactivation of hMLH1 expression in COC1/DDP cells | [88] |
| Purchased | HCT116 and HT29 cells | Annexin V-FITC PI staining Flow cytometry analysis Bax siRNA transfection Western blot analysis | Induced cell cycle arrest and apoptosis in colorectal cancer cells via p53-dependent and -independent pathways | [89] |
| Not reported | 16 NSCLC cell lines | MTT assay RNA extraction and RT-PCR | Displayed strong antitumor activities in 50% of NSCLC cell lines | [90] |
| Purchased | Human pancreatic cancer cell lines | Oligonucleotide array hybridization Western blot analysis qRT-PCR | Altered the expression of pro- and anti-apoptotic genes in pancreatic adenocarcinoma cells | [91] |
| Not reported | CD4+ T cells isolated from erythrocyte-depleted spleen cell preparations from C57BL/6 mice | RNA extraction and qRT-PCR Flow cytometry analysis Western blot analysis Determination of ROS generation Annexin V-FITC staining | Induced a rapid decline in cytokine expression and accumulation of cells in the G1 phase of the cell cycle Induced apoptotic cell death Altered the expression of a subset of genes involved in T cell responses | [92] |
| Purchased | Human NSCLC lines (Calu-1, NCI-H520, NCI-H23, and NCI-H441) | Flow cytometry analysis Annexin-V staining Immunoprecipitation Western blot analysis | Inhibited cellular growth Induced apoptosis Reduced the percentage of cells in the S phase (10% to 23%) and increased G1 populations (10% to 40%) Increased the expression of p21 without significant effect on p16, p27, CDK2, and cyclin D1 | [93] |
| Purchased | Canine mast cell tumor (MCT) | Trypan blue staining Acridine orange/ethidium bromide staining MTT assay Cell cycle analysis | Reduced the viable cell numbers Increased cell death by apoptosis Increased hypodiploid cells Reduced the G0/G1 and G2/M–phases | [94] |
| Purchased | A549 cells | MTT assay Cell morphology analysis Wound healing assay Western blot analysis RNA extraction and RT-q-PCR assay Docking methodology | Effectively inhibited radiation-induced EMT by: Altered epithelial and mesenchymal markers Modulated signaling molecules of TGFb1 pathway Inhibited cancer cell migratory potential in A549 cells Effectively bound to Snail, an enhancer of EMT | [95] |
| Purchased | HeLa cells | Flow cytometry analysis Immunofluorescence staining RT-PCR | Induced a delay at the G2/M transition, chromosome missegregation, and multi-nucleation Induced cell death Induced a transcriptional modulation of key regulator genes of the cell cycle (Cyclin B1, Plk1, Survivin, and p21WAF1/Cip1) | [96] |
| Purchased | MCF-7 cells | Western blot analysis qRT-PCR Transfection and luciferase reporter assays | Augmented ESR1 gene repression at the transcriptional level Downregulated ERα protein expression under hypoxic conditions through a proteasome-mediated pathway Inhibited cell proliferation under both normoxia and hypoxia conditions Enhanced hypoxia-induced repression of ESR1 and degradation of ERα | [97] |
| Purchased | Human TK6 lymphoblastoid cell line | Cell cycle analysis Annexin V staining Cytogenetic assays Immunoblot analysis | Induced apoptosis and G1 cell cycle arrest Induced chromosomal breakage Induced DNA breaks Induced aneuploidy | [98] |
| Purchased | Human ESCC cells, EC109 and KYSE150 | qRT-PCR Immonochemistry Western blot analysis ChIP-qPCR Annexin-V/FITC staining | Significantly induced DNA damage in ESCC cells Induced Rad9 gene expression both at transcriptional and translational levels in EC109 cells alone Enhanced DNA damage and cell death | [99] |
| Not reported | Primary hepatocytes Hepatoma cells | Western blot analysis Northern blot analysis LDH release assay Caspase-3 activation assay | Inhibited hepatocyte proliferation No induction of apoptosis in primary hepatocytes Induced apoptosis in hepatoma cells Upregulated the expression of the anti-apoptotic protein Bcl(xL) | [100] |
| Not reported | 267B1 human prostate epithelial cells | Fluorescence microscopy Agarose gel electrophoresis Flow cytometry analysis | Inhibited cell growth Induced apoptosis Inhibited the levels of IAP family members Activated caspases and NF-κB | [101] |
| Purchased | MCF10A and MCF10A-ras cell lines | Ras activation assay MTT assay DAPI staining of nuclei Flow cytometry analysis Western blot analysis | Induced morphological changes, apoptotic cell death and modulation of the cell cycle regulatory proteins in the MCF10A-ras cells Downregulated the expression of cyclin D1 and CDK4 Upregulated the expression of p21WAF1 and p53 Induced cell cycle arrest at the G1 phase in MCF10A-ras cells Decreased hyperphosphorylation levels of the Rb protein | [101] |
| Not reported | Chronic lymphocytic leukemia (CLL) cells | Flow cytometry analysis ATP assay Immunoblotting qPCR | Acted via a dual anti-HDAC/Wnt mechanism with a high selectivity and efficacy in CLL | [102] |
| Purchased | Human SCLC DMS53 cells | Light microscopy Western blot analysis MTT assay | Induced morphological differentiation and inhibition of cell growth via cell cycle arrest and subsequent apoptosis | [103] |
| Not reported | Apoptotic-resistant MCF-7TN-R cells derived from MCF-7 cells | Clonogenicity assay microRNA microarray analysis | Altered the microRNA expression profiles in apoptosis-resistant breast cancer cells | [29] |
| Purchased | Human gastric epithelial cell line BGC-823 | MTT assay Hoechst 33342 staining Western blot analysis RT-qPCR Immunohistochemistry | Inhibited cell proliferation Induced cell apoptosis Inhibited non-metastatic melanoma protein B (GPNMB) expression | [104] |
| Purchased | Plasmacytoid dendritic cells (PDC) | Cytokine ELISA RT-PCR Confocal microscopy | Inhibited the production of IFN-I, TRAIL and of the pro-inflammatory cytokines TNF-α and IL-6 by CpG-activated PDC Inhibited the production of IFNα by PDC cultured in vitro in the presence of serum obtained from systemic lupus erythematosus patients | [105] |
| Purchased | SW480 cells | AnnexinV-FITC PI staining qRT-PCR MTT assay Flow cytometry analysis | Inhibited cell growth Induced apoptosis IC50 = 1.5 μM Upregulated p21, p27, and p57 genes expression | |
| Not reported | Human hepatocellular carcinoma Hepa 1-6 cells | MTT assay qRT-PCR AnnexinV- FITC and PI staining | Inhibited cellular proliferation Induced apoptosis Increased ERα gene expression quantity | [106] |
| Not reported | Hepatocellular carcinoma HCCLM3, MHCC97H, and MHCC97L cell lines | MTT assay Cell apoptosis assay qRT-PCR | Induced apoptosis and inhibited cell growth through both mitochondrial/intrinsic and cytoplasmic/extrinsic apoptotic pathways | [47] |
| Purchased | U87 glioblastoma cells and tumorsphere-derived cells | Tumorsphere formation assay Colony formation assay RT-PCR Western blot analysis Cell migration assay Cell cycle analysis | Inhibited proliferation and altered cell cycle in U87 human GBM cells Induced senescence-like alterations in nuclear morphology in U87 cells Increased mRNA levels of C-Myc and reduced Oct4 mRNA in cells Reduced tumorsphere formation and sizes in U87 cell cultures | [52] |
| Purchased | B lymphoblastoid cell lines (LCLs), SNU-20 and SNU-1103 Epstein-Barr virus-negative Burkitt’s lymphoma cell line, BJAB | Flow cytometry analysis Trypan blue staining RNase protection assay RT-PCR Western blot analysis Immunofluorescence assay | Enhanced anti-tumor effect for EBV-associated tumors by inducing a cell cycle arrest, apoptosis, and by triggering an EBV lytic cycle | [107] |
| Purchased | HeLa and SiHa cells | Western blot analysis RNA extraction and RT-PCR ChIP assay Transfection and luciferase reporter assay Tumorigenicity in mice xenograft model | Suppressed the PMA-induced OPN gene expression Suppressed the PMA-induced c-Jun recruitment to the OPN promoter by inhibiting c-Jun expression Suppressed cervical tumor growth in response to PMA in NOD/SCID mice xenograft model | [108] |
| Purchased | SW480 and SW620 cells | Western blot analysis Immunofluorescence analysis Reporter assays ChIP assay | Modulated claudin-1 mRNA stability through the modulation of Hu antigen R and tristetraprolin in colon cancer cells | [109] |
| Purchased | Human nasopharyngeal carcinoma (NPC) cell line CNE2 and undifferentiated C666–1 | CCK-8 assay RNA extraction and RT-PCR Western blot analysis Flow cytometric analysis Transwell migration assay Scratch wound healing assay | Inhibited cell proliferation and arrested the cell cycle at G1 phases Reduced PCNA, cyclin D1, cyclin E1, CDK2, p16, and p21 expressions and stimulated CDK6 levels Promoted Vimentin and Snail1 expression Induced the EMT in CNE2 and C666–1 cells | [110] |
| Purchased | Human lung adenocarcinoma A549 cells and normal lung epithelial cells | RNA extraction and RT-PCR Immunocytochemical staining Western blot analysis Migration assay Cell cycle assay Fluorescein isothiocyanate (FITC) permeability assay | Increased anguin-1/LSR, decreased CLDN-2, promoted G1 arrest, and prevented the migration of A549 cells Increased the expression of LSR and CLDN-2 and decreased that of CLDN-1 with or without TGF-β in normal human lung epithelial cells | [111] |
| Not reported | Male Kunming mice | Testis weighing and sperm collection Histological processing Immunofluorescence Fluorescence microscopy | Increased genetic recombination frequency of spermatocyte meiosis | [112] |
| Purchased | A2780 cells | Histopathology analysis Immunohistochemistry Flow cytometry analysis | Induced morphological cell transformation, with increased cytoplasm Inhibited cell proliferation Reduced mitotic activity Induced epithelial-like differentiation with increased cytokeratin expression | [113] |
| Not reported | Human neuroblastoma (NB) cell lines | MTT assay siRNA-mediated silencing Western blot analysis | Induced cell death in neuroblastic-type NB cells by increasing the acetylation of Ku70, a Bax-binding protein CBP, Bax, and Ku70 contribute to therapeutic response to TSA against NB | [114] |
| Not reported | Raji cells and normal peripheral blood mononuclear cells | Flow cytometry analysis TUNEL assay Annexin V/PI staining | Inhibited cell proliferation Induced apoptosis Induced accumulation of cells in G0/G1 or G2/M Decreased cell population in the S phase | [115] |
| Not reported | MCF-7, MDA-MB-231 and MCF-10A cell lines | MTT assay Colony-forming assay Western blot analysis Annexin V- FITC and PI staining Cytochrome C release assay | Inhibited cell viability and proliferation without affecting MCF-10A cell Induced cell apoptosis which was initiated by G2-M arrest and depending on mitochondrial ROS produced after reduced mitochondrial respiratory chain activity | [116] |
| Purchased | Human rhabdomyosarcoma cell lines RH30 and RD | Annexin V-FITC and PI staining Flow cytometry analysis Immunohistochemical staining RQ-PCR miRNA transfection | Inhibited rhabdomyosarcoma proliferation and induced differentiation through myomir reactivation | [117] |
| Purchased | MCF-7 and MB-MDA-231 cells | MTT assay Annexin V- FITC and PI staining Flow cytometry analysis | Induced cell growth inhibition via 15-Lox-1 associated with the elevation of 15-Lox-1 metabolite (13 (S)-HODE) Induced cell cycle arrest Induced apoptosis | [118] |
| Not reported | Female wild-type BALB/c mice | Flow cytometry analysis ELISA test Cell differential counting Histopathology analysis | Suppressed murine innate allergic inflammation by blocking group 2 innate lymphoid cell (ILC2) activation | [119] |
| Purchased | MCF-7, T47-D, SKBr-3, and MDA-MB-231 cell lines Tumor xenograft model | Flow cytometry analysis Immunoblotting RT-PCR In vivo liposome uptake Immunohistochemistry of tumor sections | Induced a long-term degradation of cyclin A and a proteasome-dependent loss of ERα and cyclin D1, allowed derepression of p21WAF1/CIP1 and RhoB GTPase Induced G2/M cell cycle arrest Induced apoptosis Increased ERα mRNA and p21WAF1/CIP1 protein expression Decreased cyclin A with a G2/M blockade and cleavage of PARP | [120] |
| Purchased | MCF-7, T-47D, ZR-75-1, BT-474, MDA-MB-231, MDA-MB-453, CAL 51, and SK-BR-3 cells | Cell proliferation assay Immunoprecipitation and western blot analysis Histopathology analysis | Inhibited cell proliferation Exerted antitumor activity in vivo when administered daily (500 μg/kg) by s.c. injection for 4 weeks | [121] |
| Purchased | Human tongue squamous cell carcinoma SCC-6 cell lines | MTT assay Cell cycle analysis Cell invasion assay Western blot analysis Annexin V-FITC PI staining | Inhibited cellular proliferation Induced apoptosis Blocked the cell cycle at S and G2/M phase Inhibited cellular invasion Inhibited hypoxia-induced accumulation of HIF-1α protein and VEGF expression under hypoxic conditions | [122] |
| Purchased | Fresh tissues of ESCC were obtained from six patients | Western blot analysis Immunohistochemistry Cell Invasion Assay | Inhibited ESCC cell invasion by approximately 75% Decreased MMP-2 and MMP-9 protein levels in ESCC cells | [123] |
| Purchased | AGS gastric cancer cells | CCK-8 experiment Flow cytometry analysis RT-PCR Western blot analysis | Inhibited cell proliferation and promoted cell apoptosis, leading to AGS cell cycle arrest in G0/G1 and G2/M phases, especially G0/ G1 phase Increased p21, p53, and Bax gene expression levels Decreased Bcl-2, CDK2, and CyclinD1 gene expression levels | [123] |
| Purchased | SW480 and PC3 cells | Transwell invasion and migration assay Western blotting analysis qRT-PCR ChIP assay | Induced the reversal process of EMT in SW480 and PC3 cells, resulting in attenuated cell invasion and migration abilities Decreased the expression of transcription factor Slug | [124] |
| Not reported | 5,637 Urinary bladder cancer cells | MTT assay Cell cycle analysis Annexin V-FITC and PI staining Measurement of mitochondrial membrane potential Western blot analysis | Altered cell morphology and reduced cell viability Induced cell cycle arrest Induced cell death via apoptosis Induced apoptosis via the mitochondrial pathway by promoting MMP dissipation and caspase-9 Suppressed the PI3K-Akt signaling pathway Induced Sp1 downregulation and suppressed survivin expression | [125] |
| Purchased | MCF-7 cells | Transwell invasion and migration assay Wound healing assay RT-qPCR Western blot analysis Overexpression of SLUG | Reversed EMT and attenuated the invasive and migratory abilities of MCF-7 breast cancer cells | |
| Not reported | U937 human leukemic cells | Flow cytometry analysis Cell cycle analysis MTT assay | Induced the growth inhibition and morphological changes in a concentration-dependent manner Increased G1 cell population of the cell cycle of U937 cells Induced the population of apoptotic sub-G1 cells Inhibited cyclins, PCNA, and Cdks expression Induced Cdk inhibitors such as p16, p21, and p27 | [126] |
| Purchased | Human endometrial stromal cell line | MTT assay Real-time RT-PCR Western blot analysis | Inhibited cell proliferation Increased PR-α, PR-β, AR, and FasL expression | [127] |
| Purchased | HL-60 cells | MTT assay Annexin V-FITC PI staining Flow cytometry analysis lmmunocytochemical assay | Inhibited cell proliferation IC50 = 100 ng/mL, at the 36th Induced apoptosis | [128] |
| Not reported | HeLa cells | RNA isolation and RT-qPCR | Negatively regulated the expression of ubiquitin-specific protease 22 (USP22) Interfered with the binding of RNA polymerase II to the USP22 promoter, directly suppressing its transcription TSA-induced apoptosis was attenuated by the overexpression of USP22 in HeLa cells | |
| Not reported | HeLa cells | MTT assay Hoechst 33258 staining Flow cytometry analysis qRT-PCR | Inhibited cell growth Induced apoptosis Decreased the proportion of cells in S phase and increased the proportion of cells in G0/G1 and/or G2/M phases Induced the overexpression of genes related to malignant phenotype, including an increase in p53, p21Waf1 and p27Kipl | [129] |
| Purchased | MG-63, 786-0, HT1080 and HeLa cells | Western blot analysis Immunoprecipitation RNA isolation and qPCR Tumor xenograft (BALB/c nude mice) | Inhibited the HIF-2α protein expression Inhibited tumor growth and HIF-2α expression in vivo Destabilized HIF-2α in a proteasome dependent manner, which is unrelated to VHL | [14] |
| Purchased | HeLa cells | MTT Assay Flow Cytometric Analyses Measurement of the MMP Immunostaining Annexin V-FITC and PI staining | Reduced cell survival Induced an MMP collapse Apoptotic cell death and the MMP collapse induced by TSA were decreased by the co-treatment of cells with CytoD and LatB | [130] |
| Purchased | p815 murine mastocytoma cell line | Trypan blue staining Hoechst 33342 staining Western blot analysis Flow cytometry analysis Immunofluorescent staining | Induced apoptosis Reduced cell viability, and many apoptotic manifestations such as generation of DNA fragmentation, activation of caspase-3, cleavage of PARP, and increased of DNA hypoploidy Increased the expression level of Bad Decreased the level of Bcl-2, Bcl-xL, and X-linked inhibitor of apoptosis protein | |
| Purchased | Mature osteoclasts | Flow cytometry analysis RNA extraction and semi-quantitative RT-PCR Western blot analysis In vivo mouse calvarial resorption analyses | Induced osteoclast apoptosis Induced upregulation of p21WAF1 in osteoclasts Inhibited RANKL-directed bone destruction in vivo | [131] |
| Purchased | HeLa cells | MTT assay Western blot analysis Annexin V staining Measurement of MMP Detection of intracellular O2•− levels | Inhibited cell growth Induced apoptosis, caspase-3 activation, and the loss of mitochondrial membrane potential Increased O2•− level and induced GSH depletion in HeLa cells The administration of Bcl-2 siRNA intensified TSA-induced HeLa cell death | [132] |
| Not reported | Prostate cancer cell line DU145 | MTT assay Flow cytometry analysis Immunofluorescence staining Western blot analysis | Induced mitotic catastrophe of DU145 cells, including morphological changes, cell cycle arrest at G0/G1 phase, and abnormalities of mitosis Increased the multinuclear cells Inhibited survivin protein expression Increased the expression of P21 protein | [133] |
| Purchased | Human pancreatic cancer cell line BxPC-3 | MTT assay Cell cycle analysis Annexin V staining miRNA microarray analysis Northern blot analysis | Inhibited pancreatic cancer cell viability Arrested cells in G0/G1 phase Induced apoptosis, accompanied by differential expression of microRNAs | [134] |
| Purchased | AML-12, 3T3-L1, MDCK, Hep-3B, A549, HeLa, and MCF-7 cells | Flow cytometry analysis Immunoblotting | Suppressed TGF-β1-induced apoptosis in normal hepatocytes but not in hepatoma cells Suppressed serum starvation-induced apoptosis in non-cancer cells but not in cancer cells Induced apoptosis in cancer cells but not in non-cancer cells Activated ERK1/2 in non-cancer cells but not cancer cells | [135] |
| Not reported | OVCAR-3 cells | MTT assay Western blot analysis Caspase assay kits | Inhibited cell viability Increased the expression of cytochrome c and P53 and the expression of caspases-3, -8, and -9 Enhanced the mitochondria-mediated apoptotic pathways | [136] |
| Purchased | HeLa cells | MTT assay Fourier transform infrared spectroscopy (FT-IR) Immunofluorescence Analysis FT-IR spectroscopic measurements and analysis | Inhibited cell proliferation Induced an elevated level of cellular acetylation and conformational/structural changes of proteins in the cells Induced a higher percent of α-helix structure accompanied by an increment of acetylation level in both histones and cytoskeleton proteins | [137] |
| Not reported | HeLa and HepG2 cells | Clonogenic assay | Improved radiation resistance by activating Akt/Nrf2-dependent antioxidation pathway in cancer cells | [138] |
| Not reported | MCF-7 cells | MTT assay Annexin-V/PI staining Cell cycle analysis RT-PCR | Inhibited cell proliferation Induced apoptosis Downregulated the expression of ERα, myc-c, cyclin-D, and Bcl-2 | [139] |
| Purchased | SKOV-3 and A549 cells | MTT assay RNA extraction and qRT-PCR Vybrant apoptosis assay kit Flow cytometry analysis | Exerted dose and time dependent cytotoxicity effect on both cells Upregulated klf4 expression Induced apoptosis | [140] |
| Origin | Used Model | Experimental Approach | Key Results | References |
|---|---|---|---|---|
| Purchased | Hepatoma cells (HepG2) | MTT assay Annexin V assay DNA extraction and qRT-PCR | Sensitized hepatoma cells to Taxol (an anticancer drug) more than 5-Aza-dC and dexamethasone | [167] |
| Not reported | Head and neck squamous cell carcinoma cell line UT-SCC-77 | MTT assay Immunofluorescence Immunoblot analysis Measurement of lysosomal pH | Enhanced cisplatin-induced apoptosis by decreasing lysosomal pH | [36] |
| Purchased | MM1S and ARP-1 cells | XTT cell proliferation assay Propidium iodide staining Flow cytometry analysis RT-PCR analysis | Sensitized TNF-related apoptosis inducing ligand (TRAIL)-resistant myeloma cells by downregulating the expression of Bcl-2 and Bcl-XL Modulated the expression of Bcl-2 proapoptotic members | [162] |
| Not reported | HeLa cells | MTT assay Wound healing assay Immunocytochemistry Western blot analysis | Enhanced the DNA targeting capacity and apoptosis inducing efficacy of silver nanoparticles (AgNPs) most probably due to its effect on chromatin condensation | [170] |
| Purchased | Human breast cancer cell lines MDA-MB-231, Hs578T and ZR75-1 | RNA isolation and RT-PCR Immunoblotting MTT assay ChIP assay | Sensitized estrogen receptor (ER) α-negative, antihormone-unresponsive breast cancer cells to tamoxifen treatment | [164] |
| Purchased | Human leukemia cell lines (HL60 and U937 cells) | AlamarBlue assay Hoechst 33342 staining Apo-one homogeneous caspase-3/7 assay RNA isolation and RT-PCR | Potentiated the etoposide-induced cytotoxicity and apoptosis Activated caspases and induced the loss of the mitochondrial membrane potential | [161] |
| Purchased | Human erythroleukemic K562 cells | MTT assay Clonogenic survival assay | Increased the radio- and chemo-sensitization of K562 cells Inhibited cell proliferation Reduced clonogenic survival Induced apoptosis | [38] |
| Purchased | K562 cells | Flow cytometry analysis Western blot assay Caspase-3 and caspase-7 activity assays Clonogenic survival assay | Enhanced radiation sensitivity and accumulation of γH2A.X Inhibited cell proliferation Reduced clonogenic survival Induced apoptosis | [37] |
| Not reported | HN-3 and HN-9, human head, and neck cancer cell lines | Clonogenic assay | Radiosensitized HN-3 and HN-9 cell lines | [168] |
| Purchased | A549, HeLa, and Caski cell lines | Clonogenic assay Western blot analysis Flow cytometry analysis | Enhanced radiosensitivity by abrogating G2/M arrest in all three cell lines | [169] |
| Purchased | MDA-MB-231, MCF7, and MCF10A | MTT assay Flow cytometry analysis Western blot analysis | Sensitized the human breast adenocarcinoma MDA-MB-231 cells towards TRAIL-induced apoptosis | [156] |
| Not reported | Wild-type (A2780WT) and cisplatin-resistant (A2780RES) human ovarian cancer cells | MTT assay Immunohistochemistry qRT-PCR | Instigated apoptosis, autophagy, inhibition of cell cycle progression, and consequently loss of cell viability in A2780 cells Improved cisplatin-induced apoptosis, cell cycle arrest, and autophagy in A2780 cells Boosted the cisplatin-induced, p53-dependent apoptosis | [39] |
| Purchased | Gastric cancer cell lines AGS, NCI-N87, SNU-1, and SNU-16 cells | Annexin V/PI staining Cell Counting Kit-8 qRT-PCR Western blot analysis | Potentiated TRAIL-induced antitumor effects via the inhibition of ERK/FOXM1 pathway | [80] |
| Purchased | Human urothelial carcinoma cell lines, NTUB1and T24 | Western blot analysis MTT assay Annexin V staining In vivo xenograft mouse model | Reduced cell viability and enhanced cytotoxicity of three chemotherapeutic agents (cisplatin, gemcitabine, and doxorubicin) Potentiated the apoptotic effects of the three chemotherapeutic agents Suppressed the activation of Raf/MEK/ERK pathway associated with chemotherapeutic agent treatment TSA + chemotherapeutic agents synergistically inhibited cell viability Enhanced chemotherapy-induced antitumor effects (in vivo) | [173] |
| Purchased | Human ovarian cancer SKOV3 cells Hey8 cells | Methylene blue analysis Flow cytometric analysis Annexin V staining RNA extraction and qRT-PCR Western blot analysis | Sensitized ovarian cancer cells to TRAIL-induced apoptosis by the downregulation of c-FLIPL via the inhibition of EGFR pathway TSA + TRAIL induced apoptosis and inhibited cell viability in SKOV3 and Hey8 cells | [101] |
| Purchased | Human pancreatic cancer cell lines | Crystal Violet method Determination of IC50 values | Enhanced the response of chemotherapeutic agents in inhibiting pancreatic cancer cell proliferation | [40] |
| Purchased | Osteosarcoma cell line HOS | Western blot analysis Immunoprecipitation Flow cytometry analysis MMP assay | Reduced HOS cell viability Induced HOS cell apoptosis Reduced MMP and cytochrome c release to the cytosol Sensitized HOS cells to the action of genistein (an antitumor agent) | [172] |
| Purchased | 786-O, ACHN, and Caki-1 RCC cell lines | MTT assay under normoxic or hypoxic conditions Flow cytometry analysis Western blot analysis | Enhanced cytotoxic effects of sunitinib on RCC cells | [165] |
| Purchased | 786-O, ACHN, and Caki-1 RCC cell lines | Western blot analysis Capillary electrophoresis time-of-flight mass spectrometry (CE-TOF-MS) MTT assay Flow cytometry analysis | Reduced sunitinib resistance by triggering intracellular metabolome shifts regarding energy metabolism | [166] |
| Purchased | Athymic BALB/c nude mice HepG2 cells | Microarray and data analysis NK cell infiltration RNA extraction and RT-PCR NK cell cytotoxic assay AnnexinV-FITC PI staining | Sensitized the hepatocellular carcinoma cells to enhanced NK cell-mediated killing by regulating immune-related genes | [171] |
| Purchased | Normal ovarian surface epithelium (OSE) cells | Flow cytometry analysis Immunoblotting In vitro caspases-3 and -9 assay | Restored Apaf-1 function independent of alterations in Apaf-1 expression Restored Apaf-1 function and sensitized cells to cisplatin-induced apoptosis | [157] |
| Not reported | Lung cancer cell line A549 and the CDDP- resistant derivative A549/CDDP | Hoechst 33258 staining Flow cytometry analysis Western blot analysis | Induced apoptosis in both A549 cells and A549/CDDP cells Enhanced the sensitivity of A549/CDDP cells to cisplatin, along with concomitant DAPK upregulation | [158] |
| Purchased | Bladder cancer cells (HTB9, J82, SW1710, T24, HTB5, UMUC14, and 253J) | Clonogenic assay Flow cytometry analysis Hoechst 33342 staining Colorimetric caspase activity assay | Synergistically enhanced the antitumor effect of cisplatin and resensitized cisplatin resistant bladder cancer cells | [41] |
| Purchased | Human gastric cancer cell lines OCUM-8 and MKN-74 | MTT assay RT-PCR | Increased the efficiency of anticancer drugs Increased the expression of p21, p53, DAPK-1, and the DAPK-2 gene in both cancer cells Increased the expression of caspase-3 in OCUM-8, but not in MKN-74 | [163] |
| Purchased | NSCLC cell lines A549 and H1650 | Cell cycle analysis Clonogenic assay Annexin V-FITC and PI staining TUNEL assay Flow cytometry analysis Western blot analysis | Induced cell cycle arrest and apoptosis in A549 cells Enhanced radio-sensitivity of NSCLC cells Enhanced IR-induced G2/M arrest and apoptosis Increased IR-induced apoptosis via mitochondrial pathway Promoted IR-induced caspase-3 activation in association with repression of XIAP expression Radio-sensitized A549 cells through the downregulation of DNA repair proteins | [174] |
| Purchased | HCC cells | MTT assay Immunoblotting TUNEL assay Hoechst 33342 staining | Sensitized HBx-expressing liver cancer cells to etoposide treatment | [135] |
| Purchased | Human lung cancer cells (A549 and H1299) | Western blot assay Caspases-3 and -9 activities Tumor cell xenograft mouse model | TSA-induced apoptosis was enhanced by quercetin through the mitochondrial pathway in A549 cells The anticancer effect of TSA was enhanced by quercetin in the xenograft tumor model | [175] |
| Purchased | Male nude mice injected with A549 cells | TNF-α and IL-1β determination Immunoblot analysis Immunohistochemical staining | The anticancer effect of TSA was enhanced by quercetin | [176] |
| Not reported | Human epithelial carcinoma cell lines OVCAR-3 and SK-OV-3 | MTT assay Quantitative analysis of DNA fragmentation Western blot analysis Flow cytometry analysis ELISA test | TSA-induced apoptosis was reduced by inhibition of casein kinase 2 | [177] |
| Not reported | NIH-OVCAR-3 and SK-OV-3 cell lines | MTT assay Western blot analysis Flow cytometry analysis | TSA-induced apoptosis was potentiated by 18β-glycyrrhetinic acid | [178] |
| Purchased | A549 cells | Flow cytometry analysis RT-PCR Western blot analysis Caspase activity | The effect of TSA on inhibiting A549 cell growth was enhanced using genistein by increasing the expression of TNF receptor-1 | [179] |
| Origin | Used Model | Experimental Approach | Key Results | References |
|---|---|---|---|---|
| Purchased | SW620 human colon cancer cell line | RNA isolation and Northern analysis Apoptosis analysis Anti-ac-histone H4 Western blot | TSA + butyrate induced the growth arrest and DNA damage gene 45α (GADD45α) and GADD45β TSA + cycloheximide super-induced the expression of GADD45α and β | [180] |
| Purchased | Male Sprague Dawley rats and ddY mice RAW264 cells | Flow cytometry analysis RT-PCR Luciferase assays Western blot analysis | Inhibited osteoclast differentiation in bone marrow cultures TSA + sodium butyrate inhibited osteoclast formation and osteoclast-specific mRNA expression in RAW264 cells stimulated with receptor activator of NF-κB ligand (RANKL) TSA + sodium butyrate reduced the sRANKL-stimulated or TNF-α–stimulated trans-activation of NF-κB–dependent reporter genes | [44] |
| Not reported | MCF-7 and MDA-MB-468 cells | DAPI staining Flow cytometry analysis | TSA + HC-toxin induced antiproliferative activity in both cell lines Induced cell cycle arrest at G2/M phases Induced apoptosis | [182] |
| Purchased | Human leukemia cells (HL-60) | Trypan blue staining Measurement of intracellular ROS generation | Increased cytotoxic activity in a time- and dose-dependent manner TSA + antioxidants decreased ROS generation | [184] |
| Purchased | Human hepatoma cells (Hep3B) | Trypan blue staining Flow cytometry analysis Measurement of the intracellular ROS generation | TSA + antioxidants synergistically protected against in vitro cytotoxicity of Ni2+ in Hep3B cells | [183] |
| Purchased | Human leukemia cells (HL-60) | Cell proliferation and viability assays MDA assay | TSA + quercetin increased cytotoxicity in a time- and dose-dependent manner | [144] |
| Purchased | Lung and esophageal cancer cells | MTT assay Transfection and luciferase assay Western Blot analysis | Increased the transcriptional activity of NF-κB and p21 TSA + calphostin C decreased TSA-mediated upregulation of NF-κB and p21 activation | [186] |
| Purchased | SK-RC-39 and SK-RC-45 RCC cell lines Tumor xenograft model (forty Swiss nu/nu mice) | Western blot analysis RNA extraction and RT-PCR Annexin V- FITC and PI staining | TSA + all-trans retinoic acid (ATRA) inhibited the proliferation of RCC cell lines and the tumor growth in a xenograft model TSA alone or/+ ATRA reactivated RARh2 mRNA expression in RCC cells TSA + ATRA induced the apoptosis and partial G0-G1 arrest in SK-RC-39 cells | [45] |
| Purchased | Eight diverse human pancreatic cancer cell lines | Cell viability assay RT-PCR | TSA + proteasome inhibitor PS-341 synergistically induced apoptosis in pancreatic cancer cells | [195] |
| Not reported | Human astrocytoma A172 cells | TSA + hyperthermia (heat shock) effectively induced apoptotic cell death | [196] | |
| Not reported | Human glioblastoma A172 cells | MTT assay Flow cytometry analysis Western blot analysis RT-PCR | TSA + hyperthermia increased the thermos-sensitivity of A172 cells, resulting in cellular apoptosis | [197] |
| Purchased | Human leukemia cells (HL-60) | Cell proliferation and viability assays | TSA + curcumin increased cytotoxicity in a time- and dose-dependent manner | [198] |
| Purchased | Ten human pancreatic cancer cell lines Female nude mice | Cell cycle analysis Cell proliferation assay Immunoblot analysis RNA extraction and RT-PCR In vivo study: T3M4 human pancreatic cancer cells were s.c. injected into animals | TSA + gemcitabine synergistically inhibited the proliferation of human pancreatic adenocarcinoma cell lines (in vitro) Enhanced the apoptosis TSA + gemcitabine synergistically inhibited growth of human pancreatic adenocarcinoma cells (in vivo) | [42] |
| Purchased | Hut-78 T- and Raji B-lymphoma cell lines | RQ-PCR Western blot analysis | TSA + sodium butyrate + 5-aza-2′-deoxycytidine altered the expression of glucocorticoid receptor α and β isoforms | [199] |
| Purchased | Non-small cell lung carcinomas (NSCLC) | Immunoblot analysis Caspase activity assay | TSA + etoposide-induced apoptotic cell death in drug-resistant NSCLC cells TSA + etoposide induced apoptosis in a caspase-dependent manner accompanied by a crucial decrease in Bcl-xL expression | [160] |
| Purchased | Human neuroblastoma cell lines NB-1691 and NB-1643 Retroperitoneal human neuroblastoma xenografts | Western blot analysis Tumor volume measurement (in vivo) | TSA + interferon β induced a reduction in cell count compared to controls in NB-1691 and NB-1643 cell lines Increased the expression of p21Waf1 in NB-1691 cells TSA alone or/+ interferon β significantly restricted tumor growth | [188] |
| Not reported | Human endometrial carcinoma cells of the line Ark2, KLE, and AN3 | Trypan blue staining Annexin V and Hoechst staining Flow cytometry analysis Western blot analysis | TSA alone or/+ paclitaxel inhibited cell growth TSA alone or/+ paclitaxel increased apoptotic rates | [200] |
| Purchased | Human cancer cell lines U87 and T98 (both glioblastoma), SW480, MCF-7, HeLa | MTT assay Western blot analysis ELISA test Tumor-bearing mice | TSA + G47Δ synergistically induced cell proliferation TSA + G47Δ enhanced cyclin D1 and VEGF inhibition TSA + G47Δ enhanced anti-angiogenesis and enhanced antitumoral efficacy in animal models | [201] |
| Purchased | LNCaP prostate cancer cell line | RNA extraction and RT-PCR | TSA + somatostatin + 5-aza decitabine upregulated the somatostatin receptor expression | [202] |
| Purchased | Human leukemia HL 60 cells | MTT assay Flow cytometry analysis Trypan blue staining Western blot analysis NF-κB transcription factor assay | TSA (100 nM) + EEAC (100 μg/mL) caused synergistic inhibition of cell growth and an increase of apoptotic induction EEAC could effectively increase the cytotoxic sensitivity of TSA through the upregulation of DR5 and NF-κB activation | [203] |
| Not reported | Human neuroblastoma lines | MTT assay 32P-postlabeling assay | TSA + valproic acid increased the cytotoxicity of ellipticine (an anticancer drug) | [204] |
| Purchased | UKF-NB-3 and UKF-NB-4 neuroblastoma cell lines | MTT assay Western blot analysis | TSA + valproic acid inhibited the growth of neuroblastoma cells (IC50 values ranging from 69.8 to 129.4 nM) TSA + valproic acid induced CYP1A1 expression and depressed CYP1B1 levels, in UKF-NB-4 | [189] |
| Purchased | Human pancreatic endocrine tumor cell lines (CM, BON, and QGP-1) | Cell proliferation assay Flow cytometry analysis Annexin V- FITC and PI staining | Induced cell cycle arrest TSA + 5-aza-2′-deoxycytidine synergistically inhibited cell proliferation TSA + 5-aza-2′-deoxycytidine synergistically induced apoptotic cell death Regulated 19 proteins in both ductal and endocrine pancreatic cancer cells | [205] |
| Purchased | OVCAR-3 and SK-OV-3 cells | Colorimetric assay Clonogenic assays | TSA + Apicidin enhanced the radiosensitivity of ovarian carcinoma cells | [206] |
| Not reported | A549 cells | MTT assay Flow cytometry analysis Caspase-3 activity Comet assay | Genistein + β-carotene enhanced the cell- growth-arrest effect of TSA IN A549 cells | [207] |
| Purchased | UKF-NB-3 and UKF-NB-4 neuroblastoma cell lines | MTT assay Flow cytometry analysis Western blot analysis | TSA + valproic acid increased cytotoxicity of ellipticine and DNA adduct formation by ellipticine in human neuroblastoma cells | [208] |
| Not reported | Human bladder cancer cell lines HTB5, HTB9, T24, J82, UMUC14 and SW1710 | Clonogenic assay Flow cytometry analysis Western blot analysis | Synergistically potentiated the antitumor effect of gemcitabine TSA + gemcitabine repressed NF-κB signaling pathway activation | [187] |
| Purchased | Human laryngeal carcinoma cell line Hep-2 Male BALB/c mice | Cell Counting Kit-8 (CCK-8) assay Hep-2 transplanted tumor growth in nude mice | TSA + 5-aza-2′-deoxycytidine suppressed cell proliferation on Hep-2 in vivo and in vitro | [209] |
| Not reported | MCF-7 cells | MTT Flow cytometry analysis | Inhibited E2-induced proliferation of MCF-7 cells TSA + raloxifene enhanced the antiproliferative activity of each other by promoting cell death via apoptosis and cell cycle arrest TSA alone or/+ raloxifene increased the expression level of estrogen receptor b (ERβ) | [210] |
| Purchased | Human breast cancer cell lines, MDA- MB435eB, and SkBr3 | MTT assay Annexin V-FITC Staining Cell cycle analysis Western blot analysis | TSA + curcumin decreased the viability of SkBr3 and 435eB cells TSA + curcumin enhanced the growth inhibitory effects of either compound alone TSA + curcumin decreased phosphorylation of ERK and Akt TSA + curcumin induces a G0/G1 arrest in SkBr3 cells and a G2M arrest in 435eB cells TSA + curcumin induced apoptosis TSA + curcumin induce phosphorylation of p38 and JNK in SkBr3 cells | [192] |
| Purchased | A549 cells | MTT assay Hoechst 33258 staining Flow cytometry analysis Immunofluorescence analysis Western blot analysis | TSA + docetaxel or erlotinib produced synergistic inhibition on A549 cells TSA + docetaxel or erlotinib induced apoptosis of A549 cells TSA + docetaxel or erlotinib induced a delay at G2/M transition TSA + docetaxel or erlotinib increased the expression of cleaved-caspase-3 TSA + docetaxel increased acetylation of α-tubulin | [136] |
| Purchased | A549 cells | Trypan blue staining Hoechst 33258 staining Flow cytometry analysis Western blot analysis | TSA alone or/+ paclitaxel reduced cell proliferation TSA + paclitaxel induced apoptosis and more cells arrested in G2/M phase TSA + paclitaxel synergistically increased acetylated tubulin, caspase-3, and PARP, TSA + paclitaxel reduced surviving expression | [211] |
| Purchased | Patients with AML Human leukemia HL60, KG1, Kasumi, K562, and THP1 cells | Western analysis Flow cytometry analysis ChIP assay | TSA + chaetocin dramatically induced apoptosis and enhanced tumor suppressor gene re-expression TSA + chaetocin enhanced antileukemic activity in leukemia cells derived from patients with AML | [212] |
| Purchased | Human osteosarcoma cell lines (MG-63, HOS, SaOS-2, and U20S) and murine osteosarcoma cell line LM8 | MTT assay Cell cycle analysis Annexin V staining Quantitative PCR Western blot analysis In vivo xenograft study | TSA (0.3 μM) + metformin (10 mM) decreased the viability of osteosarcoma cell lines TSA + metformin arrested the cell cycle of MG-63 and LM8 in G1/G2 phase Suppressed in vivo tumor proliferation | [213] |
| Purchased | Human ovarian cancer cell lines HEY, SKOV3 | MTS assay Cell migration assay Western blot analysis Mouse xenografts | TSA + 5-aza-20-deoxycytidine + cisplatin (low-dose) significantly suppressed cell viability, migration, and spheroid formation and growth TSA (0.3 mg/kg) significantly suppressed tumorigenicity of HEY xenografts through inhibition of EMT and decreased pluripotency of ovarian cancer cells | [89] |
| Purchased | LNCaP and PC3 cells | WST-1 assay Western blot analysis RT-PCR | TSA + bortezomib synergistically induced apoptosis in both cancer cells TSA + bortezomib effectively inactivated NF-κB signaling TSA + bortezomib upregulated the predominant endogenous apoptotic factor caspase-3, and disrupted the NF-κB pathway in the androgen-independent PC3 cell line | [214] |
| Purchased | Human lung adenocarcinoma A549 cells | MTT assay and Hochst33258 staining Western blot analysis | TSA + cisplatin induced synergistic anti-tumor effects (induced apoptosis, inhibited cell proliferation, increased the inhibition rate, decreased pro-caspase-8, and increased caspase-8) | [137] |
| Purchased | Two CCA cell lines (poorly differentiated KKU-100 and well-differentiated KKU-M214 adenocarcinoma cells) | MTT assay Flow cytometry analysis Western blot analysis | Induced G0/G1 phase arrest in KKU-100 cells Hydroxamic acid + TSA dose-dependently reduced the viability of both cells Hydroxamic acid + TSA exerted higher cytotoxicity than drugs alone Hydroxamic acid + TSA induced more apoptotic cell death of both cells than the single drug | [215] |
| Not reported | HEp2 human laryngeal cancer cell line | Annexin V/propidium iodide staining Western blot analysis TUNEL assay MTT assay | TSA + genistein inhibited cell growth and cell migration, and promoted apoptosis in the HEp-2 cells TSA + genistein reversed endothelial growth factor-induced epithelial-mesenchymal transition (EMT) in the HEp-2 cells | [216] |
| Purchased | MCF-7 and HeLa cells | WST-8 assay Measurement of oxidative stress markers Measurement of MMP TUNEL assay Caspase-3 assay | TSA alone or/+ palladium nanoparticles (PdNPs) inhibited cell viability TSA + PdNPs had a more pronounced effect on cytotoxicity, oxidative stress, MMP, caspases-3/9 activity, and expression of pro- and anti-apoptotic genes | [217] |
| Not reported | A549 and H460 human lung cancer cell lines | Wound healing assay Flow cytometry Hoechst 33342 staining Western blot analysis A549 xenografts (female BALB-C/nude mice) and metastases tissues collection | TSA + BEZ235 synergistically inhibited NSCLC cell proliferation and induced apoptosis Synergistically suppressed NSCLC migration and invasion Decreased xenograft growth and metastasis rates and ki-67 protein expression in vivo | [193] |
| Purchased | Six human breast cancer cells | MTT assay Flow cytometry analysis Colony formation assay Immunofluorescence staining Western blot analysis Female nude mice aged 4~6 week | TSA + BEZ235 induced significant synergistic growth inhibition of multiple breast cancer cell lines TSA + BEZ235 induced apoptosis in a caspase-dependent manner TSA + BEZ235 enhanced autophagic cell death TSA + BEZ235 blocked tumour growth without noticeable side effects | [194] |
| Purchased | HeLa cells | Fluorometric activity assay Enzymatic-linked immuno-captured ELISA Affymetrix miRNA 4.1-panel arrays | TSA + Vitis vinifera extract induced the overexpression of similar miRNAs predicted to destroy certain influential oncogenes | [218] |
| Purchased | Ovarian cancer A2780 cell line | Flow cytometry analysis Western blot analysis Immunofluorescence assay Annexin V assay | TSA + PS-341 increased apoptosis and G2/M arrest TSA + PS-341 enhanced the expression of cyclin B1, resulting in the proliferation inhibition and apoptosis in A2780 and A2780T cells | [190] |
| Not reported | Panc1 and PaCa44 pancreatic cancer-derived cells | Trypan blue staining MTT assay BrdU assay Western blot analysis | TSA + valproic acid induced apoptosis in both cancer cells Increased the pro-apoptotic Bim level, reduced the anti-apoptotic Mcl-1 level and increased ROS production and autophagy in PaCA44 cells | [219] |
| Purchased | Human Huh7 hepatocellular carcinoma cell line | MTT assay Western blot analysis | TSA + sorafenib inhibited cell viability TSA + sorafenib increased cytotoxicity of human hepatocellular carcinoma cells | [220] |
| Not reported | Human Burkitt’s lymphoma (BL) cell lines Ramos and Namalwa cells | MTT assay Trypan blue staining Cell cycle analysis Annexin V staining Western blot analysis BALB/c nude mice | Reduced cell viability, induced apoptosis, and cell arrest at G0/G1 Attenuated EPS8 and downstream Phospho-Erk1/2 pathway Knockdown of EPS8 + TSA had a synergistic suppression effect on BALB/c nude mice | [221] |
| Not reported | Human urothelial carcinoma (UC) cell lines (T24 and NTUB1) Xenograft nude mouse model | MTT assay Flow cytometry analysis Western blot analysis | TSA + one of the chemotherapeutic agents induced synergistic cytotoxicity and concomitantly suppressed chemotherapeutic drug-induced activation of Raf-MEK- ERK pathway TSA + chemotherapy elicited a synergistic cytotoxic response via targeting the Raf/MEK/ ERK pathway | [159] |
| Purchased | ESCC tissue from esophageal cancer patients Human ESCC cells Eca-109 and TE-1 | Immunohistochemistry MTT assay Western blot analysis | Decreased the expression of both Beclin-1 and LC3 proteins in ESCCs TSA + BEZ235 inhibited synergistically ESCC cell viability and induced autophagy with increasing expressions of Beclin-1, LC3-II, and the ratio of LC3-II/LC3-I | [222] |
| Purchased | Human lung cancer H1299 cells | MTT assay Annexin V-FITC and PI assay Western blot analysis Quantitative RT-PCR | TSA-induced apoptosis was increased by 88% by quercetin in H1299 cells TSA-induced death receptor 5 (DR5) mRNA was increased by quercetin in H1299 cells TSA + quercetin significantly increased p300 expression | [223] |
| Purchased | Two human urothelial carcinoma (UC) cell lines (BFTC-905 and BFTC-909) | Western blot analysis MTT assay Western blot analysis In vivo xenograft Tumor size measurement | Enhanced the cytotoxicity of paclitaxel and reduced viability in human UC cells Potentiated the apoptotic effect of paclitaxel on UC cells TSA + paclitaxel synergistically inhibited viability in human UC cells | [224] |
| Not reported | SiHa and HeLa cells | Flow cytometry analysis MTT assay CCK-8 assays Colony formation assays Xenograft experiment Western blot analysis | TSA + cisplatin inhibited cell viability and colony formation ability TSA + cisplatin downregulated the protein expression of HPV16/18E7 and upregulated that of RB1 | [225] |
| Not reported | MDA-MB- 231 and MCF-7 cells | RT-PCR Flow cytometry analysis MTT assay Cell cycle analysis Cell migration assay | TSA + Zebularine sensitized breast cancer towards TRAIL treatment in 231-EGFP cells, validating the potentiality of E-cadherin as a biomarker of TRAIL treatment efficacy in the invasive breast cancer | [226] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouyahya, A.; El Omari, N.; Bakha, M.; Aanniz, T.; El Menyiy, N.; El Hachlafi, N.; El Baaboua, A.; El-Shazly, M.; Alshahrani, M.M.; Al Awadh, A.A.; et al. Pharmacological Properties of Trichostatin A, Focusing on the Anticancer Potential: A Comprehensive Review. Pharmaceuticals 2022, 15, 1235. https://doi.org/10.3390/ph15101235
Bouyahya A, El Omari N, Bakha M, Aanniz T, El Menyiy N, El Hachlafi N, El Baaboua A, El-Shazly M, Alshahrani MM, Al Awadh AA, et al. Pharmacological Properties of Trichostatin A, Focusing on the Anticancer Potential: A Comprehensive Review. Pharmaceuticals. 2022; 15(10):1235. https://doi.org/10.3390/ph15101235
Chicago/Turabian StyleBouyahya, Abdelhakim, Nasreddine El Omari, Mohamed Bakha, Tarik Aanniz, Naoual El Menyiy, Naoufal El Hachlafi, Aicha El Baaboua, Mohamed El-Shazly, Mohammed Merae Alshahrani, Ahmed Abdullah Al Awadh, and et al. 2022. "Pharmacological Properties of Trichostatin A, Focusing on the Anticancer Potential: A Comprehensive Review" Pharmaceuticals 15, no. 10: 1235. https://doi.org/10.3390/ph15101235