
Alpha-Synuclein PET Tracer Development—An Overview about Current Efforts

 , , , ,
, , , ,
Abstract
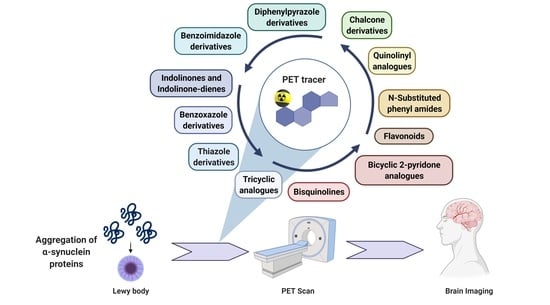
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Alpha-Synuclein and PET Imaging
2.1. Alpha-Synuclein within the Central Nervous System
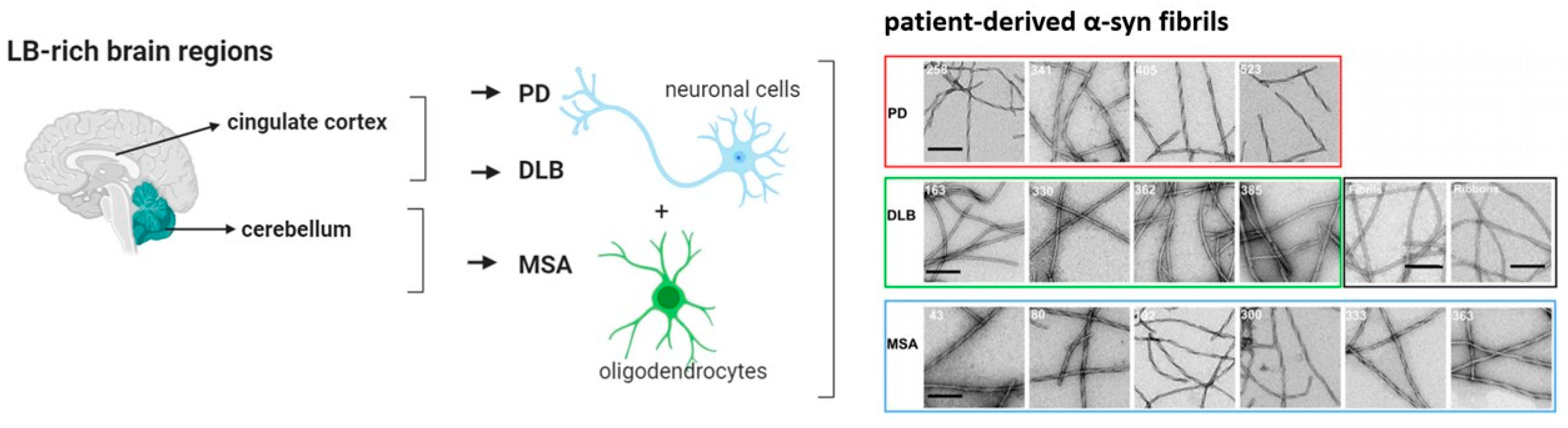
Alpha-Synucleinopathies in PD, DLB and MSA
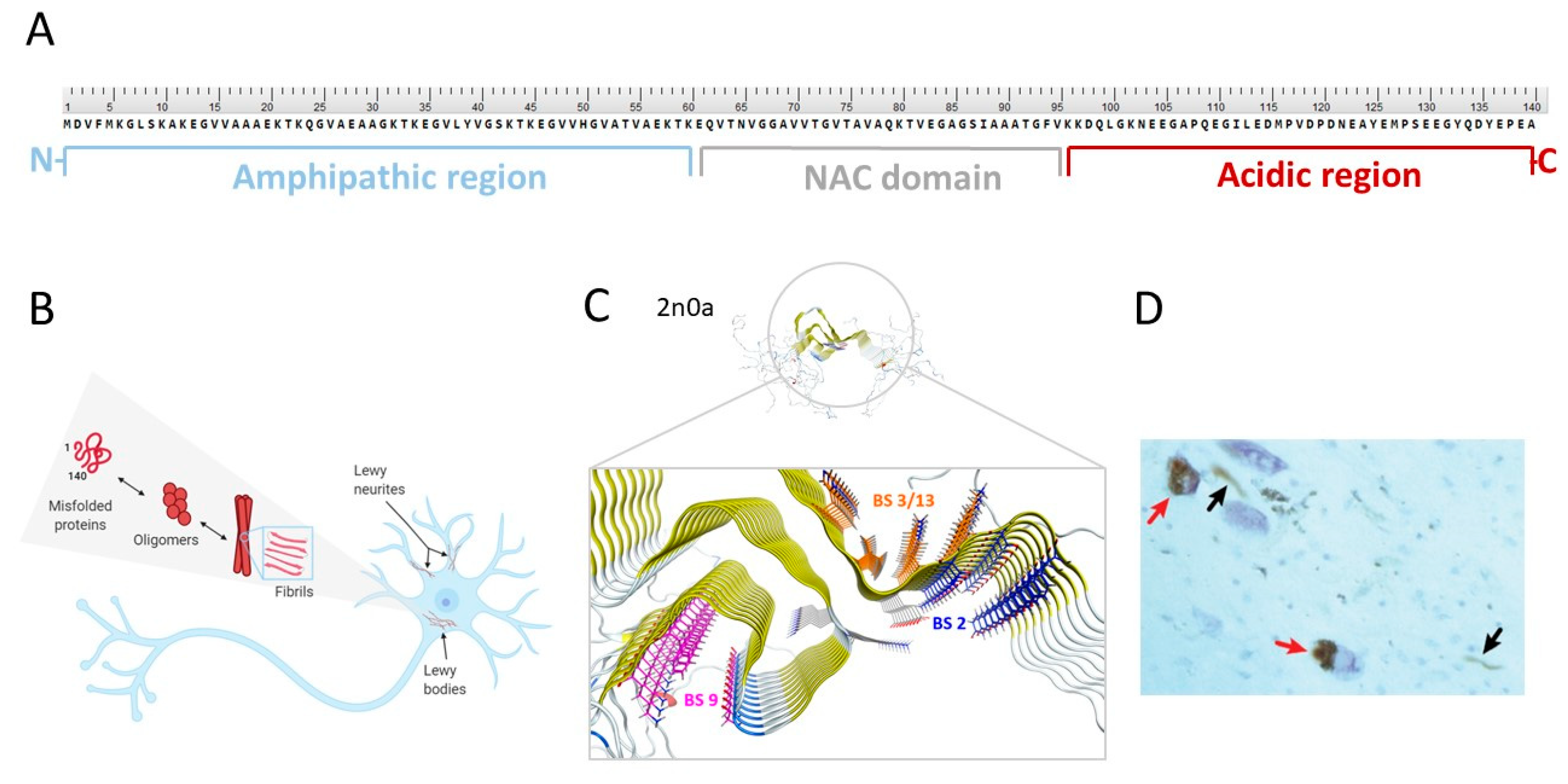
2.2. The Structure of α-Syn
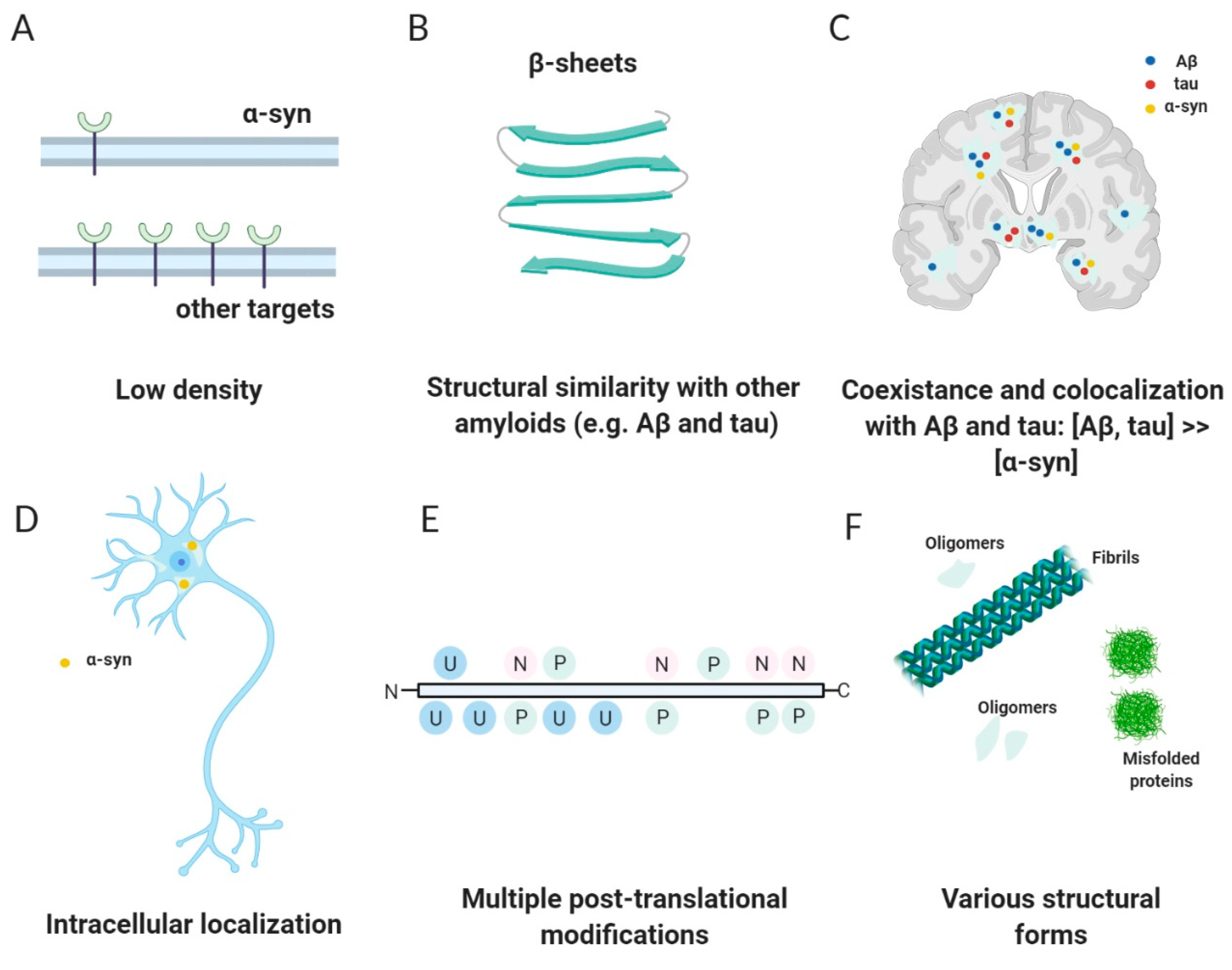
2.3. Challenges—Why Is It Difficult to Develop an α-Syn PET Tracer?
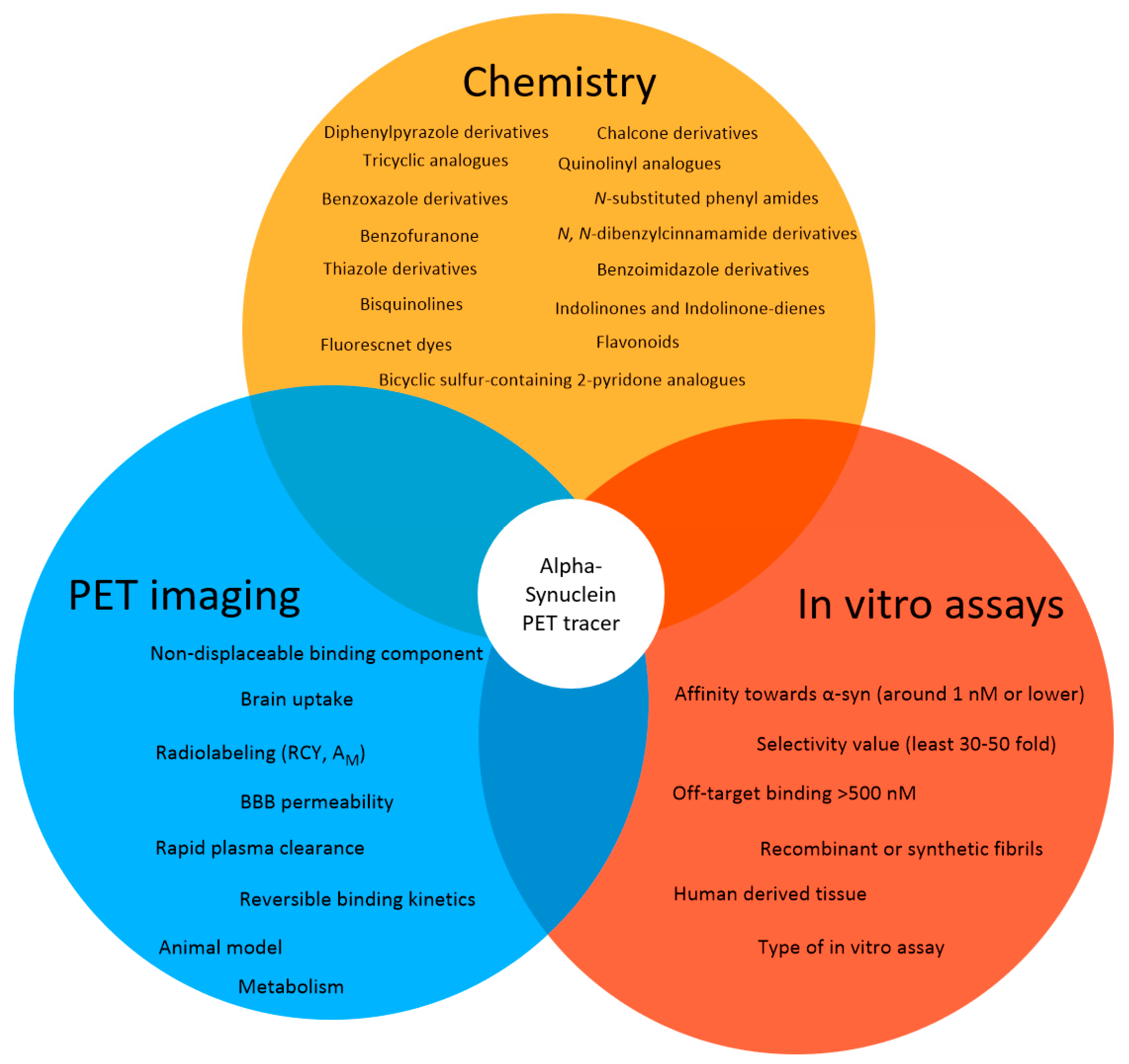
2.4. Success Criteria for a Small Molecule α-Syn PET Tracer
Suggested Success Criteria for a α-Syn PET Ligand in the Preclinic Setting
- Radiolabeling with 11C or 18F should be achievable in a RCY of >10% (activity yield >500 MBq) and in a high molar activity (>50 GBq/µmol);
- Affinity toward α-syn around 1 nM or lower;
- The radioligand enables quantification of lower density α-syn targets in the presence of higher density Aβ or tau proteins;
- The binding affinity or density selectivity for α-syn over Aβ or tau should be at least 30–50 fold (~1 nM vs. 50 nM);
- Off-target binding > 500 nM;
- Selective binding to α-synuclein-rich brain homogenates from PD patients (versus Aβ, tau rich homogenates);
- Binds to LBs/LNs in human tissue;
- BBB permeability, with early peak uptake of SUV >1.5;
- ≥0.4% ID/g in rat brain or ≥4.0% ID/g in mouse brain;
- No or minimal radiometabolites within the brain;
- Low non-displaceable binding component (rule of thumb: logD7.4 < 3 [80];
- Reversible binding kinetics with rapid plasma clearance (<30 min).
3. Alpha-Synuclein PET Tracer Development—From a Molecular Development Point of View
3.1. Tricyclic Analogues (TCAs)
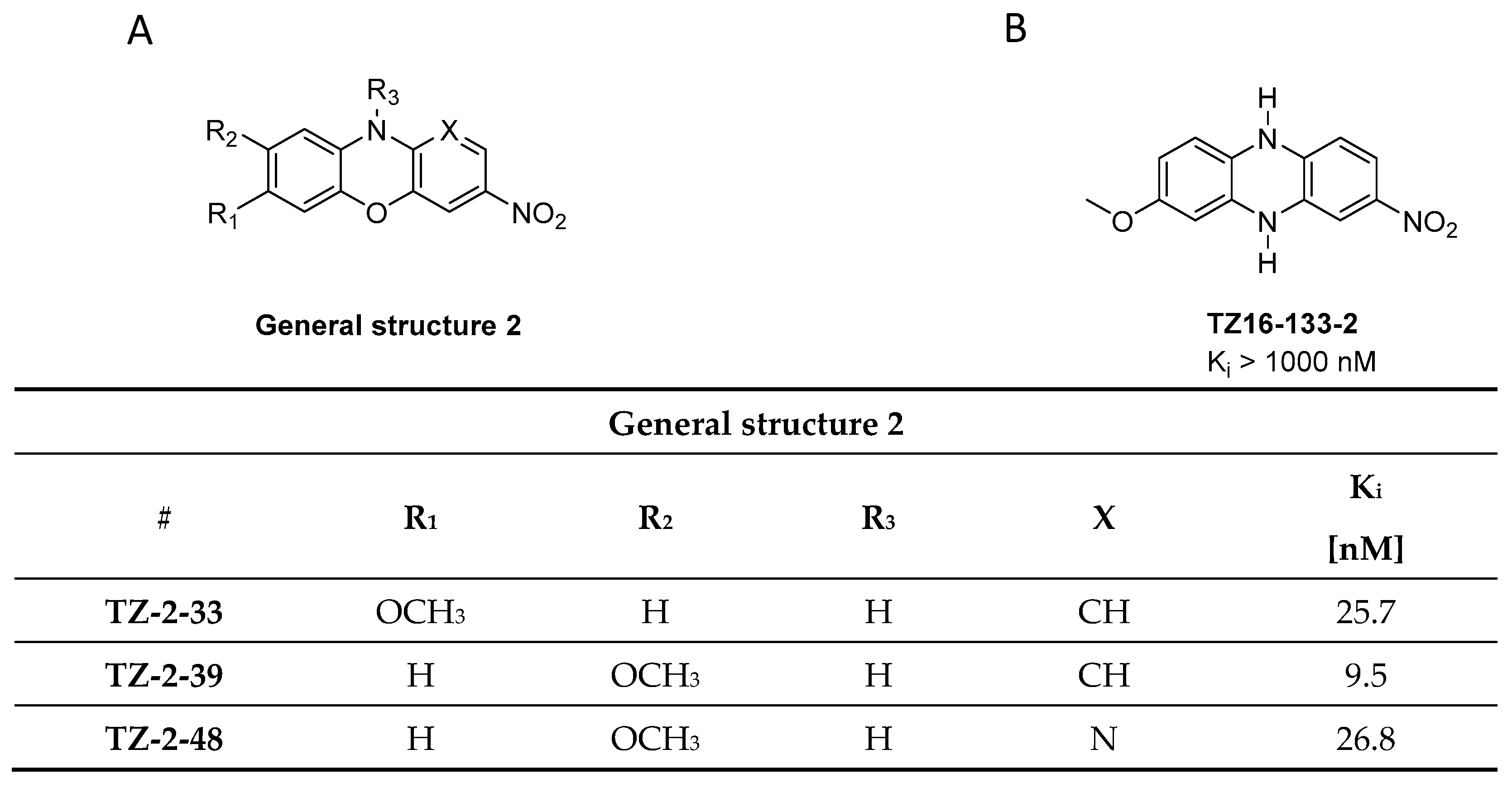
3.1.1. Phenothiazine Analogues
3.1.2. Phenoxazine and Phenazine Analogues
3.2. Benzoxazole Derivatives
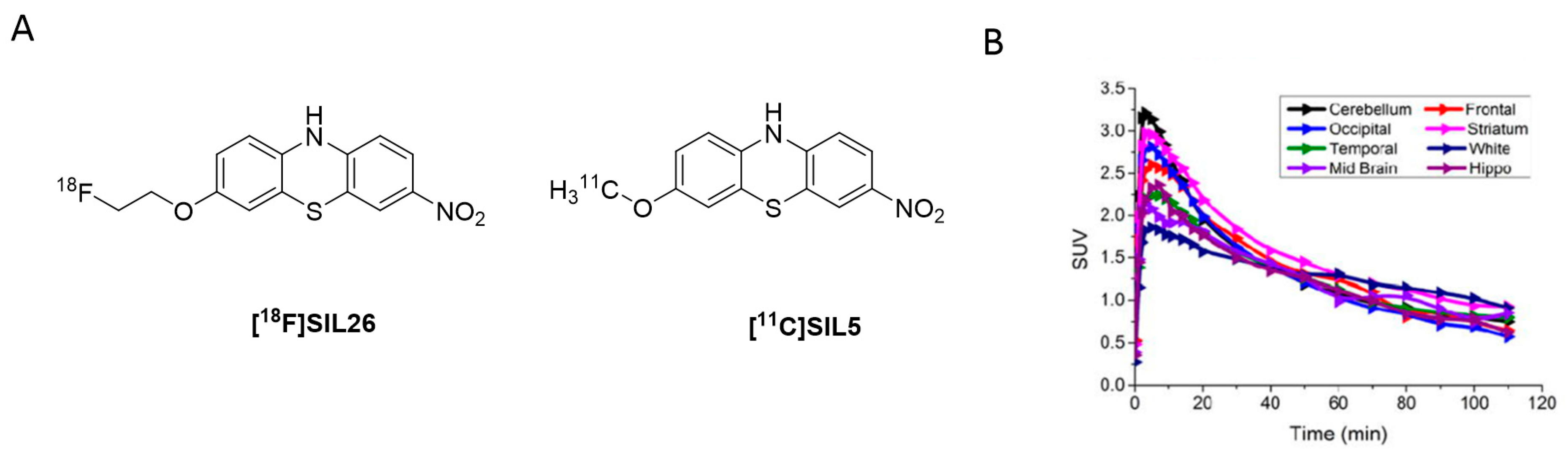
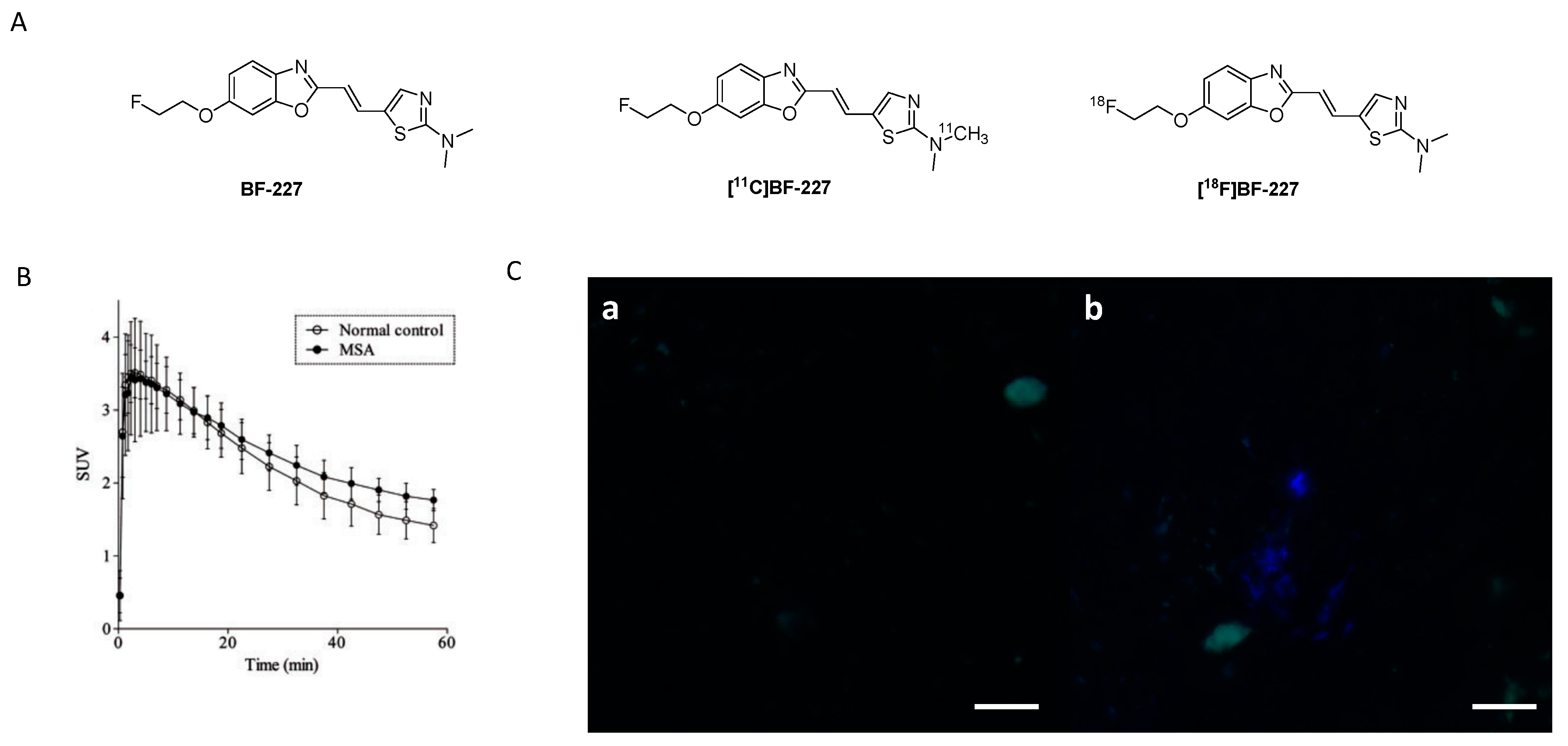
3.2.1. BF-227
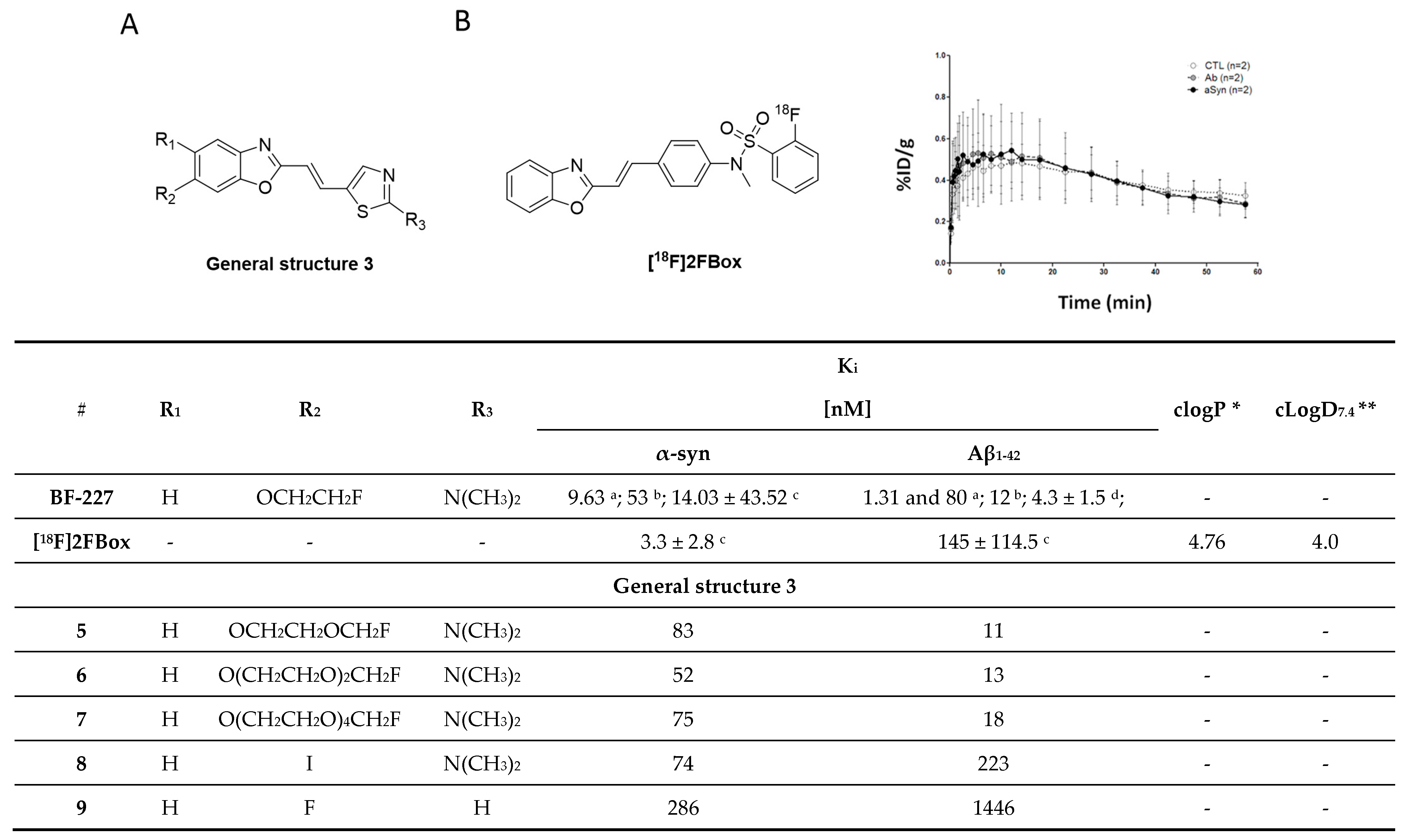
3.2.2. SAR Studies of BF-227
3.2.3. Rational Drug Design around BF-227
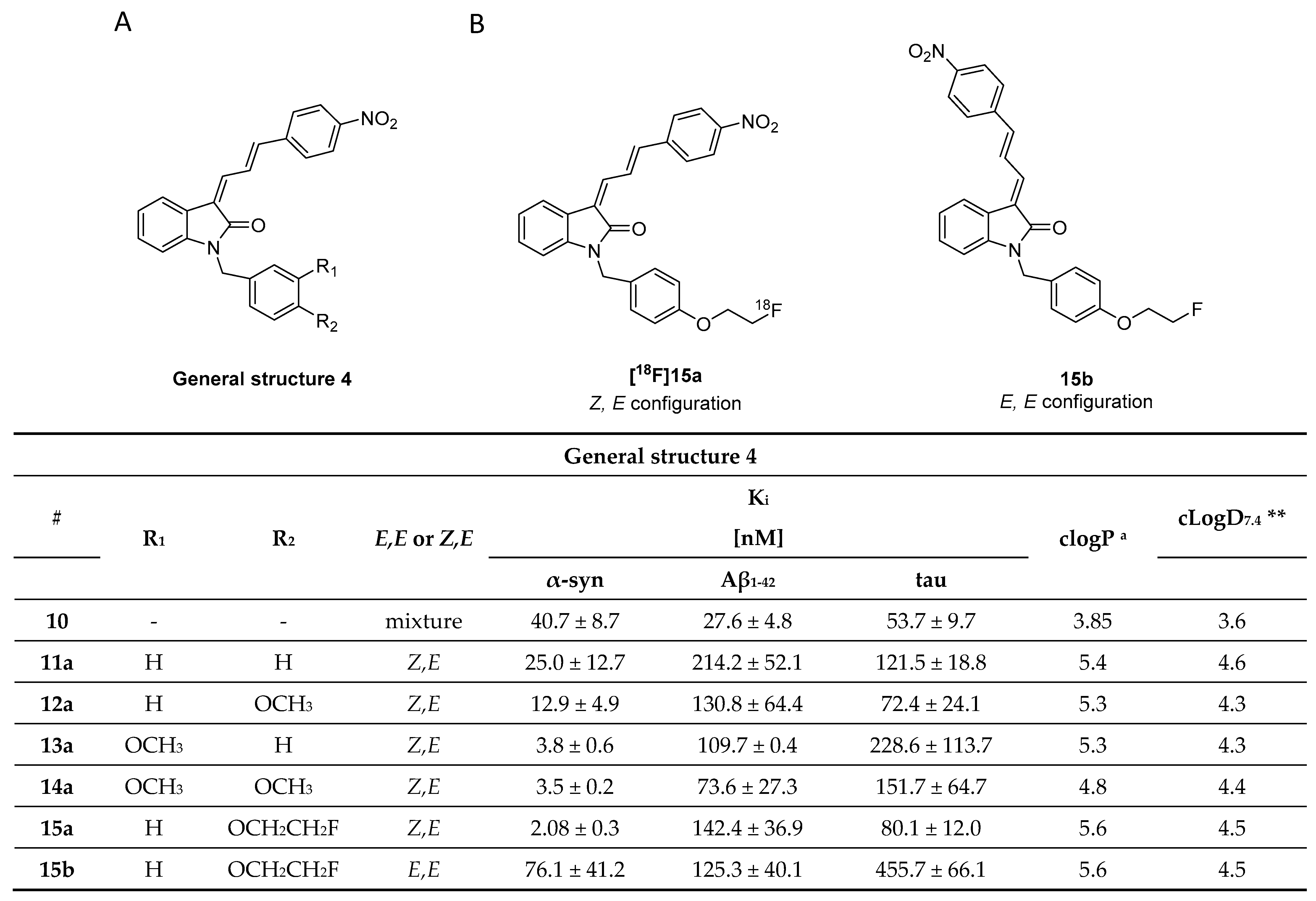
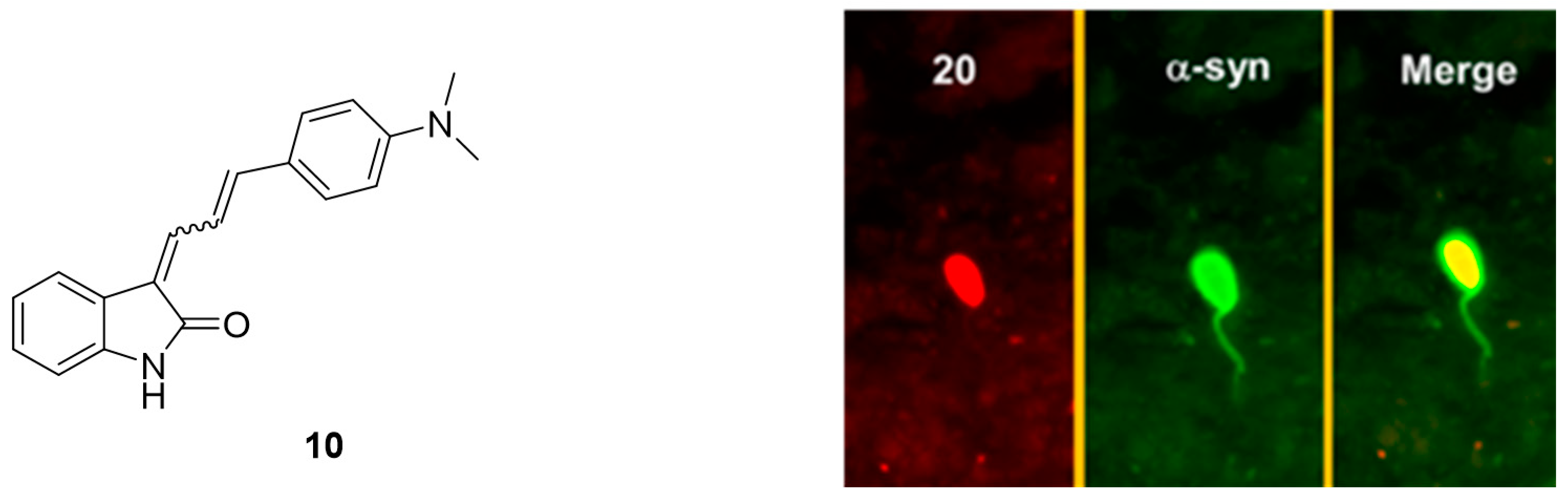
3.3. Indolinones and Indolinone-Dienes
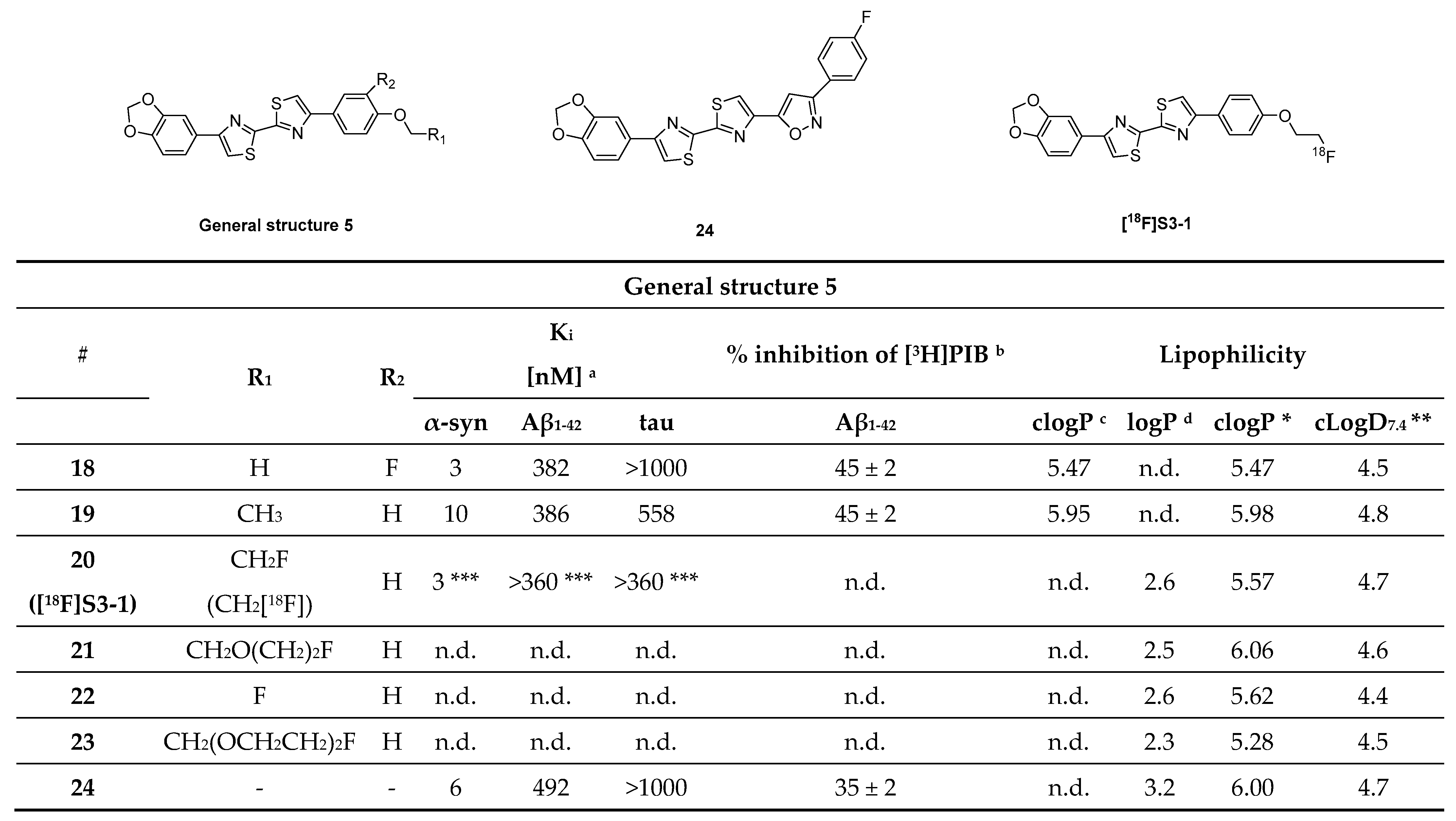
3.4. Thiazole Derivatives
3.4.1. Diphenylthiazole Derivatives
3.4.2. Bithiazole Derivatives
3.4.3. Benzothiazole Derivatives
3.4.4. Thienopyridine, Thiazolo Pyridine Derivatives
3.5. Benzoimidazole Derivatives
3.5.1. Styryl Benzodiazole Derivatives
3.5.2. Azaindole Derivatives
3.6. Diphenylpyrazole Derivatives
Furan-2-yl-1H-pyrazoles
3.7. Chalcone Derivatives
3.7.1. Radioiodinated Diphenyl Derivatives
3.7.2. Chalcone and Five-Membered Heterocyclic Isosteres
3.8. Quinolinyl Analogues
3.9. N-Substituted Phenyl Amides
3.9.1. N-Phenylbenzamide Analogues
3.9.2. N-Substituted Phenyl Amides Analogues
3.9.3. 2-Phenoxy-N-(3-phenylisoxazol-5-yl)acetamide Analogues
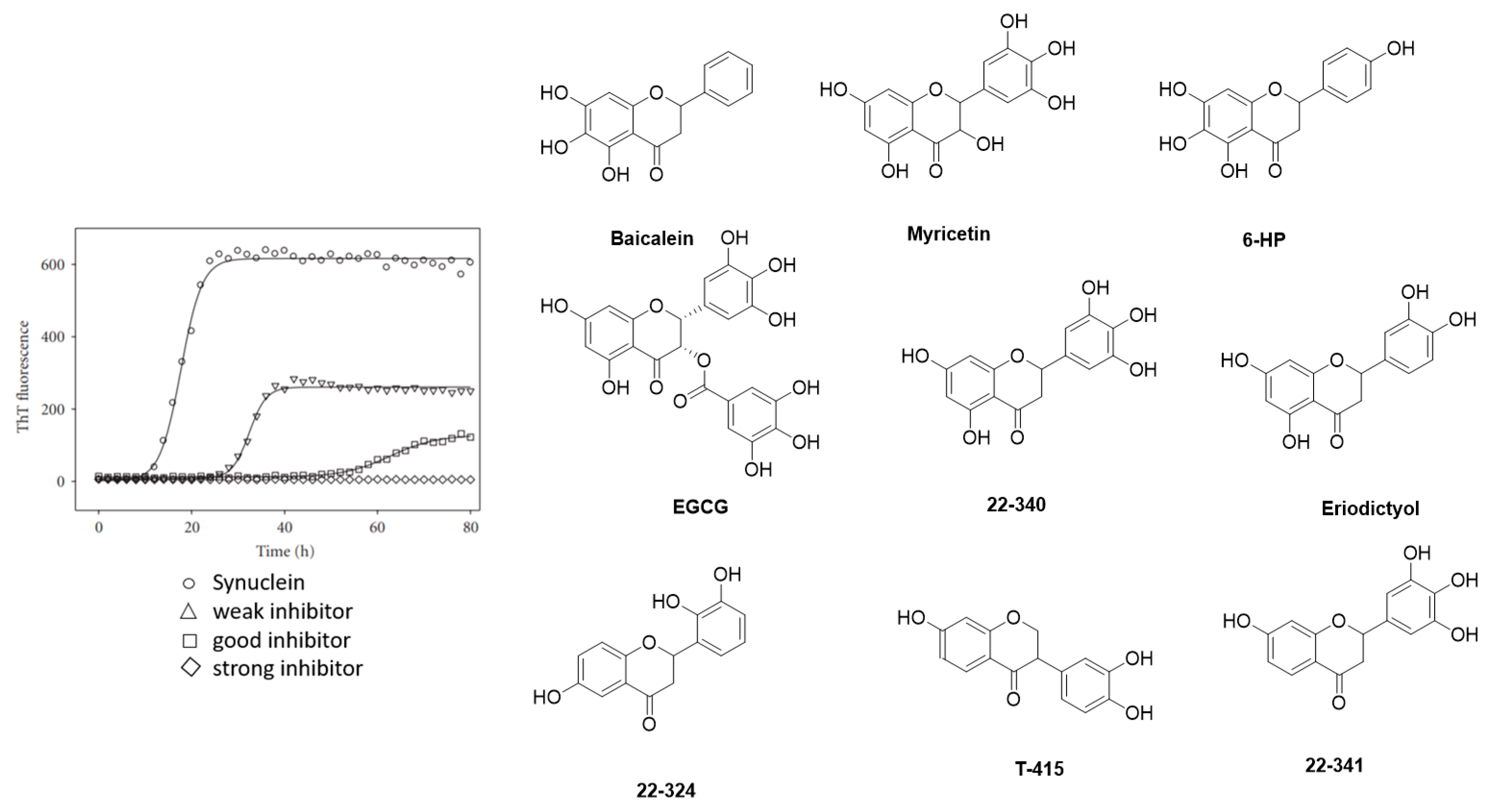
3.10. Flavonoids
3.11. Bicyclic Sulfur-Containing 2-Pyridone Analogues
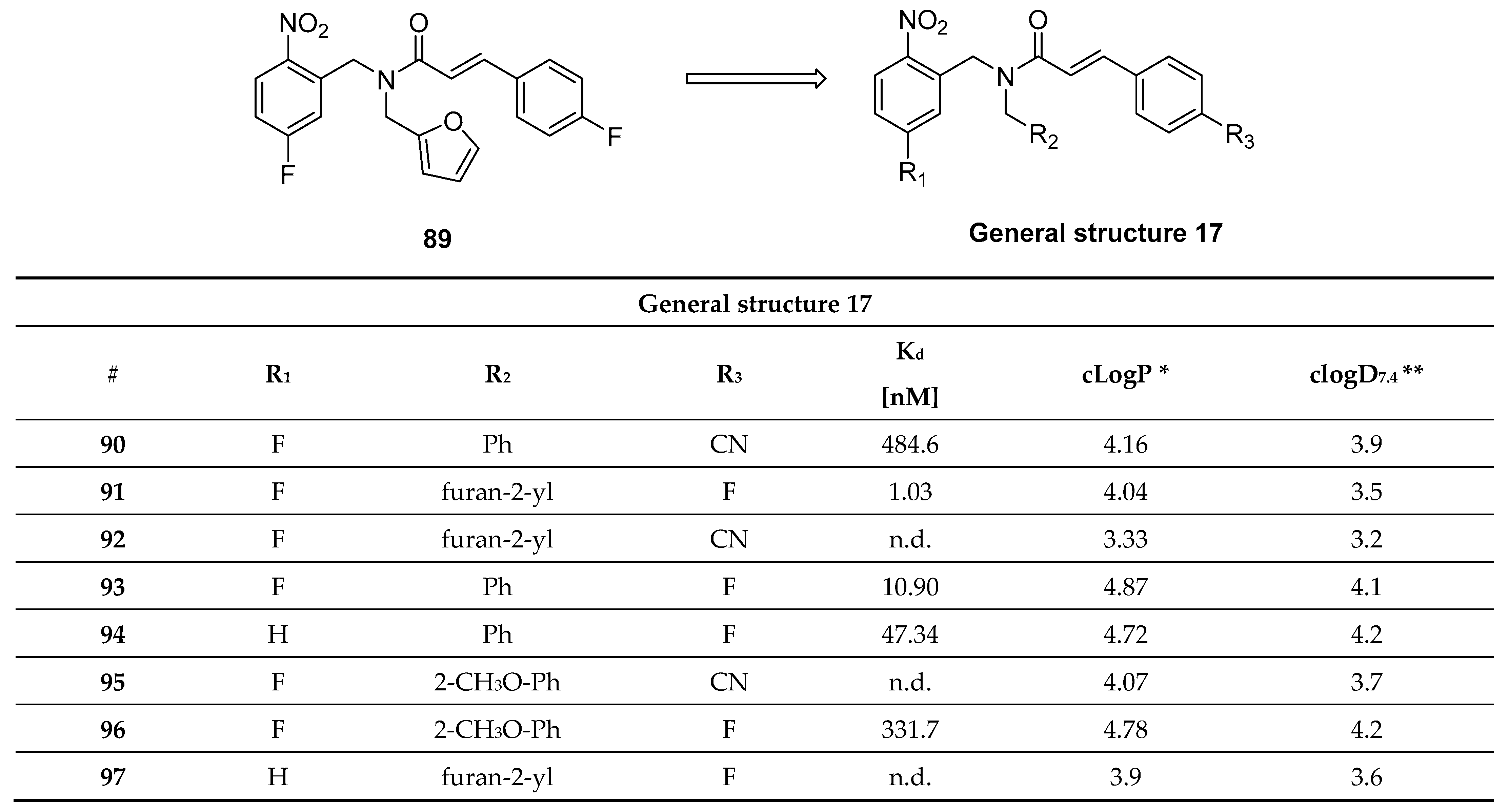
3.12. N,N-Dibenzylcinnamamide Derivatives
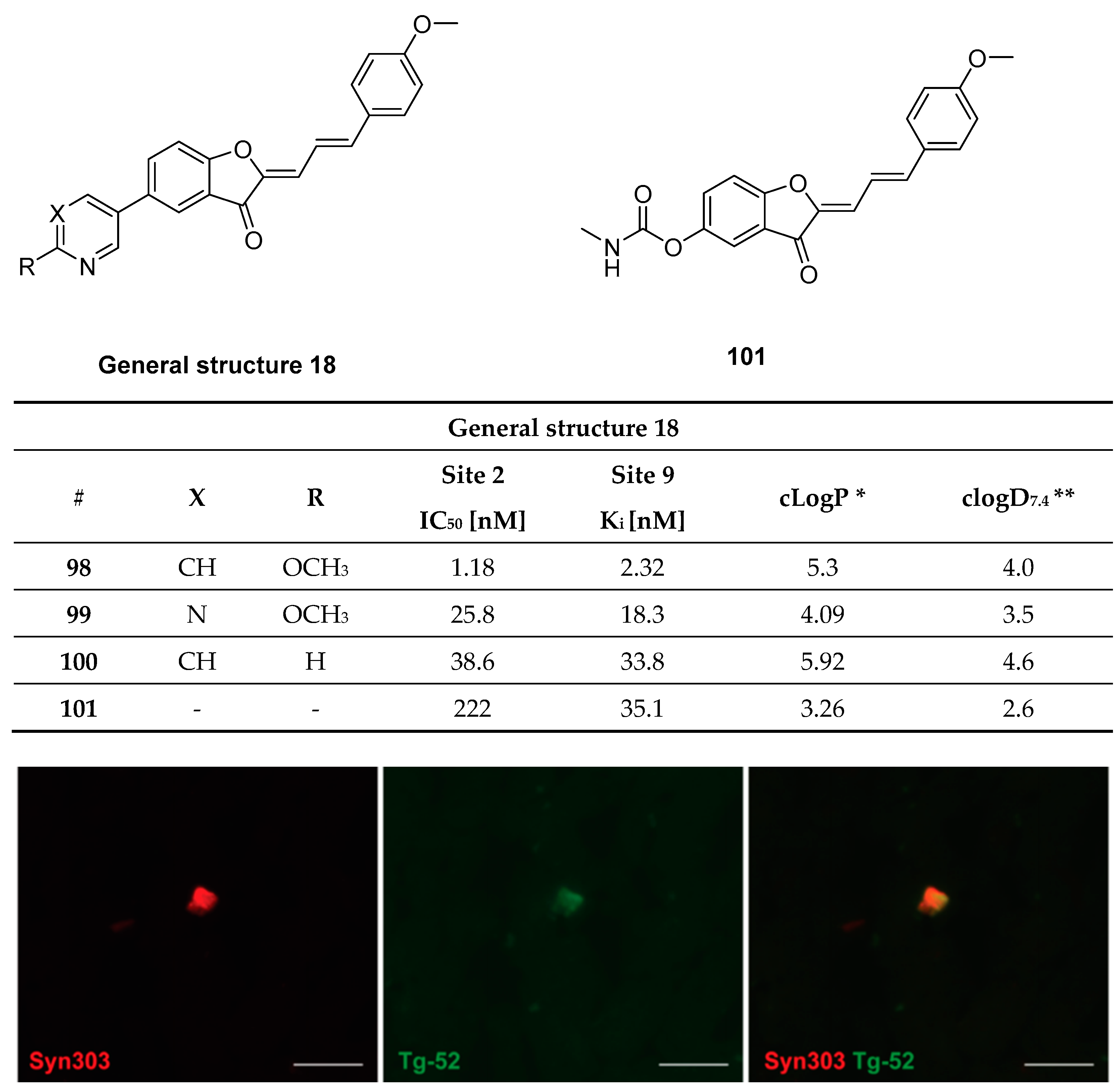
3.13. Benzofuranone
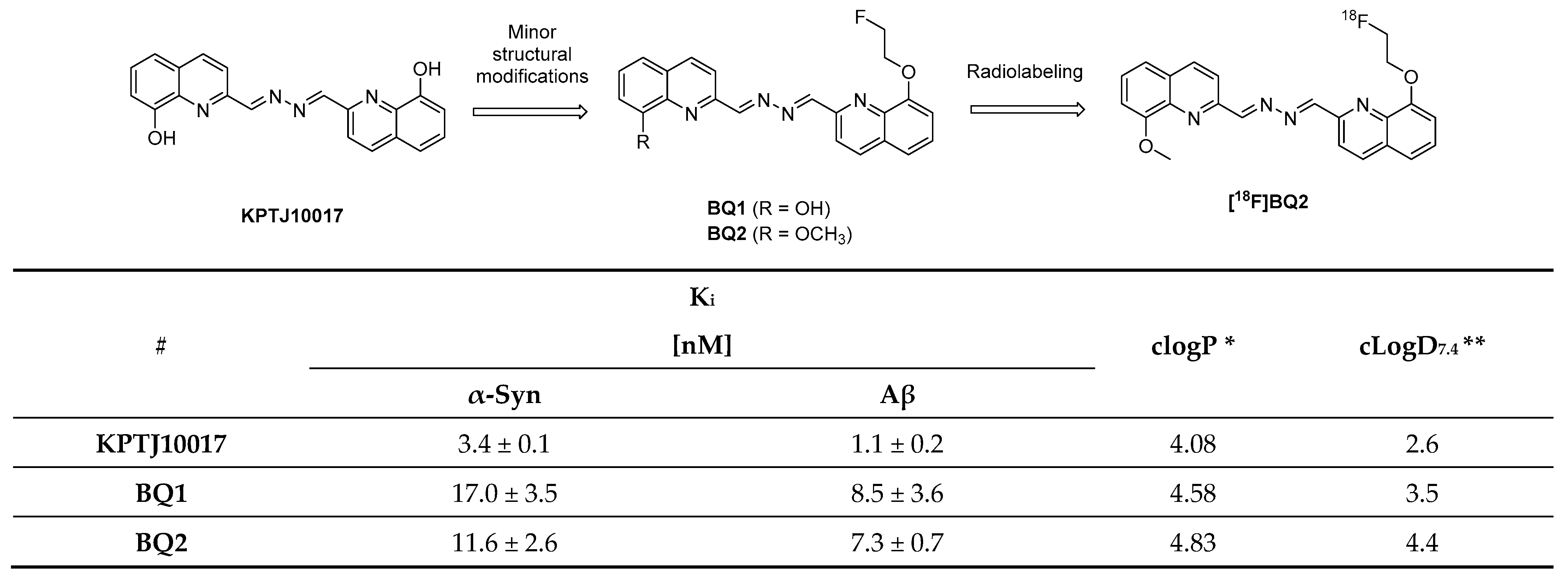
3.14. Bisquinolines
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McCann, H.; Stevens, C.; Cartwright, H.; Halliday, G. α-Synucleinopathy phenotypes. Park. Relat. Disord. 2014, 20, S62–S67. [Google Scholar] [CrossRef] [Green Version]
- De Rijk, M.C.; Tzourio, C.; Breteler, M.M.; Dartigues, J.F.; Amaducci, L.; Lopez-Pousa, S.; Manubens-Bertran, J.M.; Alperovitch, A.; Rocca, W.A. Prevalence of parkinsonism and Parkinson’s disease in Europe: The EUROPARKINSON Collaborative Study. European Community Concerted Action on the Epidemiology of Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 1997, 62, 10–15. [Google Scholar] [CrossRef] [Green Version]
- Tanner, C.M.; Goldman, S. EPIDEMIOLOGY OF PARKINSON’S DISEASE. Neurol. Clin. 1996, 14, 317–335. [Google Scholar] [CrossRef]
- Dorsey, E.R.; Constantinescu, R.; Thompson, J.P.; Biglan, K.M.; Holloway, R.G.; Kieburtz, K.; Marshall, F.J.; Ravina, B.M.; Schifitto, G.; Siderowf, A.; et al. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology 2007, 68, 384. [Google Scholar] [CrossRef] [PubMed]
- Statistics. Parkinson’s Foundation. Available online: https://www.parkinson.org/Understanding-Parkinsons/Statistics (accessed on 1 May 2021).
- Loane, C.; Politis, M. Positron emission tomography neuroimaging in Parkinson’s disease. Am. J. Transl. Res. 2011, 3, 323–341. [Google Scholar]
- Eberling, J.L.; Dave, K.D.; Frasier, M.A. α-synuclein Imaging: A Critical Need for Parkinson’s Disease Research. J. Park. Dis. 2013, 3, 565–567. [Google Scholar] [CrossRef] [Green Version]
- Winge, K.; Friberg, L.; Werdelin, L.; Nielsen, K.K.; Stimpel, H. Relationship between nigrostriatal dopaminergic degeneration, urinary symptoms, and bladder control in Parkinson’s disease. Eur. J. Neurol. 2005, 12, 842–850. [Google Scholar] [CrossRef]
- Parkinson Study Group. Dopamine Transporter Brain Imaging to Assess the Effects of Pramipexole vs Levodopa on Parkinson Disease Progression. JAMA 2002, 287, 1653–1661. [Google Scholar] [CrossRef]
- Iwai, A.; Masliah, E.; Yoshimoto, M.; Ge, N.; Flanagan, L.; de Silva, H.R.; Kittel, A.; Saitoh, T. The precursor protein of non-Aβ component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995, 14, 467–475. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Li, X.; Liu, G.; Han, J.; Zhang, C.; Li, Y.; Xu, S.; Liu, C.; Gao, Y.; Yang, H.; et al. Extensive nuclear localization of α-synuclein in normal rat brain neurons revealed by a novel monoclonal antibody. Neuroscience 2007, 145, 539–555. [Google Scholar] [CrossRef]
- Lee, H.-J.; Choi, C.; Lee, S.-J. Membrane-bound α-Synuclein Has a High Aggregation Propensity and the Ability to Seed the Aggregation of the Cytosolic Form. J. Biol. Chem. 2002, 277, 671–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, P.J.; Kawamata, H.; Ribich, S.; Hyman, B.T. Membrane association and protein conformation of alpha-synuclein in intact neurons. Effect of Parkinson’s disease-linked mutations. J. Biol. Chem. 2000, 275, 8812–8816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendor, J.; Logan, T.P.; Edwards, R.H. The Function of α-Synuclein. Neuron 2013, 79, 1044–1066. [Google Scholar] [CrossRef] [Green Version]
- George, J.M.; Jin, H.; Woods, W.S.; Clayton, D.F. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron 1995, 15, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Zeniou-Meyer, M.; Zabari, N.; Ashery, U.; Chasserot-Golaz, S.; Haeberlé, A.-M.; Demais, V.; Bailly, Y.; Gottfried, I.; Nakanishi, H.; Neiman, A.; et al. Phospholipase D1 Production of Phosphatidic Acid at the Plasma Membrane Promotes Exocytosis of Large Dense-core Granules at a Late Stage. J. Biol. Chem. 2007, 282, 21746–21757. [Google Scholar] [CrossRef] [Green Version]
- Lotharius, J.; Brundin, P. Pathogenesis of parkinson’s disease: Dopamine, vesicles and α-synuclein. Nat. Rev. Neurosci. 2002, 3, 932–942. [Google Scholar] [CrossRef]
- Schaser, A.J.; Osterberg, V.R.; Dent, S.E.; Stackhouse, T.L.; Wakeham, C.M.; Boutros, S.W.; Weston, L.J.; Owen, N.; Weissman, T.A.; Luna, E.; et al. Alpha-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci. Rep. 2019, 9, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M.; Jakes, R.; Spillantini, M.G. The Synucleinopathies: Twenty Years On. J. Park. Dis. 2017, 7, S51–S69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arima, K.; Uéda, K.; Sunohara, N.; Arakawa, K.; Hirai, S.; Nakamura, M.; Tonozuka-Uehara, H.; Kawai, M. NACP/α-synuclein immunoreactivity in fibrillary components of neuronal and oligodendroglial cytoplasmic inclusions in the pontine nuclei in multiple system atrophy. Acta Neuropathol. 1998, 96, 439–444. [Google Scholar] [CrossRef] [PubMed]
- Crowther, R.A.; Jakes, R.; Spillantini, M.G.; Goedert, M. Synthetic filaments assembled from C-terminally truncated α-synuclein. FEBS Lett. 1998, 436, 309–312. [Google Scholar] [CrossRef] [Green Version]
- Valera, E.; Masliah, E. The neuropathology of multiple system atrophy and its therapeutic implications. Auton. Neurosci. 2018, 211, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compagnoni, G.M.; Di Fonzo, A. Understanding the pathogenesis of multiple system atrophy: State of the art and future perspectives. Acta Neuropathol. Commun. 2019, 7, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Perren, A.; Gelders, G.; Fenyi, A.; Bousset, L.; Brito, F.; Peelaerts, W.; Van den Haute, C.; Gentleman, S.; Melki, R.; Baekelandt, V. The structural differences between patient-derived α-synuclein strains dictate characteristics of Parkinson’s disease, multiple system atrophy and dementia with Lewy bodies. Acta Neuropathol. 2020, 139, 977–1000. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Pu, J. Alpha-Synuclein in Parkinson’s Disease: From Pathogenetic Dysfunction to Potential Clinical Application. Park. Dis. 2016, 2016, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In vivo demonstration that -synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef] [Green Version]
- Stefanis, L. α-Synuclein in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009399. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.C.; Krainc, D. α-synuclein toxicity in neurodegeneration: Mechanism and therapeutic strategies. Nat. Med. 2017, 23, 1–13. [Google Scholar] [CrossRef]
- Danzer, K.M.; Haasen, D.; Karow, A.R.; Moussaud, S.; Habeck, M.; Giese, A.; Kretzschmar, H.; Hengerer, B.; Kostka, M. Different Species of α-Synuclein Oligomers Induce Calcium Influx and Seeding. J. Neurosci. 2007, 27, 9220–9232. [Google Scholar] [CrossRef]
- Scott, D.A.; Tabarean, I.; Tang, Y.; Cartier, A.; Masliah, E.; Roy, S. A Pathologic Cascade Leading to Synaptic Dysfunction in α-Synuclein-Induced Neurodegeneration. J. Neurosci. 2010, 30, 8083–8095. [Google Scholar] [CrossRef]
- Ingelsson, M. Alpha-Synuclein Oligomers—Neurotoxic Molecules in Parkinson’s Disease and Other Lewy Body Disorders. Front. Neurosci. 2016, 10, 408. [Google Scholar] [CrossRef] [Green Version]
- Bengoa-Vergniory, N.; Roberts, R.F.; Wade-Martins, R.; Alegre-Abarrategui, J. Alpha-synuclein oligomers: A new hope. Acta Neuropathol. 2017, 134, 819–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, P.; Bousset, L.; Melki, R.; Otzen, D.E. α-synuclein oligomers and fibrils: A spectrum of species, a spectrum of toxicities. J. Neurochem. 2019, 150, 522–534. [Google Scholar] [CrossRef] [Green Version]
- Fauvet, B.; Mbefo, M.K.; Fares, M.B.; Desobry, C.; Michael, S.; Ardah, M.T.; Tsika, E.; Coune, P.; Prudent, M.; Lion, N.; et al. α-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J. Biol. Chem. 2012, 287, 15345–15364. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Perovic, I.; Chittuluru, J.; Kaganovich, A.; Nguyen, L.T.T.; Liao, J.; Auclair, J.R.; Johnson, D.; Landeru, A.; Simorellis, A.K.; et al. A soluble α-synuclein construct forms a dynamic tetramer. Proc. Natl. Acad. Sci. USA 2011, 108, 17797–17802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miraglia, F.; Ricci, A.; Rota, L.; Colla, E. Subcellular localization of alpha-synuclein aggregates and their interaction with membranes. Neural Regen. Res. 2018, 13, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavedan, C. The synuclein family. Genome Res. 1998, 8, 871–880. [Google Scholar] [CrossRef] [Green Version]
- Beyer, K. α-Synuclein structure, posttranslational modification and alternative splicing as aggregation enhancers. Acta Neuropathol. 2006, 112, 237–251. [Google Scholar] [CrossRef]
- Zhang, J.; Li, X.; Li, J.-D. The Roles of Post-translational Modifications on α-Synuclein in the Pathogenesis of Parkinson’s Diseases. Front. Neurosci. 2019, 13, 381. [Google Scholar] [CrossRef] [Green Version]
- Menéndez-González, M.; Padilla-Zambrano, H.S.; Tomás-Zapico, C.; García, B.F. Clearing Extracellular Alpha-Synuclein from Cerebrospinal Fluid: A New Therapeutic Strategy in Parkinson’s Disease. Brain Sci. 2018, 8, 52. [Google Scholar] [CrossRef] [Green Version]
- Twohig, D.; Nielsen, H.M. α-synuclein in the pathophysiology of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 23. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, C.-J.; Ferrie, J.J.; Xu, K.; Lee, I.; Graham, T.J.A.; Tu, Z.; Yu, J.; Dhavale, D.; Kotzbauer, P.; Petersson, E.J.; et al. Alpha Synuclein Fibrils Contain Multiple Binding Sites for Small Molecules. ACS Chem. Neurosci. 2018, 9, 2521–2527. [Google Scholar] [CrossRef]
- Wassouf, Z.; Schulze-Hentrich, J.M. Alpha-synuclein at the nexus of genes and environment: The impact of environmental enrichment and stress on brain health and disease. J. Neurochem. 2019, 150, 591–604. [Google Scholar] [CrossRef]
- Conway, K.A.; Lee, S.-J.; Rochet, J.-C.; Ding, T.T.; Harper, J.D.; Williamson, R.E.; Lansbury, P.T. Accelerated Oligomerization by Parkinson’s Disease Linked α-Synuclein Mutants. Ann. N. Y. Acad. Sci. 2006, 920, 42–45. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Murray, I.V.; Trojanowski, J.Q.; Lee, V.M. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J. Biol. Chem. 2001, 276, 2380–2386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, H.-N.; Tang, L.; Luo, X.-Y.; Li, H.-T.; Hu, J.; Zhou, J.-W.; Hu, H.-Y. A Peptide Motif Consisting of Glycine, Alanine, and Valine Is Required for the Fibrillization and Cytotoxicity of Human α-Synuclein. Biochemistry 2003, 42, 8870–8878. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Meade, R.M.; Fairlie, D.P.; Mason, J.M. Alpha-synuclein structure and Parkinson’s disease—Lessons and emerging principles. Mol. Neurodegener. 2019, 14, 29. [Google Scholar] [CrossRef] [Green Version]
- Tuttle, M.D.; Comellas, G.; Nieuwkoop, A.J.; Covell, D.J.; Berthold, D.A.; Kloepper, K.D.; Courtney, J.M.; Kim, J.K.; Barclay, A.M.; Kendall, A.; et al. Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat. Struct. Mol. Biol. 2016, 23, 409–415. [Google Scholar] [CrossRef]
- Guerrero-Ferreira, R.; Taylor, N.M.I.; Mona, D.; Ringler, P.; Lauer, M.E.; Riek, R.; Britschgi, M.; Stahlberg, H. Cryo-EM structure of alpha-synuclein fibrils. eLife 2018, 7, e36402. [Google Scholar] [CrossRef]
- Li, B.; Ge, P.; Murray, K.A.; Sheth, P.; Zhang, M.; Nair, G.; Sawaya, M.R.; Shin, W.S.; Boyer, D.R.; Ye, S.; et al. Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel. Nat. Commun. 2018, 9, 3609. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, C.; Luo, F.; Liu, Z.; Gui, X.; Luo, Z.; Zhang, X.; Li, D.; Liu, C.; Li, X. Amyloid fibril structure of α-synuclein determined by cryo-electron microscopy. Cell Res. 2018, 28, 897–903. [Google Scholar] [CrossRef] [Green Version]
- Mathis, C.A.; Lopresti, B.J.; Ikonomovic, M.D.; Klunk, W.E. Small-molecule PET Tracers for Imaging Proteinopathies. Semin. Nucl. Med. 2017, 47, 553–575. [Google Scholar] [CrossRef]
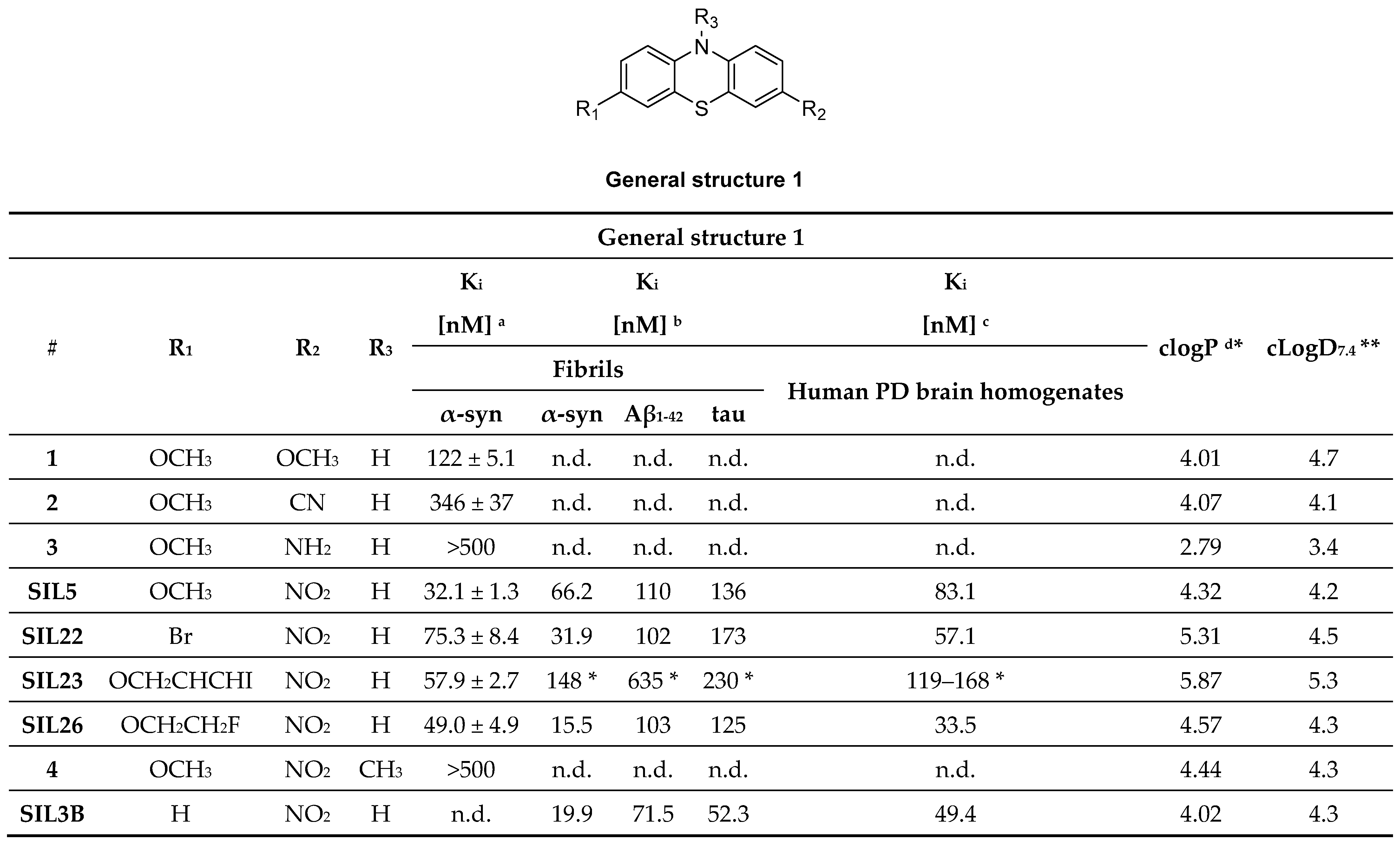
- Bagchi, D.P.; Yu, L.; Perlmutter, J.S.; Xu, J.; Mach, R.H.; Tu, Z.; Kotzbauer, P.T. Binding of the Radioligand SIL23 to α-Synuclein Fibrils in Parkinson Disease Brain Tissue Establishes Feasibility and Screening Approaches for Developing a Parkinson Disease Imaging Agent. PLoS ONE 2013, 8, e55031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, M.; Seibyl, J.; Cartier, A.; Bhatt, R.; Catafau, A.M. Molecular Imaging Insights into Neurodegeneration: Focus on α-Synuclein Radiotracers. J. Nucl. Med. 2014, 55, 1397–1400. [Google Scholar] [CrossRef] [Green Version]
- Herth, M.M.; Knudsen, G.M. PET imaging of the 5-HT2A receptor system: A tool to study the receptor’s in vivo brain function. In 5-HT2A Receptors in the Central Nervous System; Guiard, B.P., Di Giovanni, G., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 85–134. [Google Scholar]
- L’Estrade, E.T.; Erlandsson, M.; Edgar, F.G.; Ohlsson, T.; Knudsen, G.M.; Herth, M.M. Towards selective CNS PET imaging of the 5-HT7 receptor system: Past, present and future. Neuropharmacology 2020, 172, 107830. [Google Scholar] [CrossRef]
- Sun, W.; Ginovart, N.; Ko, F.; Seeman, P.; Kapur, S. In Vivo Evidence for Dopamine-Mediated Internalization of D2-Receptors after Amphetamine: Differential findings with [3H] raclopride versus [3H] spiperone. Mol. Pharmacol. 2003, 63, 456–462. [Google Scholar] [CrossRef] [PubMed]
- Kuang, G.; Murugan, N.A.; Zhou, Y.; Nordberg, A.; Ågren, H. Computational Insight into the Binding Profile of the Second-Generation PET Tracer PI2620 with Tau Fibrils. ACS Chem. Neurosci. 2020, 11, 900–908. [Google Scholar] [CrossRef]
- Murugan, N.A.; Nordberg, A.; Ågren, H. Different Positron Emission Tomography Tau Tracers Bind to Multiple Binding Sites on the Tau Fibril: Insight from Computational Modeling. ACS Chem. Neurosci. 2018, 9, 1757–1767. [Google Scholar] [CrossRef] [Green Version]
- Strohäker, T.; Jung, B.C.; Liou, S.-H.; Fernandez, C.O.; Riedel, D.; Becker, S.; Halliday, G.M.; Bennati, M.; Kim, W.S.; Lee, S.-J.; et al. Structural heterogeneity of α-synuclein fibrils amplified from patient brain extracts. Nat. Commun. 2019, 10, 5535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahnawaz, M.; Mukherjee, A.; Pritzkow, S.; Mendez, N.; Rabadia, P.; Liu, X.; Hu, B.; Schmeichel, A.; Singer, W.; Wu, G.; et al. Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 2020, 578, 273–277. [Google Scholar] [CrossRef]
- Peelaerts, W.; Bousset, L.; Van der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; Van den Haute, C.; Melki, R.; Baekelandt, V. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2015, 522, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Thakur, P.; Breger, L.S.; Lundblad, M.; Wan, O.W.; Mattsson, B.; Luk, K.C.; Lee, V.M.Y.; Trojanowski, J.Q.; Björklund, A. Modeling Parkinson’s disease pathology by combination of fibril seeds and α-synuclein overexpression in the rat brain. Proc. Natl. Acad. Sci. USA 2017, 114, E8284–E8293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espa, E.; Clemensson, E.K.H.; Luk, K.C.; Heuer, A.; Björklund, T.; Cenci, M.A. Seeding of protein aggregation causes cognitive impairment in rat model of cortical synucleinopathy. Mov. Disord. 2019, 34, 1699–1710. [Google Scholar] [CrossRef] [PubMed]
- Verdurand, M.; Levigoureux, E.; Zeinyeh, W.; Berthier, L.; Mendjel-Herda, M.; Cadarossanesaib, F.; Bouillot, C.; Iecker, T.; Terreux, R.; Lancelot, S.; et al. In Silico, in Vitro, and in Vivo Evaluation of New Candidates for α-Synuclein PET Imaging. Mol. Pharm. 2018, 15, 3153–3166. [Google Scholar] [CrossRef]
- Recasens, A.; Dehay, B.; Bové, J.; Carballo-Carbajal, I.; Dovero, S.; Pérez-Villalba, A.; Fernagut, P.-O.; Blesa, J.; Parent, A.; Perier, C.; et al. Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Ann. Neurol. 2014, 75, 351–362. [Google Scholar] [CrossRef]
- Shimozawa, A.; Ono, M.; Takahara, D.; Tarutani, A.; Imura, S.; Masuda-Suzukake, M.; Higuchi, M.; Yanai, K.; Hisanaga, S.-I.; Hasegawa, M. Propagation of pathological α-synuclein in marmoset brain. Acta Neuropathol. Commun. 2017, 5, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirik, D.; Annett, L.E.; Burger, C.; Muzyczka, N.; Mandel, R.J.; Björklund, A. Nigrostriatal alpha-synucleinopathy induced by viral vector-mediated overexpression of human alpha-synuclein: A new primate model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 2884–2889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holm, I.E.; Alstrup, A.K.O.; Luo, Y. Genetically modified pig models for neurodegenerative disorders. J. Pathol. 2016, 238, 267–287. [Google Scholar] [CrossRef]
- Lillethorup, T.P.; Glud, A.N.; Landeck, N.; Alstrup, A.K.O.; Jakobsen, S.; Vang, K.; Doudet, D.J.; Brooks, D.J.; Kirik, D.; Hinz, R.; et al. In vivo quantification of glial activation in minipigs overexpressing human α-synuclein. Synapse 2018, 72, e22060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristensen, J.L.; Herth, M.M. In vivo imaging in drug discovery. In Textbook of Drug Design and Discovery; Stromgaard, K., Krogsgaard-Larsen, P., Madsen, U., Eds.; CRC Press: Boca Raton, FL, USA, 2017. [Google Scholar]
- Pike, V.W. Considerations in the Development of Reversibly Binding PET Radioligands for Brain Imaging. Curr. Med. Chem. 2016, 23, 1818–1869. [Google Scholar] [CrossRef]
- Herth, M.M.; Ametamey, S.; Antuganov, D.; Bauman, A.; Berndt, M.; Brooks, A.F.; Bormans, G.; Choe, Y.S.; Gillings, N.; Häfeli, U.O.; et al. On the consensus nomenclature rules for radiopharmaceutical chemistry – Reconsideration of radiochemical conversion. Nucl. Med. Biol. 2020, 93, 19–21. [Google Scholar] [CrossRef]
- McCluskey, S.P.; Plisson, C.; Rabiner, E.A.; Howes, O. Advances in CNS PET: The state-of-the-art for new imaging targets for pathophysiology and drug development. Eur. J. Nucl. Med. Mol. Imaging 2019, 47, 451–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, L.; Lu, S.; Pike, V.W. Chemistry with [18 F]Fluoride Ion. Eur. J. Org. Chem. 2008, 2008, 2853–2873. [Google Scholar] [CrossRef]
- Zhang, L.; Villalobos, A.; Beck, E.M.; Bocan, T.; Chappie, T.A.; Chen, L.; Grimwood, S.; Heck, S.D.; Helal, C.J.; Hou, X.; et al. Design and Selection Parameters to Accelerate the Discovery of Novel Central Nervous System Positron Emission Tomography (PET) Ligands and Their Application in the Development of a Novel Phosphodiesterase 2A PET Ligand. J. Med. Chem. 2013, 56, 4568–4579. [Google Scholar] [CrossRef] [PubMed]
- Alpha-Synuclein Imaging Prize|Parkinson’s Disease. Available online: https://www.michaeljfox.org/news/alpha-synuclein-imaging-prize (accessed on 16 May 2021).
- Fridén, M.; Wennerberg, M.; Antonsson, M.; Sandberg-Ställ, M.; Farde, L.; Schou, M. Identification of positron emission tomography (PET) tracer candidates by prediction of the target-bound fraction in the brain. EJNMMI Res. 2014, 4, 50. [Google Scholar] [CrossRef] [Green Version]
- Kotzbauer, P.T.; Tu, Z.; Mach, R.H. Current status of the development of PET radiotracers for imaging alpha synuclein aggregates in Lewy bodies and Lewy neurites. Clin. Transl. Imaging 2016, 5, 3–14. [Google Scholar] [CrossRef]
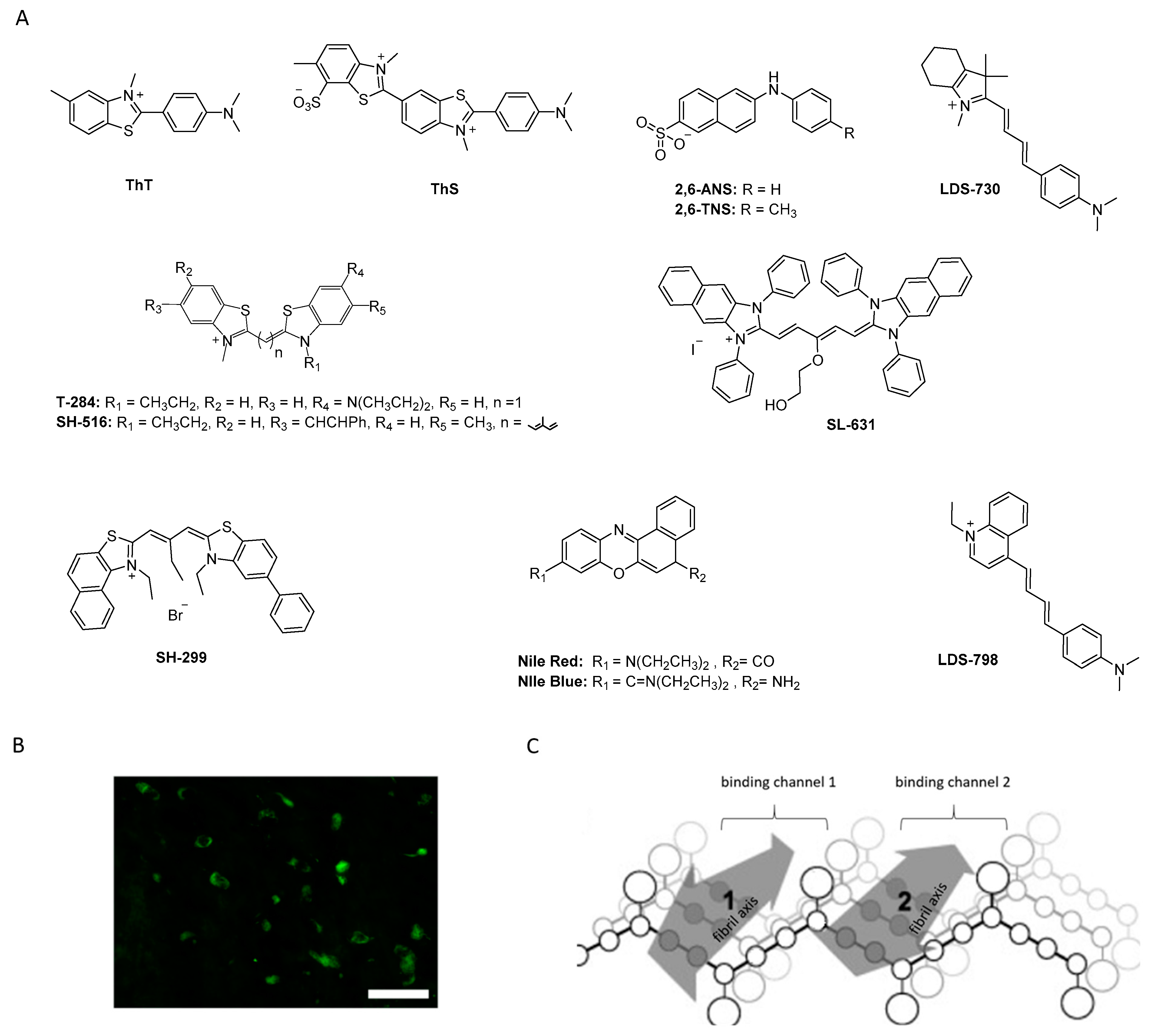
- Celej, M.S.; Jares-Erijman, E.A.; Jovin, T.M. Fluorescent N-Arylaminonaphthalene Sulfonate Probes for Amyloid Aggregation of α-Synuclein. Biophys. J. 2008, 94, 4867–4879. [Google Scholar] [CrossRef] [Green Version]
- Volkova, K.D.; Kovalska, V.B.; Balanda, A.O.; Losytskyy, M.Y.; Golub, A.G.; Vermeij, R.J.; Subramaniam, V.; Tolmachev, O.I.; Yarmoluk, S.M. Specific fluorescent detection of fibrillar α-synuclein using mono- and trimethine cyanine dyes. Bioorg. Med. Chem. 2008, 16, 1452–1459. [Google Scholar] [CrossRef] [PubMed]
- Krebs, M.R.H.; Bromley, E.H.C.; Donald, A.M. The binding of thioflavin-T to amyloid fibrils: Localisation and implications. J. Struct. Biol. 2005, 149, 30–37. [Google Scholar] [CrossRef]
- Volkova, K.D.; Kovalska, V.B.; Losytskyy, M.Y.; Veldhuis, G.; Segers-Nolten, G.M.J.; Tolmachev, O.I.; Subramaniam, V.; Yarmoluk, S.M. Studies of Interaction Between Cyanine Dye T-284 and Fibrillar Alpha-Synuclein. J. Fluoresc. 2010, 20, 1267–1274. [Google Scholar] [CrossRef]
- Kovalska, V.B.; Losytskyy, M.Y.; Tolmachev, O.I.; Slominskii, Y.L.; Segers-Nolten, G.M.J.; Subramaniam, V.; Yarmoluk, S.M. Tri- and Pentamethine Cyanine Dyes for Fluorescent Detection of α-Synuclein Oligomeric Aggregates. J. Fluoresc. 2012, 22, 1441–1448. [Google Scholar] [CrossRef]
- Neal, K.L.; Shakerdge, N.B.; Hou, S.S.; Klunk, W.E.; Mathis, C.A.; Nesterov, E.E.; Swager, T.M.; McLean, P.J.; Bacskai, B.J. Development and Screening of Contrast Agents for In Vivo Imaging of Parkinson’s Disease. Mol. Imaging Biol. 2013, 15, 585–595. [Google Scholar] [CrossRef] [Green Version]
- Patterson, J.R.; Polinski, N.K.; Duffy, M.F.; Kemp, C.J.; Luk, K.C.; Volpicelli-Daley, L.A.; Kanaan, N.M.; Sortwell, C.E. Generation of Alpha-Synuclein Preformed Fibrils from Monomers and Use In Vivo. J. Vis. Exp. 2019, 148, e59758. [Google Scholar] [CrossRef]
- Hajieva, P.; Mocko, J.B.; Moosmann, B.; Behl, C. Novel imine antioxidants at low nanomolar concentrations protect dopaminergic cells from oxidative neurotoxicity. J. Neurochem. 2009, 110, 118–132. [Google Scholar] [CrossRef]
- Masuda, M.; Suzuki, N.; Taniguchi, S.; Oikawa, T.; Nonaka, T.; Iwatsubo, T.; Hisanaga, S.-i.; Goedert, M.; Hasegawa, M. Small Molecule Inhibitors of α-Synuclein Filament Assembly. Biochemistry 2006, 45, 6085–6094. [Google Scholar] [CrossRef]
- Schwab, K.; Frahm, S.; Horsley, D.; Rickard, J.E.; Melis, V.; Goatman, E.A.; Magbagbeolu, M.; Douglas, M.; Leith, M.G.; Baddeley, T.C.; et al. A Protein Aggregation Inhibitor, Leuco-Methylthioninium Bis(Hydromethanesulfonate), Decreases α-Synuclein Inclusions in a Transgenic Mouse Model of Synucleinopathy. Front. Mol. Neurosci. 2018, 10, 447. [Google Scholar] [CrossRef]
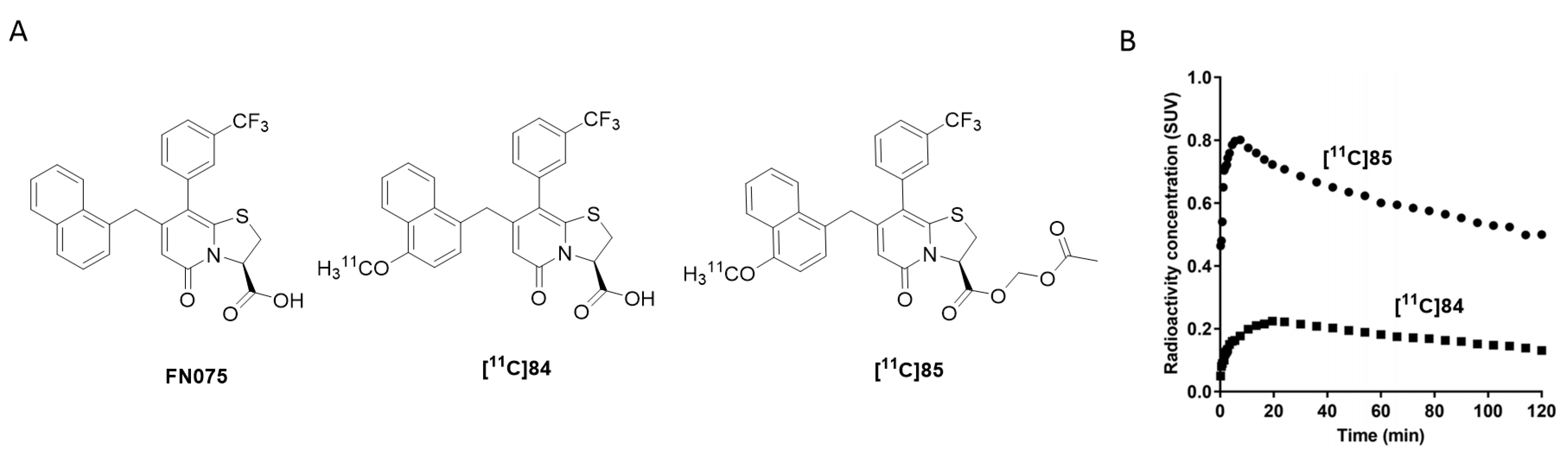
- Yu, L.; Cui, J.; Padakanti, P.K.; Engel, L.; Bagchi, D.P.; Kotzbauer, P.T.; Tu, Z. Synthesis and in vitro evaluation of α-synuclein ligands. Bioorg. Med. Chem. 2012, 20, 4625–4634. [Google Scholar] [CrossRef] [Green Version]
- Fisher, E.M. Development of PET Radiotracers for Imaging Neurodegeneration: Targeting Alpha-Synuclein Fibrils and TSPO. Ph.D. Thesis, University of Cambridge, Cambridge, UK, 2017. [Google Scholar] [CrossRef]
- Tu, Z.; Li, J.; Yue, X.; Kotzbauer, P. Alpha-Synuclein Ligands. U.S. Patent 2017/0189566 A1, 29 December 2016. [Google Scholar]
- Zhang, X.; Jin, H.; Padakanti, P.K.; Li, J.; Yang, H.; Fan, J.; Mach, R.H.; Kotzbauer, P.; Tu, Z. Radiosynthesis and in Vivo Evaluation of Two PET Radioligands for Imaging α-Synuclein. Appl. Sci. 2014, 4, 66–78. [Google Scholar] [CrossRef] [Green Version]
- Pike, V.W. PET radiotracers: Crossing the blood-brain barrier and surviving metabolism. Trends Pharmacol. Sci. 2009, 30, 431–440. [Google Scholar] [CrossRef] [Green Version]
- Tu, Z.; Mach, R.; Yu, L.; Kotzbauer, P. Tricyclic Heteroaromatic Compounds as Alpha-Synuclein Ligands. U.S. Patent Application 2013/0315825 A1, 3 May 2013. [Google Scholar]
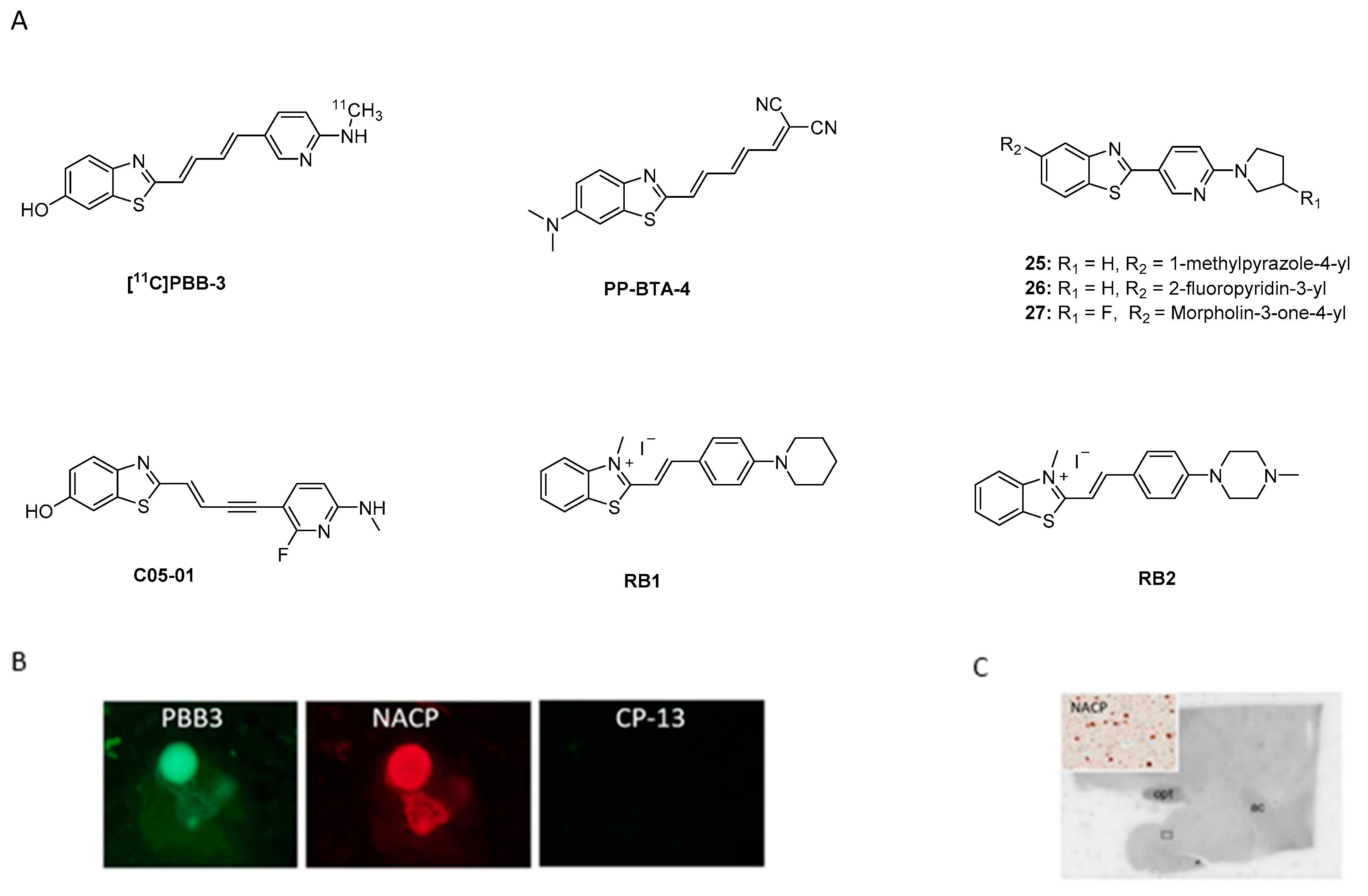
- Kudo, Y.; Okamura, N.; Furumoto, S.; Tashiro, M.; Furukawa, K.; Maruyama, M.; Itoh, M.; Iwata, R.; Yanai, K.; Arai, H. 2-(2-[2-Dimethylaminothiazol-5-yl]Ethenyl)-6- (2-[Fluoro]Ethoxy)Benzoxazole: A Novel PET Agent for In Vivo Detection of Dense Amyloid Plaques in Alzheimer’s Disease Patients. J. Nucl. Med. 2007, 48, 553–561. [Google Scholar] [CrossRef] [Green Version]
- Fodero-Tavoletti, M.T.; Mulligan, R.S.; Okamura, N.; Furumoto, S.; Rowe, C.C.; Kudo, Y.; Masters, C.L.; Cappai, R.; Yanai, K.; Villemagne, V.L. In vitro characterisation of BF227 binding to alpha-synuclein/Lewy bodies. Eur. J. Pharmacol. 2009, 617, 54–58. [Google Scholar] [CrossRef]
- Kikuchi, A.; Takeda, A.; Okamura, N.; Tashiro, M.; Hasegawa, T.; Furumoto, S.; Kobayashi, M.; Sugeno, N.; Baba, T.; Miki, Y.; et al. In vivo visualization of α-synuclein deposition by carbon-11-labelled 2-[2-(2-dimethylaminothiazol-5-yl)ethenyl]-6-[2-(fluoro)ethoxy]benzoxazole positron emission tomography in multiple system atrophy. Brain 2010, 133, 1772–1778. [Google Scholar] [CrossRef] [Green Version]
- Verdurand, M.; Levigoureux, E.; Lancelot, S.; Zeinyeh, W.; Billard, T.; Quadrio, I.; Perret-Liaudet, A.; Zimmer, L.; Chauveau, F. Amyloid-Beta Radiotracer [18F]BF-227 Does Not Bind to Cytoplasmic Glial Inclusions of Postmortem Multiple System Atrophy Brain Tissue. Contrast Med. Mol. Imaging 2018, 2018, 9165458. [Google Scholar] [CrossRef] [Green Version]
- Levigoureux, E.; Lancelot, S.; Bouillot, C.; Chauveau, F.; Verdurand, M.; Verchère, J.; Billard, T.; Baron, T.; Zimmer, L. 18F]BF227 Does not Reflect the Presence of Alpha-Synuclein Aggregates in Transgenic Mice. Curr. Alzheimer’s Res. 2014, 11, 955–960. [Google Scholar] [CrossRef]
- Josephson, L.; Stratman, N.; Liu, Y.; Qian, F.; Liang, S.H.; Vasdev, N.; Patel, S. The Binding of BF-227-Like Benzoxazoles to Human α-Synuclein and Amyloid β Peptide Fibrils. Mol. Imaging 2018, 17, 1536012118796297. [Google Scholar] [CrossRef]
- Honson, N.S.; Johnson, R.L.; Huang, W.; Inglese, J.; Austin, C.P.; Kuret, J. Differentiating Alzheimer disease-associated aggregates with small molecules. Neurobiol. Dis. 2007, 28, 251–260. [Google Scholar] [CrossRef] [Green Version]
- Chu, W.; Zhou, D.; Gaba, V.; Liu, J.; Li, S.; Peng, X.; Xu, J.; Dhavale, D.; Bagchi, D.P.; d’Avignon, A.; et al. Design, Synthesis, and Characterization of 3-(Benzylidene)indolin-2-one Derivatives as Ligands for α-Synuclein Fibrils. J. Med. Chem. 2015, 58, 6002–6017. [Google Scholar] [CrossRef] [Green Version]
- Hefti Franz, F.; Golding, G.; Li, X.; Choi, S.-R.; Esposito, L.; Yadon, M.-C.; Cummings, J.; Hudson, F.M.; Lake, T.; Snow Alan, D. Compounds for Use in the Detection of Neurodegenerative Diseases. U.S. Patent 2012/0251448 A1, 2 March 2012. [Google Scholar]
- Wester, H.-J.; Yousefi Behrooz, H. Compounds Binding to Neuropathological Aggregates. U.S. Patent WO 2016/001422 A1, 3 July 2015. [Google Scholar]
- Annual Congress of the European Association of Nuclear Medicine October 12–16, 2019, Barcelona, Spain. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1–952. [CrossRef]
- Maruyama, M.; Shimada, H.; Suhara, T.; Shinotoh, H.; Ji, B.; Maeda, J.; Zhang, M.-R.; Trojanowski, J.Q.; Lee, V.M.-Y.; Ono, M.; et al. Imaging of Tau Pathology in a Tauopathy Mouse Model and in Alzheimer Patients Compared to Normal Controls. Neuron 2013, 79, 1094–1108. [Google Scholar] [CrossRef] [Green Version]
- Ni, R.; Ji, B.; Ono, M.; Sahara, N.; Zhang, M.R.; Aoki, I.; Nordberg, A.; Suhara, T.; Higuchi, M. Comparative In Vitro and In Vivo Quantifications of Pathologic Tau Deposits and Their Association with Neurodegeneration in Tauopathy Mouse Models. J. Nucl. Med. 2018, 59, 960–966. [Google Scholar] [CrossRef] [Green Version]
- Koga, S.; Ono, M.; Sahara, N.; Higuchi, M.; Dickson, D.W. Fluorescence and autoradiographic evaluation of tau PET ligand PBB3 to α-synuclein pathology. Mov. Disord. 2017, 32, 884–892. [Google Scholar] [CrossRef]
- Watanabe, H.; Ono, M.; Ariyoshi, T.; Katayanagi, R.; Saji, H. Novel Benzothiazole Derivatives as Fluorescent Probes for Detection of β-Amyloid and α-Synuclein Aggregates. ACS Chem. Neurosci. 2017, 8, 1656–1662. [Google Scholar] [CrossRef]
- Molette, J.; Gabellieri, E.; Darmency, V. Bicyclic Compounds for Diagnosis and Therapy. U.S. Patent 2019/0071450 A1, 10 March 2017. [Google Scholar]
- Miranda-Azpiazu, P.; Svedberg, M.; Higuchi, M.; Ono, M.; Jia, Z.; Sunnemark, D.; Elmore, C.S.; Schou, M.; Varrone, A. Identification and in vitro characterization of C05-01, a PBB3 derivative with improved affinity for alpha-synuclein. Brain Res. 2020, 1749, 147131. [Google Scholar] [CrossRef]
- Gaur, P.; Galkin, M.; Kurochka, A.; Ghosh, S.; Yushchenko, D.A.; Shvadchak, V.V. Fluorescent Probe for Selective Imaging of α-Synuclein Fibrils in Living Cells. ACS Chem. Neurosci. 2021, 12, 1293–1298. [Google Scholar] [CrossRef]
- Morais, G.R.; Miranda, H.V.; Santos, I.C.; Outeiro, T.F.; Paulo, A.; Santos, I. Synthesis and in vitro evaluation of fluorinated styryl benzazoles as amyloid-probes. Bioorg. Med. Chem. 2011, 19, 7698–7710. [Google Scholar] [CrossRef]
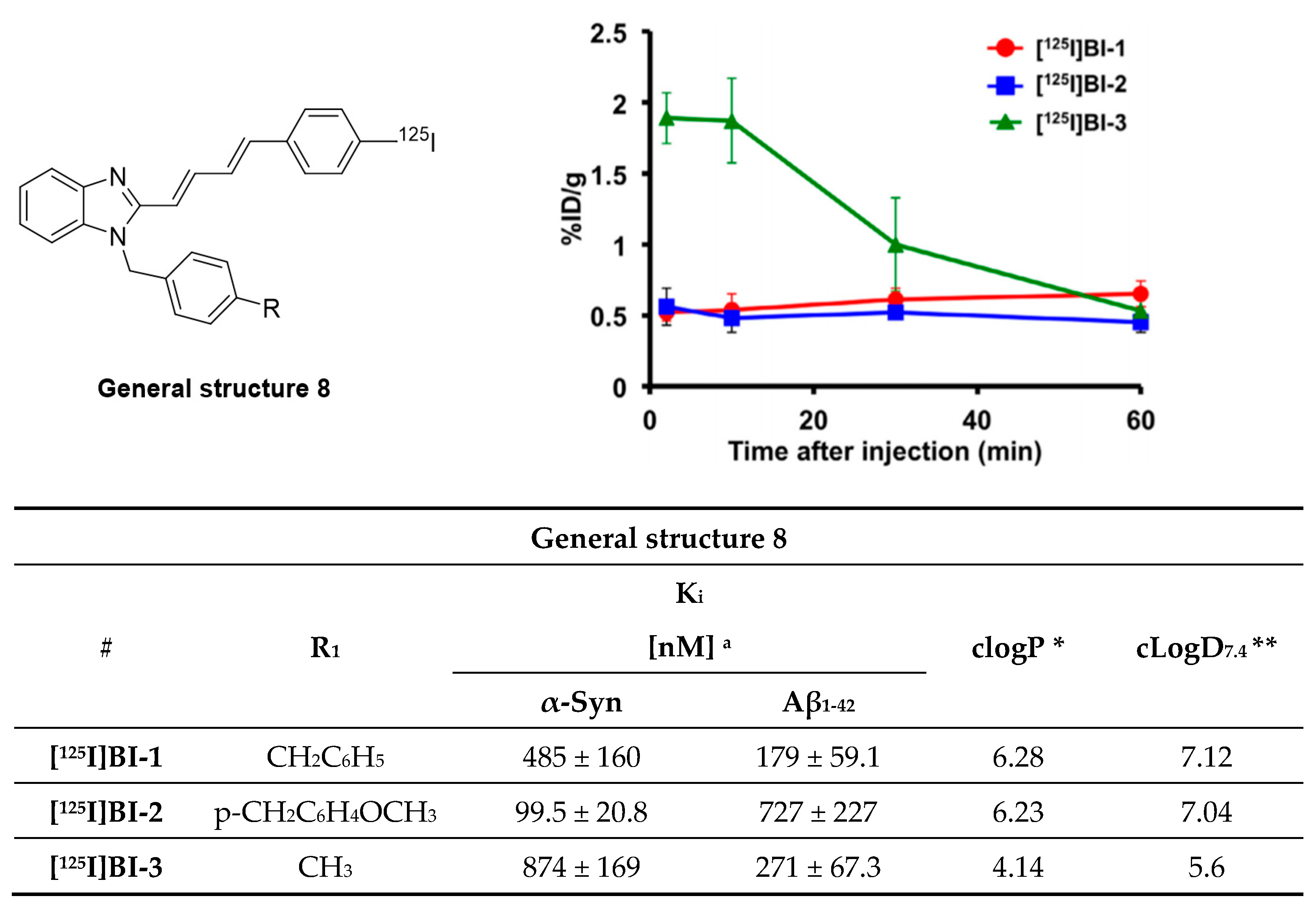
- Watanabe, H.; Ariyoshi, T.; Ozaki, A.; Ihara, M.; Ono, M.; Saji, H. Synthesis and biological evaluation of novel radioiodinated benzimidazole derivatives for imaging α-synuclein aggregates. Bioorg. Med. Chem. 2017, 25, 6398–6403. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, X.; He, Y.; Ding, R.; Liu, H.; Xu, J.; Feng, M.; Li, G.; Wang, M.; Peng, C.; et al. 18F Labeled benzimidazole derivatives as potential radiotracer for positron emission tomography (PET) tumor imaging. Bioorg. Med. Chem. 2010, 18, 2394–2401. [Google Scholar] [CrossRef]
- Routier, S.; Suzenet, F.; Chalon, S.; Buron, F.; Vercouillie, J.; Melki, R.; Boiaryna, L.; Guilloteau, D.; Pieri, L. Compounds For Using In Imaging And Particularly For The Diagnosis Of Neurodegenerative Diseases. U.S. Patent WO 2018/055316 A1, 26 September 2017. [Google Scholar]
- Kaide, S.; Watanabe, H.; Shimizu, Y.; Iikuni, S.; Nakamoto, Y.; Hasegawa, M.; Itoh, K.; Ono, M. Identification and Evaluation of Bisquinoline Scaffold as a New Candidate for α-Synuclein-PET Imaging. ACS Chem. Neurosci. 2020, 11, 4254–4261. [Google Scholar] [CrossRef]
- Wagner, J.; Ryazanov, S.; Leonov, A.; Levin, J.; Shi, S.; Schmidt, F.; Prix, C.; Pan-Montojo, F.; Bertsch, U.; Mitteregger-Kretzschmar, G.; et al. Anle138b: A novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson’s disease. Acta Neuropathol. 2013, 125, 795–813. [Google Scholar] [CrossRef] [Green Version]
- Deeg, A.A.; Reiner, A.M.; Schmidt, F.; Schueder, F.; Ryazanov, S.; Ruf, V.C.; Giller, K.; Becker, S.; Leonov, A.; Griesinger, C.; et al. Anle138b and related compounds are aggregation specific fluorescence markers and reveal high affinity binding to α-synuclein aggregates. Biochim. Biophys. Acta 2015, 1850, 1884–1890. [Google Scholar] [CrossRef] [Green Version]
- Leidel, F.; Eiden, M.; Geissen, M.; Kretzschmar, H.A.; Giese, A.; Hirschberger, T.; Tavan, P.; Schätzl, H.M.; Groschup, M.H. Diphenylpyrazole-derived compounds increase survival time of mice after prion infection. Antimicrob. Agents Chemother. 2011, 55, 4774–4781. [Google Scholar] [CrossRef]
- Wagner, J.; Krauss, S.; Shi, S.; Ryazanov, S.; Steffen, J.; Miklitz, C.; Leonov, A.; Kleinknecht, A.; Göricke, B.; Weishaupt, J.H.; et al. Reducing tau aggregates with anle138b delays disease progression in a mouse model of tauopathies. Acta Neuropathol. 2015, 130, 619–631. [Google Scholar] [CrossRef] [Green Version]
- Fellner, L.; Kuzdas-Wood, D.; Levin, J.; Ryazanov, S.; Leonov, A.; Griesinger, C.; Giese, A.; Wenning, G.K.; Stefanova, N. Anle138b Partly Ameliorates Motor Deficits Despite Failure of Neuroprotection in a Model of Advanced Multiple System Atrophy. Front. Neurosci. 2016, 10, 99. [Google Scholar] [CrossRef]
- Martinez Hernandez, A.; Urbanke, H.; Gillman, A.L.; Lee, J.; Ryazanov, S.; Agbemenyah, H.Y.; Benito, E.; Jain, G.; Kaurani, L.; Grigorian, G.; et al. The diphenylpyrazole compound anle138b blocks Aβ channels and rescues disease phenotypes in a mouse model for amyloid pathology. EMBO Mol. Med. 2018, 10, 32–47. [Google Scholar] [CrossRef] [PubMed]
- Brendel, M.; Deussing, M.; Blume, T.; Kaiser, L.; Probst, F.; Overhoff, F.; Peters, F.; von Ungern-Sternberg, B.; Ryazanov, S.; Leonov, A.; et al. Late-stage Anle138b treatment ameliorates tau pathology and metabolic decline in a mouse model of human Alzheimer’s disease tau. Alzheimer’s Res. Ther. 2019, 11, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
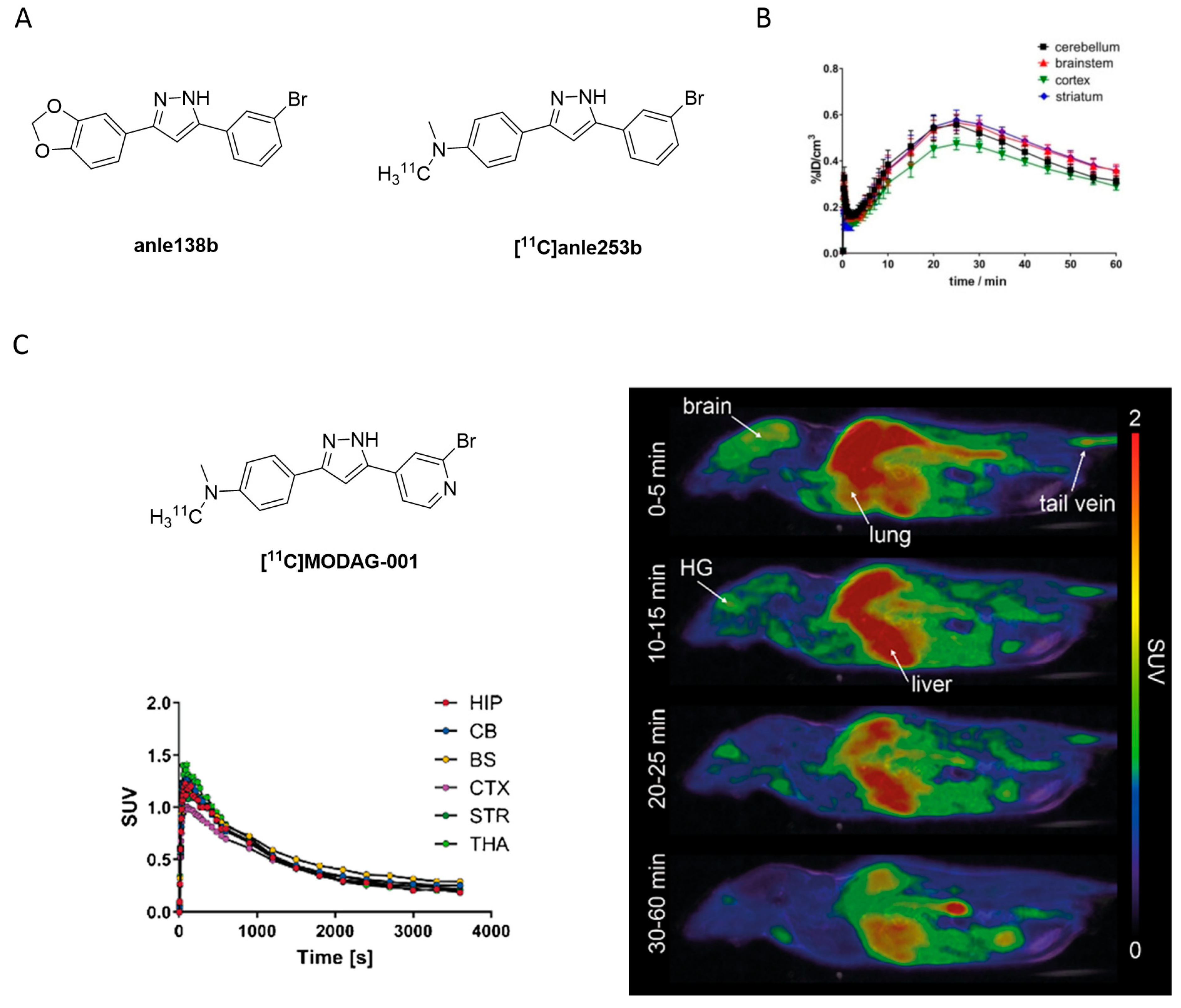
- Maurer, A.; Leonov, A.; Ryazanov, S.; Herfert, K.; Kuebler, L.; Buss, S.; Schmidt, F.; Weckbecker, D.; Linder, R.; Bender, D.; et al. 11C Radiolabeling of anle253b: A Putative PET Tracer for Parkinson’s Disease That Binds to α-Synuclein Fibrils in vitro and Crosses the Blood-Brain Barrier. ChemMedChem 2020, 15, 411–415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuebler, L.; Buss, S.; Leonov, A.; Ryazanov, S.; Schmidt, F.; Maurer, A.; Weckbecker, D.; Landau, A.M.; Lillethorup, T.P.; Bleher, D.; et al. [11C]MODAG-001—towards a PET tracer targeting α-synuclein aggregates. Eur. J. Nucl. Med. Mol. Imaging 2020, 48, 1759–1772. [Google Scholar] [CrossRef]
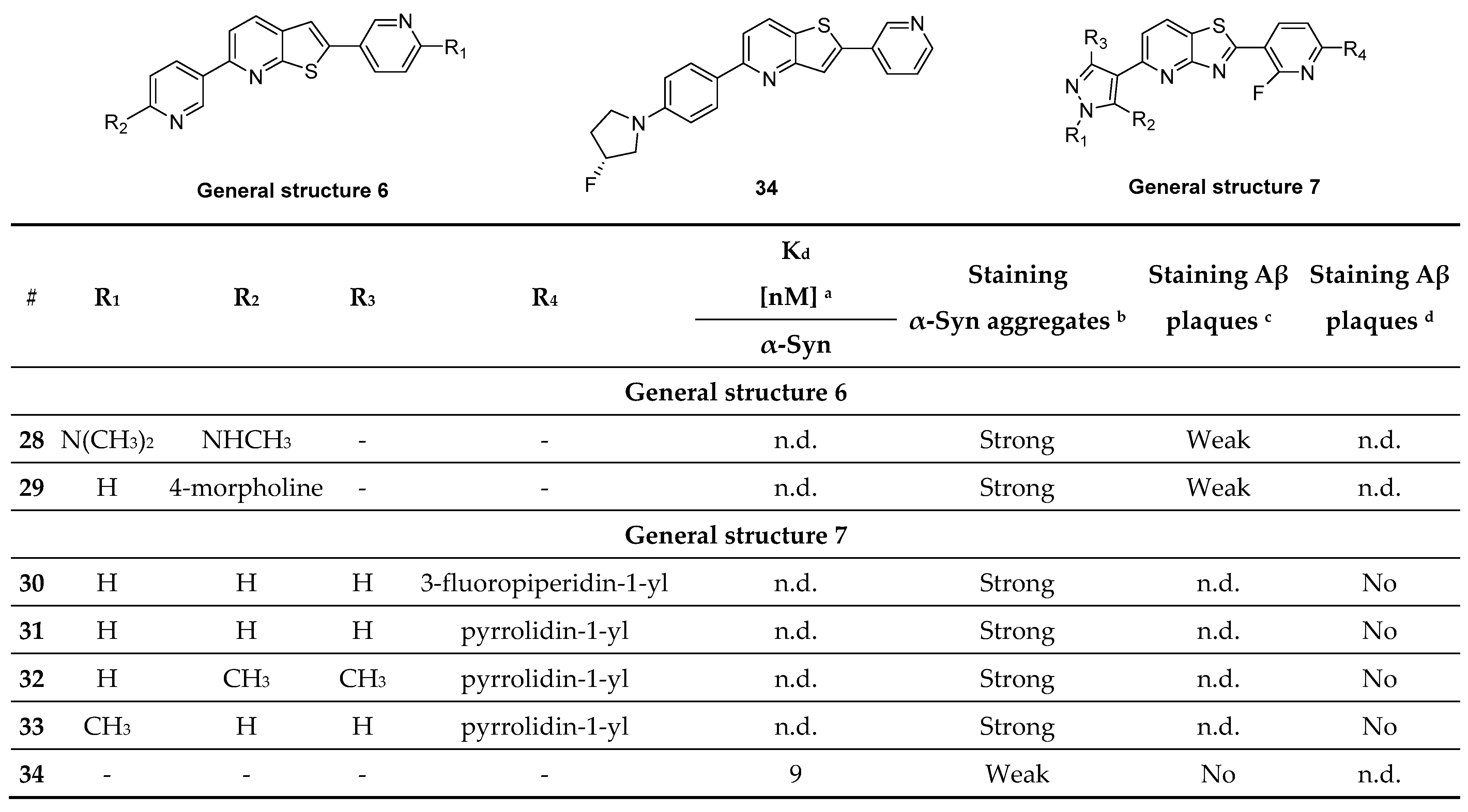
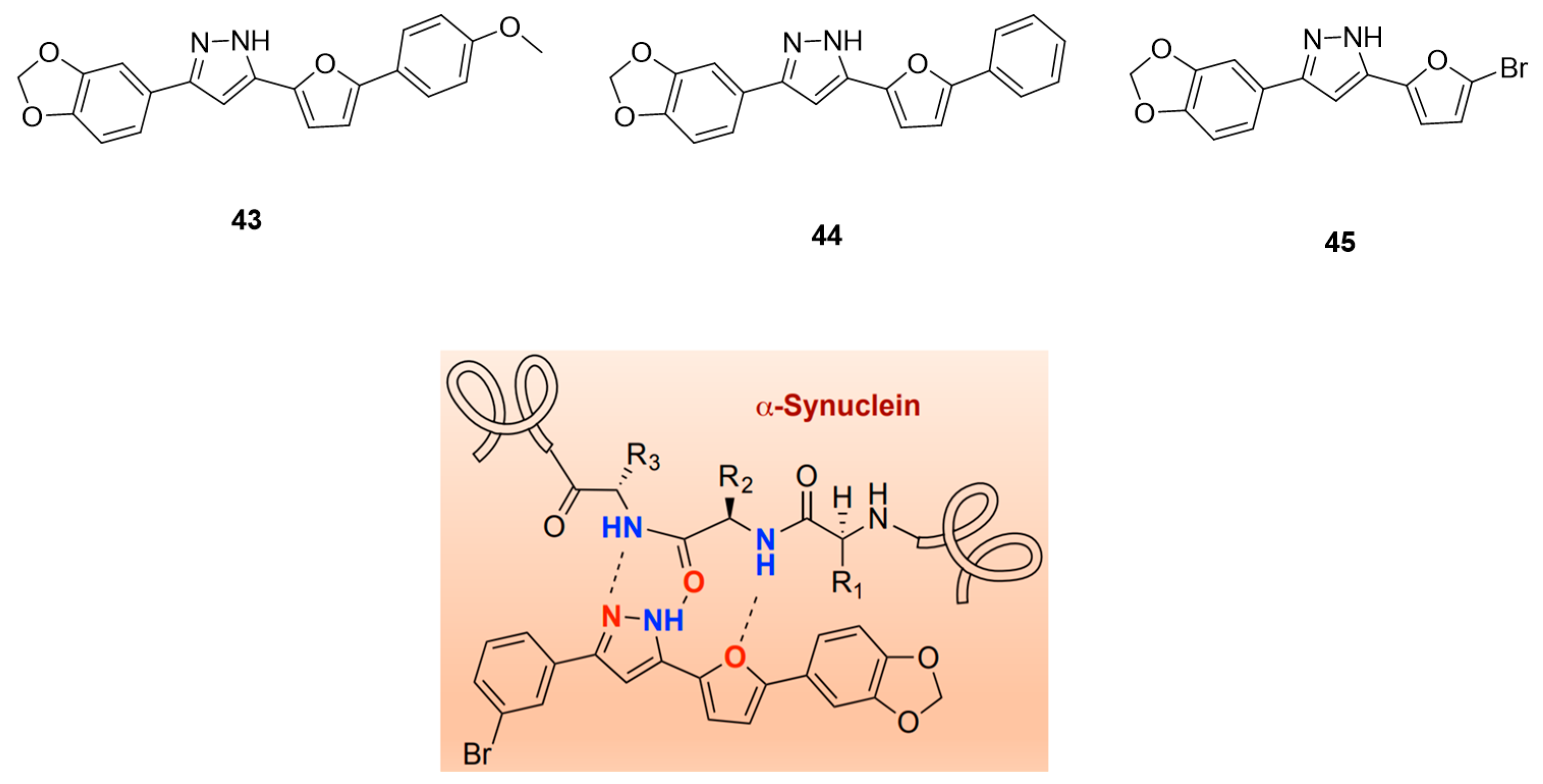
- Ryan, P.; Xu, M.; Jahan, K.; Davey, A.K.; Bharatam, P.V.; Anoopkumar-Dukie, S.; Kassiou, M.; Mellick, G.D.; Rudrawar, S. Novel Furan-2-yl-1H-pyrazoles Possess Inhibitory Activity against α-Synuclein Aggregation. ACS Chem. Neurosci. 2020, 11, 2303–2315. [Google Scholar] [CrossRef]
- Ono, M.; Haratake, M.; Mori, H.; Nakayama, M. Novel chalcones as probes for in vivo imaging of beta-amyloid plaques in Alzheimer’s brains. Bioorg. Med. Chem. 2007, 15, 6802–6809. [Google Scholar] [CrossRef]
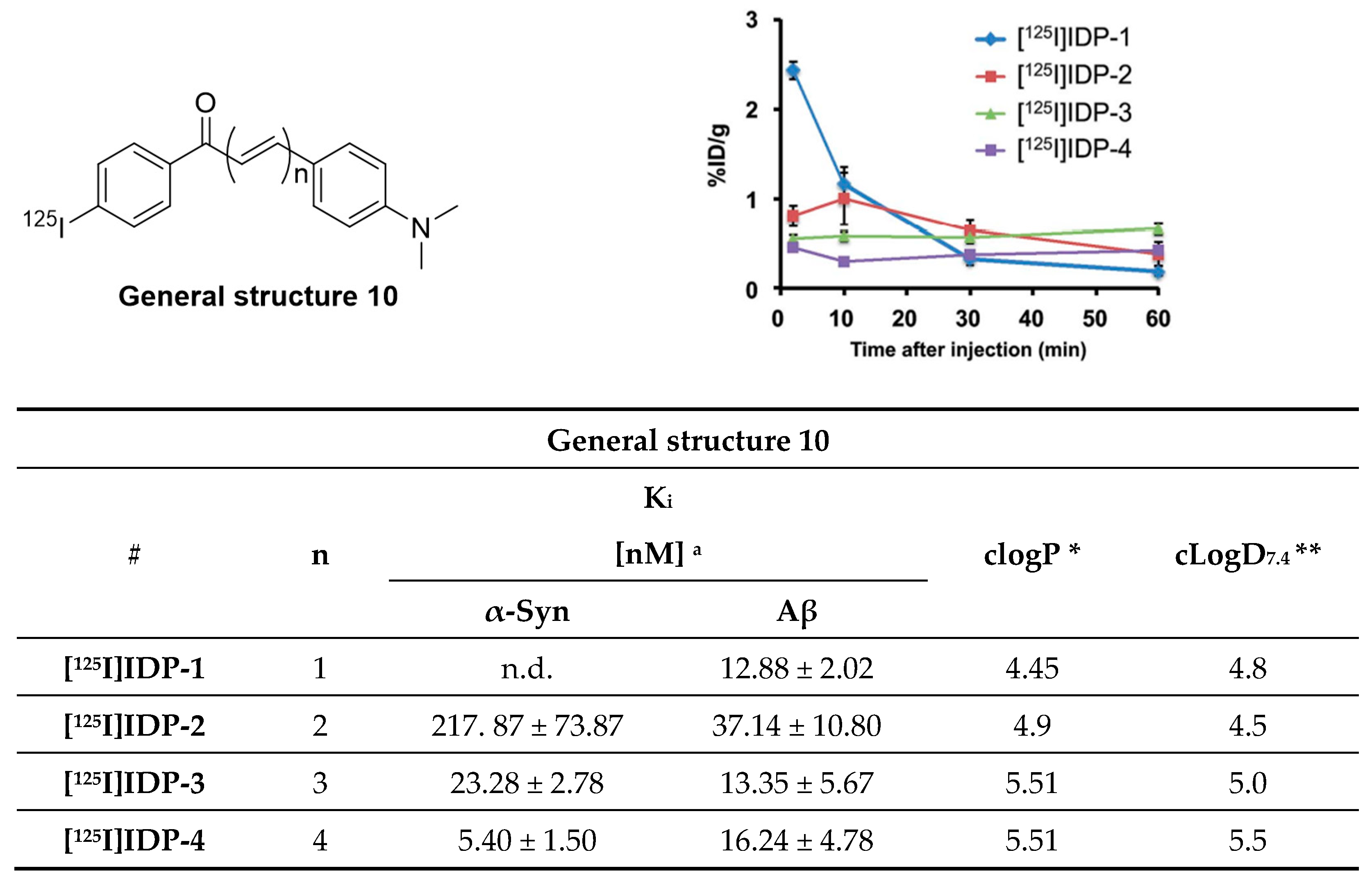
- Ono, M.; Doi, Y.; Watanabe, H.; Ihara, M.; Ozaki, A.; Saji, H. Structure–activity relationships of radioiodinated diphenyl derivatives with different conjugated double bonds as ligands for α-synuclein aggregates. RSC Adv. 2016, 6, 44305–44312. [Google Scholar] [CrossRef]
- Hsieh, C.-J.; Xu, K.; Lee, I.; Graham, T.J.A.; Tu, Z.; Dhavale, D.; Kotzbauer, P.; Mach, R.H. Chalcones and Five-Membered Heterocyclic Isosteres Bind to Alpha Synuclein Fibrils in Vitro. ACS Omega 2018, 3, 4486–4493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
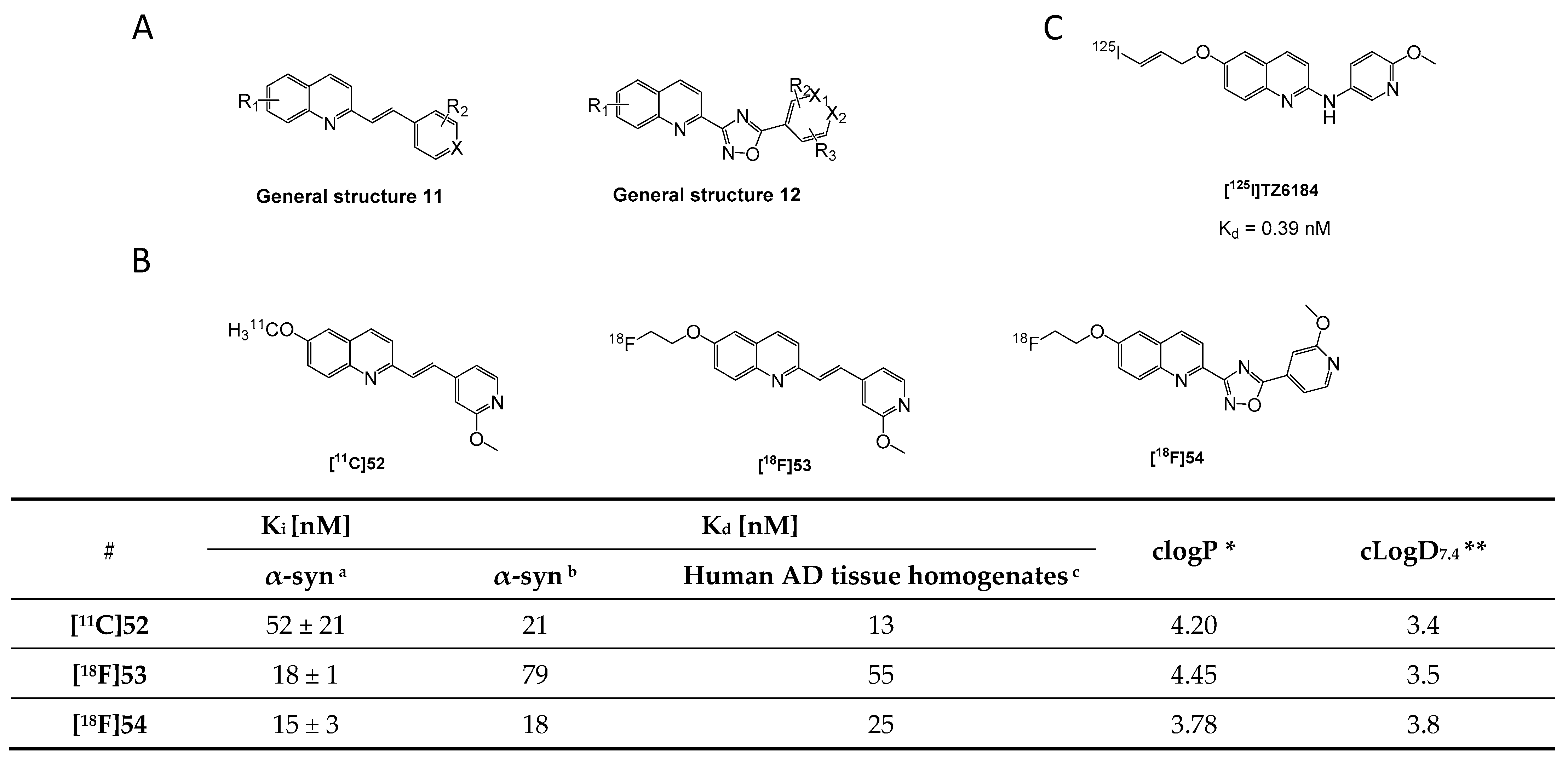
- Yue, X.; Dhavale, D.D.; Li, J.; Luo, Z.; Liu, J.; Yang, H.; Mach, R.H.; Kotzbauer, P.T.; Tu, Z. Design, synthesis, and in vitro evaluation of quinolinyl analogues for α-synuclein aggregation. Bioorg. Med. Chem. Lett. 2018, 28, 1011–1019. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Wang, M.; Glick-Wilson, B.; Meyer, J.; Peters, J.; Territo, P.; Green, M.; Hutchins, G.; Zarrinmayeh, H.; Zheng, Q.-H. Poster Presentation. Synthesis and initial in vitro characterization of [18F]fluoroalkyl derivatives of GSK1482160 as new candidate P2X7R radioligands. J. Label. Compd. Radiopharm. 2019, 62, S123–S588. [Google Scholar] [CrossRef] [Green Version]
- Borroni, E.; Gobbi, L.; Honer, M.; Edelmann, M.; Mitchell, D.; Hardick, D.; Schmidt, W.; Steele, C.; Mulla, M. Radiolabeled Compounds. U.S. Patent WO 2019/121661 A1, 18 December 2018. [Google Scholar]
- Xu, M.; Loa-Kum-Cheung, W.; Zhang, H.; Quinn, R.J.; Mellick, G.D. Identification of a New α-Synuclein Aggregation Inhibitor via Mass Spectrometry Based Screening. ACS Chem. Neurosci. 2019, 10, 2683–2691. [Google Scholar] [CrossRef]
- Johnson, D.K.; Karanicolas, J. Ultra-High-Throughput Structure-Based Virtual Screening for Small-Molecule Inhibitors of Protein–Protein Interactions. J. Chem. Inf. Model. 2016, 56, 399–411. [Google Scholar] [CrossRef] [Green Version]
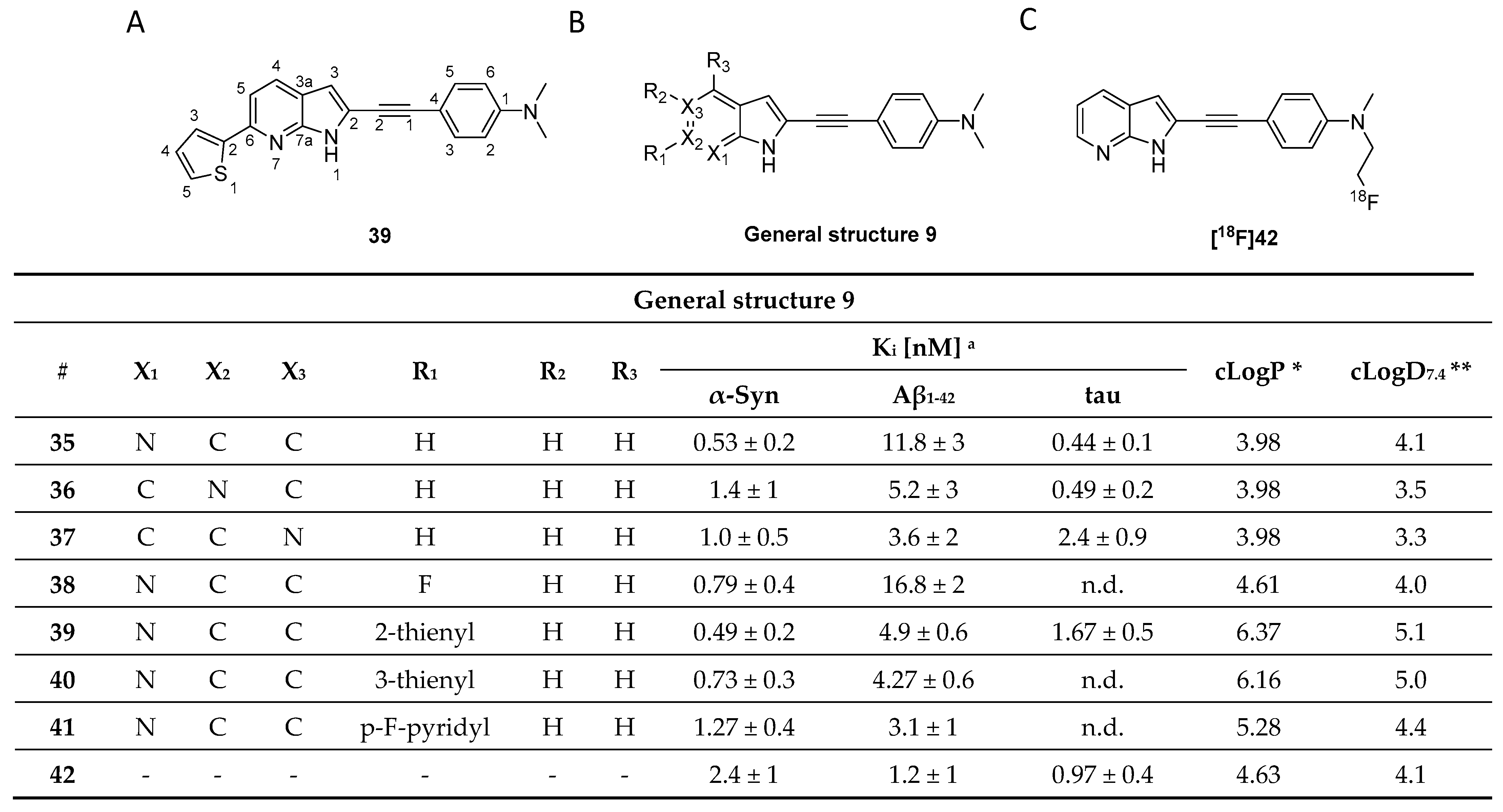
- Ferrie, J.J.; Lengyel-Zhand, Z.; Janssen, B.; Lougee, M.G.; Giannakoulias, S.; Hsieh, C.-J.; Pagar, V.V.; Weng, C.-C.; Xu, H.; Graham, T.J.A.; et al. Identification of a nanomolar affinity α-synuclein fibril imaging probe by ultra-high throughput in silico screening. Chem. Sci. 2020, 11, 12746–12754. [Google Scholar] [CrossRef]
- Meng, X.; Munishkina, L.A.; Fink, A.L.; Uversky, V.N. Effects of Various Flavonoids on the α-Synuclein Fibrillation Process. Park. Dis. 2010, 2010, 650794. [Google Scholar] [CrossRef] [Green Version]
- Snow, A.D.; Castillo, G.M.; Choi, P.Y.; Nguyen, B.P. Proanthocyanidins for the Treatment of Amyloid and Alpha-synuclein Diseases. U.S. Patent WO 2002/076381 A3, 15 February 2002. [Google Scholar]
- Horvath, I.; Weise, C.F.; Andersson, E.K.; Chorell, E.; Sellstedt, M.; Bengtsson, C.; Olofsson, A.; Hultgren, S.J.; Chapman, M.; Wolf-Watz, M.; et al. Mechanisms of Protein Oligomerization: Inhibitor of Functional Amyloids Templates α-Synuclein Fibrillation. J. Am. Chem. Soc. 2012, 134, 3439–3444. [Google Scholar] [CrossRef]
- Åberg, V.; Norman, F.; Chorell, E.; Westermark, A.; Olofsson, A.; Sauer-Eriksson, A.E.; Almqvist, F. Microwave-assisted decarboxylation of bicyclic 2-pyridone scaffolds and identification of Aβ-peptide aggregation inhibitors. Org. Biomol. Chem. 2005, 3, 2817–2823. [Google Scholar] [CrossRef]
- Singh, P.; Chorell, E.; Krishnan, K.S.; Kindahl, T.; Åden, J.; Wittung-Stafshede, P.; Almqvist, F. Synthesis of Multiring Fused 2-Pyridones via a Nitrene Insertion Reaction: Fluorescent Modulators of α-Synuclein Amyloid Formation. Org. Lett. 2015, 17, 6194–6197. [Google Scholar] [CrossRef]
- Horvath, I.; Sellstedt, M.; Weise, C.; Nordvall, L.-M.; Krishna Prasad, G.; Olofsson, A.; Larsson, G.; Almqvist, F.; Wittung-Stafshede, P. Modulation of α-synuclein fibrillization by ring-fused 2-pyridones: Templation and inhibition involve oligomers with different structure. Arch. Biochem. Biophys. 2013, 532, 84–90. [Google Scholar] [CrossRef]
- Cairns, A.G.; Vazquez-Romero, A.; Mahdi Moein, M.; Ådén, J.; Elmore, C.S.; Takano, A.; Arakawa, R.; Varrone, A.; Almqvist, F.; Schou, M. Increased Brain Exposure of an Alpha-Synuclein Fibrillization Modulator by Utilization of an Activated Ester Prodrug Strategy. ACS Chem. Neurosci. 2018, 9, 2542–2547. [Google Scholar] [CrossRef]
- Singh, P.; Adolfsson, D.E.; Ådén, J.; Cairns, A.G.; Bartens, C.; Brännström, K.; Olofsson, A.; Almqvist, F. Pyridine-Fused 2-Pyridones via Povarov and A3 Reactions: Rapid Generation of Highly Functionalized Tricyclic Heterocycles Capable of Amyloid Fibril Binding. J. Org. Chem. 2019, 84, 3887–3903. [Google Scholar] [CrossRef]
- Chen, Y.-F.; Bian, J.; Zhang, P.; Bu, L.-L.; Shen, Y.; Yu, W.-B.; Lu, X.-H.; Lin, X.; Ye, D.-Y.; Wang, J.; et al. Design, synthesis and identification of N, N-dibenzylcinnamamide (DBC) derivatives as novel ligands for α-synuclein fibrils by SPR evaluation system. Bioorg. Med. Chem. 2020, 28, 115358. [Google Scholar] [CrossRef]
- Lengyel-Zhand, Z.; Ferrie, J.J.; Janssen, B.; Hsieh, C.-J.; Graham, T.; Xu, K.-y.; Haney, C.M.; Lee, V.M.Y.; Trojanowski, J.Q.; Petersson, E.J.; et al. Synthesis and characterization of high affinity fluorogenic α-synuclein probes. Chem. Commun. 2020, 56, 3567–3570. [Google Scholar] [CrossRef] [PubMed]
- Schweighauser, M.; Shi, Y.; Tarutani, A.; Kametani, F.; Murzin, A.G.; Ghetti, B.; Matsubara, T.; Tomita, T.; Ando, T.; Hasegawa, K.; et al. Structures of α-synuclein filaments from multiple system atrophy. Nature 2020, 585, 464–469. [Google Scholar] [CrossRef]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castaño-Díez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sehlin, D.; Fang, X.T.; Cato, L.; Antoni, G.; Lannfelt, L.; Syvänen, S. Antibody-based PET imaging of amyloid beta in mouse models of Alzheimer’s disease. Nat. Commun. 2016, 7, 10759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sehlin, D.; Syvänen, S.; Ballanger, B.; Barthel, H.; Bischof, G.N.; Boche, D.; Boecker, H.; Bohn, K.P.; Borghammer, P.; Cross, D.; et al. Engineered antibodies: New possibilities for brain PET? Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 2848–2858. [Google Scholar] [CrossRef] [Green Version]
- Syvänen, S.; Fang, X.T.; Faresjö, R.; Rokka, J.; Lannfelt, L.; Olberg, D.E.; Eriksson, J.; Sehlin, D. Fluorine-18-Labeled Antibody Ligands for PET Imaging of Amyloid-β in Brain. ACS Chem. Neurosci. 2020, 11, 4460–4468. [Google Scholar] [CrossRef] [PubMed]


































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korat, Š.; Bidesi, N.S.R.; Bonanno, F.; Di Nanni, A.; Hoàng, A.N.N.; Herfert, K.; Maurer, A.; Battisti, U.M.; Bowden, G.D.; Thonon, D.; et al. Alpha-Synuclein PET Tracer Development—An Overview about Current Efforts. Pharmaceuticals 2021, 14, 847. https://doi.org/10.3390/ph14090847
Korat Š, Bidesi NSR, Bonanno F, Di Nanni A, Hoàng ANN, Herfert K, Maurer A, Battisti UM, Bowden GD, Thonon D, et al. Alpha-Synuclein PET Tracer Development—An Overview about Current Efforts. Pharmaceuticals. 2021; 14(9):847. https://doi.org/10.3390/ph14090847
Chicago/Turabian StyleKorat, Špela, Natasha Shalina Rajani Bidesi, Federica Bonanno, Adriana Di Nanni, Anh Nguyên Nhât Hoàng, Kristina Herfert, Andreas Maurer, Umberto Maria Battisti, Gregory David Bowden, David Thonon, and et al. 2021. "Alpha-Synuclein PET Tracer Development—An Overview about Current Efforts" Pharmaceuticals 14, no. 9: 847. https://doi.org/10.3390/ph14090847