
The Many Applications of Engineered Bacteriophages—An Overview



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- Phages are typically very specific in the types of bacterial cells they target
- Phages are relatively easy and inexpensive to propagate on bacterial hosts using well established protocols
- Phage capsids are highly stable, often resistant to changes in pH and temperature
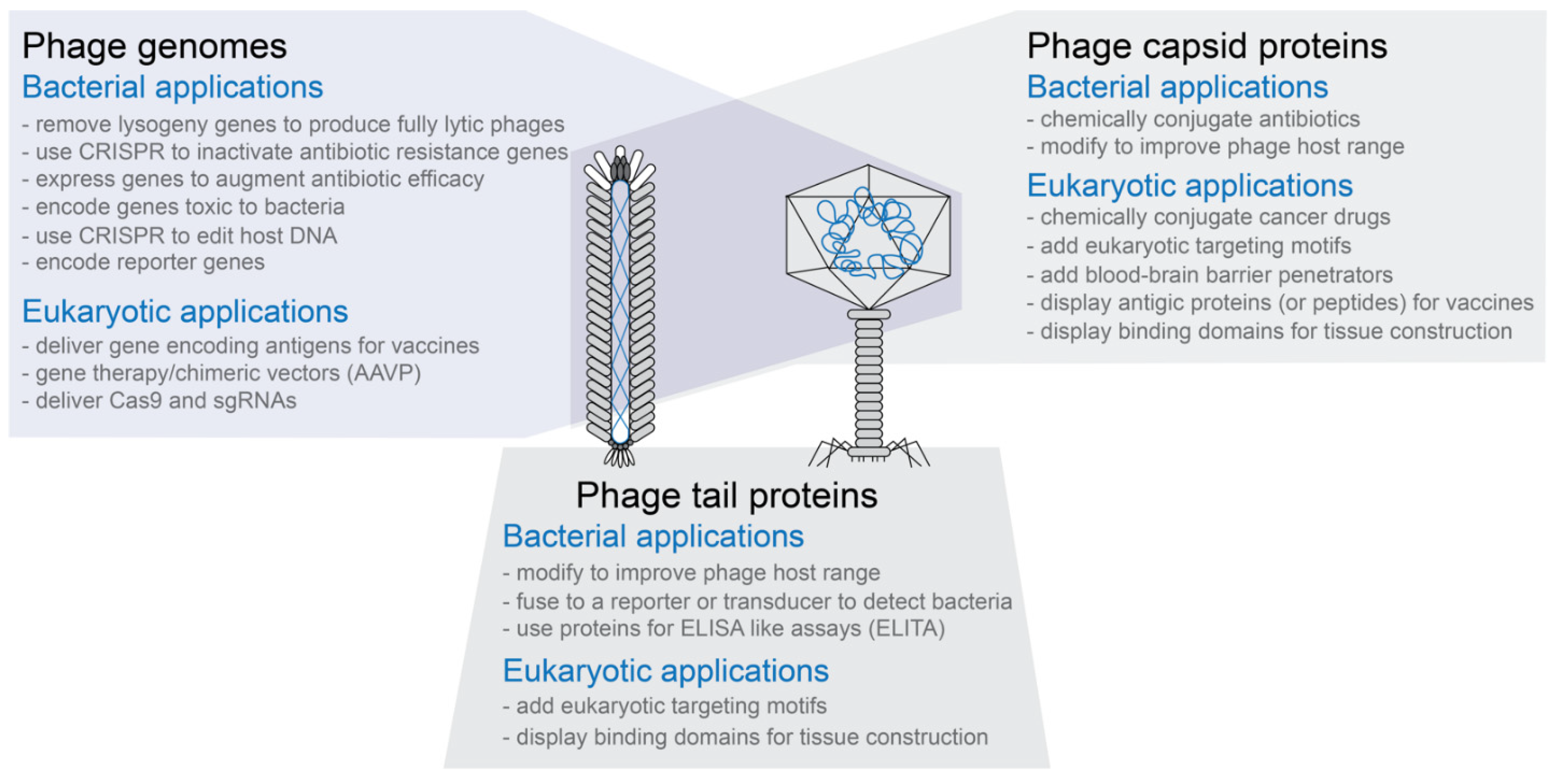
- Phage capsids have multiple proteins that can be targeted for modification without necessarily inactivating the phage capsid’s functions (see Figure 1)
- Phage capsids protect the DNA or RNA packaged in them
- Phage capsids or modified phage capsids can be used to package non-phage nucleic acid, protein, or other types of materials (see Figure 1)
- Phage genomes are small compared to bacterial or eukaryotic genomes and are often relatively easy to modify using genetic engineering techniques
- In vitro packaging systems have been created to move genetically modified genomes into capsids as well as transformation protocols to move genomes directly into cells for expression
- Phage display technology can be used to create phages with novel binding properties even to eukaryotic targets
- For human treatment applications, phages are generally recognized as safe (GRAS) agents
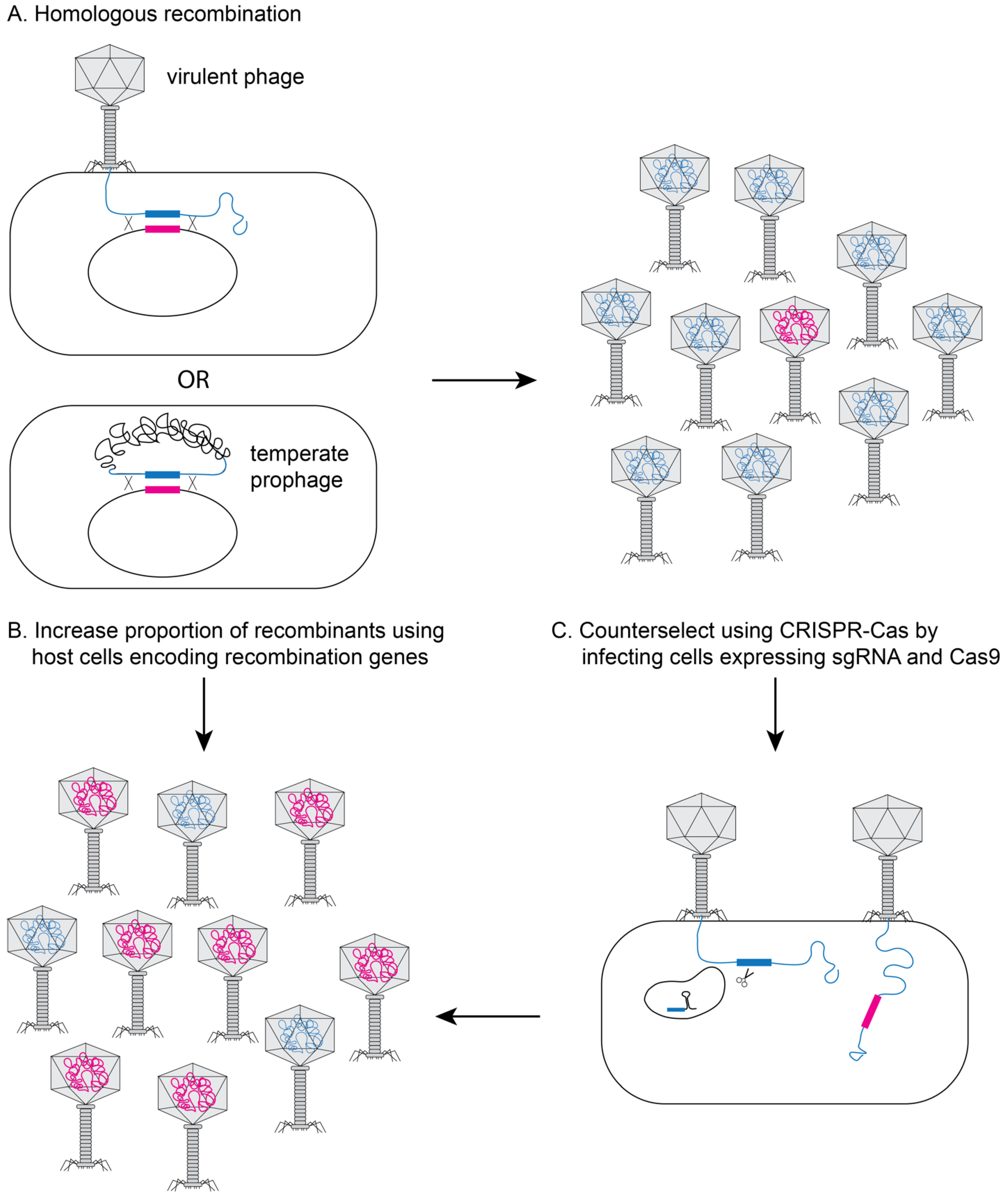
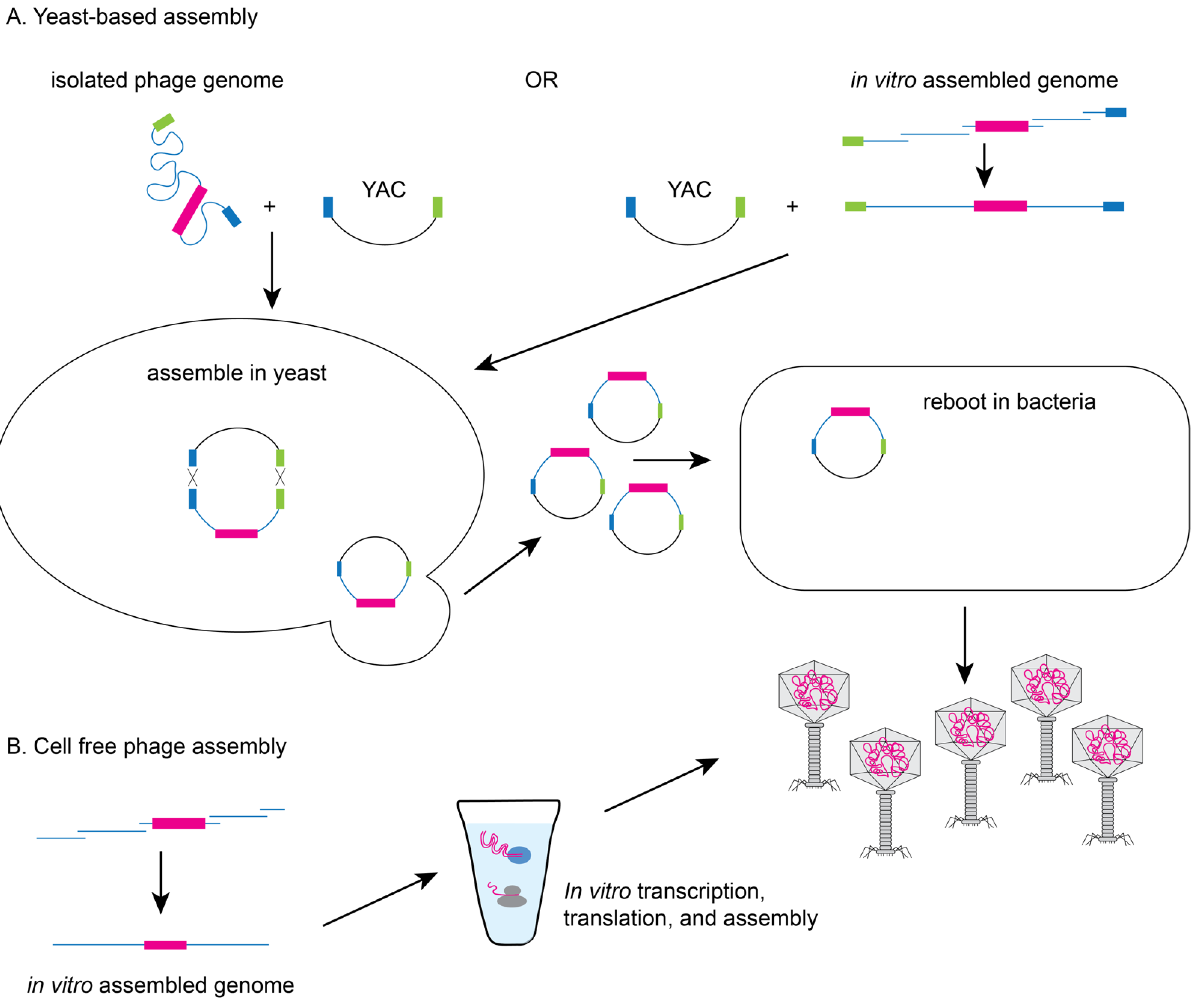
2. Methods to Genetically Engineer Phages
3. Genetically Engineered Phages for Anti-Bacterial Applications
3.1. Enhancing Phage Therapy
3.2. Modifying the Gut Microbiome
3.3. Altering Phage Host Range
3.4. Altering Antibiotic Sensitivity
3.5. Delivering Antimicrobials
3.6. Deploying Targeted CRISPR Editing
3.7. Disrupting Biofilms
3.8. Killing Bacteria with Endolysins
4. Genetically Engineered Phages for Eukaryotic Applications
4.1. Targeting Eukaryotic Cells
4.2. Delivering Drugs to Eukaryotic Cells
4.3. Delivering Genes to Eukaryotic Cells
4.4. Vaccines
5. Genetically Engineered Phages as Sensors
6. Genetically Engineered Phages to Facilitate Tissue Construction
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Salmond, G.P.; Fineran, P.C. A century of the phage: Past, present and future. Nat. Rev. Microbiol. 2015, 13, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Letarov, A.V. History of Early Bacteriophage Research and Emergence of Key Concepts in Virology. Biochemistry 2020, 85, 1093–1112. [Google Scholar] [CrossRef] [PubMed]
- Moelling, K.; Broecker, F.; Willy, C. A Wake-Up Call: We Need Phage Therapy Now. Viruses 2018, 10, 688. [Google Scholar] [CrossRef] [Green Version]
- Cooper, C.J.; Mirzaei, M.K.; Nilsson, A.S. Adapting Drug Approval Pathways for Bacteriophage-Based Therapeutics. Front. Microbiol. 2016, 7, 1209. [Google Scholar] [CrossRef]
- Hargreaves, K.R.; Otieno, J.R.; Thanki, A.; Blades, M.J.; Millard, A.D.; Browne, H.P.; Lawley, T.D.; Clokie, M.R. As Clear as Mud? Determining the diversity and prevalence of prophages in the draft genomes of estuarine isolates of clostridium difficile. Genome Biol. Evol. 2015, 7, 1842–1855. [Google Scholar] [CrossRef]
- de Jonge, P.A.; Nóbrega, F.L.; Brouns, S.J.J.; Dutilh, B.E. Molecular and Evolutionary Determinants of Bacteriophage Host Range. Trends Microbiol. 2019, 27, 51–63. [Google Scholar] [CrossRef]
- Pires, D.P.; Cleto, S.; Sillankorva, S.; Azeredo, J.; Lu, T.K. Genetically Engineered Phages: A Review of Advances over the Last Decade. Microbiol. Mol. Biol. Rev. 2016, 80, 523–543. [Google Scholar] [CrossRef] [Green Version]
- Tinoco, J.M.; Buttaro, B.; Zhang, H.; Liss, N.; Sassone, L.M.; Stevens, R. Effect of a genetically engineered bacteriophage on Enterococcus faecalis biofilms. Arch. Oral Biol. 2016, 71, 80–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilcher, S.; Loessner, M.J. Engineering Bacteriophages as Versatile Biologics. Trends Microbiol. 2019, 27, 355–367. [Google Scholar] [CrossRef]
- Zhang, H.; Fouts, D.E.; DePew, J.; Stevens, R.H. Genetic modifications to temperate Enterococcus faecalis phage Ef11 that abolish the establishment of lysogeny and sensitivity to repressor, and increase host range and productivity of lytic infection. Microbiology 2013, 159, 1023–1035. [Google Scholar] [CrossRef] [Green Version]
- Ando, H.; Lemire, S.; Pires, D.P.; Lu, T.K. Engineering Modular Viral Scaffolds for Targeted Bacterial Population Editing. Cell Syst. 2015, 1, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Yosef, I.; Goren, M.G.; Globus, R.; Molshanski-Mor, S.; Qimron, U. Extending the Host Range of Bacteriophage Particles for DNA Transduction. Mol. Cell 2017, 66, 721–728.e3. [Google Scholar] [CrossRef] [Green Version]
- Loessner, M.J.; Rees, C.E.; Stewart, G.S.; Scherer, S. Construction of luciferase reporter bacteriophage A511::luxAB for rapid and sensitive detection of viable Listeria cells. Appl. Environ. Microbiol. 1996, 62, 1133–1140. [Google Scholar] [CrossRef] [Green Version]
- Le, S.; He, X.; Tan, Y.; Huang, G.; Zhang, L.; Lux, R.; Shi, W.; Hu, F. Mapping the Tail Fiber as the Receptor Binding Protein Responsible for Differential Host Specificity of Pseudomonas aeruginosa Bacteriophages PaP1 and JG004. PLoS ONE 2013, 8, e68562. [Google Scholar] [CrossRef]
- Mahichi, F.; Synnott, A.J.; Yamamichi, K.; Osada, T.; Tanji, Y. Site-specific recombination of T2 phage using IP008 long tail fiber genes provides a targeted method for expanding host range while retaining lytic activity. FEMS Microbiol. Lett. 2009, 295, 211–217. [Google Scholar] [CrossRef] [Green Version]
- Sarkis, G.J.; Jacobs, W.R., Jr.; Hatfulll, G.F. L5 luciferase reporter mycobacteriophages: A sensitive tool for the detection and assay of live mycobacteria. Mol. Microbiol. 1995, 15, 1055–1067. [Google Scholar] [CrossRef] [PubMed]
- Tanji, Y.; Furukawa, C.; Na, S.-H.; Hijikata, T.; Miyanaga, K.; Unno, H. Escherichia coli detection by GFP-labeled lysozyme-inactivated T4 bacteriophage. J. Biotechnol. 2004, 114, 11–20. [Google Scholar] [CrossRef]
- Marinelli, L.J.; Hatfull, G.F.; Piuri, M. Recombineering: A powerful tool for modification of bacteriophage genomes. Bacteriophage 2012, 2, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Datsenko, K.A.; Wanner, B.L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fehér, T.; Karcagi, I.; Blattner, F.R.; Pósfai, G. Bacteriophage recombineering in the lytic state using the lambda red recombinases. Microb. Biotechnol. 2012, 5, 466–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiro, R.; Shitrit, D.; Qimron, U. Efficient engineering of a bacteriophage genome using the type I-E CRISPR-Cas system. RNA Biol. 2014, 11, 42–44. [Google Scholar] [CrossRef] [Green Version]
- Martel, B.; Moineau, S. CRISPR-Cas: An efficient tool for genome engineering of virulent bacteriophages. Nucleic Acids Res. 2014, 42, 9504–9513. [Google Scholar] [CrossRef] [Green Version]
- Bari, S.M.N.; Walker, F.C.; Cater, K.; Aslan, B.; Hatoum-Aslan, A. Strategies for Editing Virulent Staphylococcal Phages Using CRISPR-Cas10. ACS Synth. Biol. 2017, 6, 2316–2325. [Google Scholar] [CrossRef]
- Box, A.M.; McGuffie, M.J.; O’Hara, B.J.; Seed, K.D. Functional Analysis of Bacteriophage Immunity through a Type I-E CRISPR-Cas System in Vibrio cholerae and Its Application in Bacteriophage Genome Engineering. J. Bacteriol. 2016, 198, 578–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemay, M.-L.; Tremblay, D.M.; Moineau, S. Genome Engineering of Virulent Lactococcal Phages Using CRISPR-Cas9. ACS Synth. Biol. 2017, 6, 1351–1358. [Google Scholar] [CrossRef]
- Schilling, T.; Dietrich, S.; Hoppert, M.; Hertel, R. A CRISPR-Cas9-Based Toolkit for Fast and Precise In Vivo Genetic Engineering of Bacillus subtilis Phages. Viruses 2018, 10, 241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, P.; Wu, X.; Tang, W.-C.; Zhu, J.; Rao, V. Engineering of Bacteriophage T4 Genome Using CRISPR-Cas9. ACS Synth. Biol. 2017, 6, 1952–1961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Zhou, J.; Chen, G.-Q.; Xiu, Z.-L. Efficient Genome Engineering of a Virulent Klebsiella Bacteriophage Using CRISPR-Cas9. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Jaschke, P.R.; Lieberman, E.K.; Rodriguez, J.; Sierra, A.; Endy, D. A fully decompressed synthetic bacteriophage oX174 genome assembled and archived in yeast. Virology 2012, 434, 278–284. [Google Scholar] [CrossRef] [Green Version]
- Garamella, J.; Marshall, R.; Rustad, M.; Noireaux, V. The All E. coli TX-TL Toolbox 2.0: A Platform for Cell-Free Synthetic Biology. ACS Synth. Biol. 2016, 5, 344–355. [Google Scholar] [CrossRef]
- Shin, J.; Jardine, P.; Noireaux, V. Genome Replication, Synthesis, and Assembly of the Bacteriophage T7 in a Single Cell-Free Reaction. ACS Synth. Biol. 2012, 1, 408–413. [Google Scholar] [CrossRef]
- Rustad, M.; Eastlund, A.; Marshall, R.; Jardine, P.; Noireaux, V. Synthesis of Infectious Bacteriophages in an E. coli-based Cell-free Expression System. J. Vis. Exp. 2017, e56144. [Google Scholar] [CrossRef]
- Rustad, M.; Eastlund, A.; Jardine, P.; Noireaux, V. Cell-free TXTL synthesis of infectious bacteriophage T4 in a single test tube reaction. Synth. Biol. 2018, 3, ysy002. [Google Scholar] [CrossRef]
- Stevens, R.H.; Porras, O.D.; Delisle, A.L. Bacteriophages induced from lysogenic root canal isolates ofEnterococcus faecalis. Oral Microbiol. Immunol. 2009, 24, 278–284. [Google Scholar] [CrossRef]
- Dedrick, R.M.; Guerrero-Bustamante, C.A.; Garlena, R.A.; Russell, D.A.; Ford, K.; Harris, K.; Gilmour, K.C.; Soothill, J.; Jacobs-Sera, D.; Schooley, R.T.; et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 2019, 25, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Hsu, B.B.; Gibson, T.E.; Yeliseyev, V.; Liu, Q.; Lyon, L.; Bry, L.; Silver, P.A.; Gerber, G.K. Dynamic Modulation of the Gut Microbiota and Metabolome by Bacteriophages in a Mouse Model. Cell Host Microbe 2019, 25, 803–814.e5. [Google Scholar] [CrossRef] [Green Version]
- Bioscience, E. Eligobiotics® (Re)Programming the Microbiome. Available online: https://eligo.bio/technology/ (accessed on 1 April 2021).
- Biosciences, L. crPhageTM Technology. Available online: https://www.locus-bio.com/technology/ (accessed on 1 April 2021).
- Ross, A.; Ward, S.; Hyman, P. More Is Better: Selecting for Broad Host Range Bacteriophages. Front. Microbiol. 2016, 7, 1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoichi, M.; Abe, M.; Miyanaga, K.; Unno, H.; Tanji, Y. Alteration of tail fiber protein gp38 enables T2 phage to infect Escherichia coli O157:H7. J. Biotechnol. 2005, 115, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Heilpern, A.J.; Waldor, M.K. pIIICTX, a predicted CTXphi minor coat protein, can expand the host range of coliphage fd to include Vibrio cholerae. J. Bacteriol. 2003, 185, 1037–1044. [Google Scholar] [CrossRef] [Green Version]
- Marzari, R.; Sblattero, D.; Righi, M.; Bradbury, A. Extending filamentous phage host range by the grafting of a heterologous receptor binding domain. Gene 1997, 185, 27–33. [Google Scholar] [CrossRef]
- Yehl, K.; Lemire, S.; Yang, A.C.; Ando, H.; Mimee, M.; Torres, M.D.T.; de la Fuente-Nunez, C.; Lu, T.K. Engineering Phage Host-Range and Suppressing Bacterial Resistance through Phage Tail Fiber Mutagenesis. Cell 2019, 179, 459–469.e9. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.K.; Collins, J.J. Engineered bacteriophage targeting gene networks as adjuvants for antibiotic therapy. Proc. Natl. Acad. Sci. USA 2009, 106, 4629–4634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.; Friedman, N.; Molshanski-Mor, S.; Qimron, U. Reversing Bacterial Resistance to Antibiotics by Phage-Mediated Delivery of Dominant Sensitive Genes. Appl. Environ. Microbiol. 2012, 78, 744–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bikard, D.; Euler, C.W.; Jiang, W.; Nussenzweig, P.M.; Goldberg, G.W.; Duportet, X.; Fischetti, V.A.; Marraffini, L.A. Exploiting CRISPR-Cas nucleases to produce sequence-specific antimicrobials. Nat. Biotechnol. 2014, 32, 1146–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Citorik, R.J.; Mimee, M.; Lu, T.K. Sequence-specific antimicrobials using efficiently delivered RNA-guided nucleases. Nat. Biotechnol. 2014, 32, 1141–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yacoby, I.; Bar, H.; Benhar, I. Targeted Drug-Carrying Bacteriophages as Antibacterial Nanomedicines. Antimicrob. Agents Chemother. 2007, 51, 2156–2163. [Google Scholar] [CrossRef] [Green Version]
- Yacoby, I.; Shamis, M.; Bar, H.; Shabat, D.; Benhar, I. Targeting antibacterial agents by using drug-carrying filamentous bacteriophages. Antimicrob. Agents Chemother. 2006, 50, 2087–2097. [Google Scholar] [CrossRef] [Green Version]
- Vaks, L.; Benhar, I. In vivo characteristics of targeted drug-carrying filamentous bacteriophage nanomedicines. J. Nanobiotechnol. 2011, 9, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westwater, C.; Kasman, L.M.; Schofield, D.A.; Werner, P.A.; Dolan, J.W.; Schmidt, M.G.; Norris, J.S. Use of genetically engineered phage to deliver antimicrobial agents to bacteria: An alternative therapy for treatment of bacterial infections. Antimicrob. Agents Chemother. 2003, 47, 1301–1307. [Google Scholar] [CrossRef] [Green Version]
- Selle, K.; Fletcher, J.R.; Tuson, H.; Schmitt, D.S.; McMillan, L.; Vridhambal, G.S.; Rivera, A.J.; Montgomery, S.A.; Fortier, L.-C.; Barrangou, R.; et al. In Vivo Targeting of Clostridioides difficile Using Phage-Delivered CRISPR-Cas3 Antimicrobials. mBio 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Hsu, B.B.; Plant, I.N.; Lyon, L.; Anastassacos, F.M.; Way, J.C.; Silver, P.A. In situ reprogramming of gut bacteria by oral delivery. Nat. Commun. 2020, 11, 5030. [Google Scholar] [CrossRef]
- Ferriol-González, C.; Domingo-Calap, P. Phages for Biofilm Removal. Antibiotics 2020, 9, 268. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.K.; Collins, J.J. Dispersing biofilms with engineered enzymatic bacteriophage. Proc. Natl. Acad. Sci. USA 2007, 104, 11197–11202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, R.; Lamas-Samanamud, G.R. Inhibition of biofilm formation by t7 bacteriophages producing quorum-quenching enzymes. Appl. Environ. Microbiol. 2014, 80, 5340–5348. [Google Scholar] [CrossRef] [Green Version]
- Schmelcher, M.; Tchang, V.S.; Loessner, M.J. Domain shuffling and module engineering of Listeria phage endolysins for enhanced lytic activity and binding affinity. Microb. Biotechnol. 2011, 4, 651–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Díez-Martínez, R.; De Paz, H.D.; Garcia-Fernandez, E.; Bustamante, N.; Euler, C.W.; Fischetti, V.A.; Menéndez, M.; García, P. A novel chimeric phage lysin with high in vitro and in vivo bactericidal activity against Streptococcus pneumoniae. J. Antimicrob. Chemother. 2015, 70, 1763–1773. [Google Scholar] [CrossRef] [Green Version]
- Landlinger, C.; Tisakova, L.; Oberbauer, V.; Schwebs, T.; Muhammad, A.; Latka, A.; Van Simaey, L.; Vaneechoutte, M.; Guschin, A.; Resch, G.; et al. Engineered Phage Endolysin Eliminates Gardnerella Biofilm without Damaging Beneficial Bacteria in Bacterial Vaginosis Ex Vivo. Pathogens 2021, 10, 54. [Google Scholar] [CrossRef]
- Briers, Y.; Walmagh, M.; Van Puyenbroeck, V.; Cornelissen, A.; Cenens, W.; Aertsen, A.; Oliveira, H.; Azeredo, J.; Verween, G.; Pirnay, J.-P.; et al. Engineered endolysin-based “artilysins” to combat multidrug-resistant gram-negative pathogens. mBio 2014, 5, e01379-14. [Google Scholar] [CrossRef] [Green Version]
- Heselpoth, R.D.; Euler, C.W.; Schuch, R.; Fischetti, V.A. Lysocins: Bioengineered Antimicrobials That Deliver Lysins across the Outer Membrane of Gram-Negative Bacteria. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Kwon, S.-J.; Wu, X.; Sauve, J.; Lee, I.; Nam, J.; Kim, J.; Dordick, J.S. Selective Killing of Pathogenic Bacteria by Antimicrobial Silver Nanoparticle—Cell Wall Binding Domain Conjugates. ACS Appl. Mater. Interfaces 2018, 10, 13317–13324. [Google Scholar] [CrossRef]
- Grigonyte, A.M.; Hapeshi, A.; Constantinidou, C.; Millard, A. Modification of Bacteriophages to Increase Their Association with Lung Epithelium Cells In Vitro. Pharmaceuticals 2021, 14, 308. [Google Scholar] [CrossRef] [PubMed]
- Karimi, M.; Mirshekari, H.; Basri, S.M.M.; Bahrami, S.; Moghoofei, M.; Hamblin, M.R. Bacteriophages and phage-inspired nanocarriers for targeted delivery of therapeutic cargos. Adv. Drug Deliv. Rev. 2016, 106, 45–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urquhart, T.; Daub, E.; Honek, J.F. Bioorthogonal Modification of the Major Sheath Protein of Bacteriophage M13: Extending the Versatility of Bionanomaterial Scaffolds. Bioconjugate Chem. 2016, 27, 2276–2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, B.; Yang, M.; Mao, C. Phage as a Genetically Modifiable Supramacromolecule in Chemistry, Materials and Medicine. Acc. Chem. Res. 2016, 49, 1111–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyman, P.; Denyes, J. Bacteriophages in Nanotechnology: History and Future; Springer International Publishing: Cham, Switzerland, 2021. [Google Scholar]
- Yayon, A.; Aviezer, D.; Safran, M.; Gross, J.L.; Heldman, Y.; Cabilly, S.; Givol, D.; Katchalski-Katzir, E. Isolation of peptides that inhibit binding of basic fibroblast growth factor to its receptor from a random phage-epitope library. Proc. Natl. Acad. Sci. USA 1993, 90, 10643–10647. [Google Scholar] [CrossRef] [Green Version]
- Yao, Z.-J.; Kao, M.C.; Loh, K.-C.; Chung, M.C. A serotype-specific epitope of dengue virus 1 identified by phage displayed random peptide library. FEMS Microbiol. Lett. 1995, 127, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Saggio, I.; Laufer, R. Biotin binders selected from a random peptide library expressed on phage. Biochem. J. 1993, 293 Pt 3, 613–616. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Rheem, Y.; Yoo, B.; Chong, Y.; Bozhilov, K.N.; Kim, D.; Sadowsky, M.J.; Hur, H.-G.; Myung, N.V. Peptide-mediated shape- and size-tunable synthesis of gold nanostructures. Acta Biomater. 2010, 6, 2681–2689. [Google Scholar] [CrossRef] [PubMed]
- Naik, R.R.; Brott, L.L.; Clarson, S.J.; Stone, M.O. Silica-precipitating peptides isolated from a combinatorial phage display peptide library. J. Nanosci. Nanotechnol. 2002, 2, 95–100. [Google Scholar] [CrossRef]
- Whaley, S.R.; English, D.S.; Hu, E.L.; Barbara, P.F.; Belcher, A.M. Selection of peptides with semiconductor binding specificity for directed nanocrystal assembly. Nature 2000, 405, 665–668. [Google Scholar] [CrossRef]
- Medecigo, M.; Manoutcharian, K.; Vasilevko, V.; Govezensky, T.; Munguia, M.E.; Becerril, B.; Luz-Madrigal, A.; Vaca, L.; Cribbs, D.H.; Gevorkian, G. Novel amyloid-beta specific scFv and VH antibody fragments from human and mouse phage display antibody libraries. J. Neuroimmunol. 2010, 223, 104–114. [Google Scholar] [CrossRef] [Green Version]
- Orner, B.P.; Liu, L.; Murphy, R.M.; Kiessling, L.L. Phage display affords peptides that modulate β-amyloid aggregation. J. Am. Chem. Soc. 2006, 128, 11882–11889. [Google Scholar] [CrossRef] [PubMed]
- Bannas, P.; Hambach, J.; Koch-Nolte, F. Nanobodies and Nanobody-Based Human Heavy Chain Antibodies as Antitumor Therapeutics. Front. Immunol. 2017, 8, 1603. [Google Scholar] [CrossRef]
- Gray, B.P.; Li, S.; Brown, K.C. From phage display to nanoparticle delivery: Functionalizing liposomes with multivalent peptides improves targeting to a cancer biomarker. Bioconjugate Chem. 2013, 24, 85–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hingorani, D.V.; Whitney, M.A.; Friedman, B.; Kwon, J.-K.; Crisp, J.L.; Xiong, Q.; Gross, L.; Kane, C.J.; Tsien, R.Y.; Nguyen, Q.T. Nerve-targeted probes for fluorescence-guided intraoperative imaging. Theranostics 2018, 8, 4226–4237. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Wu, H.; Zhang, X.; Huang, J.; He, X.; Chen, L.; Guo, W.; Guo, X.; Hao, B.; Li, Y. Pre-clinical study of a TNFR1-targeted 18F probe for PET imaging of breast cancer. Amino Acids 2018, 50, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Chi, Y.-H.; Hsiao, J.-K.; Lin, M.-H.; Chang, C.; Lan, C.-H.; Wu, H.-C. Lung Cancer-Targeting Peptides with Multi-subtype Indication for Combinational Drug Delivery and Molecular Imaging. Theranostics 2017, 7, 1612–1632. [Google Scholar] [CrossRef] [Green Version]
- Petrenko, V.A.; Gillespie, J.W. Paradigm shift in bacteriophage-mediated delivery of anticancer drugs: From targeted ‘magic bullets’ to self-navigated ‘magic missiles’. Expert Opin. Drug Deliv. 2017, 14, 373–384. [Google Scholar] [CrossRef] [Green Version]
- Foglizzo, V.; Marchiò, S. Bacteriophages as Therapeutic and Diagnostic Vehicles in Cancer. Pharmaceuticals 2021, 14, 161. [Google Scholar] [CrossRef]
- Bar, H.; Yacoby, I.; Benhar, I. Killing cancer cells by targeted drug-carrying phage nanomedicines. BMC Biotechnol. 2008, 8, 37. [Google Scholar] [CrossRef] [Green Version]
- Du, B.; Han, H.; Wang, Z.; Kuang, L.; Wang, L.; Yu, L.; Wu, M.; Zhou, Z.; Qian, M. Targeted drug delivery to hepatocarcinoma in vivo by phage-displayed specific binding peptide. Mol. Cancer Res. 2010, 8, 135–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, B.; Xu, H.; Yang, M.; Mao, C. Virus-Based Cancer Therapeutics for Targeted Photodynamic Therapy. Methods Mol. Biol. 2018, 1776, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef]
- Anand, P.; O’Neil, A.; Lin, E.; Douglas, T.; Holford, M. Tailored delivery of analgesic ziconotide across a blood brain barrier model using viral nanocontainers. Sci. Rep. 2015, 5, 12497. [Google Scholar] [CrossRef] [Green Version]
- Apawu, A.K.; Curley, S.M.; Dixon, A.R.; Hali, M.; Sinan, M.; Braun, R.D.; Castracane, J.; Cacace, A.T.; Bergkvist, M.; Holt, A.G. MRI compatible MS2 nanoparticles designed to cross the blood–brain-barrier: Providing a path towards tinnitus treatment. Nanomed. Nanotechnol. Biol. Med. 2018, 14, 1999–2008. [Google Scholar] [CrossRef]
- Hosseinidoust, Z. Phage-Mediated Gene Therapy. Curr. Gene Ther. 2017, 17, 120–126. [Google Scholar] [CrossRef]
- Pranjol, M.Z.; Hajitou, A. Bacteriophage-derived vectors for targeted cancer gene therapy. Viruses 2015, 7, 268–284. [Google Scholar] [CrossRef]
- Bedi, D.; Musacchio, T.; Fagbohun, O.A.; Gillespie, J.W.; Deinnocentes, P.; Bird, R.C.; Bookbinder, L.; Torchilin, V.P.; Petrenko, V.A. Delivery of siRNA into breast cancer cells via phage fusion protein-targeted liposomes. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 315–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedi, D.; Gillespie, J.W.; Petrenko, V.A., Jr.; Ebner, A.; Leitner, M.; Hinterdorfer, P.; Petrenko, V.A. Targeted Delivery of siRNA into Breast Cancer Cells via Phage Fusion Proteins. Mol. Pharm. 2013, 10, 551–559. [Google Scholar] [CrossRef] [Green Version]
- Ashley, C.E.; Carnes, E.C.; Phillips, G.K.; Durfee, P.N.; Buley, M.D.; Lino, C.A.; Padilla, D.P.; Phillips, B.; Carter, M.B.; Willman, C.L.; et al. Cell-Specific Delivery of Diverse Cargos by Bacteriophage MS2 Virus-like Particles. ACS Nano 2011, 5, 5729–5745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajitou, A.; Trepel, M.; Lilley, C.E.; Soghomonyan, S.; Alauddin, M.M.; Marini, F.C., 3rd; Restel, B.H.; Ozawa, M.G.; Moya, C.A.; Rangel, R.; et al. A hybrid vector for ligand-directed tumor targeting and molecular imaging. Cell 2006, 125, 385–398. [Google Scholar] [CrossRef]
- Przystal, J.M.; Waramit, S.; Pranjol, M.Z.I.; Yan, W.; Chu, G.; Chongchai, A.; Samarth, G.; Olaciregui, N.G.; Tabatabai, G.; Carcaboso, A.M.; et al. Efficacy of systemic temozolomide-activated phage-targeted gene therapy in human glioblastoma. EMBO Mol. Med. 2019, 11, e8492. [Google Scholar] [CrossRef]
- Qazi, S.; Miettinen, H.M.; Wilkinson, R.A.; McCoy, K.; Douglas, T.; Wiedenheft, B. Programmed Self-Assembly of an Active P22-Cas9 Nanocarrier System. Mol. Pharm. 2016, 13, 1191–1196. [Google Scholar] [CrossRef]
- Frei, J.C.; Lai, J.R. Protein and Antibody Engineering by Phage Display. Methods Enzymol. 2016, 580, 45–87. [Google Scholar] [CrossRef] [Green Version]
- Bazan, J.; Całkosiński, I.; Gamian, A. Phage display—A powerful technique for immunotherapy: 1. Introduction and potential of therapeutic applications. Hum. Vaccin. Immunother. 2012, 8, 1817–1828. [Google Scholar] [CrossRef] [Green Version]
- Bao, Q.; Li, X.; Han, G.; Zhu, Y.; Mao, C.; Yang, M. Phage-based vaccines. Adv. Drug Deliv. Rev. 2019, 145, 40–56. [Google Scholar] [CrossRef]
- Clark, J.; Abedon, S.T.; Hyman, P. Phages as Therapeutic Delivery Vehicles. In Bacteriophages in Health and Disease; Hyman, P., Abedon, S.T., Eds.; International Press: Wallingford, UK, 2012; pp. 86–100. [Google Scholar]
- Hess, K.L.; Jewell, C.M. Phage display as a tool for vaccine and immunotherapy development. Bioeng. Transl. Med. 2020, 5, e10142. [Google Scholar] [CrossRef] [PubMed]
- Hashemi, H.; Bamdad, T.; Jamali, A.; Pouyanfard, S.; Mohammadi, M.G. Evaluation of humoral and cellular immune responses against HSV-1 using genetic immunization by filamentous phage particles: A comparative approach to conventional DNA vaccine. J. Virol. Methods 2010, 163, 440–444. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.R.; Bartley, K.; Jepson, C.D.; Craik, V.; March, J.B. Comparison of a bacteriophage-delivered DNA vaccine and a commercially available recombinant protein vaccine against hepatitis B. FEMS Immunol. Med. Microbiol. 2011, 61, 197–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, T.; Nafissi, N.; Wasfi, O.; Sheldon, K.; Wettig, S.; Slavcev, R. Immunocompatibility of Bacteriophages as Nanomedicines. J. Nanotechnol. 2012, 2012. [Google Scholar] [CrossRef] [Green Version]
- Krut, O.; Bekeredjian-Ding, I. Contribution of the Immune Response to Phage Therapy. J. Immunol. 2018, 200, 3037–3044. [Google Scholar] [CrossRef] [PubMed]
- De Berardinis, P.; Sartorius, R.; Fanutti, C.; Perham, R.N.; Del Pozzo, G.; Guardiola, J. Phage display of peptide epitopes from HIV-1 elicits strong cytolytic responses. Nat. Biotechnol. 2000, 18, 873–876. [Google Scholar] [CrossRef] [PubMed]
- Sathaliyawala, T.; Rao, M.; Maclean, D.M.; Birx, D.L.; Alving, C.R.; Rao, V.B. Assembly of human immunodeficiency virus (hiv) antigens on bacteriophage t4: A novel in vitro approach to construct multicomponent hiv vaccines. J. Virol. 2006, 80, 7688–7698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shivachandra, S.B.; Li, Q.; Peachman, K.K.; Matyas, G.R.; Leppla, S.H.; Alving, C.R.; Rao, M.; Rao, V. Multicomponent anthrax toxin display and delivery using bacteriophage T4. Vaccine 2007, 25, 1225–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Z.J.; Tian, C.J.; Zhu, Q.S.; Zhao, M.Y.; Xin, A.G.; Nie, W.X.; Ling, S.R.; Zhu, M.W.; Wu, J.Y.; Lan, H.Y.; et al. Orally delivered foot-and-mouth disease virus capsid protomer vaccine displayed on T4 bacteriophage surface: 100% protection from potency challenge in mice. Vaccine 2008, 26, 1471–1481. [Google Scholar] [CrossRef] [PubMed]
- Bartolacci, C.; Andreani, C.; Curcio, C.; Occhipinti, S.; Massaccesi, L.; Giovarelli, M.; Galeazzi, R.; Iezzi, M.; Tilio, M.; Gambini, V.; et al. Phage-Based Anti-HER2 Vaccination Can Circumvent Immune Tolerance against Breast Cancer. Cancer Immunol. Res. 2018, 6, 1486–1498. [Google Scholar] [CrossRef] [Green Version]
- Bahadir, A.O.; Balcioglu, B.K.; Uzyol, K.S.; Hatipoglu, I.; Sogut, I.; Başalp, A.; Erdag, B. Phage displayed hbv core antigen with immunogenic activity. Appl. Biochem. Biotechnol. 2011, 165, 1437–1447. [Google Scholar] [CrossRef]
- Caldeira, J.; Bustos, J.; Peabody, J.; Chackerian, B.; Peabody, D.S. Epitope-Specific Anti-hCG Vaccines on a Virus Like Particle Platform. PLoS ONE 2015, 10, e0141407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mogus, A.T.; Liu, L.; Jia, M.; Ajayi, D.T.; Xu, K.; Kong, R.; Huang, J.; Yu, J.; Kwong, P.D.; Mascola, J.R.; et al. Virus-Like Particle Based Vaccines Elicit Neutralizing Antibodies against the HIV-1 Fusion Peptide. Vaccines 2020, 8, 765. [Google Scholar] [CrossRef]
- Sartorius, R.; D’Apice, L.; Trovato, M.; Cuccaro, F.; Costa, V.; De Leo, M.G.; Marzullo, V.M.; Biondo, C.; D’Auria, S.; De Matteis, M.A.; et al. Antigen delivery by filamentous bacteriophage fd displaying an anti- DEC -205 single-chain variable fragment confers adjuvanticity by triggering a TLR 9-mediated immune response. EMBO Mol. Med. 2015, 7, 973–988. [Google Scholar] [CrossRef]
- Thrane, S.; Janitzek, C.M.; Matondo, S.; Resende, M.; Gustavsson, T.; de Jongh, W.A.; Clemmensen, S.; Roeffen, W.; van de Vegte-Bolmer, M.; van Gemert, G.J.; et al. Bacterial superglue enables easy development of efficient virus-like particle based vaccines. J. Nanobiotechnol. 2016, 14, 30. [Google Scholar] [CrossRef] [Green Version]
- Ahovan, Z.A.; Hashemi, A.; De Plano, L.M.; Gholipourmalekabadi, M.; Seifalian, A.M. Bacteriophage Based Biosensors: Trends, Outcomes and Challenges. Nanomaterials 2020, 10, 501. [Google Scholar] [CrossRef] [Green Version]
- Meile, S.; Kilcher, S.; Loessner, M.J.; Dunne, M. Reporter Phage-Based Detection of Bacterial Pathogens: Design Guidelines and Recent Developments. Viruses 2020, 12, 944. [Google Scholar] [CrossRef] [PubMed]
- Hinkley, T.C.; Singh, S.; Garing, S.; Le Ny, A.-L.M.; Nichols, K.P.; Peters, J.E.; Talbert, J.N.; Nugen, S.R. A phage-based assay for the rapid, quantitative, and single CFU visualization of E. coli (ECOR #13) in drinking water. Sci. Rep. 2018, 8, 14630. [Google Scholar] [CrossRef] [PubMed]
- Meile, S.; Sarbach, A.; Du, J.; Schuppler, M.; Saez, C.; Loessner, M.J.; Kilcher, S. Engineered Reporter Phages for Rapid Bioluminescence-Based Detection and Differentiation of Viable Listeria Cells. Appl. Environ. Microbiol. 2020, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, S.; Vincentelli, R.; Mahony, J.; Nauta, A.; Ramond, L.; Lugli, G.A.; Ventura, M.; van Sinderen, D.; Cambillau, C. Functional carbohydrate binding modules identified in evolved dits from siphophages infecting various Gram-positive bacteria. Mol. Microbiol. 2018, 110, 777–795. [Google Scholar] [CrossRef]
- Poshtiban, S.; Javed, M.A.; Arutyunov, D.; Singh, A.; Banting, G.; Szymanski, C.M.; Evoy, S. Phage receptor binding protein-based magnetic enrichment method as an aid for real time PCR detection of foodborne bacteria. Analyst 2013, 138, 5619–5626. [Google Scholar] [CrossRef]
- Kunstmann, S.; Scheidt, T.; Buchwald, S.; Helm, A.; Mulard, L.A.; Fruth, A.; Barbirz, S. Bacteriophage Sf6 Tailspike Protein for Detection of Shigella flexneri Pathogens. Viruses 2018, 10, 431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, A.; Rabsch, W.; Broeker, N.K.; Barbirz, S. Bacteriophage tailspike protein based assay to monitor phase variable glucosylations in Salmonella O-antigens. BMC Microbiol. 2016, 16, 207. [Google Scholar] [CrossRef] [Green Version]
- Askoxylakis, V.; Zitzmann, S.; Mier, W.; Graham, K.; Krämer, S.; Von Wegner, F.; Fink, R.H.; Schwab, M.; Eisenhut, M.; Haberkorn, U. Preclinical Evaluation of the Breast Cancer Cell-Binding Peptide, p160. Clin. Cancer Res. 2005, 11, 6705–6712. [Google Scholar] [CrossRef] [Green Version]
- Newton, J.R.; Miao, Y.; Deutscher, S.L.; Quinn, T.P. Melanoma imaging with pretargeted bivalent bacteriophage. J. Nucl. Med. 2007, 48, 429–436. [Google Scholar]
- Deutscher, S.L. Phage display in molecular imaging and diagnosis of cancer. Chem. Rev. 2010, 110, 3196–3211. [Google Scholar] [CrossRef] [Green Version]
- Staquicini, F.I.; Sidman, R.L.; Arap, W.; Pasqualini, R. Phage display technology for stem cell delivery and systemic therapy. Adv. Drug Deliv. Rev. 2010, 62, 1213–1216. [Google Scholar] [CrossRef]
- Hyman, P. Bacteriophages and Nanostructured Materials. Adv. Appl. Microbiol. 2012, 78, 55–73. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.-S.; Kim, W.-G.; Kim, C.; Park, G.-T.; Heo, J.; Yoo, S.Y.; Oh, J.-W. M13 Bacteriophage-Based Self-Assembly Structures and Their Functional Capabilities. Mini-Rev. Org. Chem. 2015, 12, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Yang, M.; Zhu, Y.; Wang, L.; Tomsia, A.P.; Mao, C. Phage nanofibers induce vascularized osteogenesis in 3d printed bone scaffolds. Adv. Mater. 2014, 26, 4961–4966. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.-Y.; Lee, H.; Kim, Y.; Yoo, S.Y.; Chung, W.-J.; Kim, G. Phage as versatile nanoink for printing 3-D cell-laden scaffolds. Acta Biomater. 2016, 29, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.Y.; Merzlyak, A.; Lee, S.-W. Facile growth factor immobilization platform based on engineered phage matrices. Soft Matter 2011, 7, 1660–1666. [Google Scholar] [CrossRef]
- García, R.; Latz, S.; Romero, J.; Higuera, G.; García, K.; Bastías, R. Bacteriophage Production Models: An Overview. Front. Microbiol. 2019, 10, 1187. [Google Scholar] [CrossRef]
- Cao, B.; Li, Y.; Yang, T.; Bao, Q.; Yang, M.; Mao, C. Bacteriophage-based biomaterials for tissue regeneration. Adv. Drug Deliv. Rev. 2019, 145, 73–95. [Google Scholar] [CrossRef]
- Bellis, S.L. Advantages of RGD peptides for directing cell association with biomaterials. Biomaterials 2011, 32, 4205–4210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merzlyak, A.; Indrakanti, S.; Lee, S.-W. Genetically engineered nanofiber-like viruses for tissue regenerating materials. Nano Lett. 2009, 9, 846–852. [Google Scholar] [CrossRef]
- Chung, W.-J.; Merzlyak, A.; Yoo, S.Y.; Lee, S.-W. Genetically engineered liquid-crystalline viral films for directing neural cell growth. Langmuir 2010, 26, 9885–9890. [Google Scholar] [CrossRef]
- Yoo, S.Y.; Chung, W.-J.; Kim, T.H.; Le, M.; Lee, S.-W. Facile patterning of genetically engineered M13 bacteriophage for directional growth of human fibroblast cells. Soft Matter 2011, 7, 363–368. [Google Scholar] [CrossRef]
- Chung, W.-J.; Merzlyak, A.; Lee, S.-W. Fabrication of engineered M13 bacteriophages into liquid crystalline films and fibers for directional growth and encapsulation of fibroblasts. Soft Matter 2010, 6, 4454–4459. [Google Scholar] [CrossRef]
- Shao, Z.; Zhang, X.; Pi, Y.; Wang, X.; Jia, Z.; Zhu, J.; Dai, L.; Chen, W.; Yin, L.; Chen, H.; et al. Polycaprolactone electrospun mesh conjugated with an MSC affinity peptide for MSC homing in vivo. Biomaterials 2012, 33, 3375–3387. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.Y.; Kobayashi, M.; Lee, P.P.; Lee, S.-W. Early osteogenic differentiation of mouse preosteoblasts induced by collagen-derived dgea-peptide on nanofibrous phage tissue matrices. Biomacromolecules 2011, 12, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Raja, I.S.; Kim, C.; Song, S.-J.; Shin, Y.C.; Kang, M.S.; Hyon, S.-H.; Oh, J.-W.; Han, D.-W. Virus-Incorporated Biomimetic Nanocomposites for Tissue Regeneration. Nanomaterials 2019, 9, 1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paczesny, J.; Bielec, K. Application of Bacteriophages in Nanotechnology. Nanomaterials 2020, 10, 1944. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gibb, B.; Hyman, P.; Schneider, C.L. The Many Applications of Engineered Bacteriophages—An Overview. Pharmaceuticals 2021, 14, 634. https://doi.org/10.3390/ph14070634
Gibb B, Hyman P, Schneider CL. The Many Applications of Engineered Bacteriophages—An Overview. Pharmaceuticals. 2021; 14(7):634. https://doi.org/10.3390/ph14070634
Chicago/Turabian StyleGibb, Bryan, Paul Hyman, and Christine L. Schneider. 2021. "The Many Applications of Engineered Bacteriophages—An Overview" Pharmaceuticals 14, no. 7: 634. https://doi.org/10.3390/ph14070634