
SapC–DOPS as a Novel Therapeutic and Diagnostic Agent for Glioblastoma Therapy and Detection: Alternative to Old Drugs and Agents

 ,
,
Abstract
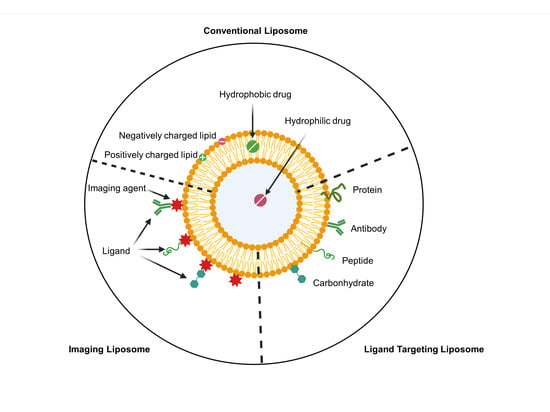
:
1. Introduction
2. Standard Diagnosis and Treatment of GBM
3. Nanocarrier as a Drug Delivery System for GBM Therapy
4. Application of Nanodrugs as Radiosensitizers for GBM Therapy
5. Radiolabeled Nanodrugs for GBM Therapy and Imaging
6. SapC–DOPS Nanovesicles for Precise Targeting of Brain Tumors
7. SapC–DOPS Nanovesicles for Tumor Detection
8. SapC–DOPS as a Carrier for Radioisotopes
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ostrom, Q.T.; Patil, N.; Cioffi, G.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017. Neuro-oncology 2020, 22, iv1–iv96. [Google Scholar] [CrossRef]
- Available online: https://www.aans.org/en/Patients/Neurosurgical-Conditions-and-Treatments/Brain-Tumors (accessed on 19 November 2021).
- Cagney, D.N.; Martin, A.M.; Catalano, P.J.; Redig, A.J.; Lin, N.U.; Lee, E.Q.; Wen, P.Y.; Dunn, I.F.; Bi, W.L.; Weiss, S.E.; et al. Incidence and prognosis of patients with brain metastases at diagnosis of systemic malignancy: A population-based study. Neuro-oncology 2017, 19, 1511–1521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uddin, M.S.; Mamun, A.A.; Alghamdi, B.S.; Tewari, D.; Jeandet, P.; Sarwar, M.S.; Ashraf, G.M. Epigenetics of glioblastoma multiforme: From molecular mechanisms to therapeutic approaches. Semin. Cancer Biol. 2020, in press. [Google Scholar] [CrossRef]
- Oronsky, B.; Reid, T.R.; Oronsky, A.; Sandhu, N.; Knox, S.J. A Review of Newly Diagnosed Glioblastoma. Front. Oncol. 2020, 10, 574012. [Google Scholar] [CrossRef] [PubMed]
- Stelzer, K.J. Epidemiology and prognosis of brain metastases. Surg. Neurol. Int. 2013, 4, S192–S202. [Google Scholar] [CrossRef]
- Alphandéry, E. Glioblastoma Treatments: An Account of Recent Industrial Developments. Front. Pharmacol. 2018, 9, 879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campos, B.; Olsen, L.R.; Urup, T.; Poulsen, H.S. A comprehensive profile of recurrent glioblastoma. Oncogene 2016, 35, 5819–5825. [Google Scholar] [CrossRef] [PubMed]
- Dréan, A.; Goldwirt, L.; Verreault, M.; Canney, M.; Schmitt, C.; Guehennec, J.; Delattre, J.-Y.; Carpentier, A.; Idbaih, A. Blood-brain barrier, cytotoxic chemotherapies and glioblastoma. Expert Rev. Neurother. 2016, 16, 1285–1300. [Google Scholar] [CrossRef]
- Harder, B.G.; Blomquist, M.R.; Wang, J.; Kim, A.J.; Woodworth, G.F.; Winkles, J.A.; Loftus, J.C.; Tran, N.L. Developments in Blood-Brain Barrier Penetrance and Drug Repurposing for Improved Treatment of Glioblastoma. Front. Oncol. 2018, 8, 462. [Google Scholar] [CrossRef] [Green Version]
- de Vries, N.A.; Beijnen, J.H.; Boogerd, W.; van Tellingen, O. Blood-brain barrier and chemotherapeutic treatment of brain tumors. Expert Rev. Neurother. 2006, 6, 1199–1209. [Google Scholar] [CrossRef]
- Zhao, M.; van Straten, D.; Broekman, M.L.D.; Préat, V.; Schiffelers, R.M. Nanocarrier-based drug combination therapy for glioblastoma. Theranostics 2020, 10, 1355–1372. [Google Scholar] [CrossRef]
- Fakhoury, M. Drug delivery approaches for the treatment of glioblastoma multiforme. Artif. Cells Nanomed. Biotechnol. 2016, 44, 1365–1373. [Google Scholar] [CrossRef] [Green Version]
- Kazda, T.; Bulik, M.; Pospisil, P.; Lakomy, R.; Smrcka, M.; Slampa, P.; Jancalek, R. Advanced MRI increases the diagnostic accuracy of recurrent glioblastoma: Single institution thresholds and validation of MR spectroscopy and diffusion weighted MR imaging. NeuroImage Clin. 2016, 11, 316–321. [Google Scholar] [CrossRef] [Green Version]
- Nam, L.; Coll, C.; Erthal, L.C.S.; De la Torre, C.; Serrano, D.; Martínez-Máñez, R.; Santos-Martínez, M.J.; Ruiz-Hernández, E. Drug Delivery Nanosystems for the Localized Treatment of Glioblastoma Multiforme. Materials 2018, 11, 779. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; He, M.Z.; Li, T.; Yang, X. MRI combined with PET-CT of different tracers to improve the accuracy of glioma diagnosis: A systematic review and meta-analysis. Neurosurg. Rev. 2019, 42, 185–195. [Google Scholar] [CrossRef] [Green Version]
- Wadajkar, A.S.; Dancy, J.G.; Hersh, D.S.; Anastasiadis, P.; Tran, N.L.; Woodworth, G.F.; Winkles, J.A.; Kim, A.J. Tumor-targeted nanotherapeutics: Overcoming treatment barriers for glioblastoma. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, e1439. [Google Scholar] [CrossRef]
- Lee, C.Y. Strategies of temozolomide in future glioblastoma treatment. OncoTargets Ther. 2017, 10, 265–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Carter, T.C.; Medina-Flores, R.; Lawler, B.E. Glioblastoma Treatment with Temozolomide and Bevacizumab and Overall Survival in a Rural Tertiary Healthcare Practice. BioMed Res. Int. 2018, 2018, 6204676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khosla, D. Concurrent therapy to enhance radiotherapeutic outcomes in glioblastoma. Ann. Transl. Med. 2016, 4, 54. [Google Scholar] [CrossRef]
- Abshire, D.; Lang, M.K. The Evolution of Radiation Therapy in Treating Cancer. Semin. Oncol. Nurs. 2018, 34, 151–157. [Google Scholar] [CrossRef]
- Shenouda, G.; Souhami, L.; Petrecca, K.; Owen, S.; Panet-Raymond, V.; Guiot, M.C.; Corredor, A.G.; Abdulkarim, B. A Phase 2 Trial of Neoadjuvant Temozolomide Followed by Hypofractionated Accelerated Radiation Therapy With Concurrent and Adjuvant Temozolomide for Patients With Glioblastoma. Int. J. Radiat. Oncol. Biol. Phys. 2017, 97, 487–494. [Google Scholar] [CrossRef]
- Sofia Vala, I.; Martins, L.R.; Imaizumi, N.; Nunes, R.J.; Rino, J.; Kuonen, F.; Carvalho, L.M.; Rüegg, C.; Grillo, I.M.; Barata, J.T.; et al. Low doses of ionizing radiation promote tumor growth and metastasis by enhancing angiogenesis. PLoS ONE 2010, 5, e11222. [Google Scholar] [CrossRef] [PubMed]
- Malaise, E.P.; Lambin, P.; Joiner, M.C. Radiosensitivity of Human Cell Lines to Small Doses. Are There Some Clinical Implications? Radiat. Res. 1994, 138, S25–S27. [Google Scholar] [CrossRef] [PubMed]
- Lambin, P.; Malaise, E.P.; Joiner, M.C. Might intrinsic radioresistance of human tumour cells be induced by radiation? Int. J. Radiat. Biol. 1996, 69, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.U.; Hosotani, R.; Wada, M.; Doi, R.; Kosiba, T.; Fujimoto, K.; Miyamoto, Y.; Tsuji, S.; Nakajima, S.; Nishimura, Y.; et al. Role of Bcl-2 family proteins (Bax, Bcl-2 and Bcl-X) on cellular susceptibility to radiation in pancreatic cancer cells. Eur. J. Cancer 1999, 35, 1374–1380. [Google Scholar] [CrossRef]
- Davis, H.W.; Vallabhapurapu, S.D.; Chu, Z.; Vallabhapurapu, S.L.; Franco, R.S.; Mierzwa, M.; Kassing, W.; Barrett, W.L.; Qi, X. Enhanced phosphatidylserine-selective cancer therapy with irradiation and SapC-DOPS nanovesicles. Oncotarget 2019, 10, 856–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taal, W.; Oosterkamp, H.M.; Walenkamp, A.M.; Dubbink, H.J.; Beerepoot, L.V.; Hanse, M.C.; Buter, J.; Honkoop, A.H.; Boerman, D.; de Vos, F.Y.; et al. Single-agent bevacizumab or lomustine versus a combination of bevacizumab plus lomustine in patients with recurrent glioblastoma (BELOB trial): A randomised controlled phase 2 trial. Lancet Oncol. 2014, 15, 943–953. [Google Scholar] [CrossRef]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef]
- Provenzale, J.M.; Silva, G.A. Uses of Nanoparticles for Central Nervous System Imaging and Therapy. Am. J. Neuroradiol. 2009, 30, 1293–1301. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [Green Version]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Cascone, T.; McKenzie, J.A.; Mbofung, R.M.; Punt, S.; Wang, Z.; Xu, C.; Williams, L.J.; Wang, Z.; Bristow, C.A.; Carugo, A.; et al. Increased Tumor Glycolysis Characterizes Immune Resistance to Adoptive T Cell Therapy. Cell Metab. 2018, 27, 977–987.e974. [Google Scholar] [CrossRef] [PubMed]
- Pelicano, H.; Martin, D.S.; Xu, R.H.; Huang, P. Glycolysis inhibition for anticancer treatment. Oncogene 2006, 25, 4633–4646. [Google Scholar] [CrossRef] [Green Version]
- Gutkin, A.; Cohen, Z.R.; Peer, D. Harnessing nanomedicine for therapeutic intervention in glioblastoma. Expert Opin. Drug Deliv. 2016, 13, 1573–1582. [Google Scholar] [CrossRef]
- Kim, S.-S.; Rait, A.; Kim, E.; DeMarco, J.; Pirollo, K.F.; Chang, E.H. Encapsulation of temozolomide in a tumor-targeting nanocomplex enhances anti-cancer efficacy and reduces toxicity in a mouse model of glioblastoma. Cancer Lett. 2015, 369, 250–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, D.; Zhang, S.; Zhong, T.; Ren, W.; Yao, X.; Guo, Y.; Duan, X.-C.; Yin, Y.-F.; Zhang, S.-S.; Zhang, X. Multi-targeting NGR-modified liposomes recognizing glioma tumor cells and vasculogenic mimicry for improving anti-glioma therapy. Oncotarget 2016, 7, 43616–43628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, Z.; Li, Y.; Pang, H.; Zheng, Y.; Zhao, Y. Pep-1 peptide-functionalized liposome to enhance the anticancer efficacy of cilengitide in glioma treatment. Colloids Surf. B Biointerfaces 2017, 158, 68–75. [Google Scholar] [CrossRef]
- Beier, C.P.; Schmid, C.; Gorlia, T.; Kleinletzenberger, C.; Beier, D.; Grauer, O.; Steinbrecher, A.; Hirschmann, B.; Brawanski, A.; Dietmaier, C.; et al. RNOP-09: Pegylated liposomal doxorubicine and prolonged temozolomide in addition to radiotherapy in newly diagnosed glioblastoma—A phase II study. BMC Cancer 2009, 9, 308. [Google Scholar] [CrossRef] [Green Version]
- A Phase I Trial of Nanoliposomal CPT-11 (NL CPT-11) in Patients with Recurrent High-Grade Gliomas. Available online: https://clinicaltrials.gov/ct2/show/NCT00734682 (accessed on 19 November 2021).
- Rixe, O.; Morris, J.C.; Puduvalli, V.K.; Villano, J.L.; Wise-Draper, T.M.; Muller, C.; Johnson, A.N.; Wesolowski, R.; Qi, X. First-in-human, first-in-class phase 1a study of BXQ-350 for solid tumors and gliomas. J. Clin. Oncol. 2018, 36, 2517. [Google Scholar] [CrossRef]
- A Study of Intraventricular Liposomal Encapsulated Ara-C (DepoCyt) in Patients with Recurrent Glioblastoma. Available online: https://clinicaltrials.gov/ct2/show/NCT01044966 (accessed on 19 November 2021).
- Brenner, A.J.; Bao, A.; Phillips, W.; Stein, G.; Awasthi, V.; Patel, T.R.; Weinberg, J.; Floyd, J. Safety and feasibility of rhenium-186 nanoliposome (186RNL) in recurrent glioma: The ReSPECT phase 1 trial. J. Clin. Oncol. 2021, 39, 2061. [Google Scholar] [CrossRef]
- Verteporfin for the Treatment of Recurrent High Grade EGFR-Mutated Glioblastoma. Available online: https://clinicaltrials.gov/ct2/show/NCT04590664 (accessed on 19 November 2021).
- A Study of RNA-lipid Particle (RNA-LP) Vaccines for Newly Diagnosed Pediatric High-Grade Gliomas (pHGG) and Adult Glioblastoma (GBM). Available online: https://clinicaltrials.gov/ct2/show/NCT04573140 (accessed on 19 November 2021).
- A Study of BXQ-350 in Children With Newly Diagnosed Diffuse Intrinsic Pontine Glioma (DIPG) or Diffuse Midline Glioma (DMG) (KONQUER). Available online: https://clinicaltrials.gov/ct2/show/NCT04771897 (accessed on 19 November 2021).
- Chédeville, A.L.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Radiotherapy Resistance. Cancers 2021, 13, 542. [Google Scholar] [CrossRef]
- Hu, R.; Saito, A.I.; Mitsuhashi, T.; Inoue, T.; Ota, T.; Ujihira, T.; Yoshida, K.; Sasai, K. Radiosensitization using hydrogen peroxide in patients with cervical cancer. Mol. Clin. Oncol. 2021, 15, 142. [Google Scholar] [CrossRef]
- Oronsky, B.T.; Knox, S.J.; Scicinski, J. Six Degrees of Separation: The Oxygen Effect in the Development of Radiosensitizers. Transl. Oncol. 2011, 4, 189–198. [Google Scholar] [CrossRef] [Green Version]
- Joh, D.Y.; Sun, L.; Stangl, M.; Al Zaki, A.; Murty, S.; Santoiemma, P.P.; Davis, J.J.; Baumann, B.C.; Alonso-Basanta, M.; Bhang, D.; et al. Selective Targeting of Brain Tumors with Gold Nanoparticle-Induced Radiosensitization. PLoS ONE 2013, 8, e62425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elechalawar, C.K.; Bhattacharya, D.; Ahmed, M.T.; Gora, H.; Sridharan, K.; Chaturbedy, P.; Sinha, S.H.; Jaggarapu, M.M.C.S.; Narayan, K.P.; Chakravarty, S.J.N.A. Dual targeting of folate receptor-expressing glioma tumor-associated macrophages and epithelial cells in the brain using a carbon nanosphere–cationic folate nanoconjugate. Nanoscale Adv. 2019, 1, 3555–3567. [Google Scholar] [CrossRef] [Green Version]
- Kefayat, A.; Ghahremani, F.; Motaghi, H.; Amouheidari, A. Ultra-small but ultra-effective: Folic acid-targeted gold nanoclusters for enhancement of intracranial glioma tumors’ radiation therapy efficacy. Nanomed. Nanotechnol. Biol. Med. 2019, 16, 173–184. [Google Scholar] [CrossRef]
- Groysbeck, N.; Stoessel, A.; Donzeau, M.; da Silva, E.C.; Lehmann, M.; Strub, J.-M.; Cianferani, S.; Dembélé, K.; Zuber, G. Synthesis and biological evaluation of 2.4 nm thiolate-protected gold nanoparticles conjugated to Cetuximab for targeting glioblastoma cancer cells via the EGFR. Nanotechnology 2019, 30, 184005. [Google Scholar] [CrossRef] [PubMed]
- Essa, B.M.; El-Mohty, A.A.; El-Hashash, M.A.; Sakr, T.M. 99mTc-citrate-gold nanoparticles as a tumor tracer: Synthesis, characterization, radiolabeling and in-vivo studies. Radiochim. Acta 2020, 108, 809–819. [Google Scholar] [CrossRef]
- Phillips, W.T.; Goins, B.; Bao, A.; Vargas, D.; Guttierez, J.E.; Trevino, A.; Miller, J.R.; Henry, J.; Zuniga, R.; Vecil, G.; et al. Rhenium-186 liposomes as convection-enhanced nanoparticle brachytherapy for treatment of glioblastoma. Neuro-oncology 2012, 14, 416–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, F.-Y.J.; Lee, T.-W.; Chang, C.-H.; Chen, L.-C.; Hsu, W.-H.; Chang, C.-W.; Lo, J.-M. Evaluation of 188Re-labeled PEGylated nanoliposome as a radionuclide therapeutic agent in an orthotopic glioma-bearing rat model. Int. J. Nanomed. 2015, 10, 463–473. [Google Scholar] [CrossRef] [Green Version]
- Mathen, P.; Rowe, L.; Mackey, M.; Smart, D.; Tofilon, P.; Camphausen, K. Radiosensitizers in the temozolomide era for newly diagnosed glioblastoma. Neuro-Oncol. Pract. 2019, 7, 268–276. [Google Scholar] [CrossRef] [Green Version]
- Wilson, G.D.; Bentzen, S.M.; Harari, P.M. Biologic Basis for Combining Drugs With Radiation. Semin. Radiat. Oncol. 2006, 16, 2–9. [Google Scholar] [CrossRef]
- Sandblom, V.; Spetz, J.; Shubbar, E.; Montelius, M.; Ståhl, I.; Swanpalmer, J.; Nilsson, O.; Forssell-Aronsson, E. Gemcitabine potentiates the anti-tumour effect of radiation on medullary thyroid cancer. PLoS ONE 2019, 14, e0225260. [Google Scholar] [CrossRef]
- Waissi, W.; Nicol, A.; Jung, M.; Rousseau, M.; Jarnet, D.; Noel, G.; Burckel, H. Radiosensitizing Pancreatic Cancer with PARP Inhibitor and Gemcitabine: An In Vivo and a Whole-Transcriptome Analysis after Proton or Photon Irradiation. Cancers 2021, 13, 527. [Google Scholar] [CrossRef]
- Augustin, M.; Wilhelm, M.; Reichert, B.; Siegler, G.M.; Dreier, J.; Rottmann, M.; Blos, M.; Kalisch, A.; Dressler, S.; Stein, H.; et al. Radiochemotherapy with gemcitabine as radiosensitizer in patients with soft tissue sarcoma. J. Clin. Oncol. 2020, 38, e23559. [Google Scholar] [CrossRef]
- Gao, J.; Fang, L.; Sun, D.; Shen, Y.; Hu, Y.; Li, N.; Chang, J.; Li, W.; Tan, J. 131I-labeled and DOX-loaded multifunctional nanoliposomes for radiotherapy and chemotherapy in brain gliomas. Brain Res. 2020, 1739, 145218. [Google Scholar] [CrossRef] [PubMed]
- Oku, N.; Yamashita, M.; Katayama, Y.; Urakami, T.; Hatanaka, K.; Shimizu, K.; Asai, T.; Tsukada, H.; Akai, S.; Kanazawa, H. PET imaging of brain cancer with positron emitter-labeled liposomes. Int. J. Pharm. 2011, 403, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Malinge, J.; Géraudie, B.; Savel, P.; Nataf, V.; Prignon, A.; Provost, C.; Zhang, Y.; Ou, P.; Kerrou, K.; Talbot, J.N.; et al. Liposomes for PET and MR Imaging and for Dual Targeting (Magnetic Field/Glucose Moiety): Synthesis, Properties, and in Vivo Studies. Mol. Pharm. 2017, 14, 406–414. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.Y.; Lee, T.W.; Kao, C.H.; Chang, C.H.; Zhang, X.; Lee, W.Y.; Chen, W.J.; Wang, S.C.; Lo, J.M. Imaging, autoradiography, and biodistribution of (188)Re-labeled PEGylated nanoliposome in orthotopic glioma bearing rat model. Cancer Biother. Radiopharm. 2011, 26, 717–725. [Google Scholar] [CrossRef]
- Bevers, E.M.; Williamson, P.L. Getting to the Outer Leaflet: Physiology of Phosphatidylserine Exposure at the Plasma Membrane. Physiol. Rev. 2016, 96, 605–645. [Google Scholar] [CrossRef]
- Connor, J.; Bucana, C.; Fidler, I.J.; Schroit, A.J. Differentiation-dependent expression of phosphatidylserine in mammalian plasma membranes: Quantitative assessment of outer-leaflet lipid by prothrombinase complex formation. Proc. Natl. Acad. Sci. USA 1989, 86, 3184–3188. [Google Scholar] [CrossRef] [Green Version]
- Riedl, S.; Rinner, B.; Asslaber, M.; Schaider, H.; Walzer, S.; Novak, A.; Lohner, K.; Zweytick, D. In search of a novel target—Phosphatidylserine exposed by non-apoptotic tumor cells and metastases of malignancies with poor treatment efficacy. Biochim. Biophys. Acta 2011, 1808, 2638–2645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolter, T.; Sandhoff, K. Lysosomal degradation of membrane lipids. FEBS Lett. 2010, 584, 1700–1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, H.W.; Vallabhapurapu, S.D.; Chu, Z.; Wyder, M.A.; Greis, K.D.; Fannin, V.; Sun, Y.; Desai, P.B.; Pak, K.Y.; Gray, B.D.; et al. Biotherapy of Brain Tumors with Phosphatidylserine-Targeted Radioiodinated SapC-DOPS Nanovesicles. Cells 2020, 9, 1960. [Google Scholar] [CrossRef] [PubMed]
- Blanco, V.M.; Chu, Z.; Vallabhapurapu, S.D.; Sulaiman, M.K.; Kendler, A.; Rixe, O.; Warnick, R.E.; Franco, R.S.; Qi, X. Phosphatidylserine-selective targeting and anticancer effects of SapC-DOPS nanovesicles on brain tumors. Oncotarget 2014, 5, 7105–7118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojton, J.; Chu, Z.; Mathsyaraja, H.; Meisen, W.H.; Denton, N.; Kwon, C.-H.; Chow, L.M.; Palascak, M.; Franco, R.; Bourdeau, T.; et al. Systemic delivery of SapC-DOPS has antiangiogenic and antitumor effects against glioblastoma. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 1517–1525. [Google Scholar] [CrossRef] [Green Version]
- Frosina, G. DNA repair and resistance of gliomas to chemotherapy and radiotherapy. Mol. Cancer Res. MCR 2009, 7, 989–999. [Google Scholar] [CrossRef] [Green Version]
- Van Meir, E.G.; Hadjipanayis, C.G.; Norden, A.D.; Shu, H.K.; Wen, P.Y.; Olson, J.J. Exciting new advances in neuro-oncology: The avenue to a cure for malignant glioma. CA Cancer J. Clin. 2010, 60, 166–193. [Google Scholar] [CrossRef]
- Wojton, J.; Meisen, W.H.; Jacob, N.K.; Thorne, A.H.; Hardcastle, J.; Denton, N.; Chu, Z.; Dmitrieva, N.; Marsh, R.; Van Meir, E.G.; et al. SapC-DOPS-induced lysosomal cell death synergizes with TMZ in glioblastoma. Oncotarget 2014, 5, 9703–9709. [Google Scholar] [CrossRef] [Green Version]
- Kaimal, V.; Chu, Z.; Mahller, Y.Y.; Papahadjopoulos-Sternberg, B.; Cripe, T.P.; Holland, S.K.; Qi, X. Saposin C coupled lipid nanovesicles enable cancer-selective optical and magnetic resonance imaging. Mol. Imaging Biol. 2011, 13, 886–897. [Google Scholar] [CrossRef]
- Blanco, V.M.; Chu, Z.; LaSance, K.; Gray, B.D.; Pak, K.Y.; Rider, T.; Greis, K.D.; Qi, X. Optical and nuclear imaging of glioblastoma with phosphatidylserine-targeted nanovesicles. Oncotarget 2016, 7, 32866–32875. [Google Scholar] [CrossRef] [PubMed]
- Winter, P.M.; Pearce, J.; Chu, Z.; McPherson, C.M.; Takigiku, R.; Lee, J.H.; Qi, X. Imaging of brain tumors with paramagnetic vesicles targeted to phosphatidylserine. J. Magn. Reson. Imaging 2015, 41, 1079–1087. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Luster, T.A.; Thorpe, P.E. Radiation-enhanced vascular targeting of human lung cancers in mice with a monoclonal antibody that binds anionic phospholipids. Clin. Cancer Res. 2007, 13, 5211–5218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.; Yin, Y.; Luster, T.A.; Watkins, L.; Thorpe, P.E. Antiphosphatidylserine antibody combined with irradiation damages tumor blood vessels and induces tumor immunity in a rat model of glioblastoma. Clin. Cancer Res. 2009, 15, 6871–6880. [Google Scholar] [CrossRef] [Green Version]
- Saha, D.; Watkins, L.; Yin, Y.; Thorpe, P.; Story, M.D.; Song, K.; Raghavan, P.; Timmerman, R.; Chen, B.; Minna, J.D.; et al. An orthotopic lung tumor model for image-guided microirradiation in rats. Radiat Res. 2010, 174, 62–71. [Google Scholar] [CrossRef] [Green Version]
- Dekempeneer, Y.; Keyaerts, M.; Krasniqi, A.; Puttemans, J.; Muyldermans, S.; Lahoutte, T.; D’Huyvetter, M.; Devoogdt, N. Targeted alpha therapy using short-lived alpha-particles and the promise of nanobodies as targeting vehicle. Expert Opin. Biol. Ther. 2016, 16, 1035–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, D.J.; Orgun, N.N.; Jones, J.C.; Hylarides, M.D.; Pagel, J.M.; Hamlin, D.K.; Wilbur, D.S.; Lin, Y.; Fisher, D.R.; Kenoyer, A.L.; et al. A preclinical model of CD38-pretargeted radioimmunotherapy for plasma cell malignancies. Cancer Res. 2014, 74, 1179–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pak, C.C.; Fidler, I.J. Molecular mechanisms for activated macrophage recognition of tumor cells. Semin. Cancer Biol. 1991, 2, 189–195. [Google Scholar]
- Liu, E.K.; Sulman, E.P.; Wen, P.Y.; Kurz, S.C. Novel Therapies for Glioblastoma. Curr. Neurol. Neurosci. Rep. 2020, 20, 19. [Google Scholar] [CrossRef]
- Shah, F.H.; Salman, S.; Idrees, J.; Idrees, F.; Shah, S.T.A.; Khan, A.A.; Ahmad, B. Current Progress of Phytomedicine in Glioblastoma Therapy. Curr. Med Sci. 2020, 40, 1067–1074. [Google Scholar] [CrossRef]



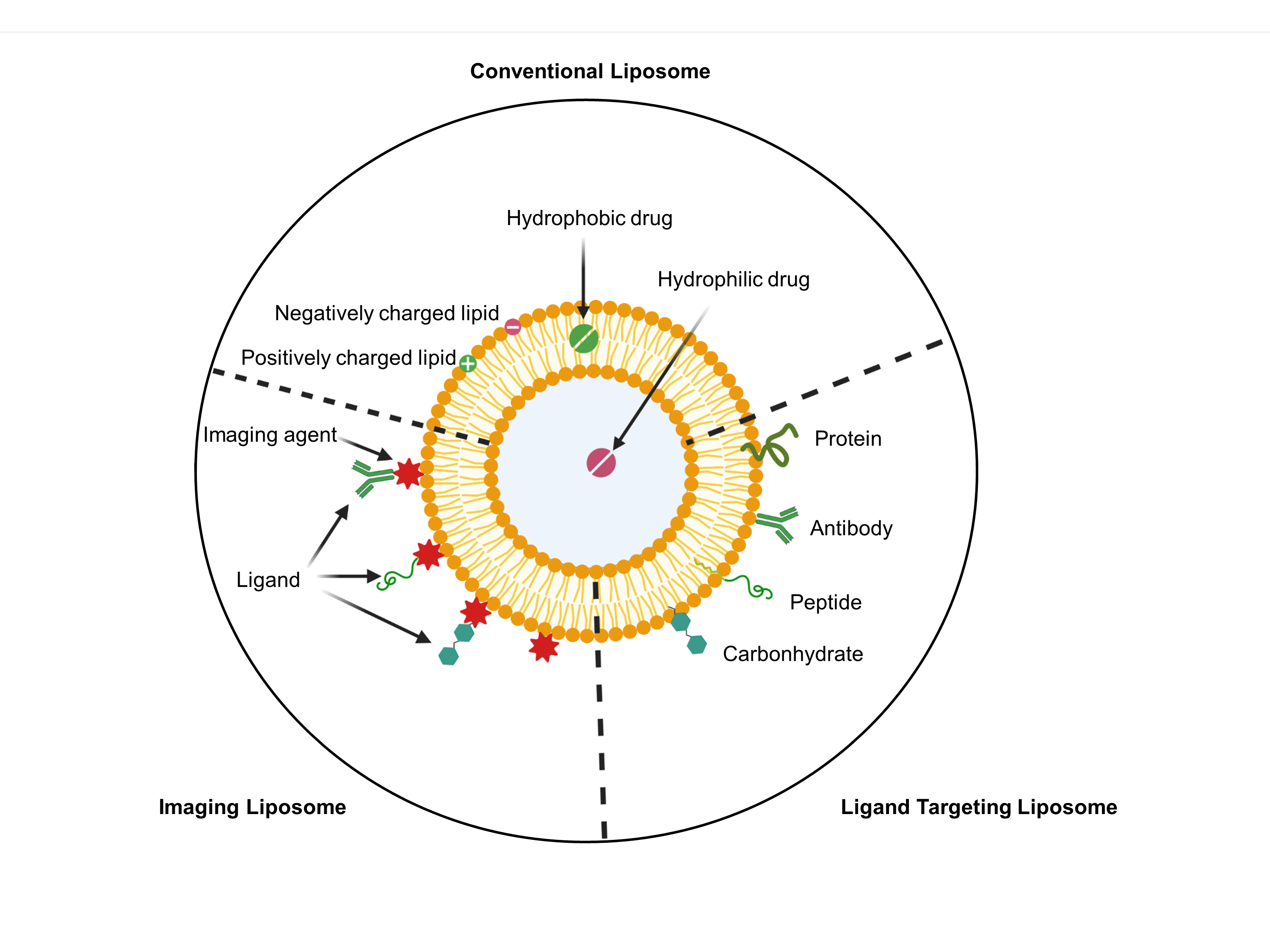{kind=link}
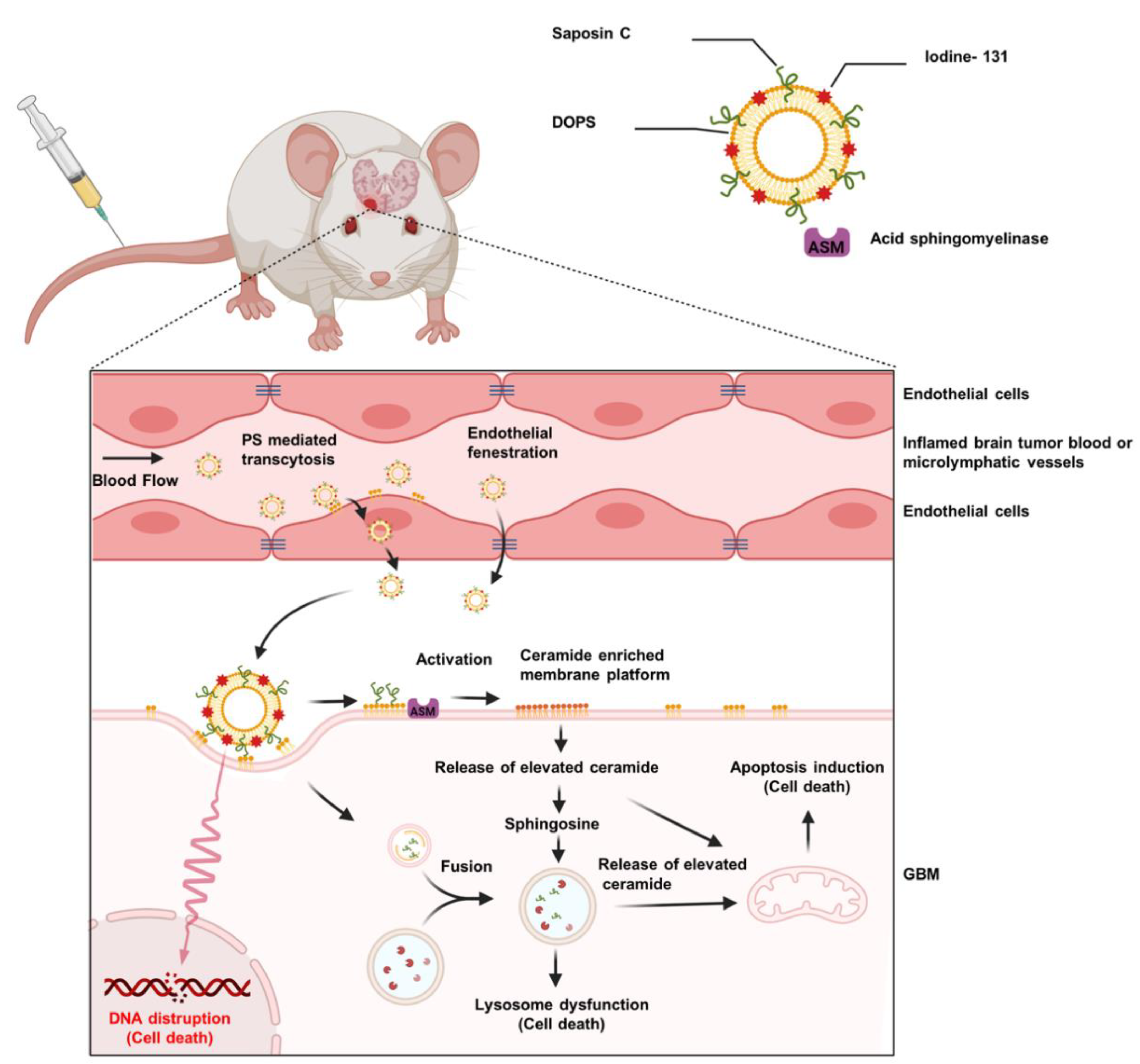{kind=link}
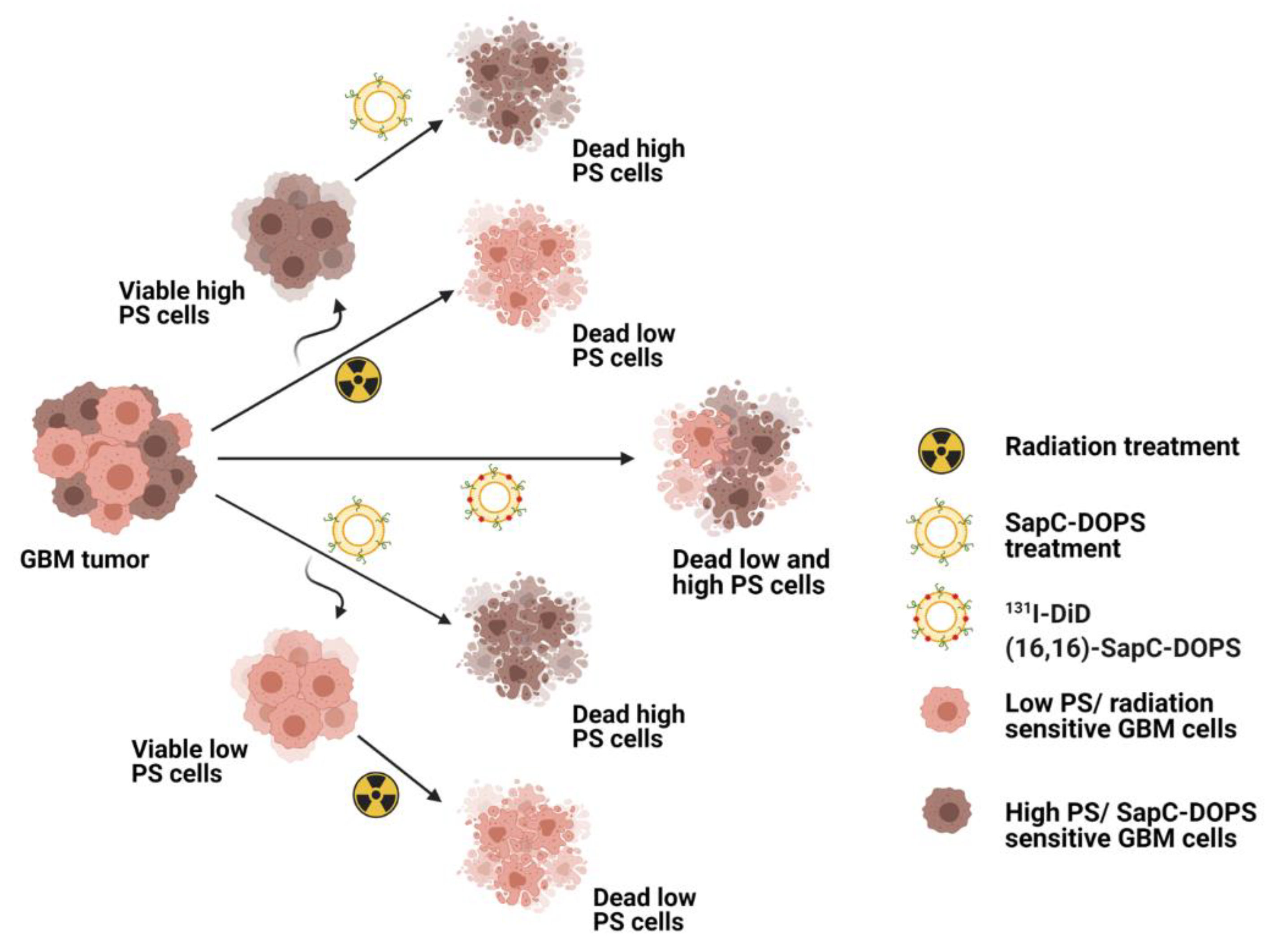{kind=link}
| Grade | Tumor |
|---|---|
| I | Tumor grows slowly and rarely spreads into adjacent brain or CNS tissue. Surgery is normally successful. |
| II | Tumor grows slowly but may spread into adjacent brain or CNS tissues and may recur following resection. |
| III | Tumor grows quickly, generally spreads, and its cells have an abnormal morphology. |
| IV | Tumor grows and spreads very quickly, and its cells have an abnormal morphology. |
| Clinical Trial Identifier | Trial Phase | Therapeutic Agent | Patient Profile | Number of Patient | Completion Status | Result | Ref. |
|---|---|---|---|---|---|---|---|
| NCT00944801 | Phase I/II | PEGylated liposomal doxorubicin (PEG-Dox) | Newly diagnosed glioblastoma | 63 | Completed | Well-tolerated toxicity. No significant survival benefit from PEG-Dox combination with RT compared to TMZ combination with RT. | [40] |
| NCT00734682 | Phase I | Nanoliposomal CPT-11 (liposomal irinotecan) | Recurrent high-grade gliomas | 34 | Completed | Not available. | [41] |
| NCT02859857 | Phase I | SapC–DOPS (BXQ-350) | Solid tumors and glioma | 86 | Completed | The therapy was well tolerated; no dose-limiting toxicity or serious adverse events were observed. | [42] |
| NCT01044966 | Phase I/II | Intraventricular liposomal encapsulated Ara-C (DepoCyt) | Recurrent glioblastoma | 12 | Terminated | The study was terminated due to lack of adequate patient enrolment into trial. | [43] |
| NCT01906385 | Phase I/II | Rhenium-186 nanoliposome (186RNL) | Recurrent glioma | 55 | Recruiting | The therapy was well tolerated; no dose-limiting toxicity or serious adverse events were observed. | [44] |
| NCT04590664 | Phase I/II | Liposomal verteporfin (Visudyne) | High-grade EGFR-mutated glioblastoma | 24 | Recruiting | Not available. | [45] |
| NCT04573140 | Phase I | mRNA lipid particle (RNA-LP) | Newly diagnosed pediatric high-grade gliomas and adult glioblastoma | 28 | Recruiting | Not available. | [46] |
| NCT04771897 | Phase I | SapC–DOPS (BXQ-350) | Newly diagnosed diffuse intrinsic pontine glioma or diffuse midline glioma | 22 | Recruiting | Not available. | [47] |
| GBM Cells | |||
|---|---|---|---|
| Fraction Affected (Fa) | X12v2 | GBM169 | Gli36EGFR |
| 0.2 | Strong synergy * | Synergy | Moderate synergy |
| 0.4 | Strong synergy | Strong synergy | Strong synergy |
| 0.6 | Strong synergy | Strong synergy | Strong synergy |
| 0.8 | Strong synergy | Synergy | Strong synergy |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaynak, A.; Davis, H.W.; Vallabhapurapu, S.D.; Pak, K.Y.; Gray, B.D.; Qi, X. SapC–DOPS as a Novel Therapeutic and Diagnostic Agent for Glioblastoma Therapy and Detection: Alternative to Old Drugs and Agents. Pharmaceuticals 2021, 14, 1193. https://doi.org/10.3390/ph14111193
Kaynak A, Davis HW, Vallabhapurapu SD, Pak KY, Gray BD, Qi X. SapC–DOPS as a Novel Therapeutic and Diagnostic Agent for Glioblastoma Therapy and Detection: Alternative to Old Drugs and Agents. Pharmaceuticals. 2021; 14(11):1193. https://doi.org/10.3390/ph14111193
Chicago/Turabian StyleKaynak, Ahmet, Harold W. Davis, Subrahmanya D. Vallabhapurapu, Koon Y. Pak, Brian D. Gray, and Xiaoyang Qi. 2021. "SapC–DOPS as a Novel Therapeutic and Diagnostic Agent for Glioblastoma Therapy and Detection: Alternative to Old Drugs and Agents" Pharmaceuticals 14, no. 11: 1193. https://doi.org/10.3390/ph14111193