Overcoming Barriers for siRNA Therapeutics: From Bench to Bedside

 , ,
, ,
Abstract
:
1. Introduction
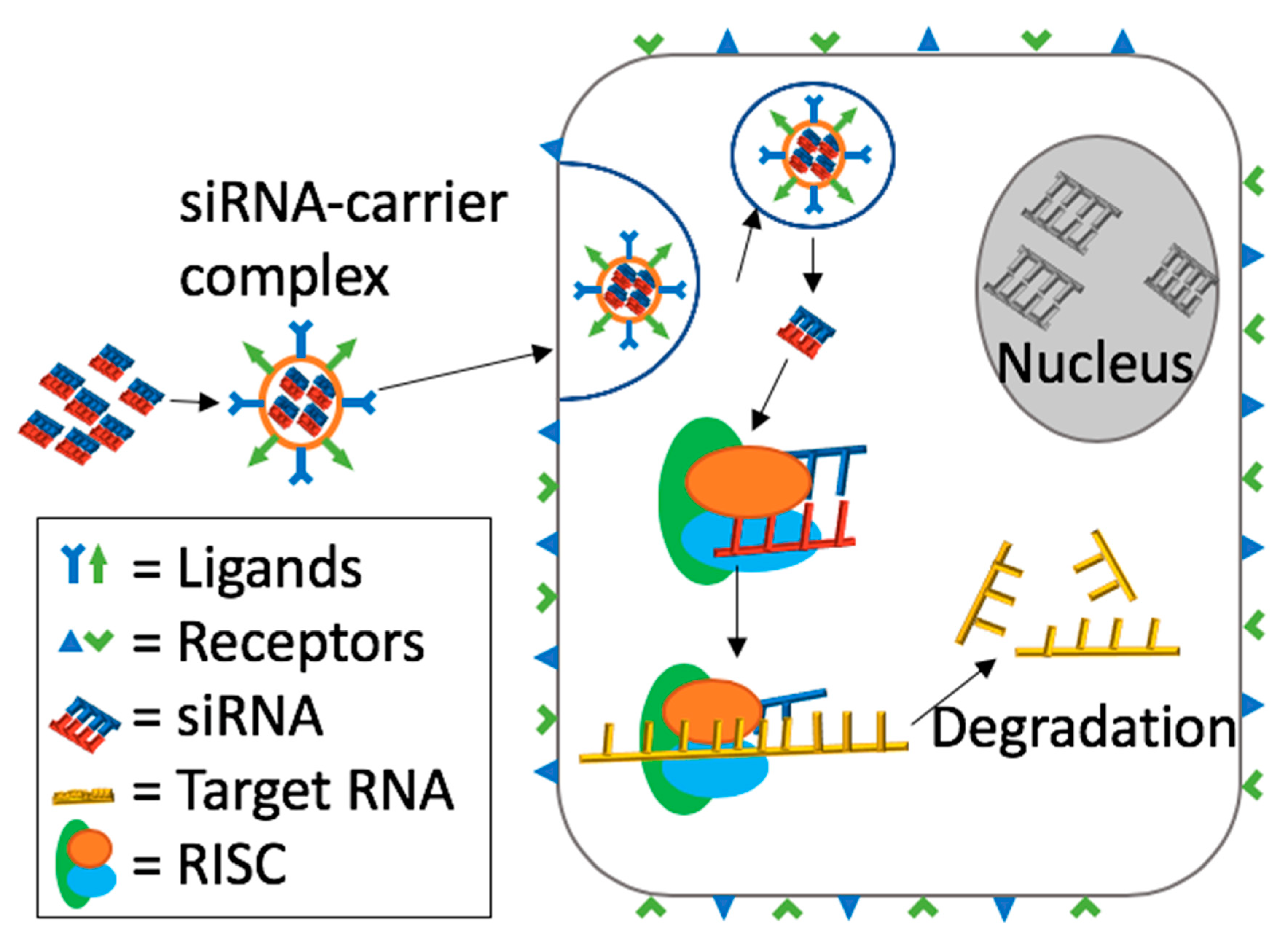
2. Mechanism of Action of siRNA Induced RNA Interference (RNAi)
3. Advantages of siRNA over Other RNAi Therapeutics
4. siRNA-Based Therapeutics in Clinical Trials
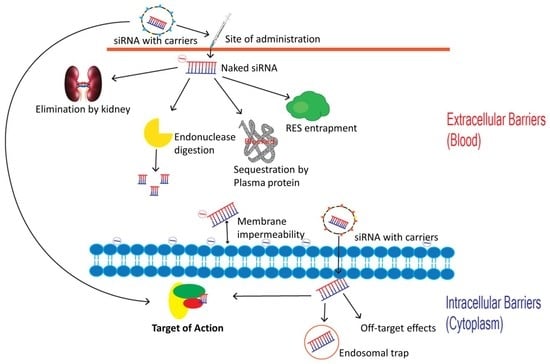
5. Barriers to siRNA Delivery
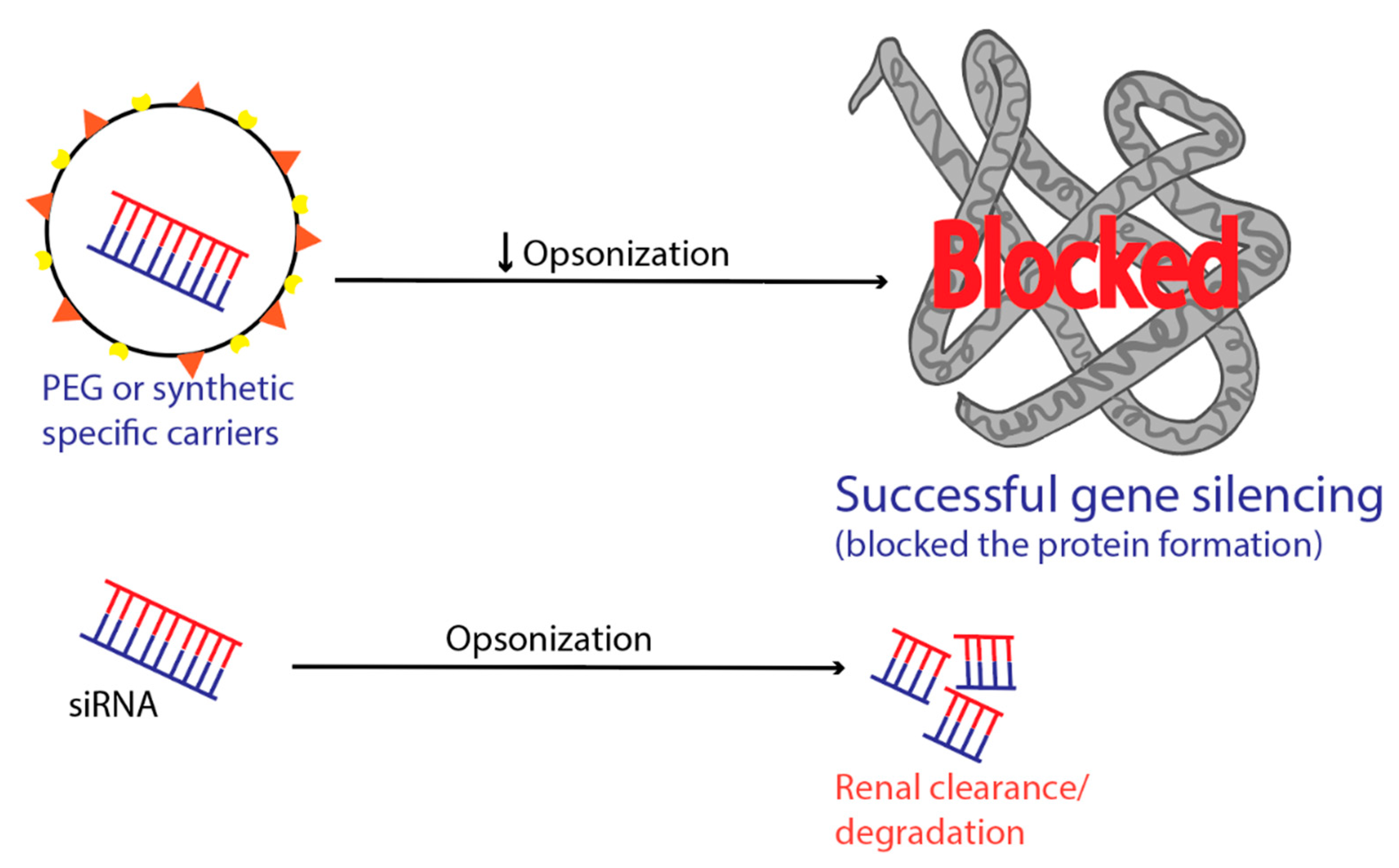
6. Intravascular Degradation and Renal Clearance
7. Activation of the Innate Immune System
7.1. TLR Dependent Pathway
7.2. TLR Independent Pathway
8. Protein Binding
9. RES Entrapment
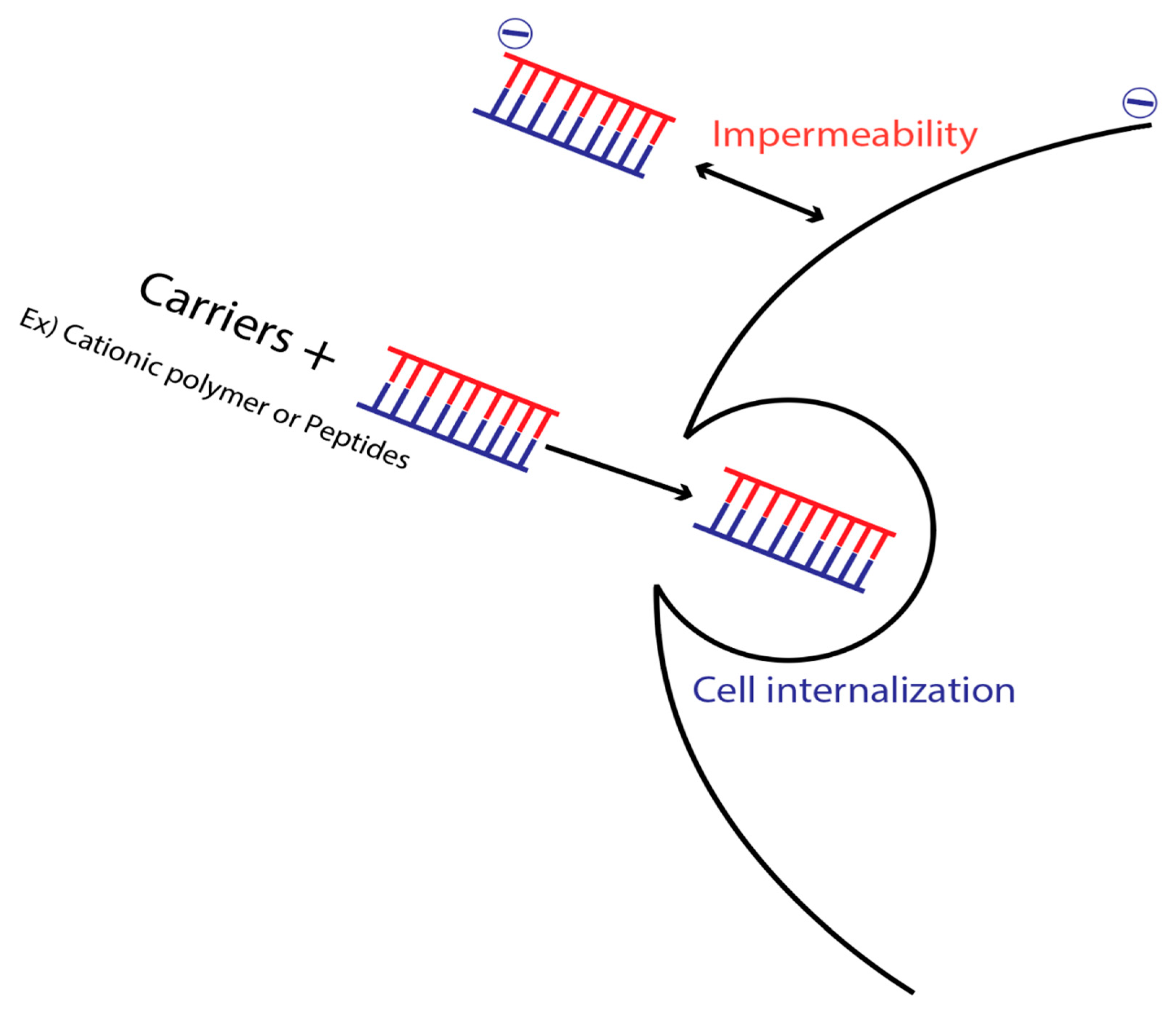
10. Membrane Impermeability
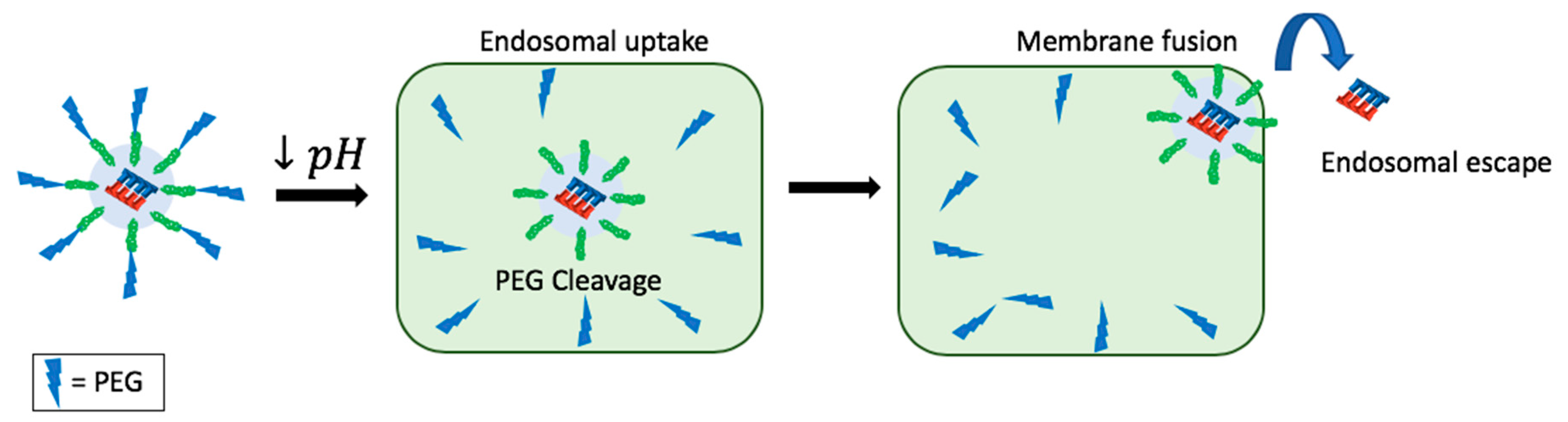
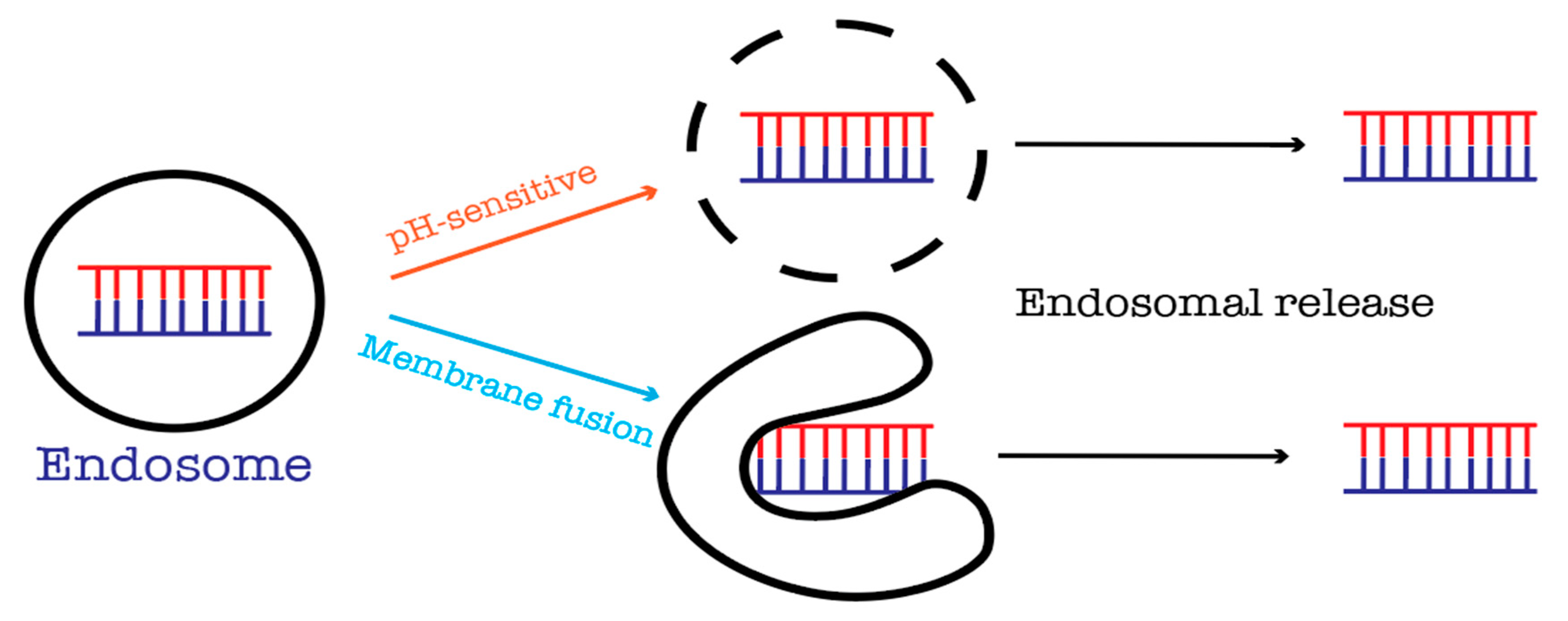
11. Endosomal Escape
12. Off-Target Effects
13. Other Strategies to Improve siRNA Effectiveness:
13.1. Aptamer-siRNA Conjugation
13.2. Exosomes for siRNA Delivery
14. Innovations and Prospects
15. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Caplen, N.J.; Parrish, S.; Imani, F.; Fire, A.; Morgan, R.A. Specific inhibition of gene expression by small double-stranded RNAs in invertebrate and vertebrate systems. Proc. Natl. Acad. Sci. USA 2001, 98, 9742–9747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Snead, N.M.; Wu, X.; Li, A.; Cui, Q.; Sakurai, K.; Burnett, J.C.; Rossi, J.J. Molecular basis for improved gene silencing by Dicer substrate interfering RNA compared with other siRNA variants. Nucleic Acids Res. 2013, 41, 6209–6221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Draz, M.S.; Fang, B.A.; Zhang, P.; Hu, Z.; Gu, S.; Weng, K.C.; Gray, J.W.; Chen, F.F. Nanoparticle-mediated systemic delivery of siRNA for treatment of cancers and viral infections. Theranostics 2014, 4, 872. [Google Scholar] [CrossRef] [PubMed]
- Weng, Y.; Xiao, H.; Zhang, J.; Liang, X.-J.; Huang, Y. RNAi therapeutic and its innovative biotechnological evolution. Biotechnol. Adv. 2019, 37, 801–825. [Google Scholar] [CrossRef]
- Scott, L.J. Givosiran: First Approval. Drugs 2020, 80, 335–339. [Google Scholar] [CrossRef]
- O’Driscoll, C.M.; Bernkop-Schnürch, A.; Friedl, J.D.; Préat, V.; Jannin, V. Oral delivery of non-viral nucleic acid-based therapeutics-do we have the guts for this? Eur. J. Pharm. Sci. 2019, 133, 190–204. [Google Scholar] [CrossRef]
- AJMC. Lumasiran Meets Primary End Point in Phase 3 Study in Patients With PH1. Available online: https://www.ajmc.com/newsroom/lumasiran-meets-primary-endpoint-in-phase-3-study-in-patients-with-ph1 (accessed on 25 December 2019).
- ClinicalTrials.gov. A Study to Evaluate Lumasiran in Children and Adults With Primary Hyperoxaluria Type 1 (ILLUMINATE-A). Available online: https://clinicaltrials.gov/ct2/show/NCT03681184 (accessed on 15 September 2020).
- Machin, N.; Ragni, M.V. An investigational RNAi therapeutic targeting antithrombin for the treatment of hemophilia A and B. J. Blood Med. 2018, 9, 135–140. [Google Scholar] [CrossRef] [Green Version]
- ClinicalTrials.gov. A Study of Fitusiran (ALN-AT3SC) in Severe Hemophilia A and B Patients Without Inhibitors. Available online: https://clinicaltrials.gov/ct2/show/NCT03417245 (accessed on 3 September 2020).
- ClinicalTrials.gov. HELIOS-B: A Study to Evaluate Vutrisiran in Patients With Transthyretin Amyloidosis With Cardiomyopathy. Available online: https://clinicaltrials.gov/ct2/show/NCT04153149 (accessed on 15 September 2020).
- Wang, J.; Lu, Z.; Wientjes, M.G.; Au, J.L.-S. Delivery of siRNA therapeutics: Barriers and carriers. AAPS J. 2010, 12, 492–503. [Google Scholar] [CrossRef]
- Setten, R.L.; Rossi, J.J.; Han, S.-P. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 2019, 18, 421–446. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; An, K.; Duan, X.; Xu, H.; Li, F.; Xu, F. Recent advances in siRNA delivery for cancer therapy using smart nanocarriers. Drug Discov. Today 2018, 23, 900–911. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-H.; Behlke, M.A.; Rose, S.D.; Chang, M.-S.; Choi, S.; Rossi, J.J. Synthetic dsRNA Dicer substrates enhance RNAi potency and efficacy. Nat. Biotechnol. 2005, 23, 222–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuckerman, J.E.; Davis, M.E. Clinical experiences with systemically administered siRNA-based therapeutics in cancer. Nat. Rev. Drug Discov. 2015, 14, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Cheloufi, S.; Dos Santos, C.O.; Chong, M.M.; Hannon, G.J. A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature 2010, 465, 584–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Carmell, M.A.; Rivas, F.V.; Marsden, C.G.; Thomson, J.M.; Song, J.-J.; Hammond, S.M.; Joshua-Tor, L.; Hannon, G.J. Argonaute2 is the catalytic engine of mammalian RNAi. Science 2004, 305, 1437–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, K.V.; Chan, S.W.-L.; Jacobsen, S.E.; Looney, D.J. Small interfering RNA-induced transcriptional gene silencing in human cells. Science 2004, 305, 1289–1292. [Google Scholar] [CrossRef] [Green Version]
- Eystathioy, T.; Chan, E.K.; Tenenbaum, S.A.; Keene, J.D.; Griffith, K.; Fritzler, M.J. A phosphorylated cytoplasmic autoantigen, GW182, associates with a unique population of human mRNAs within novel cytoplasmic speckles. Mol. Biol. Cell 2002, 13, 1338–1351. [Google Scholar] [CrossRef]
- Van Dijk, E.; Cougot, N.; Meyer, S.; Babajko, S.; Wahle, E.; Séraphin, B. Human Dcp2: A catalytically active mRNA decapping enzyme located in specific cytoplasmic structures. EMBO J. 2002, 21, 6915–6924. [Google Scholar] [CrossRef]
- Esau, C.C.; Monia, B.P. Therapeutic potential for microRNAs. Adv. Drug Deliv. Rev. 2007, 59, 101–114. [Google Scholar] [CrossRef]
- McAnuff, M.A.; Rettig, G.R.; Rice, K.G. Potency of siRNA versus shRNA mediated knockdown in vivo. J. Pharm. Sci. 2007, 96, 2922–2930. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Yamaoka, K.; Nishikawa, M.; Takakura, Y. Quantitative and temporal analysis of gene silencing in tumor cells induced by small interfering RNA or short hairpin RNA expressed from plasmid vectors. J. Pharm. Sci. 2009, 98, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E.; Zuckerman, J.E.; Choi, C.H.J.; Seligson, D.; Tolcher, A.; Alabi, C.A.; Yen, Y.; Heidel, J.D.; Ribas, A. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 2010, 464, 1067–1070. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, J.E.; Gritli, I.; Tolcher, A.; Heidel, J.D.; Lim, D.; Morgan, R.; Chmielowski, B.; Ribas, A.; Davis, M.E.; Yen, Y. Correlating animal and human phase Ia/Ib clinical data with CALAA-01, a targeted, polymer-based nanoparticle containing siRNA. Proc. Natl. Acad. Sci. USA 2014, 111, 11449–11454. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.E. The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: From concept to clinic. Mol. Pharm. 2009, 6, 659–668. [Google Scholar] [CrossRef]
- Heidel, J.D.; Liu, J.Y.-C.; Yen, Y.; Zhou, B.; Heale, B.S.; Rossi, J.J.; Bartlett, D.W.; Davis, M.E. Potent siRNA inhibitors of ribonucleotide reductase subunit RRM2 reduce cell proliferation in vitro and in vivo. Clin. Cancer Res. 2007, 13, 2207–2215. [Google Scholar] [CrossRef] [Green Version]
- Tabernero, J.; Shapiro, G.I.; LoRusso, P.M.; Cervantes, A.; Schwartz, G.K.; Weiss, G.J.; Paz-Ares, L.; Cho, D.C.; Infante, J.R.; Alsina, M. First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov. 2013, 3, 406–417. [Google Scholar] [CrossRef] [Green Version]
- Barros, S.A.; Gollob, J.A. Safety profile of RNAi nanomedicines. Adv. Drug Deliv. Rev. 2012, 64, 1730–1737. [Google Scholar] [CrossRef]
- Strumberg, D.; Schultheis, B.; Traugott, U.; Vank, C.; Santel, A.; Keil, O.; Giese, K.; Kaufmann, J.; Drevs, J. Phase I clinical development of Atu027, a siRNA formulation targeting PKN3 in patients with advanced solid tumors. Int. J. Clin. Pharmacol. Ther. 2012, 50, 76. [Google Scholar] [CrossRef]
- Schultheis, B.; Strumberg, D.; Santel, A.; Vank, C.; Gebhardt, F.; Keil, O.; Lange, C.; Giese, K.; Kaufmann, J.; Khan, M. First-in-human phase I study of the liposomal RNA interference therapeutic Atu027 in patients with advanced solid tumors. J. Clin. Oncol. 2014, 32, 4141–4148. [Google Scholar] [CrossRef]
- Northfelt, D.W.; Hamburg, S.I.; Borad, M.J.; Seetharam, M.; Curtis, K.K.; Lee, P.; Crowell, B.; Vocila, L.; Fredlund, P.; Gilbert, M.J. A phase I dose-escalation study of TKM-080301, a RNAi therapeutic directed against polo-like kinase 1 (PLK1), in patients with advanced solid tumors: Expansion cohort evaluation of biopsy samples for evidence of pharmacodynamic effects of PLK1 inhibition. J. Clin. Oncol. Am. Soc. Clin. Oncol. 2013. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Rodrigueza, W.V.; Rasco, D.W.; Patnaik, A.; Papadopoulos, K.P.; Amaya, A.; Moore, T.D.; Gaylor, S.K.; Bisgaier, C.L.; Sooch, M.P. A phase 1 study of the BCL2-targeted deoxyribonucleic acid inhibitor (DNAi) PNT2258 in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 363–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Aimetti, A.A.; Langer, R.; Gu, Z. Bioresponsive materials. Nat. Rev. Mater. 2016, 2, 1–17. [Google Scholar] [CrossRef]
- Sioud, M. RNA interference: Mechanisms, technical challenges, and therapeutic opportunities. In RNA Interference; Springer: Berlin/Heidelberg, Germany, 2015; pp. 1–15. [Google Scholar]
- Whitehead, K.A.; Langer, R.; Anderson, D.G. Knocking down barriers: Advances in siRNA delivery. Nat. Rev. Drug Discov. 2009, 8, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Grzelinski, M.; Urban-Klein, B.; Martens, T.; Lamszus, K.; Bakowsky, U.; Höbel, S.; Czubayko, F.; Aigner, A. RNA interference-mediated gene silencing of pleiotrophin through polyethylenimine-complexed small interfering RNAs in vivo exerts antitumoral effects in glioblastoma xenografts. Hum. Gene Ther. 2006, 17, 751–766. [Google Scholar] [CrossRef]
- Palliser, D.; Chowdhury, D.; Wang, Q.-Y.; Lee, S.J.; Bronson, R.T.; Knipe, D.M.; Lieberman, J. An siRNA-based microbicide protects mice from lethal herpes simplex virus 2 infection. Nature 2006, 439, 89–94. [Google Scholar] [CrossRef]
- Ballarín-González, B.; Thomsen, T.B.; Howard, K.A. Clinical translation of RNAi-based treatments for respiratory diseases. Drug Deliv. Transl. Res. 2013, 3, 84–99. [Google Scholar] [CrossRef]
- Geusens, B.; Sanders, N.; Prow, T.; Van Gele, M.; Lambert, J. Cutaneous short-interfering RNA therapy. Expert Opin. Drug Deliv. 2009, 6, 1333–1349. [Google Scholar] [CrossRef]
- Thomas, M.; Lu, J.J.; Chen, J.; Klibanov, A.M. Non-viral siRNA delivery to the lung. Adv. Drug Deliv. Rev. 2007, 59, 124–133. [Google Scholar] [CrossRef]
- Subhan, M.A.; Torchilin, V. siRNA based drug design, quality, delivery and clinical translation. Nanomed. Nanotechnol. Biol. Med. 2020, 29, 102239. [Google Scholar] [CrossRef]
- Tai, W. Current aspects of siRNA bioconjugate for in vitro and in vivo delivery. Molecules 2019, 24, 2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shegokar, R.; Al Shaal, L.; Mishra, P. SiRNA delivery: Challenges and role of carrier systems. Die Pharm. Int. J. Pharm. Sci. 2011, 66, 313–318. [Google Scholar]
- Bartlett, D.W.; Davis, M.E. Effect of siRNA nuclease stability on the in vitro and in vivo kinetics of siRNA-mediated gene silencing. Biotechnol. Bioeng. 2007, 97, 909–921. [Google Scholar] [CrossRef]
- Jackson, A.L.; Linsley, P.S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 2010, 9, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Zahir-Jouzdani, F.; Mottaghitalab, F.; Dinarvand, M.; Atyabi, F. siRNA delivery for treatment of degenerative diseases, new hopes and challenges. J. Drug Deliv. Sci. Technol. 2018, 45, 428–441. [Google Scholar] [CrossRef]
- Rana, T.M. Illuminating the silence: Understanding the structure and function of small RNAs. Nat. Rev. Mol. Cell Biol. 2007, 8, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Volkov, A.A.; Kruglova, N.Y.S.; Meschaninova, M.I.; Venyaminova, A.G.; Zenkova, M.A.; Vlassov, V.V.; Chernolovskaya, E.L. Selective protection of nuclease-sensitive sites in siRNA prolongs silencing effect. Oligonucleotides 2009, 19, 191–202. [Google Scholar] [CrossRef]
- Kim, S.-S.; Garg, H.; Joshi, A.; Manjunath, N. Strategies for targeted nonviral delivery of siRNAs in vivo. Trends Mol. Med. 2009, 15, 491–500. [Google Scholar] [CrossRef] [Green Version]
- Lewis, D.L.; Wolff, J.A. Delivery of siRNA and siRNA expression constructs to adult mammals by hydrodynamic intravascular injection. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2005; Volume 392, pp. 336–350. [Google Scholar]
- Zhou, Y.; Zhang, C.; Liang, W. Development of RNAi technology for targeted therapy—A track of siRNA based agents to RNAi therapeutics. J. Control. Release 2014, 193, 270–281. [Google Scholar] [CrossRef]
- Czauderna, F.; Fechtner, M.; Dames, S.; Aygün, H.; Klippel, A.; Pronk, G.J.; Giese, K.; Kaufmann, J. Structural variations and stabilising modifications of synthetic siRNAs in mammalian cells. Nucleic Acids Res. 2003, 31, 2705–2716. [Google Scholar] [CrossRef]
- Xia, J.; Noronha, A.; Toudjarska, I.; Li, F.; Akinc, A.; Braich, R.; Frank-Kamenetsky, M.; Rajeev, K.G.; Egli, M.; Manoharan, M. Gene silencing activity of siRNAs with a ribo-difluorotoluyl nucleotide. ACS Chem. Biol. 2006, 1, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Potenza, N.; Moggio, L.; Milano, G.; Salvatore, V.; Di Blasio, B.; Russo, A.; Messere, A. RNA interference in mammalia cells by RNA-3′-PNA chimeras. Int. J. Mol. Sci. 2008, 9, 299–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshika, S.; Minakawa, N.; Kamiya, H.; Harashima, H.; Matsuda, A. RNA interference induced by siRNAs modified with 4′-thioribonucleosides in cultured mammalian cells. FEBS Lett. 2005, 579, 3115–3118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choung, S.; Kim, Y.J.; Kim, S.; Park, H.-O.; Choi, Y.-C. Chemical modification of siRNAs to improve serum stability without loss of efficacy. Biochem. Biophys. Res. Commun. 2006, 342, 919–927. [Google Scholar] [CrossRef] [PubMed]
- Layzer, J.M.; McCaffrey, A.P.; Tanner, A.K.; Huang, Z.; Kay, M.A.; Sullenger, B.A. In vivo activity of nuclease-resistant siRNAs. RNA 2004, 10, 766–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, H.; Wang, J.H. Biomembrane-permeable and ribonuclease-resistant siRNA with enhanced activity. Oligonucleotides 2005, 15, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Dowler, T.; Bergeron, D.; Tedeschi, A.-L.; Paquet, L.; Ferrari, N.; Damha, M.J. Improvements in siRNA properties mediated by 2′-deoxy-2′-fluoro-β-D-arabinonucleic acid (FANA). Nucleic Acids Res. 2006, 34, 1669–1675. [Google Scholar] [CrossRef] [Green Version]
- Gao, K.; Huang, L. Achieving efficient RNAi therapy: Progress and challenges. Acta Pharm. Sin. B 2013, 3, 213–225. [Google Scholar] [CrossRef] [Green Version]
- Hoshika, S.; Minakawa, N.; Shionoya, A.; Imada, K.; Ogawa, N.; Matsuda, A. Study of Modification Pattern–RNAi Activity Relationships by Using siRNAs Modified with 4′-Thioribonucleosides. ChemBioChem 2007, 8, 2133–2138. [Google Scholar] [CrossRef]
- Amarzguioui, M.; Holen, T.; Babaie, E.; Prydz, H. Tolerance for mutations and chemical modifications in a siRNA. Nucleic Acids Res. 2003, 31, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Morrissey, D.V.; Blanchard, K.; Shaw, L.; Jensen, K.; Lockridge, J.A.; Dickinson, B.; McSwiggen, J.A.; Vargeese, C.; Bowman, K.; Shaffer, C.S. Activity of stabilized short interfering RNA in a mouse model of hepatitis B virus replication. Hepatology 2005, 41, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- Chernolovskaya, E.L.; Zenkova, M.A. Chemical modification of siRNA. Curr. Opin. Mol. Ther. 2010, 12, 158–167. [Google Scholar] [PubMed]
- Bramsen, J.B.; Laursen, M.B.; Damgaard, C.K.; Lena, S.W.; Ravindra Babu, B.; Wengel, J.; Kjems, J. Improved silencing properties using small internally segmented interfering RNAs. Nucleic Acids Res. 2007, 35, 5886–5897. [Google Scholar] [CrossRef] [PubMed]
- Tatiparti, K.; Sau, S.; Kashaw, S.K.; Iyer, A.K. siRNA delivery strategies: A comprehensive review of recent developments. Nanomaterials 2017, 7, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behlke, M.A. Progress towards in vivo use of siRNAs. Mol. Ther. 2006, 13, 644–670. [Google Scholar] [CrossRef]
- Chernikov, I.V.; Gladkikh, D.V.; Meschaninova, M.I.; Ven’yaminova, A.G.; Zenkova, M.A.; Vlassov, V.V.; Chernolovskaya, E.L. Cholesterol-containing nuclease-resistant siRNA accumulates in tumors in a carrier-free mode and silences MDR1 gene. Mol. Ther. Nucleic Acids 2017, 6, 209–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfrum, C.; Shi, S.; Jayaprakash, K.N.; Jayaraman, M.; Wang, G.; Pandey, R.K.; Rajeev, K.G.; Nakayama, T.; Charrise, K.; Ndungo, E.M. Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat. Biotechnol. 2007, 25, 1149–1157. [Google Scholar] [CrossRef]
- Soutschek, J.; Akinc, A.; Bramlage, B.; Charisse, K.; Constien, R.; Donoghue, M.; Elbashir, S.; Geick, A.; Hadwiger, P.; Harborth, J. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 2004, 432, 173–178. [Google Scholar] [CrossRef]
- Gaziova, Z.; Baumann, V.; Winkler, A.-M.; Winkler, J. Chemically defined polyethylene glycol siRNA conjugates with enhanced gene silencing effect. Bioorg. Med. Chem. 2014, 22, 2320–2326. [Google Scholar] [CrossRef] [Green Version]
- Iversen, F.; Yang, C.; Dagnæs-Hansen, F.; Schaffert, D.H.; Kjems, J.; Gao, S. Optimized siRNA-PEG conjugates for extended blood circulation and reduced urine excretion in mice. Theranostics 2013, 3, 201. [Google Scholar] [CrossRef]
- Dassie, J.P.; Liu, X.-Y.; Thomas, G.S.; Whitaker, R.M.; Thiel, K.W.; Stockdale, K.R.; Meyerholz, D.K.; McCaffrey, A.P.; McNamara, J.O.; Giangrande, P.H. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat. Biotechnol. 2009, 27, 839–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, A.; Choi, S.W.; Hirai, M.; Yamayoshi, A.; Moriyama, R.; Yamano, T.; Takagi, M.; Kano, A.; Shimamoto, A.; Maruyama, A. Polymer brush-stabilized polyplex for a siRNA carrier with long circulatory half-life. J. Control. Release 2007, 122, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Judge, A.; Maclachlan, I. Overcoming the innate immune response to small interfering RNA. Hum. Gene Ther. 2008, 19, 111–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Judge, A.D.; Sood, V.; Shaw, J.R.; Fang, D.; McClintock, K.; MacLachlan, I. Sequence-dependent stimulation of the mammalian innate immune response by synthetic siRNA. Nat. Biotechnol. 2005, 23, 457–462. [Google Scholar] [CrossRef]
- Robbins, M.; Judge, A.; MacLachlan, I. siRNA and innate immunity. Oligonucleotides 2009, 19, 89–102. [Google Scholar] [CrossRef]
- Meng, Z.; Lu, M. RNA interference-induced innate immunity, off-target effect, or immune adjuvant? Front. Immunol. 2017, 8, 331. [Google Scholar] [CrossRef] [Green Version]
- Forsbach, A.; Nemorin, J.-G.; Montino, C.; Müller, C.; Samulowitz, U.; Vicari, A.P.; Jurk, M.; Mutwiri, G.K.; Krieg, A.M.; Lipford, G.B. Identification of RNA sequence motifs stimulating sequence-specific TLR8-dependent immune responses. J. Immunol. 2008, 180, 3729–3738. [Google Scholar] [CrossRef]
- Yasuda, H.; Leelahavanichkul, A.; Tsunoda, S.; Dear, J.W.; Takahashi, Y.; Ito, S.; Hu, X.; Zhou, H.; Doi, K.; Childs, R. Chloroquine and inhibition of Toll-like receptor 9 protect from sepsis-induced acute kidney injury. Am. J. Physiol. Ren. Physiol. 2008, 294, F1050–F1058. [Google Scholar] [CrossRef] [Green Version]
- Kleinman, M.E.; Yamada, K.; Takeda, A.; Chandrasekaran, V.; Nozaki, M.; Baffi, J.Z.; Albuquerque, R.J.; Yamasaki, S.; Itaya, M.; Pan, Y. Sequence-and target-independent angiogenesis suppression by siRNA via TLR3. Nature 2008, 452, 591–597. [Google Scholar] [CrossRef] [Green Version]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 2003, 301, 640–643. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.T.; Tzvetkov, E.; Pereira, A.; Wu, Y.; Kasar, S.; Przetak, M.M.; Vlach, J.; Niewold, T.B.; Jensen, M.A.; Okitsu, S.L. TLR7 and TLR8 Differentially Activate the IRF and NF-κB Pathways in Specific Cell Types to Promote Inflammation. ImmunoHorizons 2020, 4, 93–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Ohto, U.; Shibata, T.; Taoka, M.; Yamauchi, Y.; Sato, R.; Shukla, N.M.; David, S.A.; Isobe, T.; Miyake, K. Structural analyses of Toll-like receptor 7 reveal detailed RNA sequence specificity and recognition mechanism of agonistic ligands. Cell Rep. 2018, 25, 3371–3381. [Google Scholar] [CrossRef] [Green Version]
- Hornung, V.; Guenthner-Biller, M.; Bourquin, C.; Ablasser, A.; Schlee, M.; Uematsu, S.; Noronha, A.; Manoharan, M.; Akira, S.; de Fougerolles, A. Sequence-specific potent induction of IFN-α by short interfering RNA in plasmacytoid dendritic cells through TLR7. Nat. Med. 2005, 11, 263–270. [Google Scholar] [CrossRef]
- Tan, X.; Jia, F.; Wang, P.; Zhang, K. Nucleic acid-based drug delivery strategies. J. Control. Release 2020, 323, 240–252. [Google Scholar] [CrossRef]
- Dermani, F.K.; Jalilian, F.A.; Hossienkhani, H.; Ezati, R.; Amini, R. SiRNA delivery technology for cancer therapy: Promise and challenges. Acta Medica Iranica 2019. [Google Scholar] [CrossRef]
- Kim, S.j.; Chen, Z.; Essani, A.B.; Elshabrawy, H.A.; Volin, M.V.; Volkov, S.; Swedler, W.; Arami, S.; Sweiss, N.; Shahrara, S. Identification of a novel toll-like receptor 7 endogenous ligand in rheumatoid arthritis synovial fluid that can provoke arthritic joint inflammation. Arthritis Rheumatol. 2016, 68, 1099–1110. [Google Scholar]
- Khan, A.A.; Pluta, L.; Yousefi, B.; Damania, B. Endosomal TLR-8 Senses microRNA-1294 Resulting in the Production of NFḱB Dependent Cytokines Running title: miRNA1294 activates TLR-8. Front. Immunol. 2019, 10, 2860. [Google Scholar]
- Gupta, K.; Saldanha, M.; Parasnis, M.; Devarajan, P.V.; Jain, R.; Dandekar, P. Toll-like receptor-mediated endocytosis in infectious disease. In Targeted Intracellular Drug Delivery by Receptor Mediated Endocytosis; Springer: Berlin/Heidelberg, Germany, 2019; pp. 323–349. [Google Scholar]
- Imaeda, A.; Tomoike, F.; Hayakawa, M.; Nakamoto, K.; Kimura, Y.; Abe, N.; Abe, H. N 6-methyl adenosine in siRNA evades immune response without reducing RNAi activity. Nucleosides Nucleotides Nucleic Acids 2019, 38, 972–979. [Google Scholar] [CrossRef]
- Hoerter, J.A.; Walter, N.G. Chemical modification resolves the asymmetry of siRNA strand degradation in human blood serum. RNA 2007, 13, 1887–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Brutkiewicz, R.R. The Toll-like receptor 9 signalling pathway regulates MR 1-mediated bacterial antigen presentation in B cells. Immunology 2017, 152, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Sioud, M. Recent advances in small interfering RNA sensing by the immune system. New Biotechnol. 2010, 27, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Hur, S. Double-stranded RNA sensors and modulators in innate immunity. Annu. Rev. Immunol. 2019, 37, 349–375. [Google Scholar] [CrossRef]
- Hull, C.M.; Bevilacqua, P.C. Discriminating self and non-self by RNA: Roles for RNA structure, misfolding, and modification in regulating the innate immune sensor PKR. Acc. Chem. Res. 2016, 49, 1242–1249. [Google Scholar] [CrossRef] [Green Version]
- Safran, S.A.; Eckert, D.M.; Leslie, E.A.; Bass, B.L. PKR activation by noncanonical ligands: A 5′-triphosphate requirement versus antisense contamination. RNA 2019, 25, 1192–1201. [Google Scholar] [CrossRef]
- Owens, D.E., III; Peppas, N.A. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm. 2006, 307, 93–102. [Google Scholar] [CrossRef]
- Sun, Y.; Zhao, Y.; Zhao, X.; Lee, R.J.; Teng, L.; Zhou, C. Enhancing the therapeutic delivery of oligonucleotides by chemical modification and nanoparticle encapsulation. Molecules 2017, 22, 1724. [Google Scholar] [CrossRef]
- Auguste, D.T.; Furman, K.; Wong, A.; Fuller, J.; Armes, S.P.; Deming, T.J.; Langer, R. Triggered release of siRNA from poly (ethylene glycol)-protected, pH-dependent liposomes. J. Control. Release 2008, 130, 266–274. [Google Scholar] [CrossRef] [Green Version]
- Salmaso, S.; Caliceti, P. Stealth properties to improve therapeutic efficacy of drug nanocarriers. J. Drug Deliv. 2013, 2013. [Google Scholar] [CrossRef]
- Peer, D.; Lieberman, J. Special delivery: Targeted therapy with small RNAs. Gene Ther. 2011, 18, 1127–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, J.; Sun, Y.; Shi, Q.-S.; Liu, P.-F.; Zhu, M.-J.; Wang, C.-H.; Du, L.-F.; Duan, Y.-R. Biodegradable nanoparticles of mPEG-PLGA-PLL triblock copolymers as novel non-viral vectors for improving siRNA delivery and gene silencing. Int. J. Mol. Sci. 2012, 13, 516–533. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Jin, C.; Long, J.; Fu, D.; Yang, F.; Xu, J.; Yu, X.; Chen, W.; Ni, Q. RNA interference of MBD1 in BxPC-3 human pancreatic cancer cells delivered by PLGA-poloxamer nanoparticles. Cancer Biol. Ther. 2009, 8, 594–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santel, A.; Aleku, M.; Keil, O.; Endruschat, J.; Esche, V.; Fisch, G.; Dames, S.; Löffler, K.; Fechtner, M.; Arnold, W. A novel siRNA-lipoplex technology for RNA interference in the mouse vascular endothelium. Gene Ther. 2006, 13, 1222–1234. [Google Scholar] [CrossRef] [PubMed]
- Musacchio, T.; Torchilin, V.P. siRNA delivery: From basics to therapeutic applications. Front. Biosci. (Landmark Ed.) 2013, 18, 58–79. [Google Scholar] [PubMed] [Green Version]
- Liu, Y.; Song, Z.; Zheng, N.; Nagasaka, K.; Yin, L.; Cheng, J. Systemic siRNA delivery to tumors by cell-penetrating α-helical polypeptide-based metastable nanoparticles. Nanoscale 2018, 10, 15339–15349. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.C.; Zhang, H.; Jakymiw, A. Peptide carriers to the rescue: Overcoming the barriers to siRNA delivery for cancer treatment. Transl. Res. 2019, 214, 92–104. [Google Scholar] [CrossRef]
- Rathnayake, P.; Gunathunge, B.; Wimalasiri, P.; Karunaratne, D.; Ranatunga, R. Trends in the binding of cell penetrating peptides to siRNA: A molecular docking study. J. Biophys. 2017, 2017. [Google Scholar] [CrossRef]
- Tanaka, K.; Kanazawa, T.; Ogawa, T.; Takashima, Y.; Fukuda, T.; Okada, H. Disulfide crosslinked stearoyl carrier peptides containing arginine and histidine enhance siRNA uptake and gene silencing. Int. J. Pharm. 2010, 398, 219–224. [Google Scholar] [CrossRef]
- Mainini, F.; Eccles, M.R. Lipid and Polymer-Based Nanoparticle siRNA Delivery Systems for Cancer Therapy. Molecules 2020, 25, 2692. [Google Scholar] [CrossRef]
- Varkouhi, A.K.; Scholte, M.; Storm, G.; Haisma, H.J. Endosomal escape pathways for delivery of biologicals. J. Control. Release 2011, 151, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Morris, V.B.; Labhasetwar, V. Effectiveness of small interfering RNA delivery via arginine-rich polyethylenimine-based polyplex in metastatic and doxorubicin-resistant breast cancer cells. J. Pharmacol. Exp. Ther. 2019, 370, 902–910. [Google Scholar] [CrossRef] [PubMed]
- Urban-Klein, B.; Werth, S.; Abuharbeid, S.; Czubayko, F.; Aigner, A. RNAi-mediated gene-targeting through systemic application of polyethylenimine (PEI)-complexed siRNA in vivo. Gene Ther. 2005, 12, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Sonawane, N.D.; Szoka, F.C.; Verkman, A. Chloride accumulation and swelling in endosomes enhances DNA transfer by polyamine-DNA polyplexes. J. Biol. Chem. 2003, 278, 44826–44831. [Google Scholar] [CrossRef] [Green Version]
- Choi, K.Y.; Correa, S.; Min, J.; Li, J.; Roy, S.; Laccetti, K.H.; Dreaden, E.; Kong, S.; Heo, R.; Roh, Y.H. Binary targeting of siRNA to hematologic cancer cells in vivo using layer-by-layer nanoparticles. Adv. Funct. Mater. 2019, 29, 1900018. [Google Scholar] [CrossRef]
- Huang, H.; Yuan, S.; Ma, Z.; Ji, P.; Ma, X.; Wu, Z.; Qi, X. Genetic recombination of poly (l-lysine) functionalized apoferritin nanocages that resemble viral capsid nanometer-sized platforms for gene therapy. Biomater. Sci. 2020, 8, 1759–1770. [Google Scholar] [CrossRef]
- Shin, J.; Shum, P.; Thompson, D.H. Acid-triggered release via dePEGylation of DOPE liposomes containing acid-labile vinyl ether PEG–lipids. J. Control. Release 2003, 91, 187–200. [Google Scholar] [CrossRef]
- Cupic, K.I.; Rennick, J.J.; Johnston, A.P.; Such, G.K. Controlling endosomal escape using nanoparticle composition: Current progress and future perspectives. Nanomedicine 2019, 14, 215–223. [Google Scholar] [CrossRef]
- Koynova, R.; Wang, L.; MacDonald, R.C. An intracellular lamellar–nonlamellar phase transition rationalizes the superior performance of some cationic lipid transfection agents. Proc. Natl. Acad. Sci. USA 2006, 103, 14373–14378. [Google Scholar] [CrossRef] [Green Version]
- Hatakeyama, H.; Akita, H.; Kogure, K.; Oishi, M.; Nagasaki, Y.; Kihira, Y.; Ueno, M.; Kobayashi, H.; Kikuchi, H.; Harashima, H. Development of a novel systemic gene delivery system for cancer therapy with a tumor-specific cleavable PEG-lipid. Gene Ther. 2007, 14, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Leng, Q.; Woodle, M.C.; Mixson, A.J. Targeted delivery of siRNA therapeutics to malignant tumors. J. Drug Deliv. 2017, 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, S.; Keiser, K.; Nair, J.K.; Charisse, K.; Manoharan, R.M.; Kretschmer, P.; Peng, C.G.; Kel’in, V.A.; Kandasamy, P.; Willoughby, J.L. siRNA conjugates carrying sequentially assembled trivalent N-acetylgalactosamine linked through nucleosides elicit robust gene silencing in vivo in hepatocytes. ACS Chem. Biol. 2015, 10, 1181–1187. [Google Scholar] [CrossRef] [PubMed]
- Nair, J.K.; Willoughby, J.L.; Chan, A.; Charisse, K.; Alam, M.R.; Wang, Q.; Hoekstra, M.; Kandasamy, P.; Kel’in, A.V.; Milstein, S. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014, 136, 16958–16961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, A.L.; Burchard, J.; Schelter, J.; Chau, B.N.; Cleary, M.; Lim, L.; Linsley, P.S. Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. RNA 2006, 12, 1179–1187. [Google Scholar] [CrossRef] [Green Version]
- Walker, R.A.; Day, S.J. Transferrin receptor expression in non-malignant and malignant human breast tissue. J. Pathol. 1986, 148, 217–224. [Google Scholar] [CrossRef]
- Gurrath, M.; Müller, G.; Kessler, H.; Aumailley, M.; Timpl, R. Conformation/activity studies of rationally designed potent anti-adhesive RGD peptides. Eur. J. Biochem. 1992, 210, 911–921. [Google Scholar] [CrossRef]
- Han, H.D.; Mangala, L.S.; Lee, J.W.; Shahzad, M.M.; Kim, H.S.; Shen, D.; Nam, E.J.; Mora, E.M.; Stone, R.L.; Lu, C. Targeted gene silencing using RGD-labeled chitosan nanoparticles. Clin. Cancer Res. 2010, 16, 3910–3922. [Google Scholar] [CrossRef] [Green Version]
- Song, E.; Zhu, P.; Lee, S.-K.; Chowdhury, D.; Kussman, S.; Dykxhoorn, D.M.; Feng, Y.; Palliser, D.; Weiner, D.B.; Shankar, P. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat. Biotechnol. 2005, 23, 709–717. [Google Scholar] [CrossRef]
- Foy, J.W.-D.; Rittenhouse, K.; Modi, M.; Patel, M. Local tolerance and systemic safety of pegaptanib sodium in the dog and rabbit. J. Ocul. Pharmacol. Ther. 2007, 23, 452–466. [Google Scholar] [CrossRef]
- Kruspe, S.; Giangrande, P.H. Aptamer-siRNA chimeras: Discovery, progress, and future prospects. Biomedicines 2017, 5, 45. [Google Scholar] [CrossRef] [Green Version]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Hicke, B.J.; Stephens, A.W.; Gould, T.; Chang, Y.-F.; Lynott, C.K.; Heil, J.; Borkowski, S.; Hilger, C.-S.; Cook, G.; Warren, S. Tumor targeting by an aptamer. J. Nucl. Med. 2006, 47, 668–678. [Google Scholar] [PubMed]
- Thiel, K.W.; Giangrande, P.H. Therapeutic applications of DNA and RNA aptamers. Oligonucleotides 2009, 19, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Chi-Hong, B.C.; Dellamaggiore, K.R.; Ouellette, C.P.; Sedano, C.D.; Lizadjohry, M.; Chernis, G.A.; Gonzales, M.; Baltasar, F.E.; Fan, A.L.; Myerowitz, R. Aptamer-based endocytosis of a lysosomal enzyme. Proc. Natl. Acad. Sci. USA 2008, 105, 15908–15913. [Google Scholar]
- Ko, H.Y.; Lee, J.H.; Kang, H.; Ryu, S.H.; Song, I.C.; Lee, D.S.; Kim, S. A nucleolin-targeted multimodal nanoparticle imaging probe for tracking cancer cells using an aptamer. J. Nucl. Med. 2010, 51, 98–105. [Google Scholar]
- Wullner, U.; Neef, I.; Eller, A.; Kleines, M.; Tur, M.K.; Barth, S. Cell-specific induction of apoptosis by rationally designed bivalent aptamer-siRNA transcripts silencing eukaryotic elongation factor 2. Curr. Cancer Drug Targets 2008, 8, 554–565. [Google Scholar] [CrossRef]
- McNamara, J.O.; Andrechek, E.R.; Wang, Y.; Viles, K.D.; Rempel, R.E.; Gilboa, E.; Sullenger, B.A.; Giangrande, P.H. Cell type–specific delivery of siRNAs with aptamer-siRNA chimeras. Nat. Biotechnol. 2006, 24, 1005–1015. [Google Scholar] [CrossRef]
- Li, N.; Larson, T.; Nguyen, H.H.; Sokolov, K.V.; Ellington, A.D. Directed evolution of gold nanoparticle delivery to cells. Chem. Commun. 2010, 46, 392–394. [Google Scholar] [CrossRef] [Green Version]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Van den Boorn, J.G.; Schlee, M.; Coch, C.; Hartmann, G. SiRNA delivery with exosome nanoparticles. Nat. Biotechnol. 2011, 29, 325–326. [Google Scholar] [CrossRef]
- Shtam, T.A.; Kovalev, R.A.; Varfolomeeva, E.Y.; Makarov, E.M.; Kil, Y.V.; Filatov, M.V. Exosomes are natural carriers of exogenous siRNA to human cells in vitro. Cell Commun. Signal. 2013, 11, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R.F. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kooijmans, S.A.; Vader, P.; van Dommelen, S.M.; van Solinge, W.W.; Schiffelers, R.M. Exosome mimetics: A novel class of drug delivery systems. Int. J. Nanomed. 2012, 7, 1525. [Google Scholar]
- Dai, S.; Wei, D.; Wu, Z.; Zhou, X.; Wei, X.; Huang, H.; Li, G. Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Mol. Ther. 2008, 16, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Xing, H.; Xun, Z.; Yang, T.; Ding, P.; Cai, C.; Wang, D.; Zhao, X. Exosome-based small RNA delivery: Progress and prospects. Asian J. Pharm. Sci. 2018, 13, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sekhar, G.N.; Watson, C.P.; Fidanboylu, M.; Sanderson, L.; Thomas, S.A. Delivery of antihuman African trypanosomiasis drugs across the blood–brain and blood–CSF barriers. In Advances in Pharmacology; Elsevier: Amsterdam, The Netherlands, 2014; Volume 71, pp. 245–275. [Google Scholar]
- Dominska, M.; Dykxhoorn, D.M. Breaking down the barriers: siRNA delivery and endosome escape. J. Cell Sci. 2010, 123, 1183–1189. [Google Scholar] [CrossRef] [Green Version]
- Fakhr, E.; Zare, F.; Teimoori-Toolabi, L. Precise and efficient siRNA design: A key point in competent gene silencing. Cancer Gene Ther. 2016, 23, 73–82. [Google Scholar] [CrossRef]
- Robbins, M.; Judge, A.; Liang, L.; McClintock, K.; Yaworski, E.; MacLachlan, I. 2′-O-methyl-modified RNAs act as TLR7 antagonists. Mol. Ther. 2007, 15, 1663–1669. [Google Scholar] [CrossRef]
- Bramsen, J.B.; Laursen, M.B.; Nielsen, A.F.; Hansen, T.B.; Bus, C.; Langkjær, N.; Babu, B.R.; Højland, T.; Abramov, M.; Van Aerschot, A. A large-scale chemical modification screen identifies design rules to generate siRNAs with high activity, high stability and low toxicity. Nucleic Acids Res. 2009, 37, 2867–2881. [Google Scholar] [CrossRef] [PubMed]
- Janas, M.M.; Schlegel, M.K.; Harbison, C.E.; Yilmaz, V.O.; Jiang, Y.; Parmar, R.; Zlatev, I.; Castoreno, A.; Xu, H.; Shulga-Morskaya, S. Selection of GalNAc-conjugated siRNAs with limited off-target-driven rat hepatotoxicity. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Shum, K.; Rossi, J. siRNA Delivery Methods; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Snead, N.M.; Escamilla-Powers, J.R.; Rossi, J.J.; McCaffrey, A.P. 5′ Unlocked nucleic acid modification improves siRNA targeting. Mol. Ther. Nucleic Acids 2013, 2, e103. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.; Leake, D.; Boese, Q.; Scaringe, S.; Marshall, W.S.; Khvorova, A. Rational siRNA design for RNA interference. Nat. Biotechnol. 2004, 22, 326–330. [Google Scholar] [CrossRef]
- Dowdy, S.F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017, 35, 222–229. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of RNAi | Silent Feature of RNAi Therapeutics | References |
|---|---|---|
| miRNA | Stem-loop structure Binds imperfectly to mRNA and degrades many sets of similar target mRNA | [24] |
| shRNA | RNA with a tight hairpin turn Requires promoter to be expressed and must be located in the nucleus to act | [25] |
| siRNA | Short stretch dsRNA capable of degrading complementary mRNA Higher transfection efficiency and fewer obstacles. | [26] |
| Extracellular Barriers | Intracellular Barriers |
|---|---|
| Endogenous Nucleases | Endosomal trap |
| Elimination through kidneys due to small size | Arrival at the correct intracellular site of action (cytosol), |
| Impermeability to biological membranes | Off-target effects. |
| Entrapment by Reticuloendothelial System | |
| Plasma protein sequestration | |
| Capillary Endothelium Crossing |
| Modification | Type of Modification | Outcome | Reference |
|---|---|---|---|
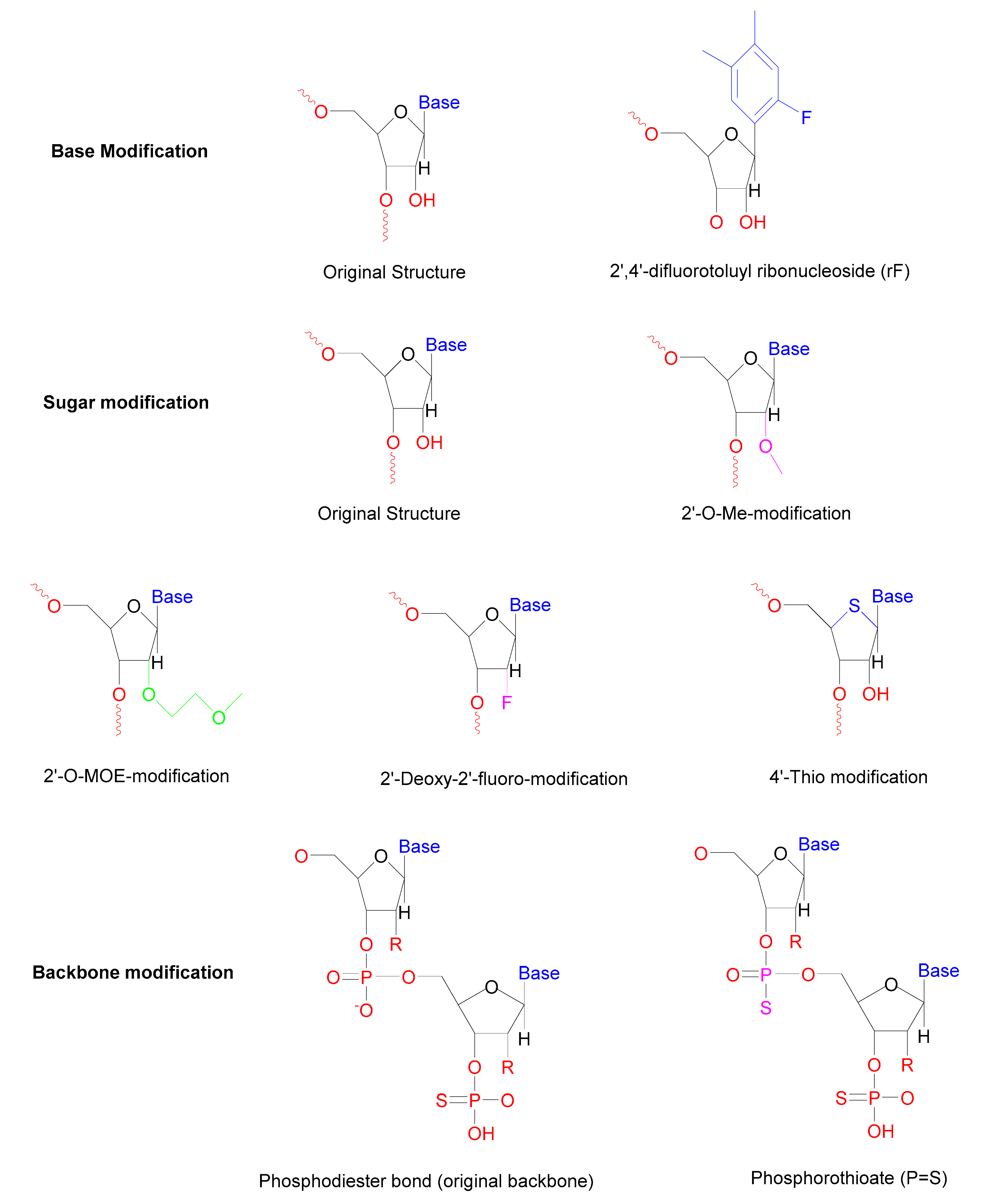
| Base | Internal uridine to rF (2,4-difluorotoluyl ribonucleoside) substitutions | Enhanced resistance towards serum nucleases | [57] |
| Sugar | 2′-Deoxy-2′-fluoro-β-D-arabino, 2′-O-MOE modification, 2′-O-Me modification | Increase the half-life in the serum | [63] |
| Locked nucleic acids | Longer half-life reduces immune activation | [64] | |
| 4′-Thio modified the ring oxygen | Enhanced resistance towards nucleases | [65] | |
| Backbone | Phosphorothioate (PS) modifications Morpholino oligomers | The longer half-life of duplex More potent | [66] |
| Sequence | Signaling Mechanism | Type of Cytokines | References |
|---|---|---|---|
| 5′-GUCCUUCAA-3′ motif | TLR8 | Interferon-α | [92,93] |
| GU-rich sequence | TLR7 | IFN-α | [94] |
| AU-rich sequence | TLR7 and TLR8 | IFN-α, tumor necrosis factor (TNF)-α | [95] |
| 5′-UGUGU-3′ | TLR8 | IFN-α | [92] |
| Repetition of uracil | TLR7 | interleukin-6 and TNF-α | [96] |
| GalNAc Conjugation | Modifications | Results | References |
|---|---|---|---|
| Triantennary | -Added PS linkages for the protection against the 5’-exonuclease. | -Reduced the off-target effects. -silenced mTTR in the liver cell (hepatocytes) | [131] |
| Monovalent | -2’ and 4’of the RNA’s ribose sugar backbone for GalNAc linker conjugation. | -Similar or better binding affinity and gene silencing efficiency compared to Triantennary GalNAc. | [130] |
| Receptor | Selection method | RNA/DNA | Delivery Applications | References |
|---|---|---|---|---|
| Transferrin receptor (TfR) | The extracellular purview of mouse TfR | RNA/DNA | Protein involved targeting lysosome | [141] |
| Nucleolin | N/A | DNA | Imaging variety of tumors | [142] |
| Tenascin C (TN-C) | Purified TN-C | RNA | Imaging variety of tumors | [139] |
| Prostate-specific membrane antigen (PSMA) | The extracellular purview of PSMA | RNA | Cellular imaging, siRNA delivery along with anticancer drug delivery | [143,144] |
| Epidermal growth factor receptor (EGFR) | The extracellular purview of EGFR | RNA | Delivery of nanoparticles | [145] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sajid, M.I.; Moazzam, M.; Kato, S.; Yeseom Cho, K.; Tiwari, R.K. Overcoming Barriers for siRNA Therapeutics: From Bench to Bedside. Pharmaceuticals 2020, 13, 294. https://doi.org/10.3390/ph13100294
Sajid MI, Moazzam M, Kato S, Yeseom Cho K, Tiwari RK. Overcoming Barriers for siRNA Therapeutics: From Bench to Bedside. Pharmaceuticals. 2020; 13(10):294. https://doi.org/10.3390/ph13100294
Chicago/Turabian StyleSajid, Muhammad Imran, Muhammad Moazzam, Shun Kato, Kayley Yeseom Cho, and Rakesh Kumar Tiwari. 2020. "Overcoming Barriers for siRNA Therapeutics: From Bench to Bedside" Pharmaceuticals 13, no. 10: 294. https://doi.org/10.3390/ph13100294