Evaluation of Structurally Distorted Split GFP Fluorescent Sensors for Cell-Based Detection of Viral Proteolytic Activity



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plasmids
2.2. Mammalian Cell Lines
2.3. 293 Cell Transduction and Selection
2.4. Adenoviral Vector Stocks
2.5. Split Sensors Characterization
2.6. Evaluation of GFP10 and GFP11 Levels in 293 Sensor Cells
2.7. Data and Statistical Analysis
3. Results
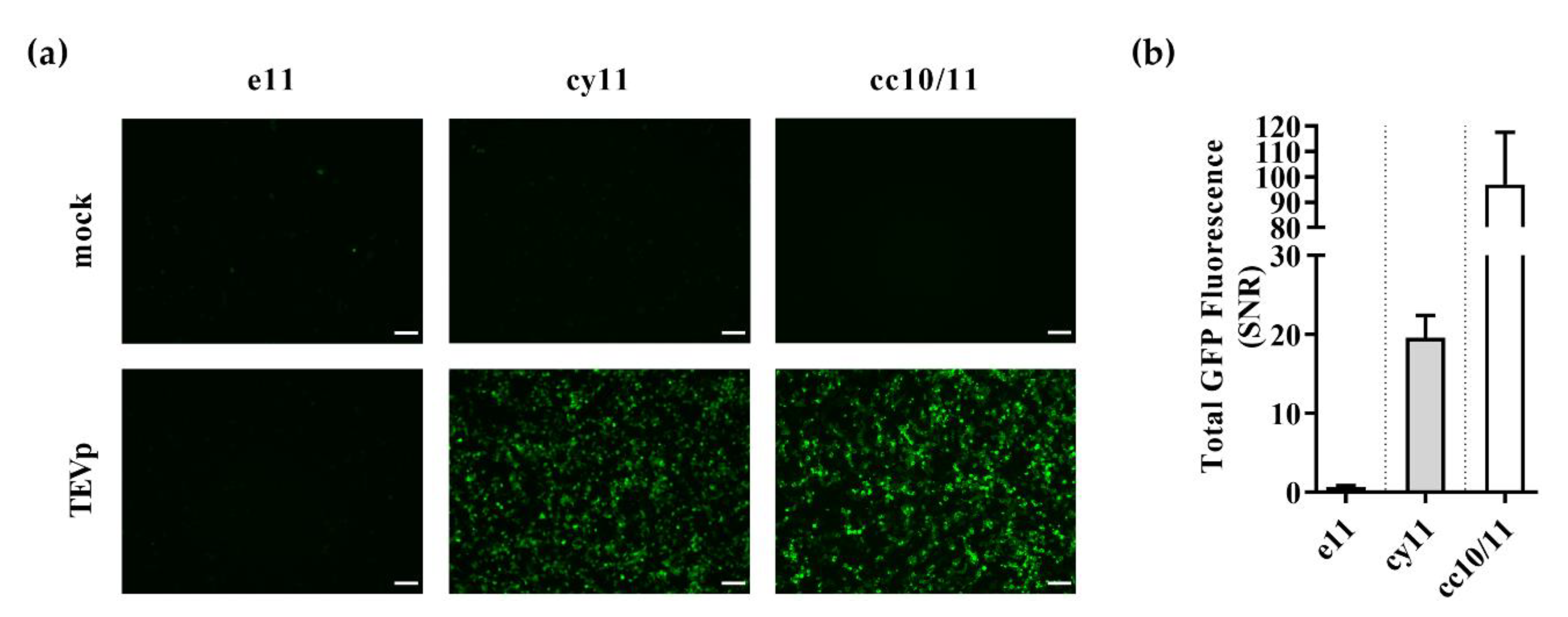
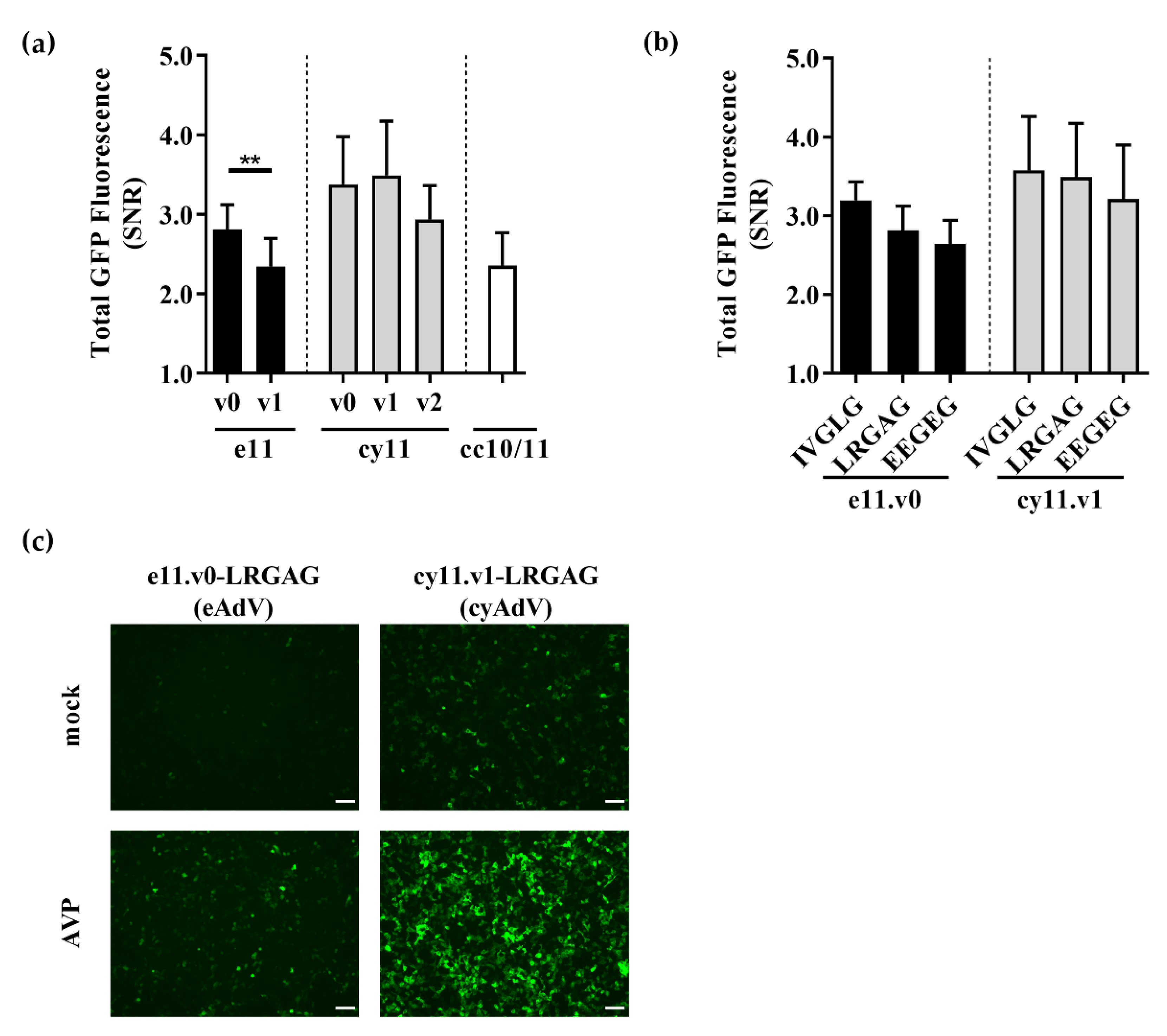
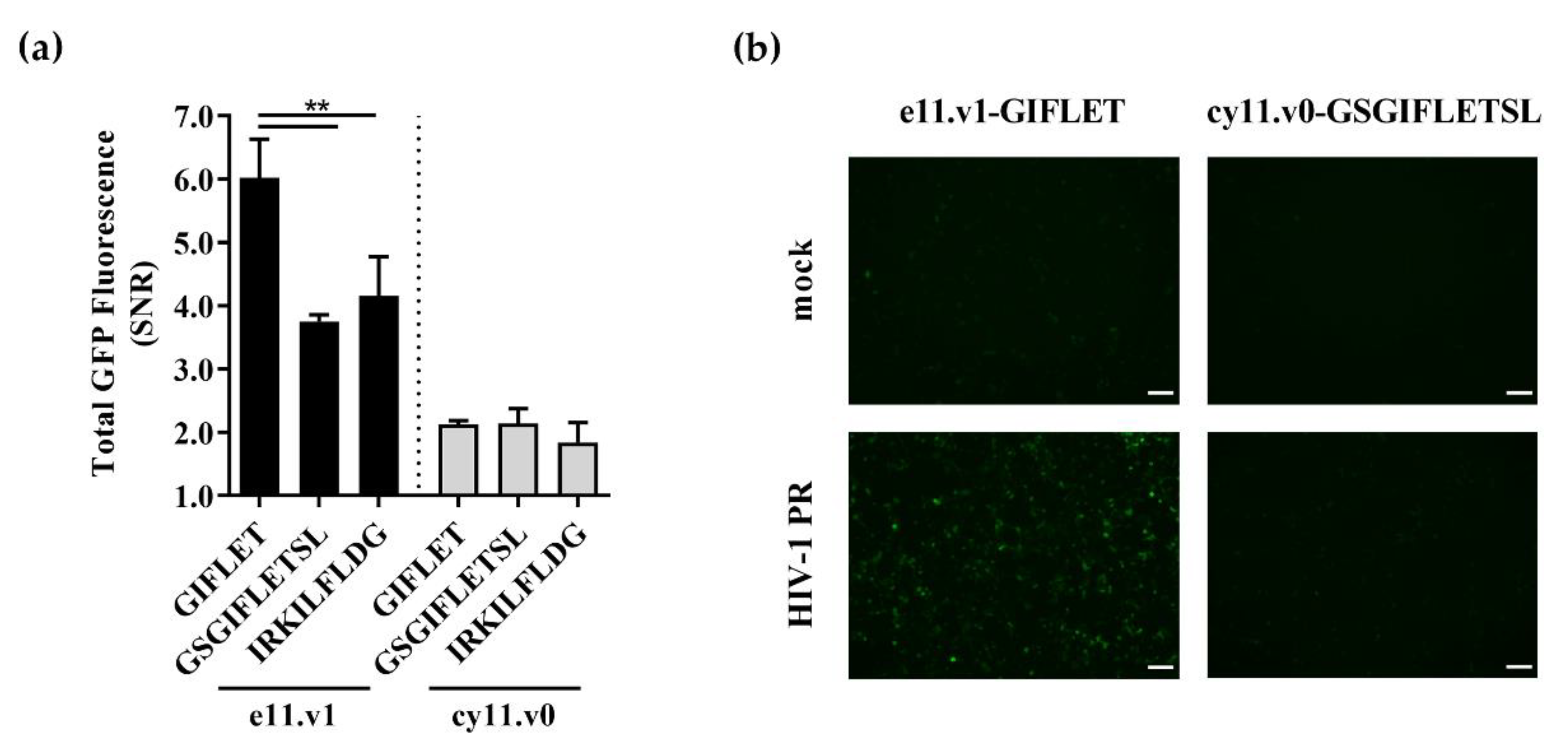
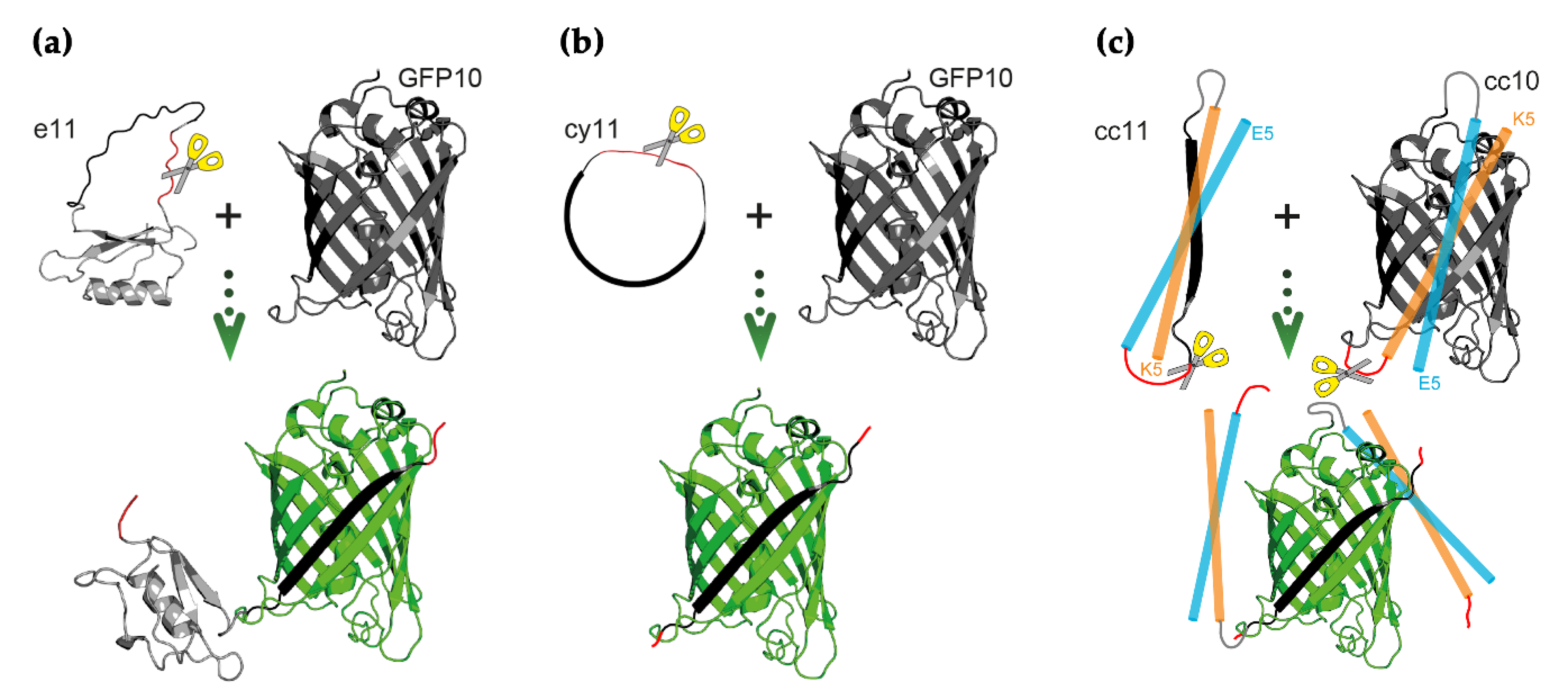
3.1. Design of Switch-On Split GFP Sensors Activated by Proteolysis
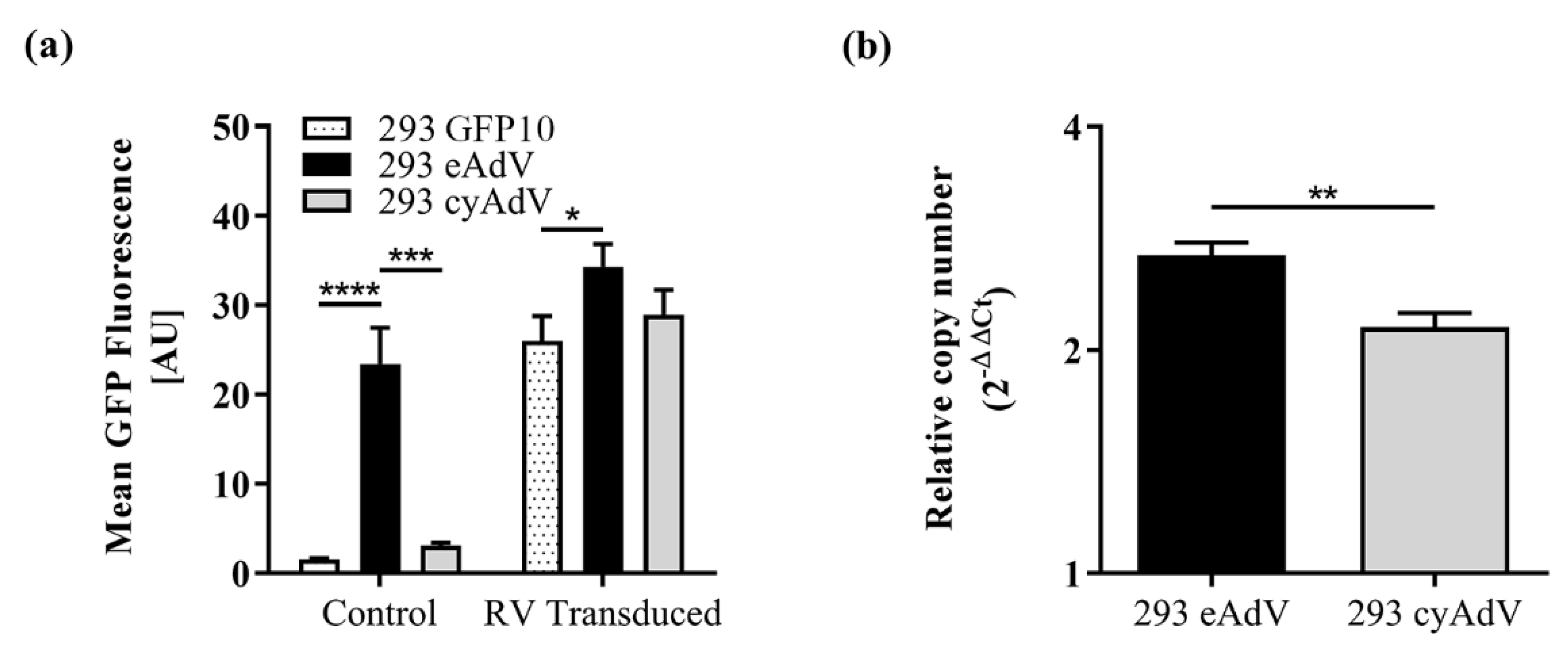
3.2. Switch-On Sensors Activated by Adenovirus and Lentivirus Proteases
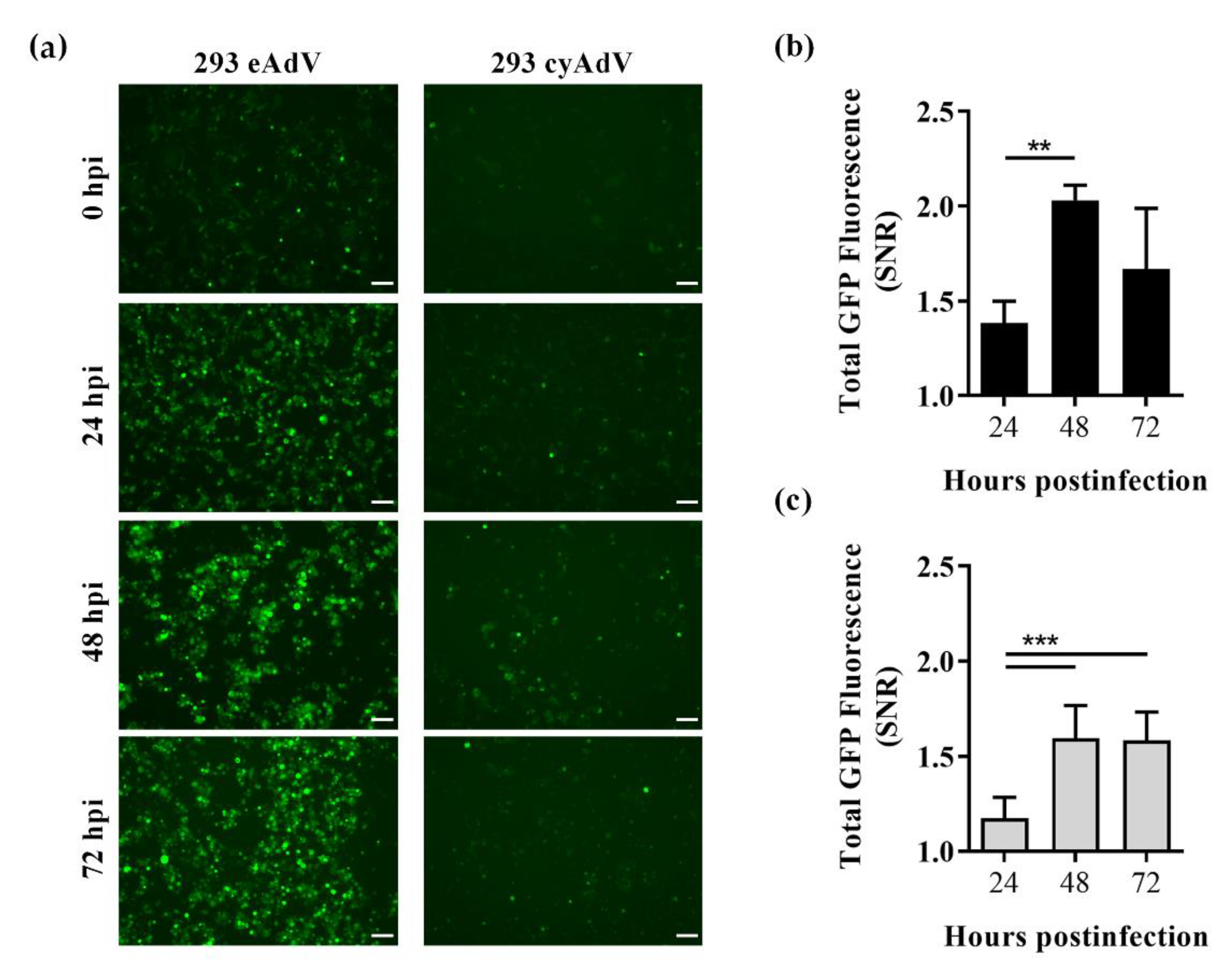
3.3. Whole Cell Biosensing Platform for Monitoring of Adenoviral Infection
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Paules, C.I.; Eisinger, R.W.; Marston, H.D.; Fauci, A.S. What recent history has taught us about responding to emerging infectious disease threats. Ann. Intern. Med. 2017, 167, 805. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E.; Li, G. Approved antiviral drugs over the past 50 years. Clin. Microbiol. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, A.F.; Soares, H.R.; Guerreiro, M.R.; Alves, P.M.; Coroadinha, A.S. Viral vaccines and their manufacturing cell substrates: New trends and designs in modern vaccinology. Biotechnol. J. 2015, 10, 1329–1344. [Google Scholar] [CrossRef] [PubMed]
- Niemann, J.; Kühnel, F. Oncolytic viruses: Adenoviruses. Virus Genes 2017, 53, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Milone, M.C.; O’Doherty, U. Clinical use of lentiviral vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Green, M.; Piña, M.; Kimes, R.C. Biochemical studies on adenovirus multiplication XII. Plaquing efficiencies of purified human adenoviruses. Virology 1967, 31, 562–565. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, L.K.; Smyth, G.K.; Greenfield, P.F. Accuracy of the endpoint assay for virus titration. Cytotechnology 1992, 8, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.-L.; Liu, S.-Q.; Zhou, D.-G.; Xu, L.-L.; Li, X.-D.; Zhang, P.-T.; Li, P.-H.; Ye, H.-Q.; Wei, H.-P.; Yuan, Z.-M.; et al. Development of neutralization assay using an eGFP Chikungunya virus. Viruses 2016, 8, 181. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Wang, L.; Cui, Q.; Li, P.; Wang, Y.; Zhang, Y.; Yang, Y.; Rong, L.; Du, R. A Mechanism underlying attenuation of recombinant influenza a viruses carrying reporter genes. Viruses 2018, 10, 679. [Google Scholar] [CrossRef] [Green Version]
- Tong, L. Viral proteases. Chem. Rev. 2002, 102, 4609–4626. [Google Scholar] [CrossRef]
- Ventoso, I.; Blanco, R.; Perales, C.; Carrasco, L. HIV-1 protease cleaves eukaryotic initiation factor 4G and inhibits cap-dependent translation. Proc. Natl. Acad. Sci. USA 2001, 98, 12966–12971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foy, E. Regulation of interferon regulatory factor-3 by the Hepatitis C virus serine protease. Science 2003, 300, 1145–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agbowuro, A.A.; Huston, W.M.; Gamble, A.B.; Tyndall, J.D.A. Proteases and protease inhibitors in infectious diseases. Med. Res. Rev. 2018, 38, 1295–1331. [Google Scholar] [CrossRef] [PubMed]
- Cabantous, S.; Terwilliger, T.C.; Waldo, G.S. Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol. 2005, 23, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.P.; Stanger, M.J.; Belfort, M. Protease activation of split green fluorescent protein. ChemBioChem 2010, 11, 2259–2263. [Google Scholar] [CrossRef]
- Zhao, F.; Zhao, T.; Deng, L.; Lv, D.; Zhang, X.; Pan, X.; Xu, J.; Long, G. Visualizing the essential role of complete virion assembly machinery in efficient Hepatitis C virus cell-to-cell transmission by a viral infection-activated split-intein-mediated reporter system. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, S.; Terauchi, M.; Hugo, A.; Kim, T.; Araki, Y.; Wada, T. Creation of a caspase-3 sensing system using a combination of split-GFP and split-intein. Chem. Commun. 2013, 49, 10323. [Google Scholar] [CrossRef]
- To, T.-L.; Schepis, A.; Ruiz-González, R.; Zhang, Q.; Yu, D.; Dong, Z.; Coughlin, S.R.; Shu, X. Rational design of a GFP-based fluorogenic caspase reporter for imaging apoptosis in vivo. Cell Chem. Biol. 2016, 23, 875–882. [Google Scholar] [CrossRef] [Green Version]
- Coroadinha, A.S.; Schucht, R.; Gama-Norton, L.; Wirth, D.; Hauser, H.; Carrondo, M.J.T. The use of recombinase mediated cassette exchange in retroviral vector producer cell lines: Predictability and efficiency by transgene exchange. J. Biotechnol. 2006, 124, 457–468. [Google Scholar] [CrossRef]
- Parks, T.D.; Leuther, K.K.; Howard, E.D.; Johnston, S.A.; Dougherty, W.G. Release of proteins and peptides from fusion proteins using a recombinant plant virus proteinase. Anal. Biochem. 1994, 216, 413–417. [Google Scholar] [CrossRef]
- Iwai, H.; Züger, S.; Jin, J.; Tam, P.-H. Highly efficient protein trans-splicing by a naturally split DnaE intein from Nostoc punctiforme. FEBS Lett. 2006, 580, 1853–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diouri, M.; Keyvani-Amineh, H.; Geoghegan, K.F.; Weber, J.M. Cleavage efficiency by adenovirus protease is site-dependent. J. Biol. Chem. 1996, 271, 32511–32514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangel, W.; San Martín, C. Structure, function and dynamics in adenovirus maturation. Viruses 2014, 6, 4536–4570. [Google Scholar] [CrossRef] [Green Version]
- Beck, Z.Q.; Hervio, L.; Dawson, P.E.; Elder, J.H.; Madison, E.L. Identification of efficiently cleaved substrates for HIV-1 protease using a phage display library and use in inhibitor development. Virology 2000, 274, 391–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tözsér, J.; Bláha, I.; Copeland, T.D.; Wondrak, E.M.; Oroszlan, S. Comparison of the HIV-1 and HIV-2 proteinases using oligopeptide substrates representing cleavage sites in Gag and Gag-Pol polyproteins. FEBS Lett. 1991, 281, 77–80. [Google Scholar] [CrossRef] [Green Version]
- Balakirev, M.Y.; Jaquinod, M.; Haas, A.L.; Chroboczek, J. Deubiquitinating function of adenovirus proteinase. J. Virol. 2002, 76, 6323–6331. [Google Scholar] [CrossRef] [Green Version]
- Kapust, R.B.; Tözsér, J.; Fox, J.D.; Anderson, D.E.; Cherry, S.; Copeland, T.D.; Waugh, D.S. Tobacco etch virus protease: Mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. Des. Sel. 2001, 14, 993–1000. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, A.F.; Formas-Oliveira, A.S.; Guerreiro, M.R.; Tomás, H.A.; Alves, P.M.; Coroadinha, A.S. Single-step cloning-screening method: A new tool for developing and studying high-titer viral vector producer cells. Gene Ther. 2015, 22, 685–695. [Google Scholar] [CrossRef] [Green Version]
- Dull, T.; Zufferey, R.; Kelly, M.; Mandel, R.J.; Nguyen, M.; Trono, D.; Naldini, L. A third-generation lentivirus vector with a conditional packaging system. J. Virol. 1998, 72, 8463–8471. [Google Scholar] [CrossRef] [Green Version]
- Guerreiro, M.R.; Freitas, D.F.; Alves, P.M.; Coroadinha, A.S. Detection and quantification of label-free infectious adenovirus using a switch-on cell-based fluorescent biosensor. ACS Sensors 2019, 4, 1654–1661. [Google Scholar] [CrossRef]
- Darling, A.J.; Boose, J.A.; Spaltro, J. Virus assay methods: Accuracy and validation. Biologicals 1998, 26, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Ormö, M.; Cubitt, A.B.; Kallio, K.; Gross, L.A.; Tsien, R.Y.; Remington, S.J. Crystal structure of the Aequorea victoria green fluorescent protein. Science 1996, 273, 1392–1395. [Google Scholar] [CrossRef] [Green Version]
- Hyberts, S.G.; Goldberg, M.S.; Havel, T.F.; Wagner, G. The solution structure of eglin c based on measurements of many NOEs and coupling constants and its comparison with X-ray structures. Protein Sci. 1992, 1, 736–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, A.; Russell, S.; Talbot, P.; Russell, W.C.; Kemp, G.D. Characterization of the adenovirus proteinase: Substrate specificity. J. Gen. Virol. 1989, 70, 3225–3234. [Google Scholar] [CrossRef]
- Ruzindana-Umunyana, A.; Imbeault, L.; Weber, J.M. Substrate specificity of adenovirus protease. Virus Res. 2002, 89, 41–52. [Google Scholar] [CrossRef]
- Cabantous, S.; Nguyen, H.B.; Pedelacq, J.-D.; Koraïchi, F.; Chaudhary, A.; Ganguly, K.; Lockard, M.A.; Favre, G.; Terwilliger, T.C.; Waldo, G.S. A new protein-protein interaction sensor based on tripartite split-GFP association. Sci. Rep. 2013, 3, 2854. [Google Scholar] [CrossRef]
- Kent, K.P.; Oltrogge, L.M.; Boxer, S.G. Synthetic control of green fluorescent protein. J. Am. Chem. Soc. 2009, 131, 15988–15989. [Google Scholar] [CrossRef] [Green Version]
- Ozalp, C.; Szczesna-Skorupa, E.; Kemper, B. Bimolecular fluorescence complementation analysis of cytochrome p450 2c2, 2e1, and NADPH-cytochrome p450 reductase molecular interactions in living cells. Drug Metab. Dispos. 2005, 33, 1382–1390. [Google Scholar] [CrossRef] [Green Version]
- Demonte, D.; Li, N.; Park, S. Postsynthetic domain assembly with NpuDnaE and SspDnaB split inteins. Appl. Biochem. Biotechnol. 2015, 177, 1137–1151. [Google Scholar] [CrossRef]
- Chen, P.H.; Ornelles, D.A.; Shenk, T. The adenovirus L3 23-kilodalton proteinase cleaves the amino-terminal head domain from cytokeratin 18 and disrupts the cytokeratin network of HeLa cells. J. Virol. 1993, 67, 3507–3514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, M.T.; Mangel, W.F. Interaction of actin and its 11-amino acid C-terminal peptide as cofactors with the adenovirus proteinase. FEBS Lett. 2004, 563, 213–218. [Google Scholar] [CrossRef] [Green Version]
- Mangel, W.F.; McGrath, W.J.; Toledo, D.L.; Anderson, C.W. Viral DNA and a viral peptide can act as cofactors of adenovirus virion proteinase activity. Nature 1993, 361, 274–275. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Lutz, S. Circular permutation: A different way to engineer enzyme structure and function. Trends Biotechnol. 2011, 29, 18–25. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerreiro, M.R.; Fernandes, A.R.; Coroadinha, A.S. Evaluation of Structurally Distorted Split GFP Fluorescent Sensors for Cell-Based Detection of Viral Proteolytic Activity. Sensors 2021, 21, 24. https://doi.org/10.3390/s21010024
Guerreiro MR, Fernandes AR, Coroadinha AS. Evaluation of Structurally Distorted Split GFP Fluorescent Sensors for Cell-Based Detection of Viral Proteolytic Activity. Sensors. 2021; 21(1):24. https://doi.org/10.3390/s21010024
Chicago/Turabian StyleGuerreiro, Miguel R., Ana R. Fernandes, and Ana S. Coroadinha. 2021. "Evaluation of Structurally Distorted Split GFP Fluorescent Sensors for Cell-Based Detection of Viral Proteolytic Activity" Sensors 21, no. 1: 24. https://doi.org/10.3390/s21010024