
Host Species and Captivity Distinguish the Microbiome Compositions of a Diverse Zoo-Resident Non-Human Primate Population
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Pipeline and Database Comparison
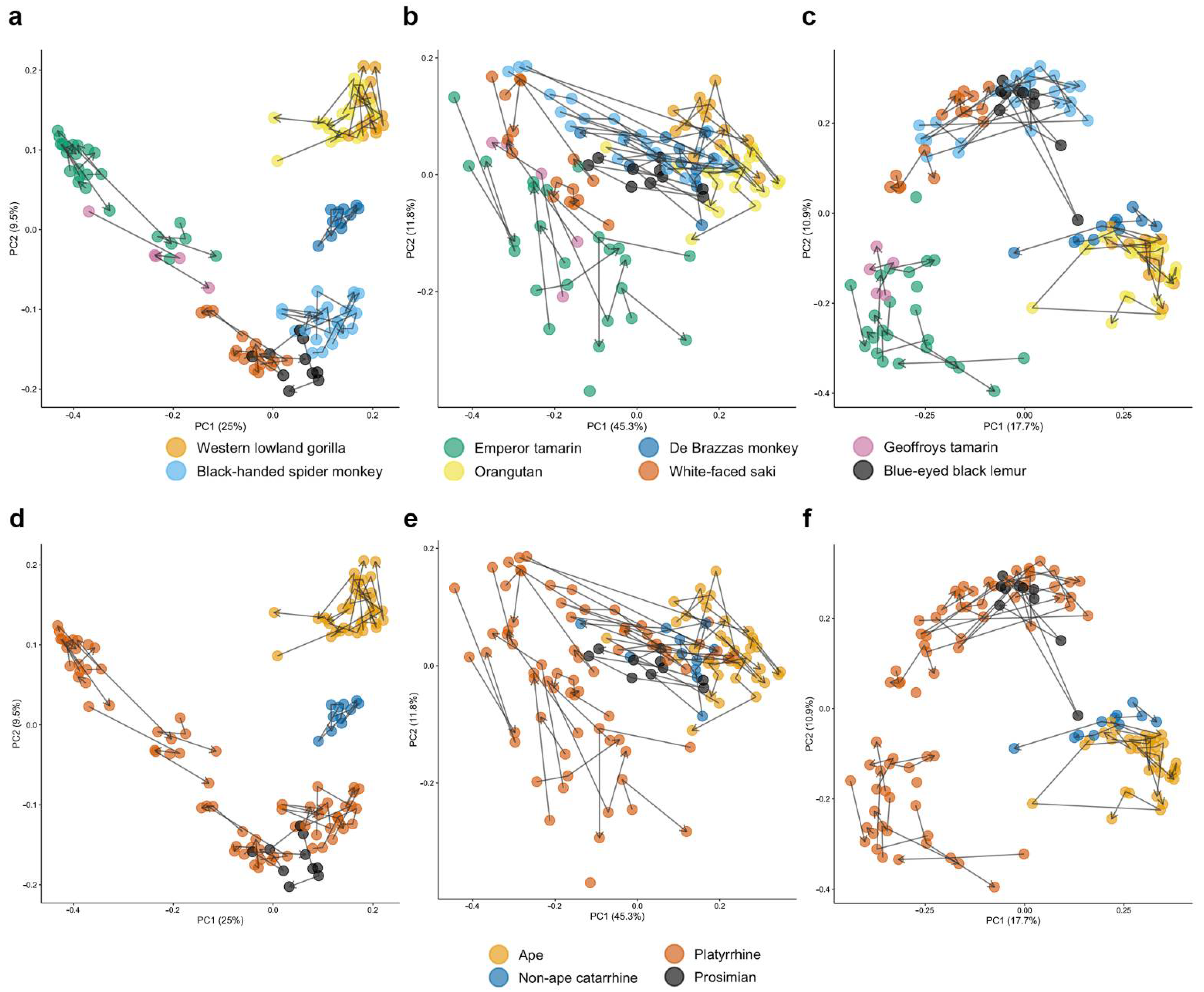
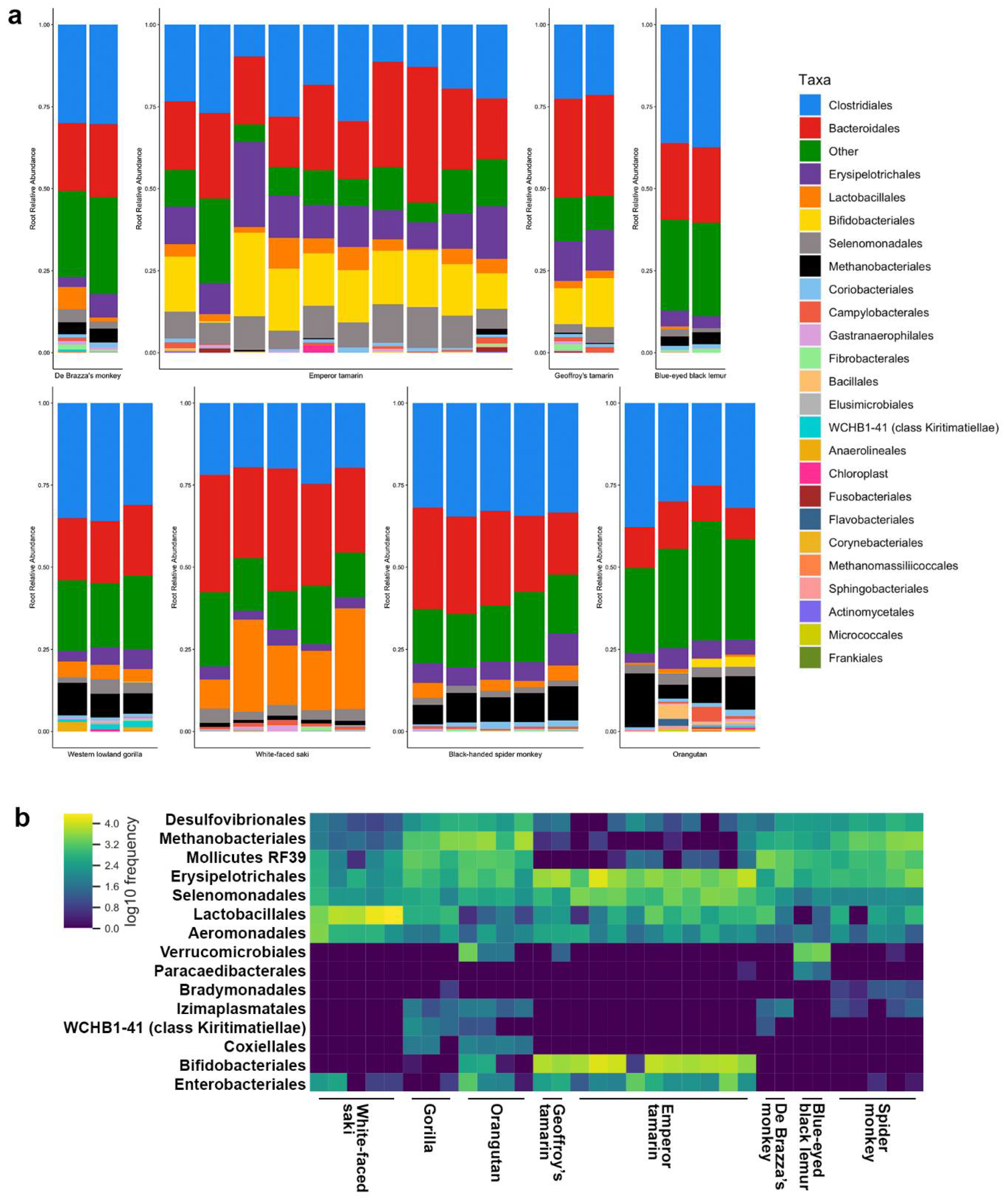
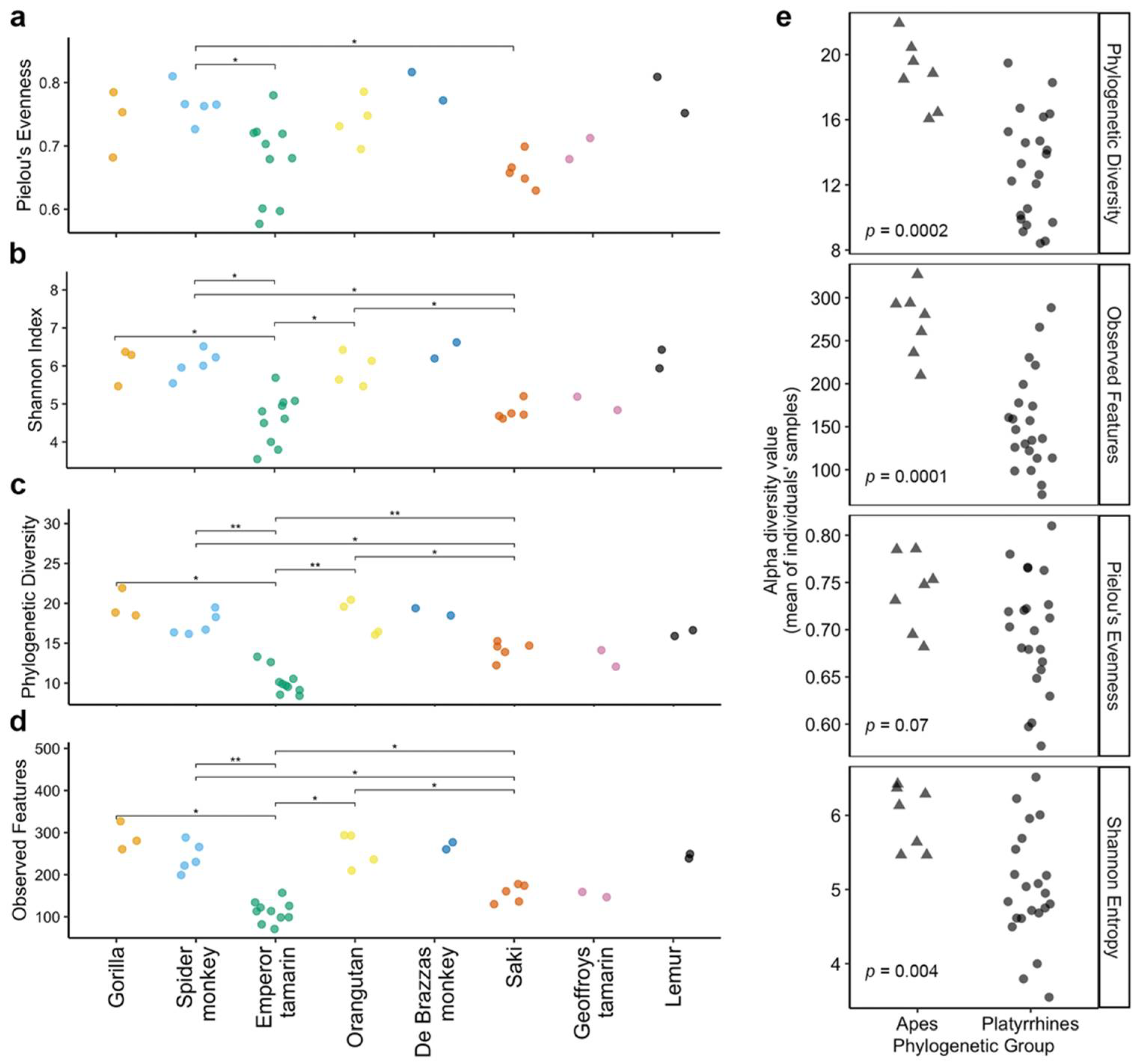
3.2. Host Species Are Distinguished by Microbiome Composition in Diverse Zoo-Resident Primates
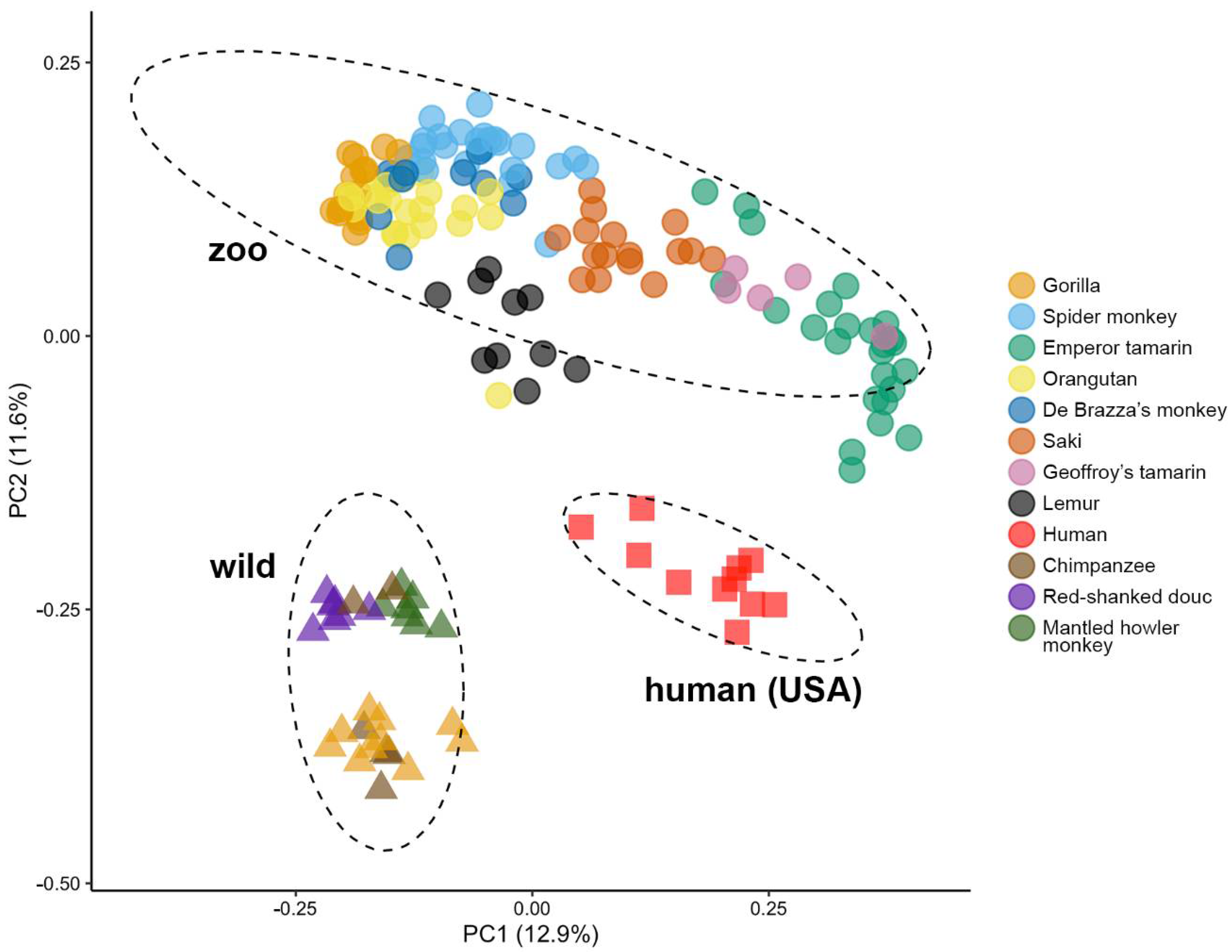
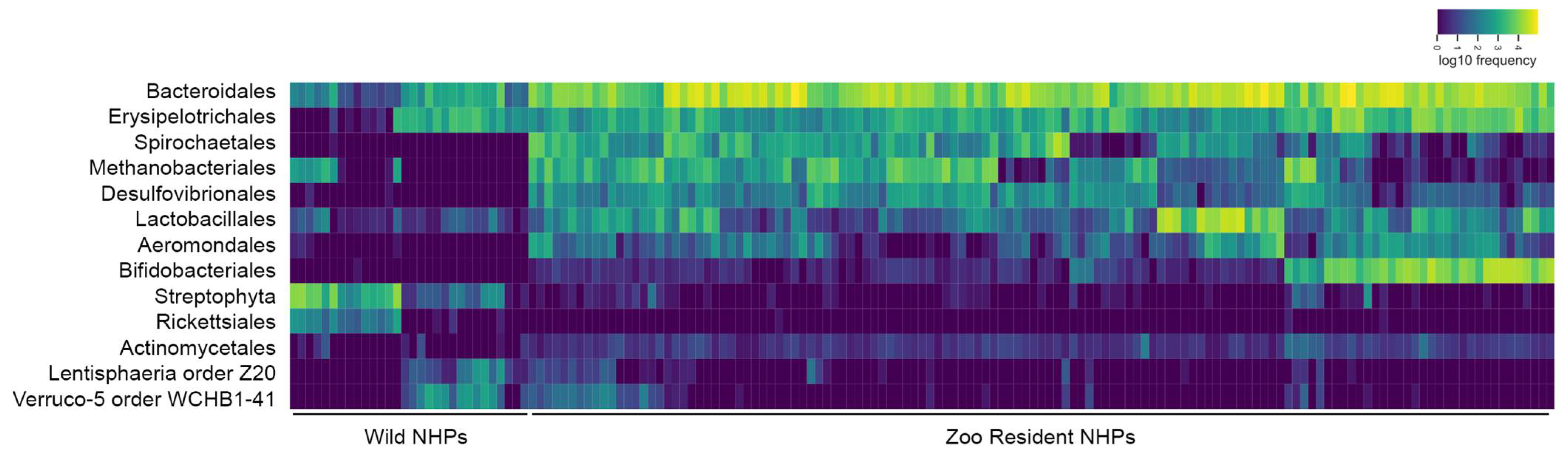
3.3. Zoo-Resident Primates Have Distinct Microbiomes Compared to Wild Counterparts
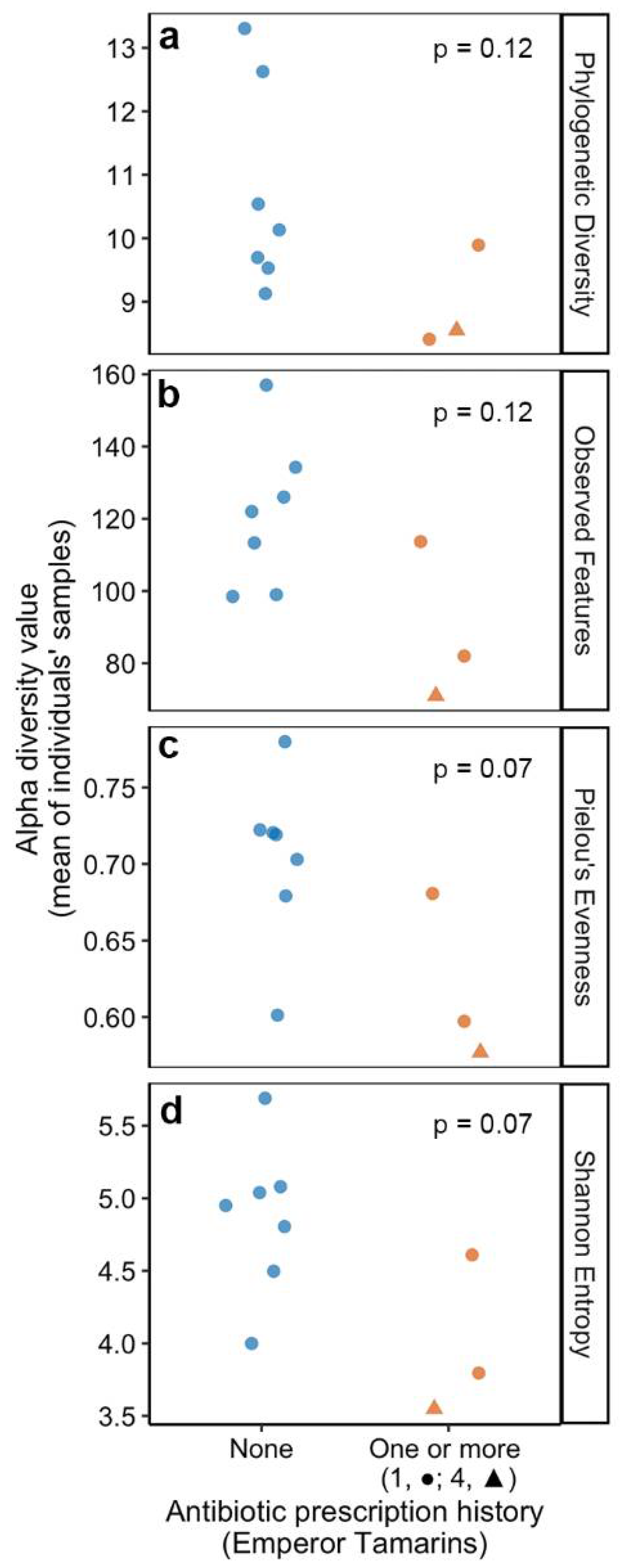
3.4. Historic Antibiotic Usage Is Linked to Lower Gut Microbiome Diversity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morgan, X.C.; Huttenhower, C. Chapter 12: Human Microbiome Analysis. PLoS Comput. Biol. 2012, 8, e1002808. [Google Scholar] [CrossRef] [PubMed]
- Björk, J.R.; Dasari, M.; Grieneisen, L.; Archie, E.A. Primate Microbiomes over Time: Longitudinal Answers to Standing Questions in Microbiome Research. Am. J. Primatol. 2019, 81, e22970. [Google Scholar] [CrossRef] [PubMed]
- Human Microbiome Project Consortium Structure, Function and Diversity of the Healthy Human Microbiome. Nature 2012, 486, 207–214. [CrossRef] [PubMed]
- Lloyd-Price, J.; Mahurkar, A.; Rahnavard, G.; Crabtree, J.; Orvis, J.; Hall, A.B.; Brady, A.; Creasy, H.H.; McCracken, C.; Giglio, M.G.; et al. Strains, Functions and Dynamics in the Expanded Human Microbiome Project. Nature 2017, 550, 61–66, advance online publication. [Google Scholar] [CrossRef]
- Petersen, C.; Round, J.L. Defining Dysbiosis and Its Influence on Host Immunity and Disease. Cell Microbiol. 2014, 16, 1024–1033. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current Understanding of the Human Microbiome. Nat. Med. 2018, 24, 392–400. [Google Scholar] [CrossRef]
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The Impact of the Gut Microbiota on Human Health: An Integrative View. Cell 2012, 148, 1258–1270. [Google Scholar] [CrossRef]
- Halfvarson, J.; Brislawn, C.J.; Lamendella, R.; Vázquez-Baeza, Y.; Walters, W.A.; Bramer, L.M.; D’Amato, M.; Bonfiglio, F.; McDonald, D.; Gonzalez, A.; et al. Dynamics of the Human Gut Microbiome in Inflammatory Bowel Disease. Nat. Microbiol. 2017, 2, 1–7. [Google Scholar] [CrossRef]
- Clayton, J.B.; Gomez, A.; Amato, K.; Knights, D.; Travis, D.A.; Blekhman, R.; Knight, R.; Leigh, S.; Stumpf, R.; Wolf, T.; et al. The Gut Microbiome of Nonhuman Primates: Lessons in Ecology and Evolution. Am. J. Primatol. 2018, 80, e22867. [Google Scholar] [CrossRef]
- Amato, K.R. Missing Links: The Role of Primates in Understanding the Human Microbiome. mSystems 2019, 4, e00165-19. [Google Scholar] [CrossRef] [Green Version]
- Amato, K.R.; Metcalf, J.L.; Song, S.J.; Hale, V.L.; Clayton, J.; Ackermann, G.; Humphrey, G.; Niu, K.; Cui, D.; Zhao, H.; et al. Using the Gut Microbiota as a Novel Tool for Examining Colobine Primate GI Health. Glob. Ecol. Conserv. 2016, 7, 225–237. [Google Scholar] [CrossRef]
- West, A.G.; Waite, D.W.; Deines, P.; Bourne, D.G.; Digby, A.; McKenzie, V.J.; Taylor, M.W. The Microbiome in Threatened Species Conservation. Biol. Conserv. 2019, 229, 85–98. [Google Scholar] [CrossRef]
- Lee, R.V.; Prowten, A.W.; Anthone, S.; Satchidanand, S.K.; Fisher, J.E.; Anthone, R. Typhlitis Due to Balantidium Coli in Captive Lowland Gorillas. Rev. Infect. Dis. 1990, 12, 1052–1059. [Google Scholar] [CrossRef]
- Madara, J.L.; Podolsky, D.K.; King, N.W.; Sehgal, P.K.; Moore, R.; Winter, H.S. Characterization of Spontaneous Colitis in Cotton-Top Tamarins (Saguinus Oedipus) and Its Response to Sulfasalazine. Gastroenterology 1985, 88, 13–19. [Google Scholar] [CrossRef]
- Clayton, J.B.; Vangay, P.; Huang, H.; Ward, T.; Hillmann, B.M.; Al-Ghalith, G.A.; Travis, D.A.; Long, H.T.; Tuan, B.V.; Minh, V.V.; et al. Captivity Humanizes the Primate Microbiome. Proc. Natl. Acad. Sci. USA 2016, 113, 10376–10381. [Google Scholar] [CrossRef] [PubMed]
- Frankel, J.S.; Mallott, E.K.; Hopper, L.M.; Ross, S.R.; Amato, K.R. The Effect of Captivity on the Primate Gut Microbiome Varies with Host Dietary Niche. Am. J. Primatol. 2019, 81, e23061. [Google Scholar] [CrossRef]
- Moeller, A.H.; Peeters, M.; Ndjango, J.-B.; Li, Y.; Hahn, B.H.; Ochman, H. Sympatric Chimpanzees and Gorillas Harbor Convergent Gut Microbial Communities. Genome Res. 2013, 23, 1715–1720. [Google Scholar] [CrossRef]
- Ochman, H.; Worobey, M.; Kuo, C.-H.; Ndjango, J.-B.N.; Peeters, M.; Hahn, B.H.; Hugenholtz, P. Evolutionary Relationships of Wild Hominids Recapitulated by Gut Microbial Communities. PLoS Biol. 2010, 8, e1000546. [Google Scholar] [CrossRef]
- Grieneisen, L.; Dasari, M.; Gould, T.J.; Björk, J.R.; Grenier, J.-C.; Yotova, V.; Jansen, D.; Gottel, N.; Gordon, J.B.; Learn, N.H.; et al. Gut Microbiome Heritability Is Nearly Universal but Environmentally Contingent. Science 2021, 373, 181–186. [Google Scholar] [CrossRef]
- Moeller, A.H.; Li, Y.; Mpoudi Ngole, E.; Ahuka-Mundeke, S.; Lonsdorf, E.V.; Pusey, A.E.; Peeters, M.; Hahn, B.H.; Ochman, H. Rapid Changes in the Gut Microbiome during Human Evolution. Proc. Natl. Acad. Sci. USA 2014, 111, 16431–16435. [Google Scholar] [CrossRef] [Green Version]
- Perelman, P.; Johnson, W.E.; Roos, C.; Seuánez, H.N.; Horvath, J.E.; Moreira, M.A.M.; Kessing, B.; Pontius, J.; Roelke, M.; Rumpler, Y.; et al. A Molecular Phylogeny of Living Primates. PLOS Genet. 2011, 7, e1001342. [Google Scholar] [CrossRef] [PubMed]
- Hicks, A.L.; Lee, K.J.; Couto-Rodriguez, M.; Patel, J.; Sinha, R.; Guo, C.; Olson, S.H.; Seimon, A.; Seimon, T.A.; Ondzie, A.U.; et al. Gut Microbiomes of Wild Great Apes Fluctuate Seasonally in Response to Diet. Nat. Commun. 2018, 9, 1786. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.A.; Meyer, F.; Antonopoulos, D.; Balaji, P.; Brown, C.T.; Brown, C.T.; Desai, N.; Eisen, J.A.; Evers, D.; Field, D.; et al. Meeting Report: The Terabase Metagenomics Workshop and the Vision of an Earth Microbiome Project. Stand. Genomic. Sci. 2010, 3, 243–248. [Google Scholar] [CrossRef]
- Al-Ghalith, G.A.; Hillmann, B.; Ang, K.; Shields-Cutler, R.; Knights, D. SHI7 Is a Self-Learning Pipeline for Multipurpose Short-Read DNA Quality Control. mSystems 2018, 3, e00202-17. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a Chimera-Checked 16S RRNA Gene Database and Workbench Compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Nearing, J.T.; Douglas, G.M.; Comeau, A.M.; Langille, M.G.I. Denoising the Denoisers: An Independent Evaluation of Microbiome Sequence Error-Correction Approaches. PeerJ 2018, 6, e5364. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-Resolution Sample Inference from Illumina Amplicon Data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic. Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009; ISBN 978-0-387-98140-6. [Google Scholar]
- Lozupone, C.; Knight, R. UniFrac: A New Phylogenetic Method for Comparing Microbial Communities. Appl. Environ. Microbiol. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [Green Version]
- Muegge, B.D.; Kuczynski, J.; Knights, D.; Clemente, J.C.; González, A.; Fontana, L.; Henrissat, B.; Knight, R.; Gordon, J.I. Diet Drives Convergence in Gut Microbiome Functions across Mammalian Phylogeny and within Humans. Science 2011, 332, 970–974. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Van Treuren, W.; White, R.A.; Eggesbø, M.; Knight, R.; Peddada, S.D. Analysis of Composition of Microbiomes: A Novel Method for Studying Microbial Composition. Microb. Ecol. Health Dis. 2015, 26, 27663. [Google Scholar] [CrossRef] [PubMed]
- Knight, R.; Vrbanac, A.; Taylor, B.C.; Aksenov, A.; Callewaert, C.; Debelius, J.; Gonzalez, A.; Kosciolek, T.; McCall, L.-I.; McDonald, D.; et al. Best Practices for Analysing Microbiomes. Nat. Rev. Microbiol. 2018, 16, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Poussin, C.; Sierro, N.; Boué, S.; Battey, J.; Scotti, E.; Belcastro, V.; Peitsch, M.C.; Ivanov, N.V.; Hoeng, J. Interrogating the Microbiome: Experimental and Computational Considerations in Support of Study Reproducibility. Drug Discov. Today 2018, 23, 1644–1657. [Google Scholar] [CrossRef]
- Vlčková, K.; Gomez, A.; Petrželková, K.J.; Whittier, C.A.; Todd, A.F.; Yeoman, C.J.; Nelson, K.E.; Wilson, B.A.; Stumpf, R.M.; Modrý, D.; et al. Effect of Antibiotic Treatment on the Gastrointestinal Microbiome of Free-Ranging Western Lowland Gorillas (Gorilla g. Gorilla). Microb. Ecol. 2016, 72, 943–954. [Google Scholar] [CrossRef]
- Sidiropoulos, D.N.; Al-Ghalith, G.A.; Shields-Cutler, R.R.; Ward, T.L.; Johnson, A.J.; Vangay, P.; Knights, D.; Kashyap, P.C.; Xian, Y.; Ramer-Tait, A.E.; et al. Wild Primate Microbiomes Prevent Weight Gain in Germ-Free Mice. Anim. Microbiome 2020, 2, 16. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet Rapidly and Reproducibly Alters the Human Gut Microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef]
- Brown, J.M.; Hazen, S.L. Targeting of Microbe-Derived Metabolites to Improve Human Health: The next Frontier for Drug Discovery. J. Biol. Chem. 2017, 292, 8560–8568. [Google Scholar] [CrossRef]
- Kuntz, T.M.; Gilbert, J.A. Introducing the Microbiome into Precision Medicine. Trends. Pharmacol. Sci. 2017, 38, 81–91. [Google Scholar] [CrossRef]
- O’Callaghan, A.; van Sinderen, D. Bifidobacteria and Their Role as Members of the Human Gut Microbiota. Front. Microbiol. 2016, 7, 925. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-R.; Zhou, L.-Z.; Fang, S.-T.; Long, H.-Y.; Chen, J.-Y.; Zhang, G.-X. Isolation of Desulfovibrio Spp. from Human Gut Microbiota Using a next-Generation Sequencing Directed Culture Method. Lett. Appl. Microbiol. 2019, 68, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Bornbusch, S.L.; Greene, L.K.; McKenney, E.A.; Volkoff, S.J.; Midani, F.S.; Joseph, G.; Gerhard, W.A.; Iloghalu, U.; Granek, J.; Gunsch, C.K. A Comparative Study of Gut Microbiomes in Captive Nocturnal Strepsirrhines. Am. J. Primatol. 2019, 81, e22986. [Google Scholar] [CrossRef] [PubMed]
- Gouba, N.; Raoult, D.; Drancourt, M. Plant and Fungal Diversity in Gut Microbiota as Revealed by Molecular and Culture Investigations. PLoS ONE 2013, 8, e59474. [Google Scholar] [CrossRef]
- Sonnenburg, E.D.; Smits, S.A.; Tikhonov, M.; Higginbottom, S.K.; Wingreen, N.S.; Sonnenburg, J.L. Diet-Induced Extinctions in the Gut Microbiota Compound over Generations. Nature 2016, 529, 212–215. [Google Scholar] [CrossRef]
- Carter, J.K.; Bhattacharya, D.; Borgerding, J.N.; Fiel, M.I.; Faith, J.J.; Friedman, S.L. Modeling Dysbiosis of Human NASH in Mice: Loss of Gut Microbiome Diversity and Overgrowth of Erysipelotrichales. PLoS ONE 2021, 16, e0244763. [Google Scholar] [CrossRef]
- Koo, B.S.; Hwang, E.H.; Kim, G.; Oh, H.; Son, Y.; Lee, D.; Lim, K.S.; Kang, P.; Lee, S.; Lee, H.Y.; et al. Evaluation of Fecal Microbiomes Associated with Obesity in Captive Cynomolgus Monkeys (Macaca Fascicularis). J. Vet. Sci. 2019, 20, e19. [Google Scholar] [CrossRef]
- Li, J.; Li, Y.; Zhou, Y.; Wang, C.; Wu, B.; Wan, J. Actinomyces and Alimentary Tract Diseases: A Review of Its Biological Functions and Pathology. Biomed. Res. Int. 2018, 2018, 3820215. [Google Scholar] [CrossRef]
- Rodiño-Janeiro, B.K.; Vicario, M.; Alonso-Cotoner, C.; Pascua-García, R.; Santos, J. A Review of Microbiota and Irritable Bowel Syndrome: Future in Therapies. Adv. Ther. 2018, 35, 289–310. [Google Scholar] [CrossRef]
- Pérez-Santiago, J.; Gianella, S.; Massanella, M.; Spina, C.A.; Karris, M.Y.; Var, S.R.; Patel, D.; Jordan, P.S.; Young, J.A.; Little, S.J.; et al. Gut Lactobacillales Are Associated with Higher CD4 and Less Microbial Translocation during HIV Infection. AIDS 2013, 27, 1921–1931. [Google Scholar] [CrossRef]
- Looft, T.; Allen, H.K. Collateral Effects of Antibiotics on Mammalian Gut Microbiomes. Gut Microbes 2012, 3, 463–467. [Google Scholar] [CrossRef]
- Hernández, E.; Bargiela, R.; Diez, M.S.; Friedrichs, A.; Pérez-Cobas, A.E.; Gosalbes, M.J.; Knecht, H.; Martínez-Martínez, M.; Seifert, J.; von Bergen, M.; et al. Functional Consequences of Microbial Shifts in the Human Gastrointestinal Tract Linked to Antibiotic Treatment and Obesity. Gut Microbes 2013, 4, 306–315. [Google Scholar] [CrossRef]
- Prince, A.L.; Pace, R.M.; Dean, T.; Takahashi, D.; Kievit, P.; Friedman, J.E.; Aagaard, K.M. The Development and Ecology of the Japanese Macaque Gut Microbiome from Weaning to Early Adolescence in Association with Diet. Am. J. Primatol. 2019, 81, e22980. [Google Scholar] [CrossRef]
- Vangay, P.; Johnson, A.J.; Ward, T.L.; Al-Ghalith, G.A.; Shields-Cutler, R.R.; Hillmann, B.M.; Lucas, S.K.; Beura, L.K.; Thompson, E.A.; Till, L.M.; et al. US Immigration Westernizes the Human Gut Microbiome. Cell 2018, 175, 962–972.e10. [Google Scholar] [CrossRef]
- Kaplan, R.C.; Wang, Z.; Usyk, M.; Sotres-Alvarez, D.; Daviglus, M.L.; Schneiderman, N.; Talavera, G.A.; Gellman, M.D.; Thyagarajan, B.; Moon, J.-Y.; et al. Gut Microbiome Composition in the Hispanic Community Health Study/Study of Latinos Is Shaped by Geographic Relocation, Environmental Factors, and Obesity. Genome Biol. 2019, 20, 219. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wills, M.O.; Shields-Cutler, R.R.; Brunmeier, E.; Weissenborn, M.; Murphy, T.; Knights, D.; Johnson, T.J.; Clayton, J.B. Host Species and Captivity Distinguish the Microbiome Compositions of a Diverse Zoo-Resident Non-Human Primate Population. Diversity 2022, 14, 715. https://doi.org/10.3390/d14090715
Wills MO, Shields-Cutler RR, Brunmeier E, Weissenborn M, Murphy T, Knights D, Johnson TJ, Clayton JB. Host Species and Captivity Distinguish the Microbiome Compositions of a Diverse Zoo-Resident Non-Human Primate Population. Diversity. 2022; 14(9):715. https://doi.org/10.3390/d14090715
Chicago/Turabian StyleWills, Maya O., Robin R. Shields-Cutler, Emily Brunmeier, Madison Weissenborn, Tami Murphy, Dan Knights, Timothy J. Johnson, and Jonathan B. Clayton. 2022. "Host Species and Captivity Distinguish the Microbiome Compositions of a Diverse Zoo-Resident Non-Human Primate Population" Diversity 14, no. 9: 715. https://doi.org/10.3390/d14090715