
(R)-(+)-3,5-Dinitro-N-(1-phenylethyl)benzothioamide

Department of Chemistry and Biochemistry, University of Southern Mississippi, Hattiesburg, MS 39406, USA
*
Author to whom correspondence should be addressed.
Molbank 2023, 2023(2), M1650; https://doi.org/10.3390/M1650
Submission received: 22 April 2023
/
Revised: 12 May 2023
/
Accepted: 17 May 2023
/
Published: 20 May 2023
(This article belongs to the Section Organic Synthesis)
Abstract
:(R)-(+)-3,5-dinitro-N-(1-phenylethyl)benzothioamide 1 is a potential chiral solvating agent (CSA) for the spectral resolution of enantiomers via 1H NMR spectroscopy. The single enantiomer of 1 was synthesized from commercially available (R)-(+)-a-methylbenzylamine 2 in two steps with 85% yield.
1. Introduction
Chiral solvating agents (CSAs) are a class of molecules utilized for the spectral resolution of enantiomers via NMR spectroscopy (Figure 1) [1]. Such a resolution is possible because the CSAs associate with analytes via non-covalent interactions (NCIs) to form diastereomeric complexes resulting in chemical shift differences ∆∆δ of the enantiomers [2]. The scaffold of the CSA must possess some stereogenic features (atom, axis, plane) embedded in its backbone along with functional groups capable of eliciting NCIs, including hydrogen bond donation/acceptance and pi acidity/basicity [3,4].
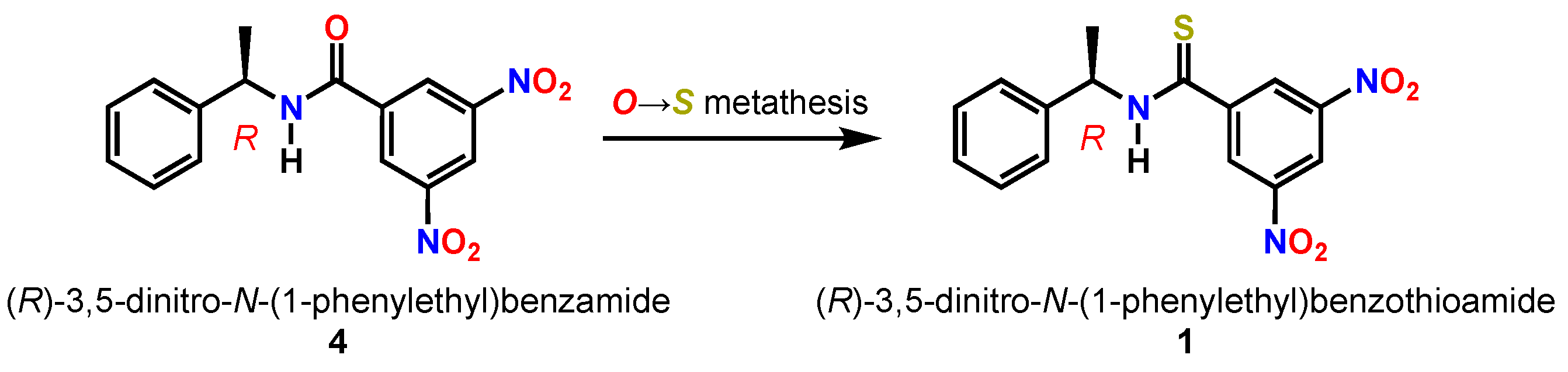
(R)-(−)-3,5-dinitro-N-(1-phenylethyl)benzamide 4, widely known as Kagan’s amide [5], is a validated CSA for the discrimination of a wide selection of analytes with functional groups including alcohols [6], amines/amides [7], carboxylic acids, phosphine oxides [8], phospholene oxides [9], and sulfoxides. To date, the thioamide variant 1 of the Kagan amide 4 has not been disclosed in the literature. However, the desnitro compound (R)-N-(1-phenylethyl)benzothioamide has been prepared [10], characterized in the solid state by single-crystal X-ray diffraction [11], and utilized synthetically [12]. Given the broad utility of the thiocarbonyl functional group [13] in validated organocatalysts such as thioureas [14] and thiosquaramides [15] that enable asymmetric transformations through NCIs [16,17], we hypothesized that thioamide 1 would be a competent CSA analogous to 4. Since sulfur has a larger van der Waals radius than oxygen (S = 1.85 Å vs. O = 1.40 Å), the C=S bond is longer than the C=O bond (1.60 Å vs. 1.40 Å) [18]. Because of these physical properties, thioamides are less prone to self-aggregation than amides [19] since they are weaker hydrogen bond acceptors. Additionally, due to the increased acidity of the N–H bond of ∆pKa = −6 [20], thioamides are stronger hydrogen bond donors [21]. With these physical factors in mind, we set out to synthesize 1 for the purposes of using it as a CSA with the goal of using it as a tool for the determination of absolute configuration [22].
2. Results and Discussion
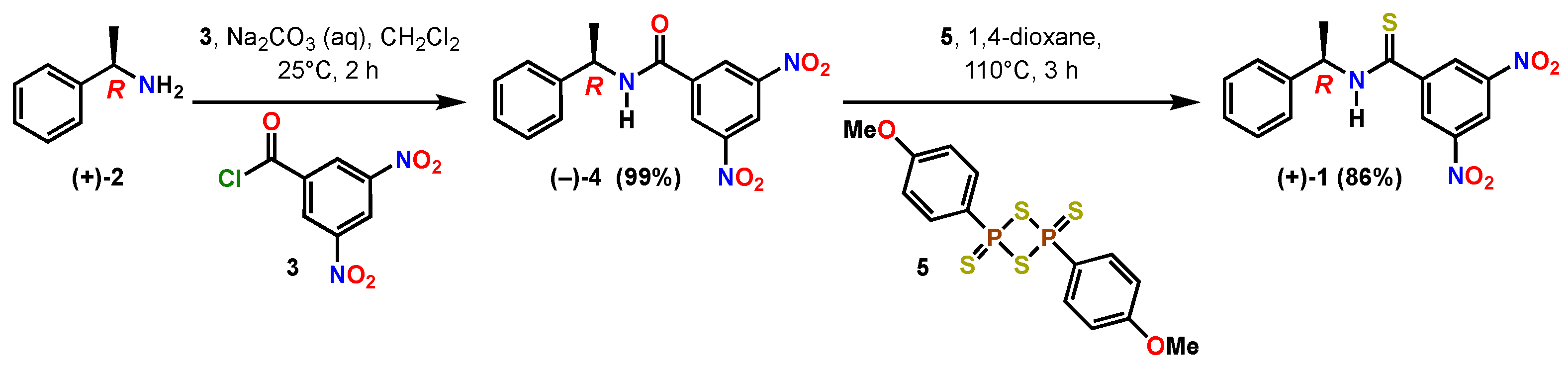
The title compound (R)-(+)-3,5-dinitro-N-(1-phenylethyl)benzothioamide 1 was prepared in one step from (R)-(−)-3,5-dinitro-N-(1-phenylethyl)benzamide 4 (Scheme 1). The Kagan amide 4 was readily prepared in quantitative yield as an off-white solid (mp 151–153 °C) in decagram quantities through the coupling of commercially available enantiopure (R)-(+)-α-methylbenzylamine 2 and 3,5-dinitrobenzoyl chloride 3 under biphasic conditions with dichloromethane in aqueous sodium carbonate. The specific rotation of 4 was measured in three different solvents to be [α] −46.781 (c 0.873, acetone), [α] −13.540 (c 1.090, ethanol) and [α] −2.986 (c 1.007, CHCl3).
Amide (−)-4 was treated with Lawesson’s thionating reagent 5 [23,24], resulting in complete conversion to the thioamide 1. The crude 1H NMR showed the presence of residual aromatic impurities that mandated a relatively straightforward purification by flash column chromatography over silica gel to yield the thioamide variant 1 as bright yellow solid with mp 79–81 °C (Supplementary Material). The molecular formula of 1 was confirmed by means of high-resolution mass spectrometry to be C15H13N3O4S with m/z 354.0520 of the sodium salt.
With the confirmation that the O→S carbonyl metathesis occurred, the structure of 1 was fully elucidated using infrared and nuclear magnetic resonance spectroscopy. The thiocarbonyl stretch C=S of 1 was noticeably absent in the infrared spectrum from the typical amide C=O stretching region as observed with 4 at 1642 cm–1. It is known that C=S stretching lies in the 1200–1100 cm–1 region and is much weaker than C=O stretching [25]. While the 13C signal for the carbonyl carbon of 4 appeared at 161.8 ppm, the thioamide 1 shifted 29.9 ppm downfield to 191.7 ppm (Table 1). The positional assignments of carbon and hydrogen were carried out using 1D and 2D NMR techniques. The specific rotation of 1 was measured to be [α] +22.91 (c 0.965, CHCl3).
3. Materials and Methods
3.1. Materials
Starting materials were purchased from commercial vendors and checked for identity and purity using IR, NMR and HPLC and were used without purification unless noted. (R)-(+)-1-phenylethylamine (CAS# 3886-69-9) was purchased from Oakwood Chemical (Product # 037431, 99.9% ee). 3,5-Dinitrobenzoyl chloride (CAS# 99-33-2) was purchased from Oakwood Chemical (Product # 493922). Lawesson’s reagent (CAS# 19172-47-5) was purchased from Aldrich (Product # 227439).
3.2. Methods
Analytical thin-layer chromatography was performed using Sorbent Technologies 250 μm glass-backed UV254 silica gel plates. The plates were first visualized by means of fluorescence upon 254 nm irradiation then using an iodine chamber and subsequently with phosphomolybdic acid with heating. Flash column chromatography was performed using Sorbent Technologies 40–63 µm, pore size 60 Å silica gel in Luknova columns on a Teledyne ISCO CombiFlash Rf with solvent systems indicated. Solvent removal was effected using a Buchi R3 rotary evaporator with a V900 diaphragm pump (~10 mmHg). Further drying of samples was conducted using a Welch vacuum pump at <0 mmHg. All isolated yields refer to material that is chromatographically (TLC or HPLC) and spectroscopically (1H NMR) homogenous.
3.3. Instrumentation and Analysis
Melting points were measured on a Laboratory Devices Mel-temp with a Thermco 0–400 °C mercury thermometer (serial number 26296) using 1.5–1.8 mm O.D. tubes (ChemGlass part number CG-1841-01) and are uncorrected. Infrared spectra were recorded on a Nicolet Nexus 470 FTIR spectrometer as neat liquids, oils, solids, or as thin films formed from the evaporation of NMR solvent over the ATR plate. Nuclear magnetic resonance spectra were measured at ambient temperature (~25 °C) on a Bruker UltraShield 400 MHz with deuterated chloroform-d (D,99.8% + 0.05% v/v TMS) from Cambridge Isotope Laboratories (Product # DLM-7TB). Proton nuclear magnetic resonance spectra were recorded at 400 MHz and were recorded in parts per million from internal residual protons on the scale and were reported as follows: chemical shift [multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet), coupling constant(s) in hertz, integration, interpretation]. 13C NMR data were recorded at 100 MHz and were reported as follows: chemical shift with multiplicity as determined from DEPT (CH, CH3 up and CH2 down) and/or HSQC experiments. Structures were fully elucidated by assigning 1H peaks to their respective 13C peaks using the 1D and 2D NMR experiments. High-resolution mass spectra were recorded at the Old Dominion University College of Science Major Instrumentation Center (COSMIC) on a Bruker 12 Tesla APEX-Qe FTICR-MS with an Apollo II ion source. Optical rotations were nominally measured between 24 and 26 °C on a Rudolph Autopol polarimeter using a cell with a path length of 1.0 dm and a volume of 2.0 mL (part number 32-5-100-2.0). Solutions were generally prepared from approximately 0.0300 g of purified material dissolved in 3.0 mL of HPLC-grade chloroform (CAS# 67-66-3, stabilized with ethanol, Oakwood item number 101614) dispensed with a VWR Labmax solvent dispenser.
3.4. (R)-(+)-3,5-Dinitro-N-(1-phenylethyl)benzothioamide (1)
A 100 mL round bottom flask with a stir bar was charged with (R)-(−)-3,5-dinitro-N-(1-phenylethyl)benzamide 4 (2.207 g, 7.0 mmol, 1 eq) and 1,4-dioxane (24 mL, 0.30 M) to give a pale-yellow solution. Lawesson’s reagent 5 (1.55 g, 3.85 mmol, 0.55 eq) was added resulting in a cloudy yellow mixture. The flask was equipped with a reflux condenser and a drying tube filled with Drierite and heated to 110 °C for three hours. Upon heating, the reaction became a clear dark gold color. In-process analysis via TLC (4:1 hexanes-ethyl acetate) showed many spots that were not the starting material. The starting material at Rf = 0.25 stained dark magenta-purple with PAA was no longer present. The solution was poured into 20 mL of cold deionized water in a 100 mL round bottom flask that was chilled in an ice water bath. The quench mixture was aged in the ice bath for 1 h. A darker gold oil separated from the mixture, but no solid formed. The solvent was concentrated in vacuo into an oil. The 1H NMR spectra of the crude material indicated complete conversion of the starting amide to the desired product, but the presence of multiple aromatic by-products. The material was purified by chromatography over 80 g of normal-phase silica gel using an isocratic elution of 4:1 hexanes-ethyl acetate. The product-rich fractions were pooled and concentrated to give 2.00 g (86% yield) of the title compound as a dark gold oily solid with the characterization data: MP: 79–81 °C; Rf = 0.35 (4:1 hexanes-ethyl acetate; uv → PAA, I2); IR (thin film): cm−1 3342 (N-H), 1535 (NO2), 1340 (NO2); [α] +22.91 (c 0.965 g/100 mL, CHCl3; T 27.6 °C); 1H NMR (CDCl3, 400 MHz): 8.99 (t, 1H, J = 2.04 Hz), 8.81 (d, 2H, J = 2.04 Hz), 8.12 (s, 1H), 7.29–7.43 (m, 5H), 5.83 (dq, 1H, J = 7.20, 7.00 Hz), 1.76 (d, 3H, J = 6.92 Hz); 13C{1H} NMR (CDCl3, 100 MHz): 191.7 (s), 148.1 (s), 144.5 (s), 140.4 (s), 129.0 (d), 128.3 (d), 126.8 (d), 126.7 (d), 119.9 (d), 56.2 (d), 19.9 (q); HRMS (ESI): Exact mass calcd for C15H13N3O4S [M+Na]+ m/z 354.0518. Found m/z 354.0520.
4. Conclusions
The treatment of the Kagan amide (−)-4 with Lawesson’s reagent 5 in 1,4-dioxane effected the smooth transformation to the thioamide (+)-1 with 85% yield on a multi-gram scale. Given the commercial availability of all of the reagents at relatively inexpensive cost, this method is a viable route to obtain the sulfur derivative of the common chiral solvating agent.
Supplementary Materials
Provides spectra data of 1 including IR, 1H, COSY, 13C, HSQC, HMBC NMR and HRMS.
Author Contributions
Conceptualization, M.G.D.; methodology, E.C. and M.G.D.; validation, E.C. and M.G.D.; formal analysis, E.C. and M.G.D.; writing—original draft preparation, E.C. and M.G.D.; writing—review and editing, M.G.D.; supervision, M.G.D.; project administration, M.G.D.; funding acquisition, M.G.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Institutes of Health through the NIGMS R15GM129751. NMR spectra were acquired with a Bruker 400 MHz which was provided by NSF CHE 0840390. The ISCO ACCQ Prep system was provided by the NIGMS P20GM103476. E.C. was supported by a traineeship through the NSF DGE 1449999.
Data Availability Statement
Data are contained within the article or Supplementary Material.
Acknowledgments
M.G.D. is a fall 2020 awardee of the College of Arts and Sciences Seed Grant sponsored by the Dean’s Office and a spring 2021 recipient of the Lucas Award for Faculty Excellence in Research sponsored by the Office of the Provost.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Silva, M.S. Recent Advances in Multinuclear NMR Spectroscopy for Chiral Recognition of Organic Compounds. Molecules 2017, 22, 247. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, T.J.; Wilcox, J.D. Chiral Reagents for the Determination of Enantiomeric Excess and Absolute Configuration Using NMR Spectroscopy. Chirality 2003, 15, 256–270. [Google Scholar] [CrossRef] [PubMed]
- Gal, J. Molecular Chirality: Language, History and Significance. In Differentiation of Enantiomers I; Springer: New York, NY, USA, 2013; pp. 1–21. [Google Scholar] [CrossRef]
- Parker, D. NMR Determination of Enantiomeric Purity. Chem. Rev. 1991, 91, 1441–1457. [Google Scholar] [CrossRef]
- Deshmukh, M.; Duñach, E.; Juge, S.; Kagan, H.B. A Convenient Family of Chiral Shift Reagents for Measurement of Enantiomeric Excesses of Sulfoxides. Tetrahedron Lett. 1984, 25, 3467–3470. [Google Scholar] [CrossRef]
- Wolf, C.; Cook, A.M.; Dannatt, J.E. Enantiodifferentiation of Multifunctional Tertiary Alcohols by NMR Spectroscopy with a Whelk-O Type Chiral Solvating Agent. Tetrahedron Asymmetry 2014, 25, 163–169. [Google Scholar] [CrossRef]
- Iwaniuk, D.P.; Wolf, C. A Versatile and Practical Solvating Agent for Enantioselective Recognition and NMR Analysis of Protected Amines. J. Org. Chem. 2010, 75, 6724–6727. [Google Scholar] [CrossRef]
- Duñach, E.; Kagan, H.B. A Simple Chiral Shift Reagent for Measurement of Enantiomeric Excesses of Phosphine Oxides. Tetrahedron Lett. 1985, 26, 2649–2652. [Google Scholar] [CrossRef]
- Pakulski, X.; Demchuk, O.M.; Kwiatosz, R.; Osinski, P.W.; Swierczynska, W.; Pietrusiewicz, K.M. The Classical Kagan’s Amides are Still Practical NMR Chiral Shift Reagents: Determination of Enantiomeric Purity of P-Chiroenic Phospholene Oxides. Tetrahedron Asymmetry 2003, 14, 1459–1462. [Google Scholar] [CrossRef]
- Kaboudin, B.; Yarahmadi, V.; Kato, J.-Y.; Yokomatsu, T. A Simple and Novel Method for the Direct Conversion of Carboxylic Acids into Thioamides. RSC Adv. 2013, 3, 6435–6441. [Google Scholar] [CrossRef]
- Lu, Z.-L.; Fun, H.-K.; Chantrapromma, S.; Brown, R.S. N-[(R)-1-Phenylethyl]thiobenzamide. Acta Cryst. 2006, E62, o1513–o1515. [Google Scholar] [CrossRef]
- Sakamoto, M.; Kawanishi, H.; Mino, T.; Kasashima, Y.; Fujita, T. Photochemical Asymmetric Synthesis of Phenyl-Bearing Quaternary Chiral Carbons Using Chiral-Memory Effect on b-Hydrogen Abstraction by Thiocarbonyl Group. Chem. Commun. 2006, 4608–4610. [Google Scholar] [CrossRef] [PubMed]
- Abboud, J.L.M.; Mo, O.; de Paz, J.L.G.; Yanez, M.; Esseffar, M.; Bouab, W.; El-Mouhtadi, M.; Mokhlisse, R.; Ballesteros, E.; Herreros, M.; et al. Thiocarbonyl versus Carbonyl Compounds: A Comparision of Intrinsic Reactivities. J. Am. Chem. Soc. 1993, 115, 12468–12476. [Google Scholar] [CrossRef]
- Madarász, A.; Dósa, Z.; Varga, S.; Soós, T.; Csámpai, A.; Pápai, I. Thiourea Derivatives as Bronsted Acid Organocatalysts. ACS Catal. 2016, 6, 4379–4387. [Google Scholar] [CrossRef]
- Rombola, M.; Sumaria, C.S.; Montgomery, T.D.; Rawal, V.H. Development of Chiral, Bifunctional Thiosquaramides: Enantioselective Michael Additions of Barbituric Acids to Nitroalkenes. J. Am. Chem. Soc. 2017, 139, 5297–5300. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, S.E.; Seguin, T.J.; Guan, Y.; Doney, A.C. Noncovalent Interactions in Organocatalysis and the Prospect of Computational Catalyst Desgin. Acc. Chem. Res. 2016, 49, 1061–1069. [Google Scholar] [CrossRef]
- Phillips, A.M.F.; Prechtl, M.H.G.; Pombeiro, A.J.L. Non-Covalent Interactions in Enantioselective Organocatalysis: Theoretical and Mechanistic Studies of Reactions Mediated by Dual H-Bond Donors, Bifunctional Squaramides, Thioureas and Related Catalysts. Catalysts 2021, 11, 569. [Google Scholar] [CrossRef]
- Truter, M.R. An Accurate Determination of the Crystal Structure of Thioacetamide. J. Chem. Soc. 1960, 997–1007. [Google Scholar] [CrossRef]
- Zieliński, T.; Jurczak, J. Thioamides versus Amides in Anion Binding. Tetrahedron 2005, 61, 4081–4089. [Google Scholar] [CrossRef]
- Bordwell, F.G. Equilibrium Acidities in Dimethyl Sulfoxide Solution. Acc. Chem. Res. 1988, 21, 456–463. [Google Scholar] [CrossRef]
- Lee, H.J.; Choi, Y.S.; Lee, K.B.; Park, J.; Yoon, C.J. Hydrogen Bonding Abilities of Thioamide. J. Phys. Chem. A 2002, 106, 7010–7017. [Google Scholar] [CrossRef]
- Seco, J.M.; Quiñoá, E.; Riguera, R. The Assignment of Absolute Configuration by NMR. Chem. Rev. 2004, 104, 17–118. [Google Scholar] [CrossRef]
- Yde, B.; Yousif, N.M.; Pedersen, U.; Thomsen, I.; Lawesson, S.O. Studies on Organophosphorus Compounds XLVII: Preparation of Thiated Synthons of Amides, Lactams and Imides by Use of Some New P,S-Containing Reagents. Tetrahedron 1984, 40, 2047–2052. [Google Scholar] [CrossRef]
- Ozturk, T.; Ertas, E.; Mert, O. Use of Lawesson’s Reagent in Organic Synthesis. Chem. Rev. 2007, 107, 5210–5278. [Google Scholar] [CrossRef] [PubMed]
- Spinner, E. Detection of Thiocarbonyl Groups by Infrared Spectroscopy. J. Org. Chem. 1958, 23, 2037–2038. [Google Scholar] [CrossRef]
Figure 1.
Kagan amide and thioamide chiral solvating agents.

Scheme 1.
Two-step chemical synthesis of (R)-(+)-3,5-dinitro-N-(1-phenylethyl)benzothioamide (1).


{kind=link}
{kind=link}
Table 1.
Structural assignments using 1H (400 MHz) and 13C (100 MHz) NMR data of (−)-4 and (+)-1 in CDCl3.
Table 1.
Structural assignments using 1H (400 MHz) and 13C (100 MHz) NMR data of (−)-4 and (+)-1 in CDCl3.
 | ||||
|---|---|---|---|---|
| 13C NMR Data 1 | 1H NMR Data 2 | |||
| Position 3 | 4 δC | 1 δC | 4 δH | 1 δH |
| a | 161.8 (s) | 191.7 (s) | - | - |
| b | 148.6 (s) | 144.5 (s) | - | - |
| c | 141.8 (s) | 148.1 (s) | - | - |
| d | 137.9 (s) | 140.4 (s) | - | - |
| e | 129.0 (d) | 129.0 (d) | 7.41–7.35 (m, 2H) | 7.43–7.29 (m, 2H) |
| f | 128.0 (d) | 128.3 (d) | 7.33–7.28 (m, 1H) | 7.43–7.29 (m, 1H) |
| g | 127.1 (d) | 126.7 (d) | 8.93 (d, J = 2.04 Hz, 2H) | 8.81 (d, J = 2.04 Hz, 2H) |
| h | 126.3 (d) | 126.8 (d) | 7.41–7.35 (m, 2H) | 7.43–7.29 (m, 2H) |
| i | 121.1 (d) | 119.9 (d) | 9.14 (t, J = 2.08 Hz, 1H) | 8.99 (t, J = 2.04 Hz, 1H) |
| j | 50.3 (d) | 56.2 (d) | 5.34 (dq, J = 7.16, 7.12 Hz, 1H) | 5.83 (dq, J = 7.20, 7.00 Hz, 1H) |
| k | 21.4 (q) | 19.9 (q) | 1.67 (d, J = 6.92 Hz, 3H) | 1.76 (d, J = 6.92 Hz, 3H) |
| l | - | - | 6.64 (s, 1H) | 8.12 (s, 1H) |
1 13C NMR signal multiplicity determined by DEPT90, DEPT135 and 1H-13C HSQC. 2 Integration and multiplicity determined by 1H NMR and J-value coupling analysis. 3 Positions assigned using 1H-1H COSY, 1H-13C HSQC, and 1H-13C HMBC analysis.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Donahue, M.G.; Crull, E. (R)-(+)-3,5-Dinitro-N-(1-phenylethyl)benzothioamide. Molbank 2023, 2023, M1650. https://doi.org/10.3390/M1650
AMA Style
Donahue MG, Crull E. (R)-(+)-3,5-Dinitro-N-(1-phenylethyl)benzothioamide. Molbank. 2023; 2023(2):M1650. https://doi.org/10.3390/M1650
Chicago/Turabian StyleDonahue, Matthew G., and Emily Crull. 2023. "(R)-(+)-3,5-Dinitro-N-(1-phenylethyl)benzothioamide" Molbank 2023, no. 2: M1650. https://doi.org/10.3390/M1650
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.