
Synthesis, Characterization and Chemistry of Tetrakis(Propargylisocyanide) Copper(I) Complex

Univ Rennes, Ecole Nationale Supérieure de Chimie de Rennes, CNRS, ISCR (Institut des Sciences Chimiques de Rennes)—UMR 6226, F-35000 Rennes, France
*
Authors to whom correspondence should be addressed.
Molbank 2023, 2023(1), M1599; https://doi.org/10.3390/M1599
Submission received: 7 February 2023
/
Revised: 24 February 2023
/
Accepted: 26 February 2023
/
Published: 2 March 2023
(This article belongs to the Section Organic Synthesis)
{kind=link}
{kind=link}
{kind=link}
Abstract
:The kinetically unstable propargylisocyanide was reacted with the tetrakis(acetonitrile) copper(I) hexafluorophosphate and the formed complex was then involved in a copper-catalyzed alkyne-azide cycloaddition reaction (CuAAC). After the decomplexation of the adduct, the isocyanide was engaged in a Ugi reaction. By such a complexation, reactions can be carried out on the CC triple bond without the constraint of the instability of the free compound or the competitive reactivity of the isocyanide group.
1. Introduction
Isocyanides are well-studied compounds, especially for their peculiar reactivity. These isomers of nitriles possess a carbenoid structure due to the atomic arrangement R-NC, which confers these compounds their unique reactivity [1]. In addition to cycloadditions, they are able to react in multi-component reactions (MCRs) like the well-known Passerini [2,3] and Ugi [4,5,6] reactions. Recently, we described the use of unsaturated isocyanides bearing CC double bonds or triple bonds in such MCRs [7]. We showed that despite their instability, they are interesting building blocks that are able to afford highly functionalized products (Scheme 1). However, their handling remains tedious due to their combined volatility, instability and vile odor. To solve this problem, Zwikker and Stephany [8] described the complexation of two of them (the propargyl and the allenyl isocyanides) to copper(I) in order to stabilize and handle them, knowing that their decomplexation with NaCN proved to be efficient. Nevertheless, except for the isomerization of the propargylic complex into the allenic one with the help of 1,5-diazabicyclo[4.3.0]non-5-ene (DBN), their chemistry was not investigated.
In this article, we focused our attention on the propargylic copper(I) complex in order to characterize it and investigate its reactivity in Copper-catalyzed Alkyne-Azide Cycloaddition (CuAAC) [9,10]. We thus showed that it was possible to make it react, decomplex the corresponding isocyanide from Cu(I) and subsequently react it in a Ugi reaction. This provides an alternative pathway to the Ugi reaction followed by CuAAC we recently reported. In addition, we showed it was possible to keep the isocyanide function after reacting the CC triple bond.
2. Results and Discussion
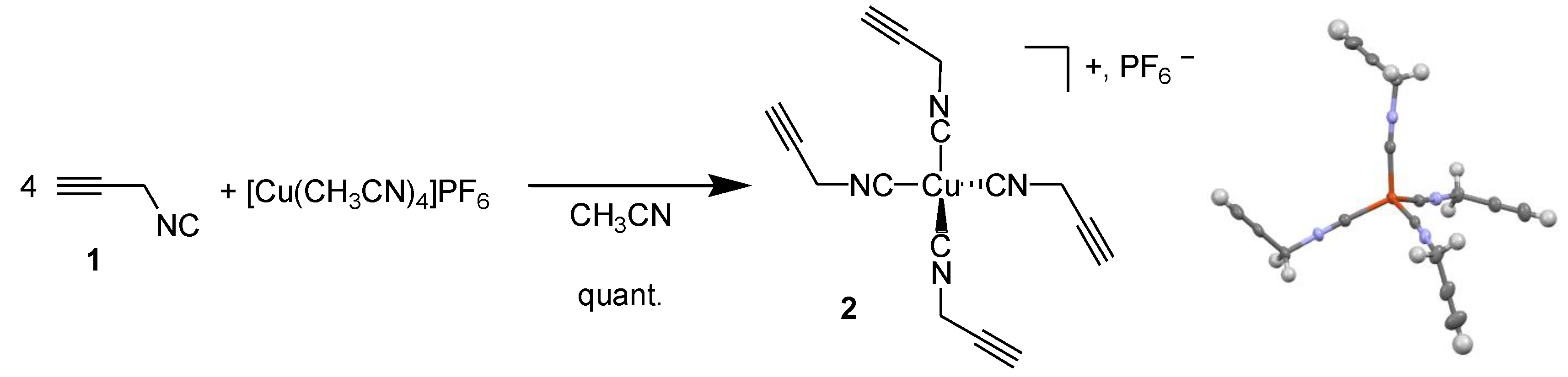
We first reproduced the synthesis of the tetrakis(propargylisocyanide) copper(I) complex 2 by mixing four equivalents of propargylisocyanide 1 with one equivalent of tetrakis(acetonitrile) copper(I) hexafluorophosphate in acetonitrile to afford the expected complex quantitatively (Scheme 2). The only difference with the original publication [8] is the counter-ion, which is PF6− here instead of BF4− or ClO4− as in the previous case. We were able to confirm the structure of this complex by X-ray crystallography, in addition to the usual techniques (1H and 13C NMR spectroscopy, see the Supplementary Materials). The geometry around the copper atom is a slightly distorted tetrahedron, with C-Cu-C comprised between 104 and 115°. The fact that crystals suitable for X-ray diffraction could be obtained at room temperature confirmed the great stability of this compound, while propargylisocyanide slowly decomposes at −30 °C in pure form. This observation encouraged us to continue this study and evaluate the reactivity of this compound.
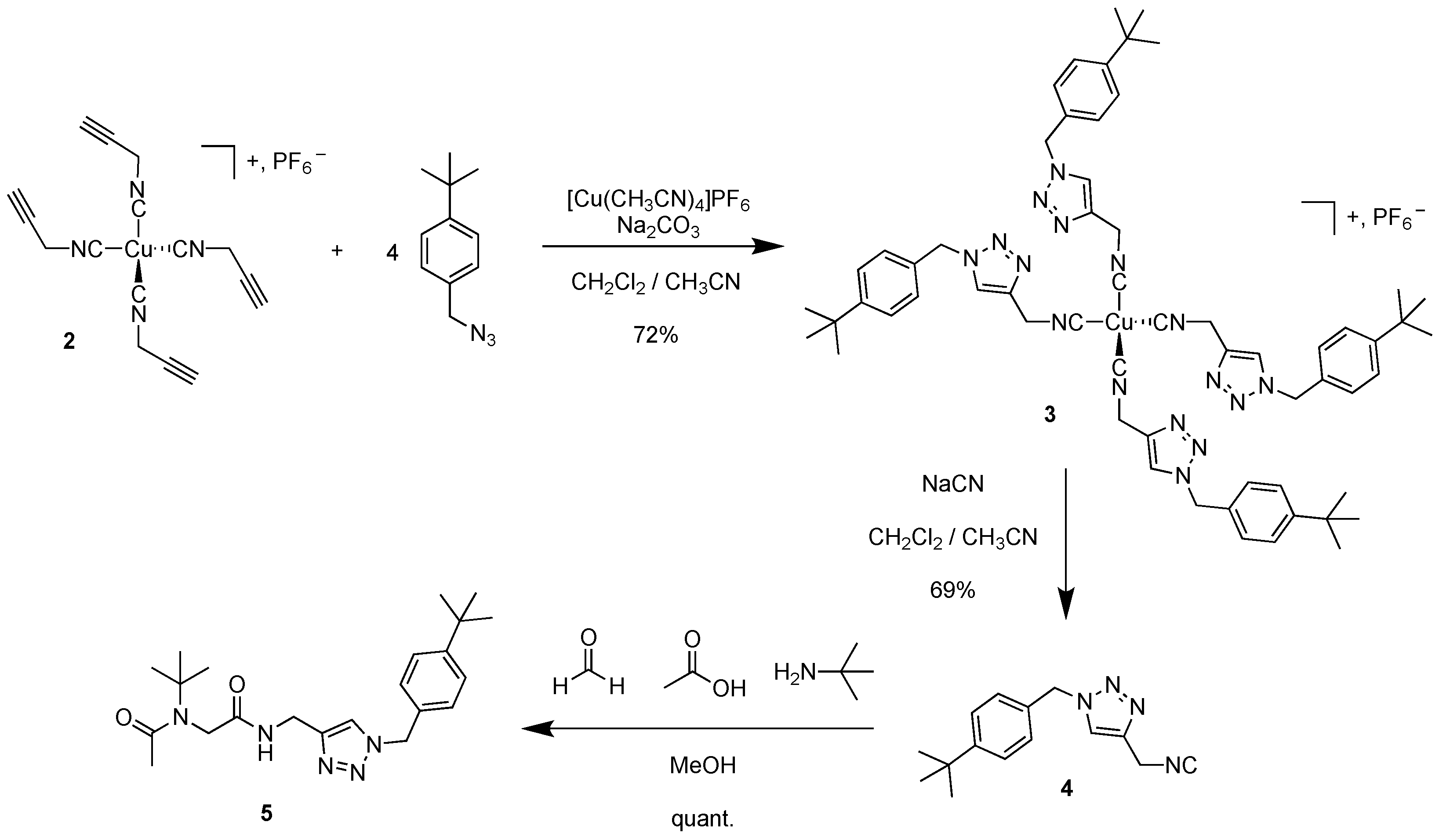
This complex 2 was reacted with an excess of 4-tert-butylbenzylazide (6.7 equiv.) in the presence of [Cu(CH3CN)4]PF6 (1.1 equiv.) and Na2CO3 (1.3 equiv.) in a mixture of dichloromethane and acetonitrile at room temperature overnight. The corresponding Cu(I) complex 3 bearing four triazole units was isolated in a satisfying yield of 72% after silica column chromatography, showing again the stability of such a complex. This also demonstrated the ability of this kind of compound to be further functionalized. The corresponding isocyanide was subsequently decomplexed from copper using an excess of NaCN (14 equiv.) to isolate the free isonitrile 4 bearing a triazole function in 69% yield. It is noteworthy that isonitrile 4 could not be isolated from direct CuAAC (2 mol% of CuI, di(isopropyl)ethylamine (3 equiv.) as a base and toluene as solvent), showing the importance of complexing it before reaction (only the starting azide was observed by NMR spectroscopy). Finally, the latter was reacted in Ugi conditions with tert-butylamine, acetic acid and formaldehyde in methanol overnight to form quantitatively the corresponding Ugi-product 5 that we already described using another pathway (Scheme 3) [7]. This shows that after functionalization, the isocyanide was able to react according to a Ugi reaction, instead of subsequently functionalizing the Ugi product.
3. Materials and Methods
Reactions were monitored by thin layer chromatography (Merck TLC 0.25 mm silica gel plates 60 F254) and visualized under UV irradiation at 245 nm and KMnO4 staining solution. Compounds were purified by flash column chromatography using Geduran silica gel 60 (43−60 μm). Melting points were measured on a Stuart® SMP10 melting point apparatus. NMR spectra were recorded on a Bruker (Wissembourg, France) Avance spectrometer at 23 °C, 400 MHz for proton and 100 MHz for carbon. Spectra were recorded in deuterochloroform referenced to CHCl3 (1H, 7.26 ppm) or CDCl3 (13C, 77.2 ppm) and in deuterodimethyl sulfoxide referenced to (CD3)(CD2H)SO (1H, 2.50 ppm) or (CD3)2SO (13C, 39.5 ppm). Chemical shifts (δ) are reported in ppm and coupling constants (J) are reported in Hertz. The following abbreviations are used to describe multiplicity: s-singlet, br s-broad singlet, d-doublet, t-triplet, q-quadruplet, hept-heptuplet and m-multiplet, quadr. moment-quadrupole moment. HRMS spectra were recorded on a Maxis+ (Bruker Daltonics, Bremen, Germany) and Q-Exactive (Thermo Fischer Scientific, Bremen, Germany), depending on the targeted structure.
Solvents such as acetonitrile (anhydrous 99.8%), methanol (99.8%) and dichloromethane (anhydrous, ≥99.8%) were directly purchased from Sigma-Aldrich (Saint-Quentin-Fallavier, France).
The commercial reagents employed in the synthesis were tetrakis(acetonitrile) copper(I) hexafluorophosphate (97%, Sigma-Aldrich), tert-butylamine (≥99.5%, Sigma-Aldrich), formaldehyde (ASC reagent, 37 wt.% in H2O, Sigma-Aldrich), acetic acid (glacial, ACS reagent, ≥99.7%, Sigma-Aldrich), Na2CO3 (anhydrous powder, Sigma-Aldrich), MgSO4 (anhydrous powder, Sigma-Aldrich) and NaCN (reagent grade, 97%, Sigma-Aldrich).
Single crystal diffraction data were collected on a D8 Venture Bruker AXS diffractometer with multilayer monochromatized Mo-Kα radiation. Structures were solved by the dual-space algorithm using the SHELXT program [11] and then refined with full-matrix least-squares methods based on F2 (SHELXL program) [12]. All non-hydrogen atoms were refined with anisotropic atomic displacement parameters. H atoms were finally included in their calculated positions and treated as riding on their parent atom with constrained thermal parameters.
4-tert-Butylbenzylazide [13] and propargylisocyanide [8] were synthesized according to published procedures.
3.1. Tetrakis(propargylisonitrile) Copper(I) Hexafluorophosphate 2
To a −20 °C cooled solution of tetrakis(acetonitrile) copper(I) hexafluorophosphate (2.24 g, 6.0 mmol, 0.25 equiv.) in acetonitrile (12 mL), was added under dinitrogen a cooled solution of propargylisonitrile (1.56 g, 24.0 mmol, 1.0 equiv.) in acetonitrile (4 mL). The resulting solution was allowed to reach room temperature and stirred for 20 min. The mixture was concentrated under high vacuum and the solid obtained was diluted with dichloromethane. The organic phase was then washed with water. The organic layers were gathered, dried over MgSO4, filtered and concentrated under reduced pressure to give 2.81 g of the desired tetrakis(propargylisonitrile) copper(I) hexafluorophosphate 2 (quantitative yield) as a white solid. Suitable crystals for X-ray diffraction were grown by slow evaporation of an acetonitrile solution of compound 2.
1H NMR (400 MHz, DMSO-d6) δ 4.93 (d, 2H, J = 2.6 Hz), 3.73 (t, 1H, J = 2.6 Hz); 13C NMR (100 MHz, DMSO-d6) δ 138.5 (m (quadr. moment)), 77.4, 73.7, 33.4; 19F NMR (376 MHz, DMSO-d6) δ-70.2 (d, J = 712 Hz); 31P NMR (162 MHz, DMSO-d6) δ-144.2 (hept, J = 711 Hz).
Crystal data: C16H12CuF6N4P, M = 468.81 g mol−1, T = 150(2) K, monoclinic, space group = C 2/c, a = 5.4651(6) Å, b = 22.311(2) Å, c = 16.4771(18) Å, β = 94.038(4)°, V = 2004.1 (4) Å3, Z = 4, Dc = 1.554 g cm−3, absorption coefficient = 1.231 mm−1, F(000) = 936, reflections collected = 13,996, independent reflections = 2290 (Rint = 0.0446), data/restraints/parameters = 2290/0/128. Final R indices (I > 2σ): R1 = 0.0513. R indices (all data): wR2 = 0.1401, goodness-of-fit on F2 of 1.049.
3.2. Tetrakis(1-(4-(tert-butyl)benzyl)-4-(isocyanomethyl)-1H-1,2,3-triazole) Copper(I) Hexafluorophosphate 3
To a solution of 2 (100 mg, 0.21 mmol, 1.0 equiv.) tetrakis(acetonitrile) copper(I) hexafluorophosphate (86 mg, 0.23 mmol, 1.1 equiv.) in dichloromethane (3.0 mL) and acetonitrile (1.0 mL) was added under dinitrogen 4-(tert-butyl)benzyl azide (270 mg, 1.43 mmol, 6.7 equiv.) and Na2CO3 (30 mg, 0.28 mmol, 1.3 equiv.). The resulting solution, stirred for 18 h at 25 °C, was monitored by TLC (dichloromethane/MeOH (97:3), Rf: 0.3) until the complete consumption of 2. The mixture was concentrated under reduced pressure to give a mixture of 3 and an excess of 4-(tert-butyl)benzyl azide. The resulting solid was purified by column chromatography (SiO2, eluent: CH2Cl2/acetone from 100:0 to 0:100) to remove the excess of 4-(tert-butyl)benzyl azide and gave 3. Yield 189 mg (72%); orange solid.
1H NMR (400 MHz, CDCl3) δ 7.86 (br s, 1H), 7.34 (d, 2H, J = 8.4 Hz), 7.23 (d, 2H, J = 8.4 Hz), 5.40 (br s, 2H), 4.86 (br s, 2H), 1.24 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 152.2, 140.9 (m (quadr. moment)), 138.9, 130.7, 128.5, 126.2, 125.1, 54.6, 38.0, 34.7, 31.3; 19F NMR (376 MHz, CDCl3) δ -72.0 (d, J = 713 Hz); 31P NMR (162 MHz, CDCl3) δ -144.2 (hept, J = 713 Hz).
HRMS (ESI) m/z [M − PF6]+ calcd. for C60H72N16Cu: 1079.54164, found: 1079.5415.
3.3. 1-(4-(tert-Butyl)benzyl)-4-(isocyanomethyl)-1H-1,2,3-triazole 4
To a solution of NaCN (73 mg, 1.48 mmol, 14 equiv.) in dichloromethane (1.0 mL), acetonitrile (0.5 mL) and water (0.5 mL) was added 3 (130 mg, 0.11 mmol, 1 equiv.) and the resulting solution was stirred for 1.5 h at 20 °C. The biphasic mixture was diluted with dichloromethane, then the organic phase was separated, dried over MgSO4, filtered and concentrated under reduced pressure and gave 4. Yield 74 mg (69%); white solid.
1H NMR (400 MHz, CDCl3) δ 7.56 (br s, 1H), 7.41 (d, 2H, J = 8.4 Hz), 7.22 (d, 2H, J = 8.4 Hz), 5.49 (s, 2H), 4.72 (br s, 2H), 1.31 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 158.3 (m (quadr. moment)), 152.3, 140.9, 131.1, 128.1, 126.2, 122.1, 54.2, 37.9 (t (quadr. moment), J = 7.32 Hz), 34.7, 31.3.
3.4. N-(tert-butyl)-N-(2-(((1-(4-(tert-butyl)benzyl)-1H-1,2,3-triazol-4-yl)methyl)amino)-2-oxoethyl)acetamide 5
In a three-neck round-bottom flask under dinitrogen containing a solution stirred at 0 °C of tert-butylamine (19 mg, 0.256 mmol, 1.0 equiv.), formaldehyde (23 mg, 37% in water, 0.256 mmol, 1.0 equiv.), acetic acid (15 mg, 0.256 mmol, 1.0 equiv.) in MeOH (2.5 mL), was added a cooled solution of isonitrile 4 (65 mg, 0.256 mmol, 1.0 equiv.) in MeOH (0.5 mL). The mixture was stirred for 18.5 h at 28 °C and monitored by TLC (CH2Cl2/MeOH (97:3), Rf: 0.20). The greenish solution was concentrated under reduced pressure and gave 5. Yield 102 mg (quantitative yield); beige solid (mp: 118 °C).
1H NMR (400 MHz, CDCl3) δ 7.45 (br s, 1H), 7.38 (d, 2H, J = 8.2 Hz), 7.19 (d, 2H, J = 8.2 Hz), 6.83 (br s, 1H), 5.46 (s, 2H), 4.54 (br s, 2H), 3.95 (s, 2H), 2.02 (s, 3H), 1.37 (s, 9H), 1.30 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 172.5, 170.1, 152.1, 144.7, 131.4, 127.9, 126.1, 122.2, 57.8, 54.1, 49.9, 34.9, 34.7, 31.3, 28.7, 25.4.
HRMS (ESI) m/z [M + Na]+ calcd. for C22H33N5O2Na: 422.2527, found: 422.2526.
4. Conclusions
In this article, we showed that it was possible to react the CC triple bond of kinetically unstable propargylisocyanide after complexation to Cu(I) and release the corresponding isocyanide to make it react in the Ugi reaction.
Supplementary Materials
The following supporting information can be downloaded online, Figure S1: 1H NMR spectrum of 2; Figure S2: 13C NMR spectrum of 2; Figure S3: 19F NMR spectrum of 2; Figure S4: 31P NMR spectrum of 2; Figure S5: 1H NMR spectrum of 3; Figure S6: 13C NMR spectrum of 3; Figure S7: 19F NMR spectrum of 3; Figure S8: 31P NMR spectrum of 3; Figure S9: 1H NMR spectrum of 4; Figure S10: 13C NMR spectrum of 4; Figure S11: 1H NMR spectrum of 5; Figure S12: 13C NMR spectrum of 5.
Author Contributions
Conceptualization, J.-C.G. and Y.T.; formal analysis, A.Q., T.R., J.-C.G. and Y.T.; investigation, J.-C.G. and Y.T.; data curation, A.Q. and T.R.; writing—original draft preparation, A.Q., J.-C.G. and Y.T.; writing—review and editing, Y.T.; supervision, J.-C.G. and Y.T.; project administration, J.-C.G. and Y.T.; funding acquisition, J.-C.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
CCDC 2238883 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif or by emailing [email protected], or by contacting the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK, fax: +44-1223-336033.
Acknowledgments
A.Q. is grateful to the Université de Rennes for his PhD fellowship.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Nenajdenko, V.G. Isocyanide Chemistry: Applications in Synthesis and Material Science; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar] [CrossRef]
- Van Berkel, S.S.; Boegels, B.G.M.; Wijdeven, M.A.; Westermann, B.; Rutjes, F.P.J.T. Recent Advances in Asymmetric Isocyanide-Based Multicomponent Reactions. Eur. J. Org. Chem. 2012, 2012, 3543–3559. [Google Scholar] [CrossRef]
- Wahby, Y.; Abdel-Hamid, H.; Salah Ayoup, M. Two decades of recent advances of Passerini reactions: Synthetic and potential pharmaceutical applications. New J. Chem. 2022, 45, 1445–1468. [Google Scholar] [CrossRef]
- Ugi, I.; Lohberger, S.; Karl, R. Comprehensive Organic Synthesis; Trost, B.M., Fleming, I., Eds.; Pergamon: Oxford, UK, 1991; Volume 2, p. 1083. [Google Scholar]
- Domling, A.; Huang, Y. Piperazine scaffolds via isocyanide-based multicomponent reactions. Synthesis 2010, 2010, 2859–2883. [Google Scholar] [CrossRef]
- Sadjadi, S.; Heravi, M.M. Recent application of isocyanides in synthesis of heterocycles. Tetrahedron 2011, 67, 2707–2752. [Google Scholar] [CrossRef]
- Quelhas, A.; Gazzeh, H.; Roisnel, T.; Trolez, Y.; Guillemin, J.-C. Passerini and Ugi Reactions Involving Kinetically Unstable Isocyanides. Eur. J. Org. Chem. 2021, 2021, 6002–6005. [Google Scholar] [CrossRef]
- Zwikker, J.W.; Stephany, R.W. Propargyl and Allenyl Isocyanide and Some of Their Copper(I)Complexes. Synth. Commun. 1973, 3, 19–23. [Google Scholar] [CrossRef]
- Tornøe, C.W.; Christensen, C.; Meldal, M. Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar] [CrossRef] [PubMed]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, S.G.; Alvarez, M.T. A Practical Procedure for the Synthesis of Alkyl Azides at Ambient Temperature in Dimethyl Sulfoxide in High Purity and Yield. Synthesis 1997, 1997, 413–414. [Google Scholar] [CrossRef]
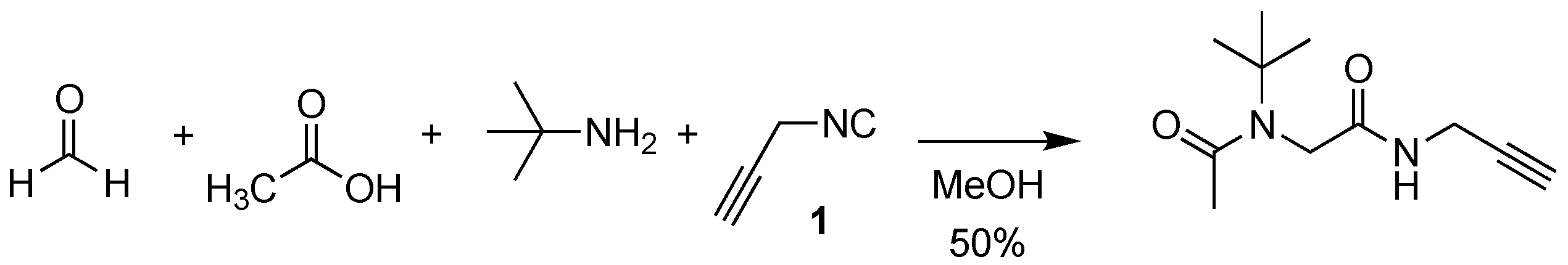
Scheme 1.
Previous work: reactivity of isocyanide 1 in Ugi reaction.

Scheme 2.
Synthesis of copper(I) complex 2 and its X-ray structure. Ellipsoids are drawn with 50% probability; PF6− counter-ion was removed for clarity. Orange: copper; grey: carbon; blue: nitrogen; white: hydrogen.
Scheme 2.
Synthesis of copper(I) complex 2 and its X-ray structure. Ellipsoids are drawn with 50% probability; PF6− counter-ion was removed for clarity. Orange: copper; grey: carbon; blue: nitrogen; white: hydrogen.

Scheme 3.
Synthesis of compound 5 from copper(I) complex 2.

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Quelhas, A.; Roisnel, T.; Guillemin, J.-C.; Trolez, Y. Synthesis, Characterization and Chemistry of Tetrakis(Propargylisocyanide) Copper(I) Complex. Molbank 2023, 2023, M1599. https://doi.org/10.3390/M1599
AMA Style
Quelhas A, Roisnel T, Guillemin J-C, Trolez Y. Synthesis, Characterization and Chemistry of Tetrakis(Propargylisocyanide) Copper(I) Complex. Molbank. 2023; 2023(1):M1599. https://doi.org/10.3390/M1599
Chicago/Turabian StyleQuelhas, Alexandre, Thierry Roisnel, Jean-Claude Guillemin, and Yann Trolez. 2023. "Synthesis, Characterization and Chemistry of Tetrakis(Propargylisocyanide) Copper(I) Complex" Molbank 2023, no. 1: M1599. https://doi.org/10.3390/M1599
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.