
1-(4-Bromo-2,3,5,6-tetrafluoropheyl)-3-(3-phenylbenzyl)-4-methylimidazolium Bromide

Abstract
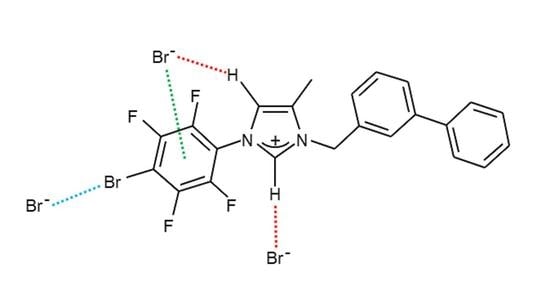
:
1. Introduction

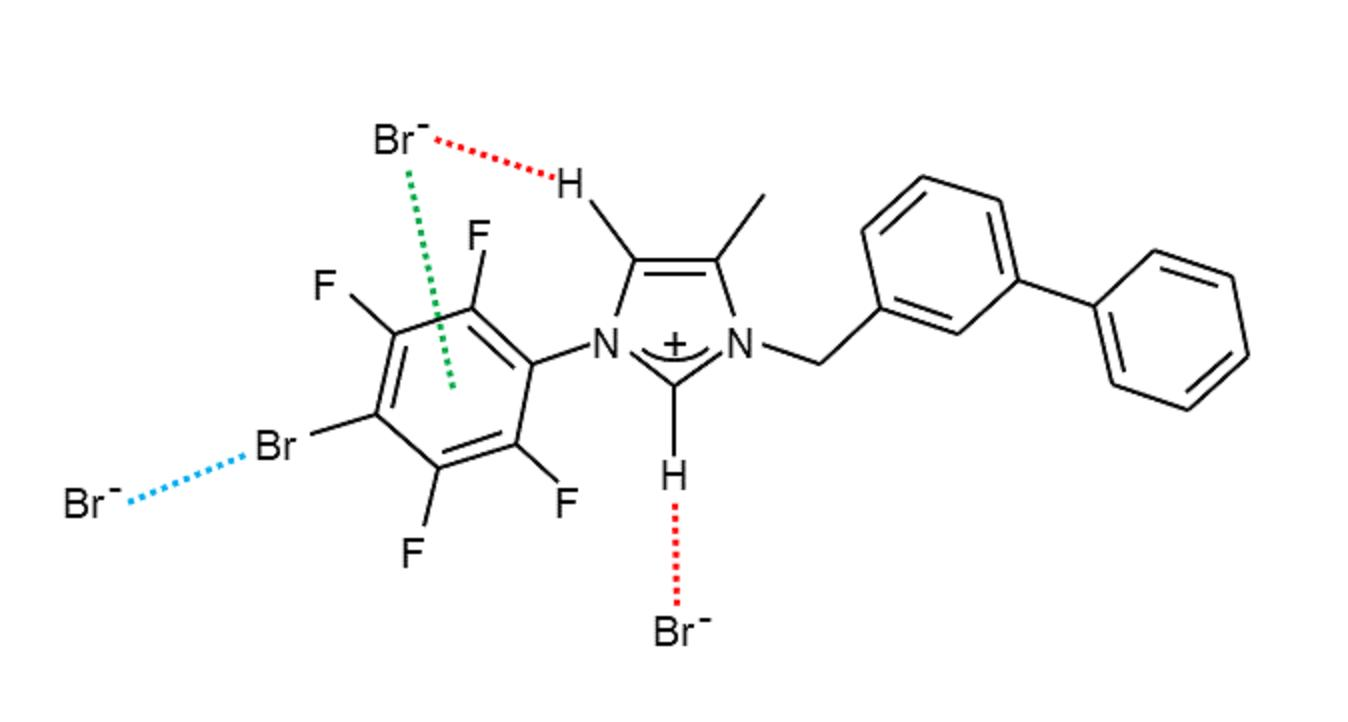
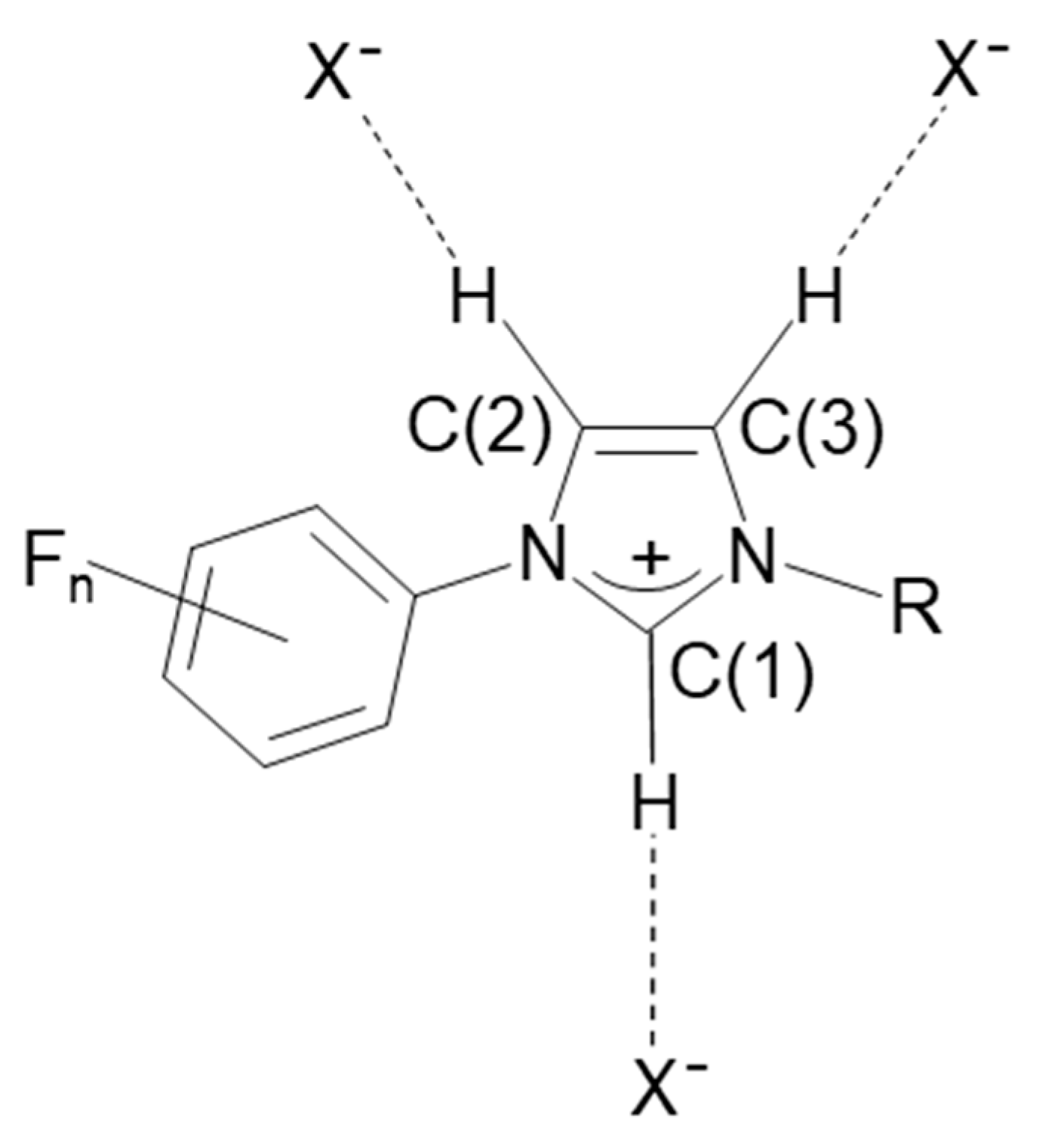
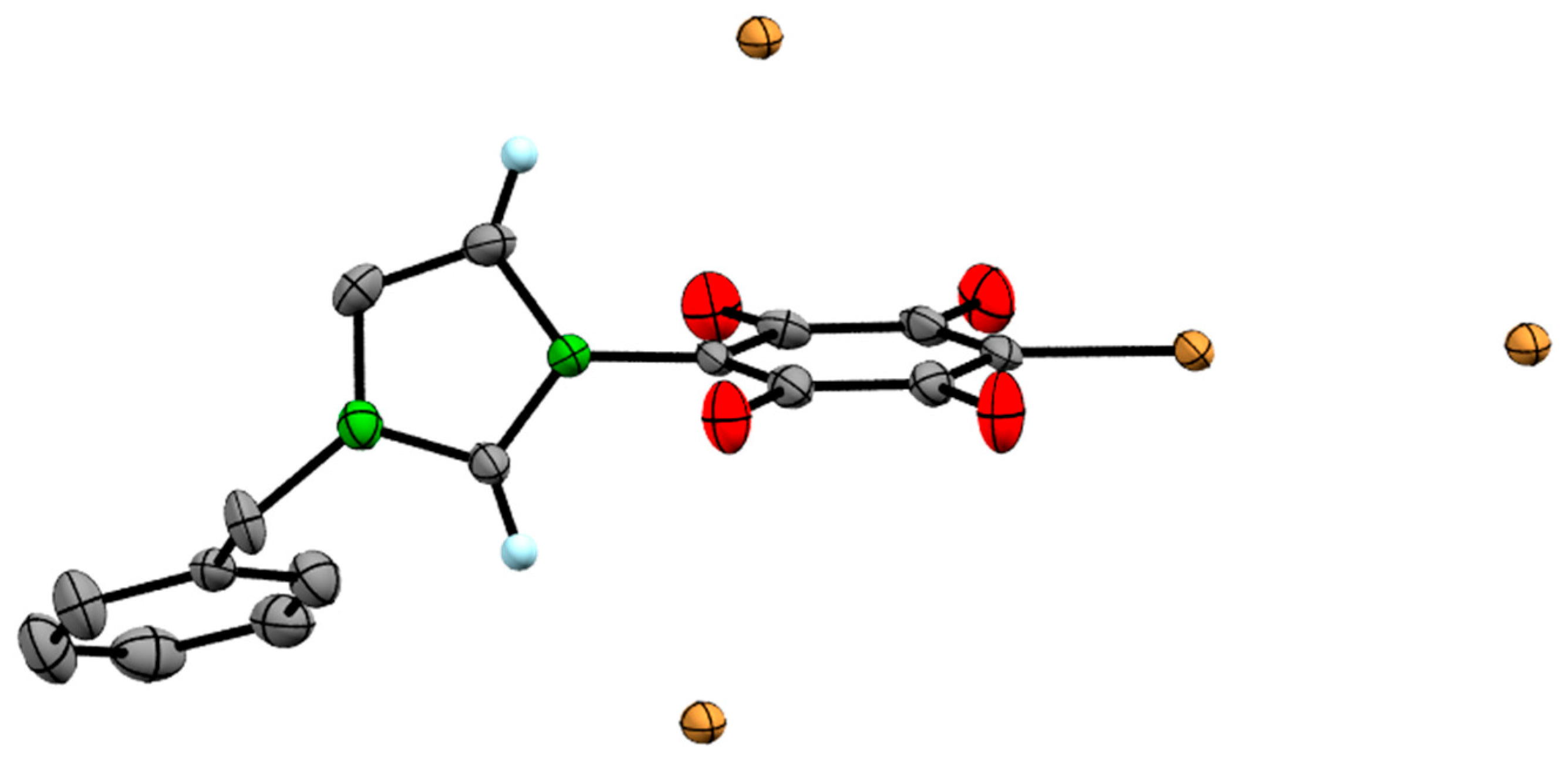
2. Results
3. Materials and Methods
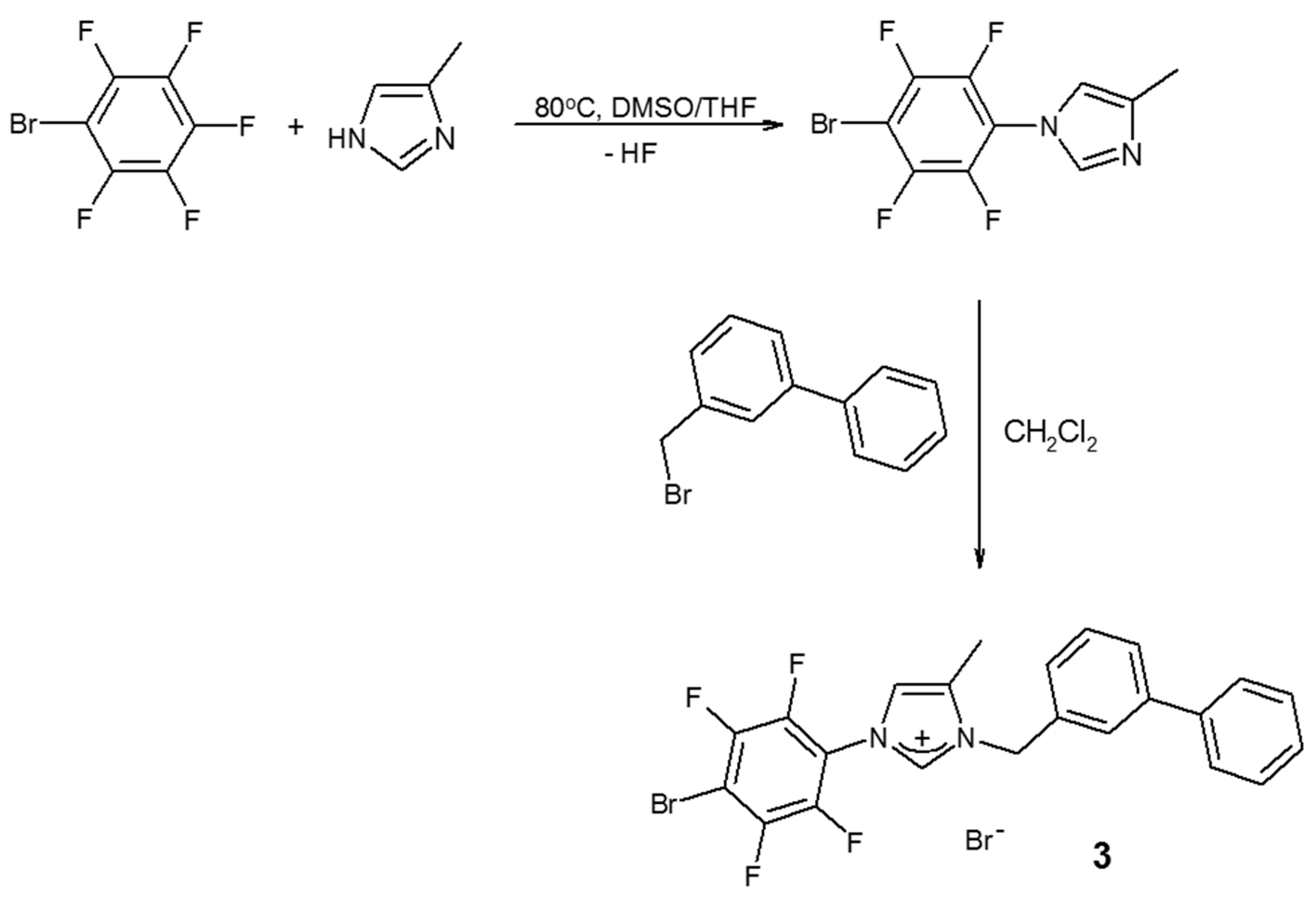
3.1. Synthesis of 1-(4-Bromo-2,3,5,6-tetrafluoropheyl)-3-(3-phenylbenzyl)-4-methylimidazolium Bromide (3)
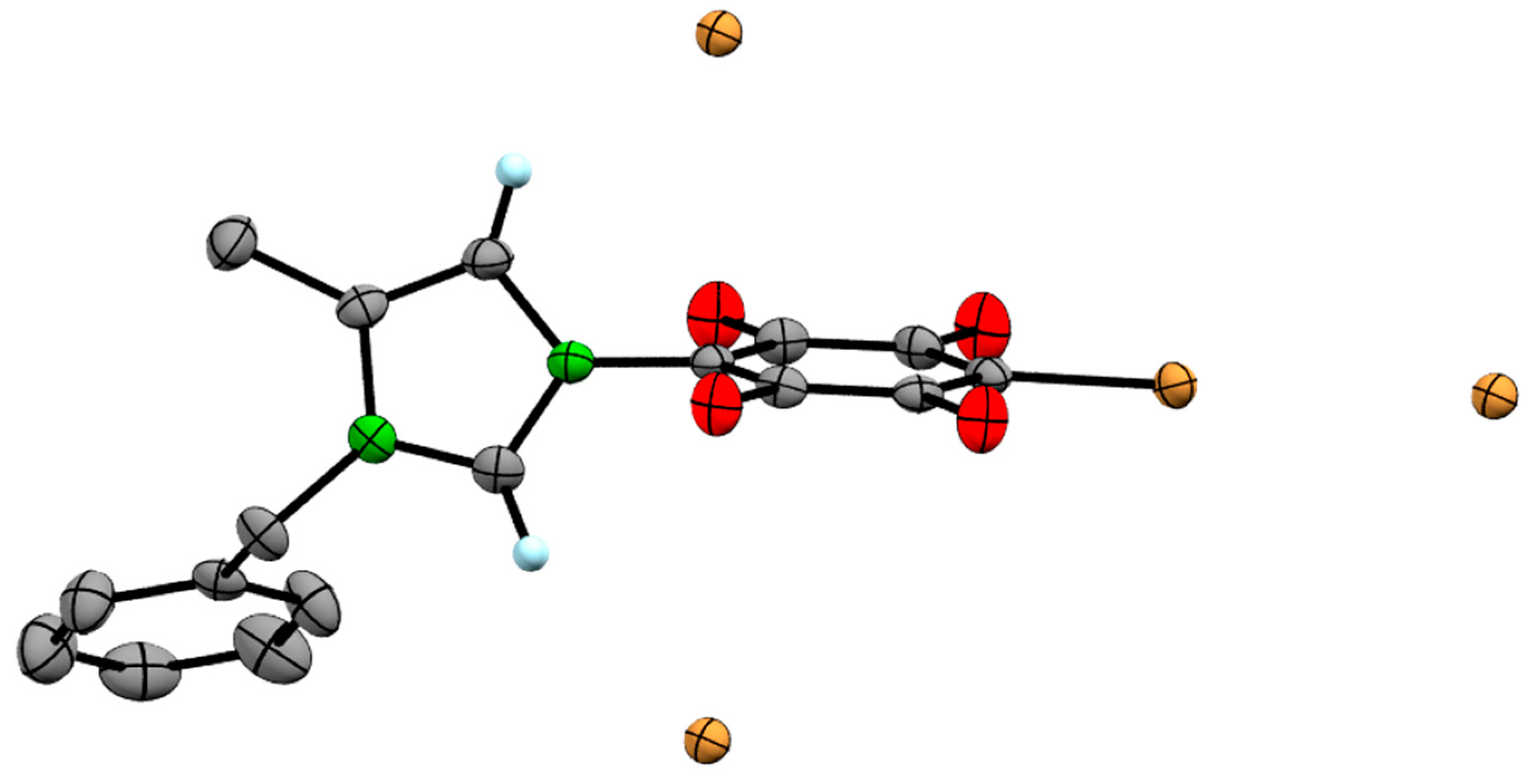
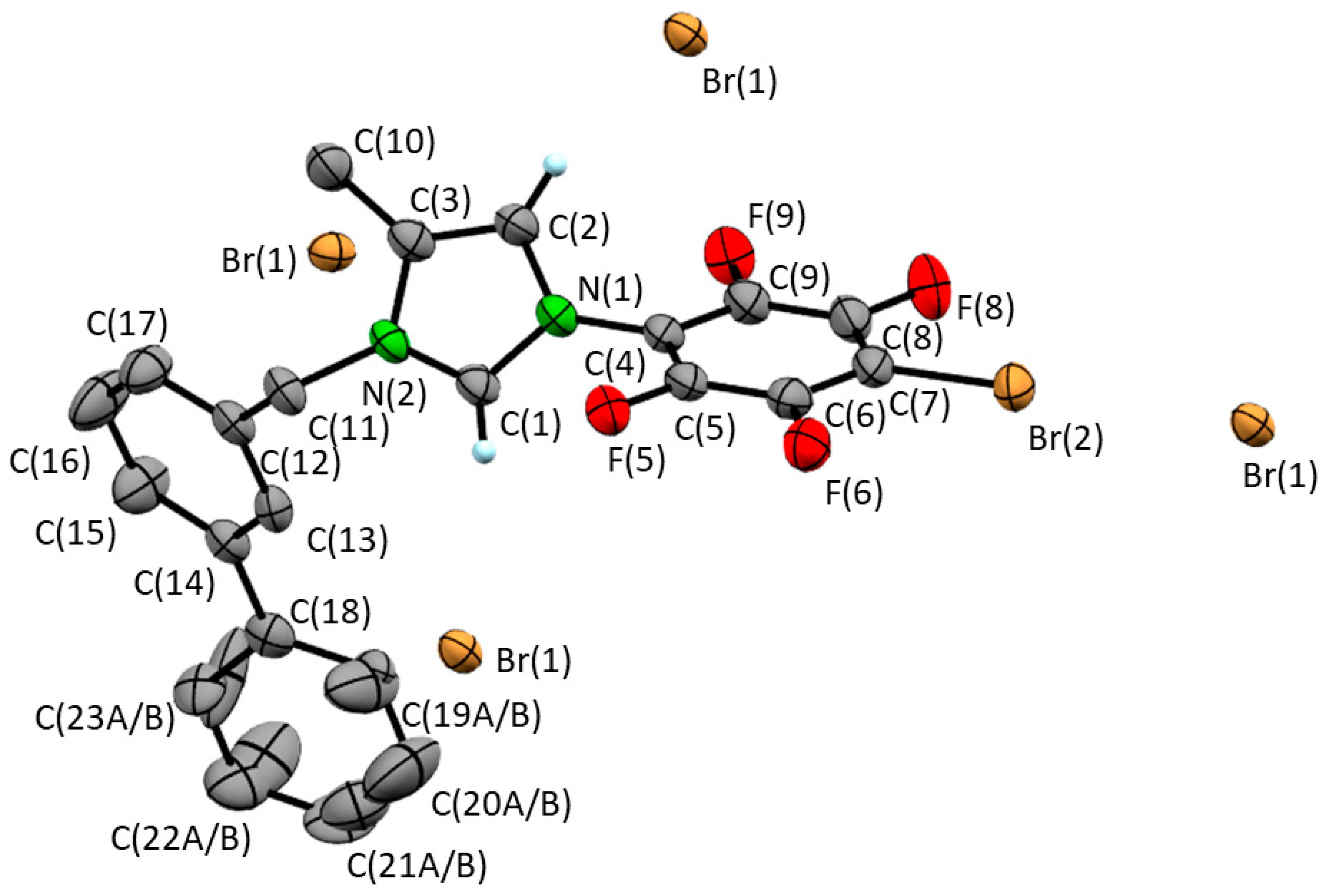
3.2. Single Crystal XRD Determination
3.3. DFT Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Tsuzuki, S.; Tokuda, H.; Mikami, M. Theoretical analysis of the hydrogen bond of imidazolium C2–H with anions. Phys. Chem. Chem. Phys. 2007, 9, 4780–4784. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Becerra, J.M.; Hernández-Ortega, S.; Morales-Morales, D.; Valdés-Martinéz, J. Bottom-up design and construction of a non-centrosymmetric network through π–π stacking interactions. CrystEngComm 2009, 11, 226–228. [Google Scholar] [CrossRef]
- Giese, M.; Albrecht, M.; Valkonen, A.; Rissanen, K. The pentafluorophenyl group as a p-acceptor for anions: A case study. Chem. Sci. 2015, 6, 354–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Althagbi, H.I.; Edwards, A.J.; Nicholson, B.K.; Reason, D.A.; Saunders, G.C.; Sim, S.A.; van der Heijden, D.A. Arene-Perfluoroarene-Anion Stacking and Hydrogen Bonding Interactions in Imidazolium Salts for the Crystal Engineering of Polarity. Cryst. Growth Des. 2016, 16, 174–188. [Google Scholar] [CrossRef]
- Saunders, G.C.; Thomas, H.P. Crystal structure and theoretical calculations of 1-(4-trifluoromethyl-2,3,5,6-tetrafluorophenyl)-3-benzylimidazolium bromide. J. Struct. Chem. 2017, 58, 211–215. [Google Scholar] [CrossRef]
- Althagbi, H.I.; Bernstein, D.R.; Crombie, W.C.; Lane, J.R.; McQuiston, D.K.; Oosterwijk, M.A.; Saunders, G.C.; Zou, W. The crystal structures of 1-(4-halo-2,3,5,6-tetrafluorophenyl)-3-benzylimidazolium bromides: The relative importance of anion-π, lone pair–π, π–π stacking and halogen bonding interactions. J. Fluorine Chem. 2018, 206, 61–71. [Google Scholar] [CrossRef]
- Althagbi, H.A.; Evans, D.N.; Saunders, G.C. The crystal structures of 1-(4-bromo-2,3,5,6-tetrafluorophenyl)-3-benzyl-methylimidazolium bromides. J. Mol. Struct. 2018, 1171, 755–761. [Google Scholar] [CrossRef]
- Arcus, V.L.; Bernstein, D.R.; Crombie, C.W.; Saunders, G.C. Infinite stacking of alternating polyfluoroaryl rings and bromide anions. CrystEngComm 2013, 15, 9841–9843. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Bourhis, L.J.; Dolomanov, O.V.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. The anatomy of a comprehensive constrained, restrained refinement program for the modern computing environment—Olex2 dissected. Acta Crystallogr. 2015, A71, 59–75. [Google Scholar] [CrossRef] [Green Version]
- Shao, Y.; Gan, Z.; Epifanovsky, E.; Gilbert, A.T.B.; Wormit, M.; Kussmann, J.; Lange, A.W.; Behn, A.; Deng, J.; Feng, X.; et al. Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol. Phys. 2015, 113, 184–215. [Google Scholar] [CrossRef] [Green Version]
- Mardirossian, N.; Head-Gordon, M. ωB97X-V: A 10-parameter, range-separated hybrid, generalized gradient approximation density functional with nonlocal correlation, designed by a survival-of-the-fittest strategy. Phys. Chem. Chem. Phys. 2014, 16, 9904–9924. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| d(X···Br−), Å | ∕ (Y—X···Br−), ° | (C3N2···Br−), Å | Energy, kJ mol−1 a | |
|---|---|---|---|---|
| C(1)—H···Br− | 3.429(2) | 135.6(1), Y = N(1) 114.5(1), Y = N(2) | 0.693 | −371 (−332) |
| C(2)—H···Br− | 3.473(2) | 100.3(1), Y = N(1) 138.3(2), Y = C(3) | 1.773 | −323 (−299) |
| N(2)···Br− | 3.365(2) | 90.3(1), Y = C(1) 93.5(1), Y = C(3) 80.4(1), Y = C(11) | 3.351 | −345 (−391) |
| Br(2)···Br− | 3.2387(3) | 178.27(7), Y = C(7) | - | −207 (−134) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acharige, U.A.I.; Parsot, P.D.; Saunders, G.C. 1-(4-Bromo-2,3,5,6-tetrafluoropheyl)-3-(3-phenylbenzyl)-4-methylimidazolium Bromide. Molbank 2023, 2023, M1594. https://doi.org/10.3390/M1594
Acharige UAI, Parsot PD, Saunders GC. 1-(4-Bromo-2,3,5,6-tetrafluoropheyl)-3-(3-phenylbenzyl)-4-methylimidazolium Bromide. Molbank. 2023; 2023(1):M1594. https://doi.org/10.3390/M1594
Chicago/Turabian StyleAcharige, Udari A. I., Priya D. Parsot, and Graham C. Saunders. 2023. "1-(4-Bromo-2,3,5,6-tetrafluoropheyl)-3-(3-phenylbenzyl)-4-methylimidazolium Bromide" Molbank 2023, no. 1: M1594. https://doi.org/10.3390/M1594