
Selective Oxidation of Halophenols Catalyzed by an Artificial Miniaturized Peroxidase


Abstract
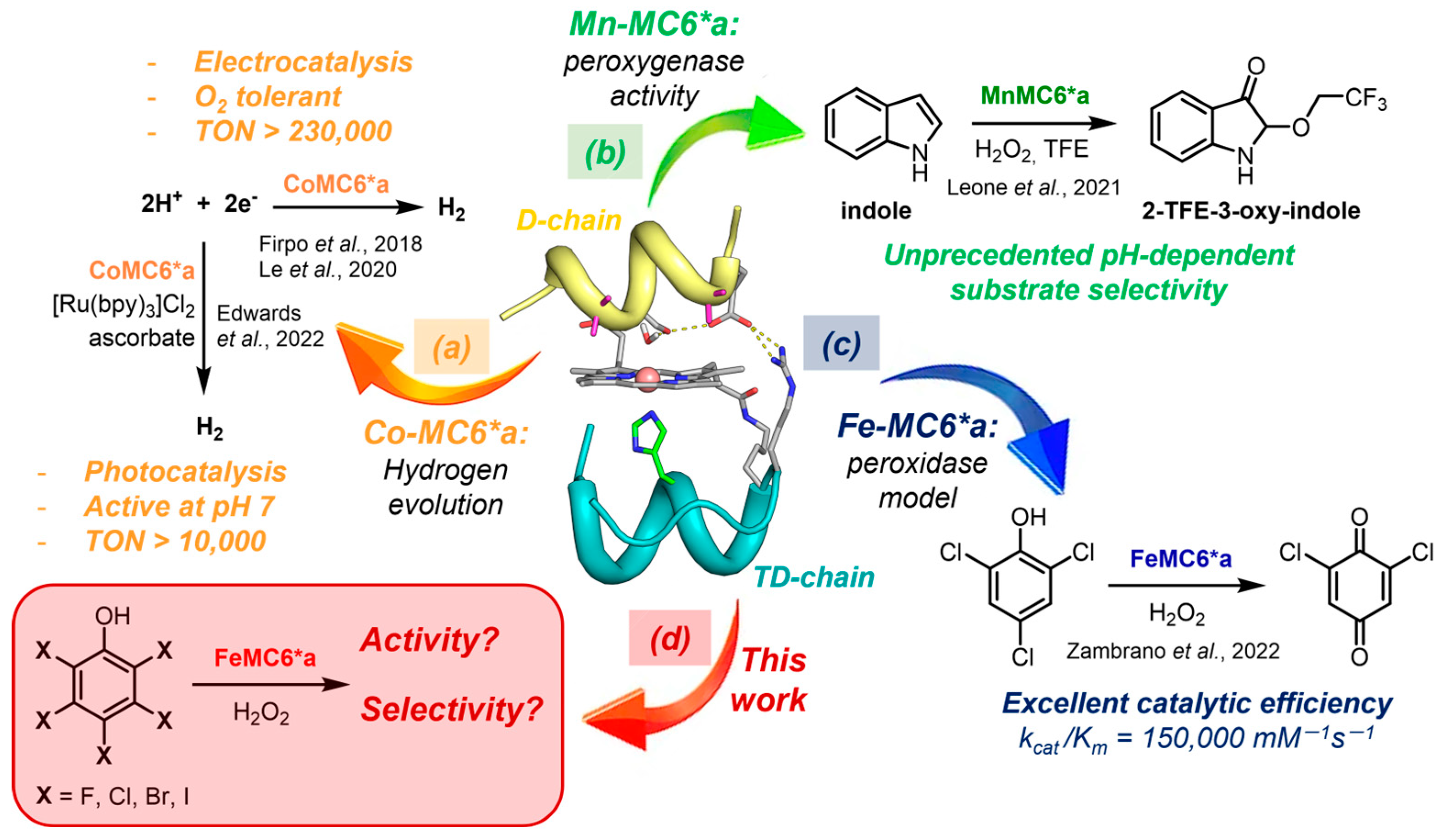
:1. Introduction
2. Results and Discussion
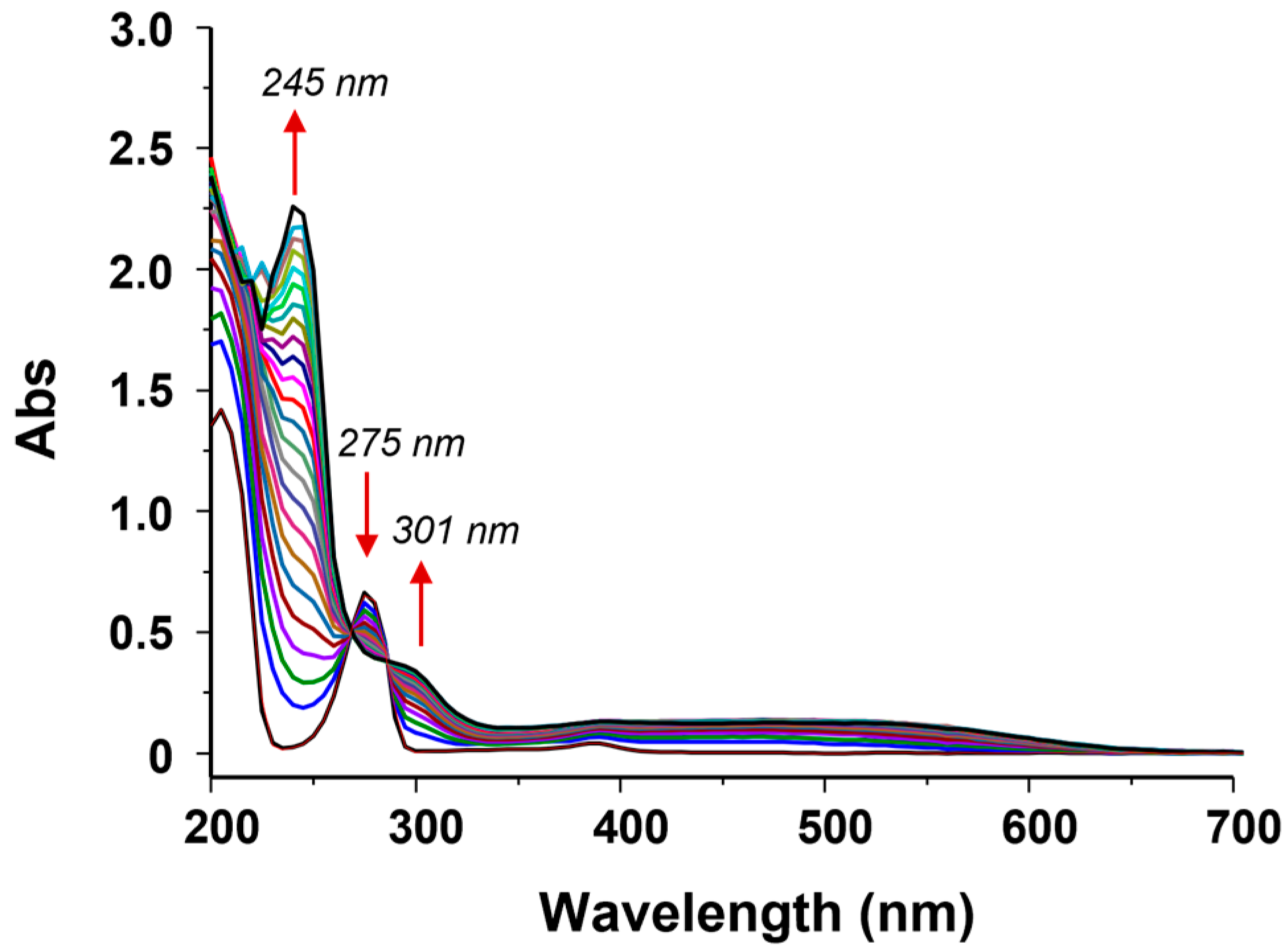
2.1. H2O2-Dependent Dehalogenation Activity of Fe(III)-MC6*a
2.2. Outcome of the Reaction: Case Study of 4-Halophenols
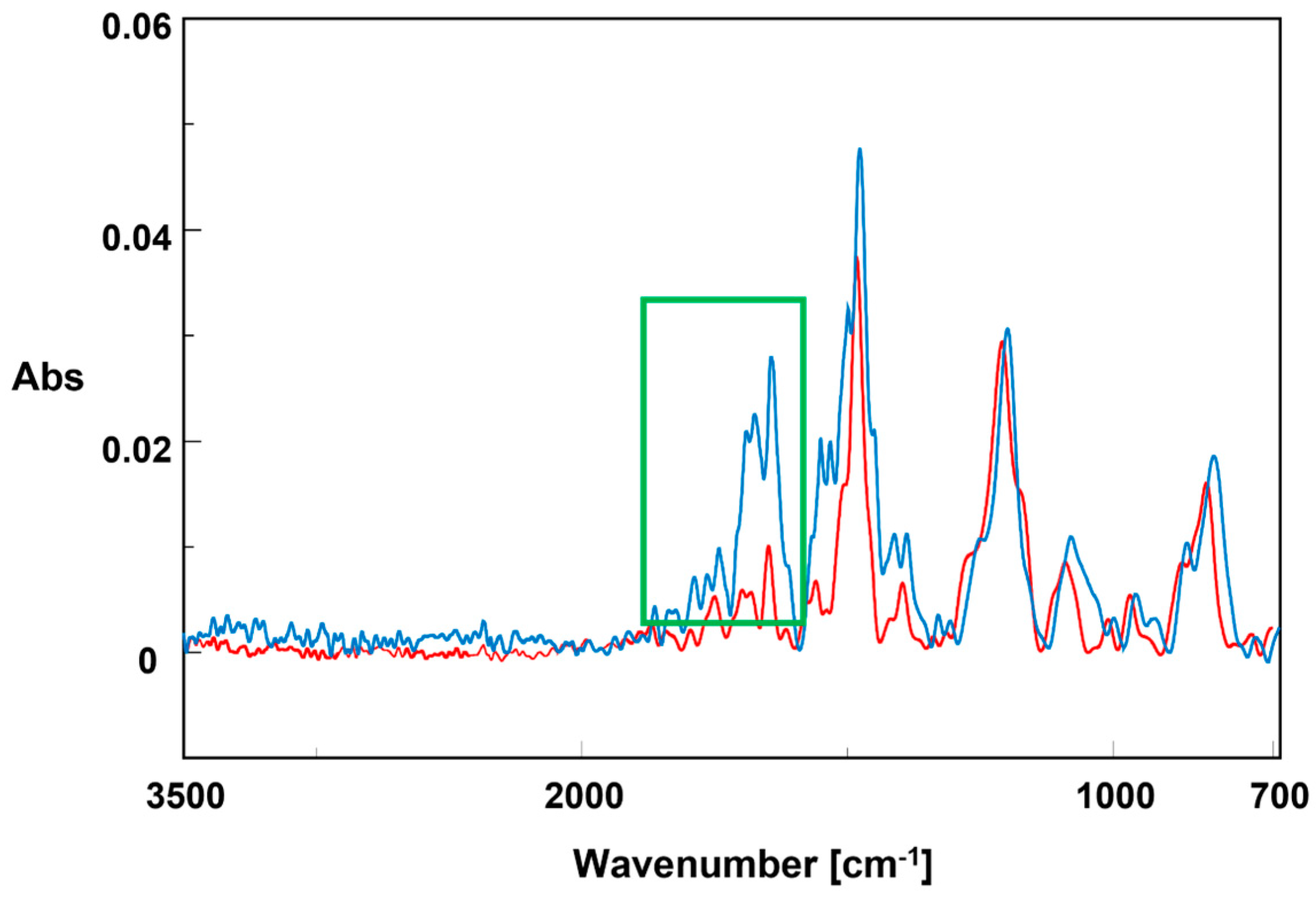
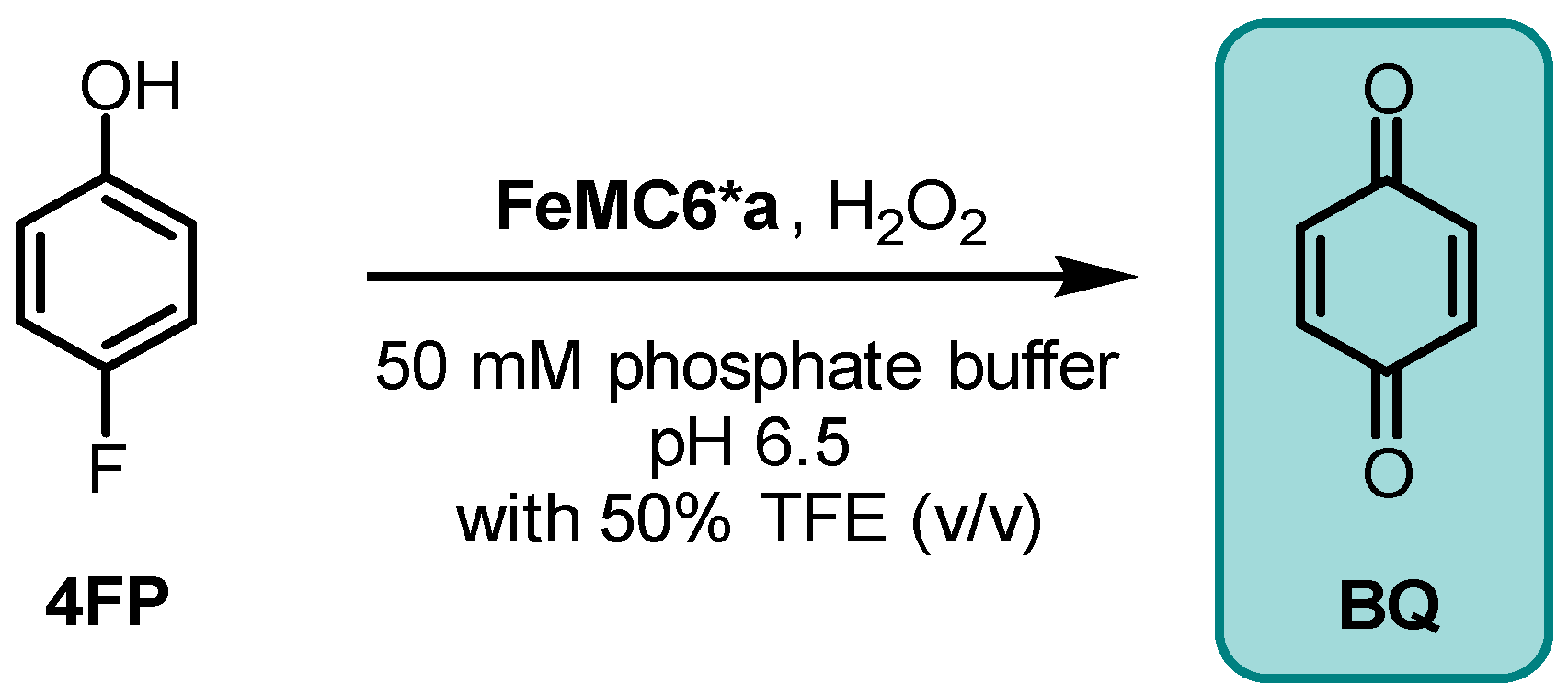
2.2.1. Benzoquinone Formation in 4-Fluorophenol Conversion
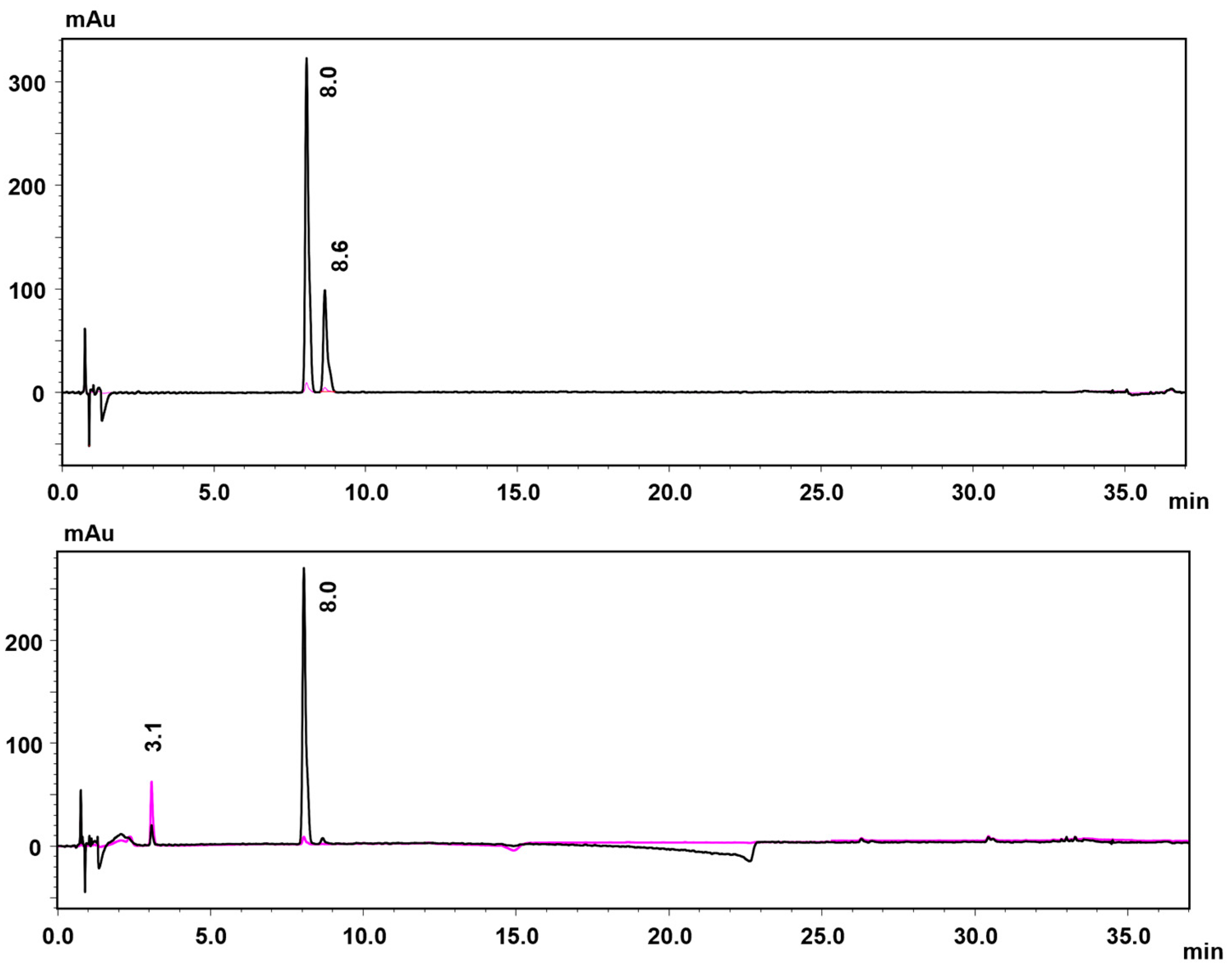
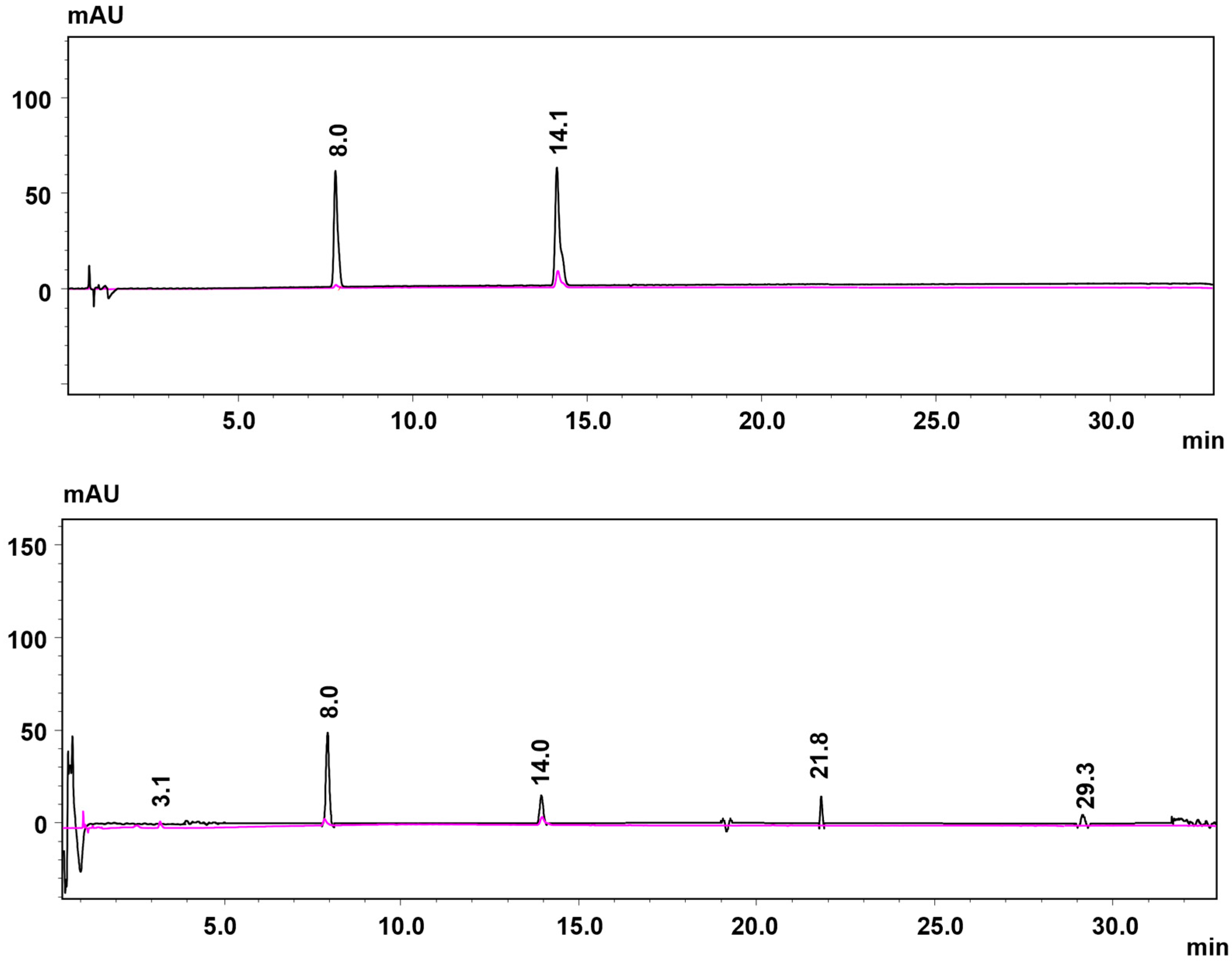
2.2.2. Coupling Products in 4-Chlorophenol Conversion
2.2.3. Identification of the Insoluble Products in the Fe-MC6*a-Catalyzed 4-CP Oxidation
2.3. Fe-MC6*a Chemodivergent Transformations in the Oxidation of Halophenols
3. Materials and Methods
3.1. Fe-MC6*a-Catalyzed Oxidations of Halophenols: General Procedure
3.2. TON Determination
3.3. Synthesis of Compounds 1a and 1b
3.4. Analysis of Polymerization Products in the Enzyme-Catalyzed 4-CP Oxidation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Poulos, T.L. Heme Enzyme Structure and Function. Chem. Rev. 2014, 114, 3919–3962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, E.I.; Heppner, D.E.; Johnston, E.M.; Ginsbach, J.W.; Cirera, J.; Qayyum, M.; Kieber-Emmons, M.T.; Kjaergaard, C.H.; Hadt, R.G.; Tian, L. Copper Active Sites in Biology. Chem. Rev. 2014, 114, 3659–3853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jasniewski, A.J.; Que, L. Dioxygen Activation by Nonheme Diiron Enzymes: Diverse Dioxygen Adducts, High-Valent Intermediates, and Related Model Complexes. Chem. Rev. 2018, 118, 2554–2592. [Google Scholar] [CrossRef]
- Nastri, F.; D’Alonzo, D.; Leone, L.; Zambrano, G.; Pavone, V.; Lombardi, A. Engineering Metalloprotein Functions in Designed and Native Scaffolds. Trends Biochem. Sci. 2019, 44, 1022–1040. [Google Scholar] [CrossRef] [PubMed]
- Wittwer, M.; Markel, U.; Schiffels, J.; Okuda, J.; Sauer, D.F.; Schwaneberg, U. Engineering and Emerging Applications of Artificial Metalloenzymes with Whole Cells. Nat. Catal. 2021, 4, 814–827. [Google Scholar] [CrossRef]
- Maglio, O.; Nastri, F.; Lombardi, A. Structural and Functional Aspects of Metal Binding Sites in Natural and Designed Metalloproteins. In Ionic Interactions in Natural and Synthetic Macromolecules; Ciferri, A., Perico, A., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 361–450. ISBN 978-1-118-16585-0. [Google Scholar]
- Huang, X.; Groves, J.T. Oxygen Activation and Radical Transformations in Heme Proteins and Metalloporphyrins. Chem. Rev. 2018, 118, 2491–2553. [Google Scholar] [CrossRef] [Green Version]
- Valasatava, Y.; Rosato, A.; Furnham, N.; Thornton, J.M.; Andreini, C. To What Extent Do Structural Changes in Catalytic Metal Sites Affect Enzyme Function? J. Inorg. Biochem. 2018, 179, 40–53. [Google Scholar] [CrossRef]
- Lu, Y.; Yeung, N.; Sieracki, N.; Marshall, N.M. Design of Functional Metalloproteins. Nature 2009, 460, 855–862. [Google Scholar] [CrossRef] [Green Version]
- Schwizer, F.; Okamoto, Y.; Heinisch, T.; Gu, Y.; Pellizzoni, M.M.; Lebrun, V.; Reuter, R.; Köhler, V.; Lewis, J.C.; Ward, T.R. Artificial Metalloenzymes: Reaction Scope and Optimization Strategies. Chem. Rev. 2018, 118, 142–231. [Google Scholar] [CrossRef] [Green Version]
- Nastri, F.; Chino, M.; Maglio, O.; Bhagi-Damodaran, A.; Lu, Y.; Lombardi, A. Design and Engineering of Artificial Oxygen-Activating Metalloenzymes. Chem. Soc. Rev. 2016, 45, 5020–5054. [Google Scholar] [CrossRef] [Green Version]
- Chino, M.; Leone, L.; Zambrano, G.; Pirro, F.; D’Alonzo, D.; Firpo, V.; Aref, D.; Lista, L.; Maglio, O.; Nastri, F.; et al. Oxidation Catalysis by Iron and Manganese Porphyrins within Enzyme-like Cages. Biopolymers 2018, 109, e23107. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, A.; Nastri, F.; Pavone, V. Peptide-Based Heme−Protein Models. Chem. Rev. 2001, 101, 3165–3190. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, A.; Pirro, F.; Maglio, O.; Chino, M.; DeGrado, W.F. De Novo Design of Four-Helix Bundle Metalloproteins: One Scaffold, Diverse Reactivities. Acc. Chem. Res. 2019, 52, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Chalkley, M.J.; Mann, S.I.; DeGrado, W.F. De Novo Metalloprotein Design. Nat. Rev. Chem. 2021, 6, 31–50. [Google Scholar] [CrossRef]
- Koebke, K.J.; Pinter, T.B.J.; Pitts, W.C.; Pecoraro, V.L. Catalysis and Electron Transfer in De Novo Designed Metalloproteins. Chem. Rev. 2022, 122, 12046–12109. [Google Scholar] [CrossRef]
- Pirro, F.; Schmidt, N.; Lincoff, J.; Widel, Z.X.; Polizzi, N.F.; Liu, L.; Therien, M.J.; Grabe, M.; Chino, M.; Lombardi, A.; et al. Allosteric Cooperation in a De Novo-Designed Two-Domain Protein. Proc. Natl. Acad. Sci. USA 2020, 117, 33246–33253. [Google Scholar] [CrossRef]
- Chino, M.; Zhang, S.-Q.; Pirro, F.; Leone, L.; Maglio, O.; Lombardi, A.; DeGrado, W.F. Spectroscopic and Metal Binding Properties of a De Novo Metalloprotein Binding a Tetrazinc Cluster. Biopolymers 2018, 109, e23339. [Google Scholar] [CrossRef]
- Petrik, I.D.; Liu, J.; Lu, Y. Metalloenzyme Design and Engineering through Strategic Modifications of Native Protein Scaffolds. Curr. Opin. Chem. Biol. 2014, 19, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-W. Biodegradation of Aromatic Pollutants by Metalloenzymes: A Structural-Functional-Environmental Perspective. Coord. Chem. Rev. 2021, 434, 213774. [Google Scholar] [CrossRef]
- Renata, H.; Wang, Z.J.; Arnold, F.H. Expanding the Enzyme Universe: Accessing Non-Natural Reactions by Mechanism-Guided Directed Evolution. Angew. Chem. Int. Ed. 2015, 54, 3351–3367. [Google Scholar] [CrossRef] [Green Version]
- Bunzel, H.A.; Anderson, J.L.R.; Mulholland, A.J. Designing Better Enzymes: Insights from Directed Evolution. Curr. Opin. Struct. Biol. 2021, 67, 212–218. [Google Scholar] [CrossRef]
- Zhu, J.; Avakyan, N.; Kakkis, A.; Hoffnagle, A.M.; Han, K.; Li, Y.; Zhang, Z.; Choi, T.S.; Na, Y.; Yu, C.-J.; et al. Protein Assembly by Design. Chem. Rev. 2021, 121, 13701–13796. [Google Scholar] [CrossRef]
- Eberly, J.O.; Ely, R.L. Thermotolerant Hydrogenases: Biological Diversity, Properties, and Biotechnological Applications. Crit. Rev. Microbiol. 2008, 34, 117–130. [Google Scholar] [CrossRef]
- Pineda-Knauseder, A.J.; Vargas, D.A.; Fasan, R. Organic Solvent Stability and Long-term Storage of Myoglobin-based Carbene Transfer Biocatalysts. Biotechnol. Appl. Biochem. 2020, 67, 516–526. [Google Scholar] [CrossRef]
- Hindson, S.A.; Bunzel, H.A.; Frank, B.; Svistunenko, D.A.; Williams, C.; van der Kamp, M.W.; Mulholland, A.J.; Pudney, C.R.; Anderson, J.L.R. Rigidifying a De Novo Enzyme Increases Activity and Induces a Negative Activation Heat Capacity. ACS Catal. 2021, 11, 11532–11541. [Google Scholar] [CrossRef]
- Chen, K.; Arnold, F.H. Engineering New Catalytic Activities in Enzymes. Nat. Catal. 2020, 3, 203–213. [Google Scholar] [CrossRef]
- Jenkins, J.M.X.; Noble, C.E.M.; Grayson, K.J.; Mulholland, A.J.; Anderson, J.L.R. Substrate Promiscuity of a de Novo Designed Peroxidase. J. Inorg. Biochem. 2021, 217, 111370. [Google Scholar] [CrossRef]
- Leone, L.; Chino, M.; Nastri, F.; Maglio, O.; Pavone, V.; Lombardi, A. Mimochrome, a Metalloporphyrin-based Catalytic Swiss Knife. Biotechnol. Appl. Biochem. 2020, 67, 495–515. [Google Scholar] [CrossRef]
- Di Costanzo, L.; Geremia, S.; Randaccio, L.; Nastri, F.; Maglio, O.; Lombardi, A.; Pavone, V. Miniaturized Heme Proteins: Crystal Structure of Co(III)-Mimochrome IV. J. Biol. Inorg. Chem. 2004, 9, 1017–1027. [Google Scholar] [CrossRef]
- Firpo, V.; Le, J.M.; Pavone, V.; Lombardi, A.; Bren, K.L. Hydrogen Evolution from Water Catalyzed by Cobalt-Mimochrome VI*a, a Synthetic Mini-Protein. Chem. Sci. 2018, 9, 8582–8589. [Google Scholar] [CrossRef] [Green Version]
- Le, J.M.; Alachouzos, G.; Chino, M.; Frontier, A.J.; Lombardi, A.; Bren, K.L. Tuning Mechanism through Buffer Dependence of Hydrogen Evolution Catalyzed by a Cobalt Mini-Enzyme. Biochemistry 2020, 59, 1289–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, E.H.; Le, J.M.; Salamatian, A.A.; Peluso, N.L.; Leone, L.; Lombardi, A.; Bren, K.L. A Cobalt Mimochrome for Photochemical Hydrogen Evolution from Neutral Water. J. Inorg. Biochem. 2022, 230, 111753. [Google Scholar] [CrossRef] [PubMed]
- Leone, L.; D’Alonzo, D.; Balland, V.; Zambrano, G.; Chino, M.; Nastri, F.; Maglio, O.; Pavone, V.; Lombardi, A. Mn-Mimochrome VI*a: An Artificial Metalloenzyme With Peroxygenase Activity. Front. Chem. 2018, 6, 590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leone, L.; D’Alonzo, D.; Maglio, O.; Pavone, V.; Nastri, F.; Lombardi, A. Highly Selective Indole Oxidation Catalyzed by a Mn-Containing Artificial Mini-Enzyme. ACS Catal. 2021, 11, 9407–9417. [Google Scholar] [CrossRef]
- Caserta, G.; Chino, M.; Firpo, V.; Zambrano, G.; Leone, L.; D’Alonzo, D.; Nastri, F.; Maglio, O.; Pavone, V.; Lombardi, A. Enhancement of Peroxidase Activity in Artificial Mimochrome VI Catalysts through Rational Design. ChemBioChem 2018, 19, 1823–1826. [Google Scholar] [CrossRef]
- Zambrano, G.; Nastri, F.; Pavone, V.; Lombardi, A.; Chino, M. Use of an Artificial Miniaturized Enzyme in Hydrogen Peroxide Detection by Chemiluminescence. Sensors 2020, 20, 3793. [Google Scholar] [CrossRef]
- Zambrano, G.; Sekretareva, A.; D’Alonzo, D.; Leone, L.; Pavone, V.; Lombardi, A.; Nastri, F. Oxidative Dehalogenation of Trichlorophenol Catalyzed by a Promiscuous Artificial Heme-Enzyme. RSC Adv. 2022, 12, 12947–12956. [Google Scholar] [CrossRef]
- Garba, Z.N.; Zhou, W.; Lawan, I.; Xiao, W.; Zhang, M.; Wang, L.; Chen, L.; Yuan, Z. An Overview of Chlorophenols as Contaminants and Their Removal from Wastewater by Adsorption: A Review. J. Environ. Manag. 2019, 241, 59–75. [Google Scholar] [CrossRef]
- Mercier, C.; Youmans, P. 4-Fluorophenol: A Key Intermediate for Agrochemicals and Pharmaceuticals. In Industrial Chemistry Library; Elsevier: Amsterdam, The Netherlands, 1996; Volume 8, pp. 293–300. ISBN 978-0-444-82434-9. [Google Scholar]
- Hamid, M. Khalil-ur-Rehman Potential Applications of Peroxidases. Food Chem. 2009, 115, 1177–1186. [Google Scholar] [CrossRef]
- Osborne, R.L.; Coggins, M.K.; Walla, M.; Dawson, J.H. Horse Heart Myoglobin Catalyzes the H2O2 -Dependent Oxidative Dehalogenation of Chlorophenols to DNA-Binding Radicals and Quinones. Biochemistry 2007, 46, 9823–9829. [Google Scholar] [CrossRef]
- Franzen, S.; Thompson, M.K.; Ghiladi, R.A. The Dehaloperoxidase Paradox. Biochim. Biophys. Acta BBA—Proteins Proteom. 2012, 1824, 578–588. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, A. Carbon–Fluorine Bond Cleavage Mediated by Metalloenzymes. Chem. Soc. Rev. 2020, 49, 4906–4925. [Google Scholar] [CrossRef]
- Osborne, R.L.; Raner, G.M.; Hager, L.P.; Dawson, J.H. C. Fumago Chloroperoxidase Is Also a Dehaloperoxidase: Oxidative Dehalogenation of Halophenols. J. Am. Chem. Soc. 2006, 128, 1036–1037. [Google Scholar] [CrossRef]
- Murphy, C.D. Fluorophenol Oxidation by a Fungal Chloroperoxidase. Biotechnol. Lett. 2006, 29, 45–49. [Google Scholar] [CrossRef]
- Pirzad, R.; Newman, J.D.; Dowman, A.A.; Cowell, D.C. Horseradish Peroxidase Assay—Radical Inactivation or Substrate Inhibition? Revision of the Catalytic Sequence Following Mass Spectral Evidence. Analyst 1994, 119, 213–218. [Google Scholar] [CrossRef]
- Kozuch, S.; Martin, J.M.L. “Turning Over” Definitions in Catalytic Cycles. ACS Catal. 2012, 2, 2787–2794. [Google Scholar] [CrossRef]
- Chen, Y.P.; Woodin, S.A.; Lincoln, D.E.; Lovell, C.R. An Unusual Dehalogenating Peroxidase from the Marine Terebellid Polychaete Amphitrite ornata. J. Biol. Chem. 1996, 271, 4609–4612. [Google Scholar] [CrossRef] [Green Version]
- De Serrano, V.; Franzen, S. Structural Evidence for Stabilization of Inhibitor Binding by a Protein Cavity in the Dehaloperoxidase-Hemoglobin from Amphitrite ornata. Biopolymers 2012, 98, 27–35. [Google Scholar] [CrossRef]
- Thompson, M.K.; Davis, M.F.; de Serrano, V.; Nicoletti, F.P.; Howes, B.D.; Smulevich, G.; Franzen, S. Internal Binding of Halogenated Phenols in Dehaloperoxidase-Hemoglobin Inhibits Peroxidase Function. Biophys. J. 2010, 99, 1586–1595. [Google Scholar] [CrossRef] [Green Version]
- Vatsis, K.P.; Coon, M.J. Ipso-Substitution by Cytochrome P450 with Conversion of p-Hydroxybenzene Derivatives to Hydroquinone: Evidence for Hydroperoxo-Iron As the Active Oxygen Species. Arch. Biochem. Biophys. 2002, 397, 119–129. [Google Scholar] [CrossRef]
- Harkey, A.; Kim, H.-J.; Kandagatla, S.; Raner, G.M. Defluorination of 4-Fluorophenol by Cytochrome P450BM3-F87G: Activation by Long Chain Fatty Aldehydes. Biotechnol. Lett. 2012, 34, 1725–1731. [Google Scholar] [CrossRef] [PubMed]
- Osman, A.M.; Boeren, S.; Veeger, C.; Rietjens, I.M.C.M. MP8-Dependent Oxidative Dehalogenation: Evidence for the Direct Formation of 1,4-Benzoquinone from 4-Fluorophenol by a Peroxidase-Type of Reaction Pathway. Chem. Biol. Interact. 1997, 104, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.A.; Katz, J.L.; West, T.R. Synthesis of Diaryl Ethers through the Copper-Promoted Arylation of Phenols with Arylboronic Acids. An Expedient Synthesis of Thyroxine. Tetrahedron Lett. 1998, 39, 2937–2940. [Google Scholar] [CrossRef]
- Bentley, K.W.; Joyce, L.A.; Sherer, E.C.; Sheng, H.; Wolf, C.; Welch, C.J. Antenna Biphenols: Development of Extended Wavelength Chiroptical Reporters. J. Org. Chem. 2016, 81, 1185–1191. [Google Scholar] [CrossRef] [PubMed]
- Osborne, R.L.; Coggins, M.K.; Raner, G.M.; Walla, M.; Dawson, J.H. The Mechanism of Oxidative Halophenol Dehalogenation by Amphitrite ornata Dehaloperoxidase Is Initiated by H2O2 Binding and Involves Two Consecutive One-Electron Steps: Role of Ferryl Intermediates. Biochemistry 2009, 48, 4231–4238. [Google Scholar] [CrossRef]
- Belyea, J.; Gilvey, L.B.; Davis, M.F.; Godek, M.; Sit, T.L.; Lommel, S.A.; Franzen, S. Enzyme Function of the Globin Dehaloperoxidase from Amphitrite ornata is Activated by Substrate Binding. Biochemistry 2005, 44, 15637–15644. [Google Scholar] [CrossRef]
- NIST/EPA Gas-Phase Infrared Database. Available online: https://webbook.nist.gov/cgi/cbook.cgi?ID=C106514&Mask=80#IR-Spec (accessed on 27 April 2023).
- Nicell, J.A.; Bewtra, J.K.; Taylor, K.E.; Biswas, N.; St Pierre, C. Enzyme Catalyzed Polymerization and Precipitation of Aromatic Compounds from Wastewater. Water Sci. Technol. 1992, 25, 157–164. [Google Scholar] [CrossRef]
- Nicell, J.A.; Bewtra, J.K.; Biswas, N.; St Pierre, C.C.; Taylor, K.E. Enzyme Catalyzed Polymerization and Precipitation of Aromatic Compounds from Aqueous Solution. Can. J. Civ. Eng. 1993, 20, 725–735. [Google Scholar] [CrossRef]
- Reihmann, M.; Ritter, H. Synthesis of Phenol Polymers Using Peroxidases. In Enzyme-Catalyzed Synthesis of Polymers; Kobayashi, S., Ritter, H., Kaplan, D., Eds.; Advances in Polymer Science; Springer: Berlin/Heidelberg, Germany, 2006; Volume 194, pp. 1–49. ISBN 978-3-540-29212-8. [Google Scholar]
- Vojinović, V.; Carvalho, R.H.; Lemos, F.; Cabral, J.M.S.; Fonseca, L.P.; Ferreira, B.S. Kinetics of Soluble and Immobilized Horseradish Peroxidase-Mediated Oxidation of Phenolic Compounds. Biochem. Eng. J. 2007, 35, 126–135. [Google Scholar] [CrossRef]
- Sarno, M.; Iuliano, M. New Nano-Biocatalyst for 4-Chlorophenols Removal from Wastewater. Mater. Today Proc. 2020, 20, 74–81. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, T.; Zhang, Y.-X.; Sang, X.-J.; Su, F.; Zhu, Z.-M.; Zhang, L.-C. A POM-Based Copper-Coordination Polymer Crystal Material for Phenolic Compound Degradation by Immobilizing Horseradish Peroxidase. Dalton Trans. 2021, 50, 15198–15209. [Google Scholar] [CrossRef]
- Yamada, K.; Shibuya, T.; Noda, M.; Uchiyama, N.; Kashiwada, A.; Matsuda, K.; Hirata, M. Influence of Position of Substituent Groups on Removal of Chlorophenols and Cresols by Horseradish Peroxidase and Determination of Optimum Conditions. Biosci. Biotechnol. Biochem. 2007, 71, 2503–2510. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Sono, M.; Du, J.; Dawson, J.H. Evidence of the direct involvement of the substrate TCP radical in functional switching from oxyferrous O2 carrier to ferric peroxidase in the dual-function hemoglobin/dehaloperoxidase from Amphitrite ornata. Biochemistry 2014, 53, 4956–4969. [Google Scholar] [CrossRef]
- Suzuki, Y.; Nakamura, M.; Otsuka, Y.; Suzuki, N.; Ohyama, K.; Kawakami, T.; Sato, K.; Kajita, S.; Hishiyama, S.; Fujii, T.; et al. Novel Enzymatic Activity of Cell Free Extract from Thermophilic Geobacillus Sp. UZO 3 Catalyzes Reductive Cleavage of Diaryl Ether Bonds of 2,7-Dichlorodibenzo-p-Dioxin. Chemosphere 2011, 83, 868–872. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Substrate | Abbreviation | TON |
|---|---|---|---|
| 1 | 2-Chlorophenol | 2-CP | 310 ± 19 |
| 2 | 4-Fluorophenol 1 | 4-FP | 503 ± 10 |
| 3 | 4-Chlorophenol | 4-CP | 325 ± 22 |
| 4 | 4-Bromophenol | 4-BP | 235 ± 10 |
| 5 | 4-Iodophenol | 4-IP | 208 ± 30 |
| 6 | 2,4-Dichlorophenol 2 | 2,4-DCP | 2588 ± 103 |
| 7 | 2,4,6-Trifluorophenol 2 | 2,4,6-TFP | 3825 ± 171 |
| 8 | 2,4,6-Trichlorophenol 2 | 2,4,6-TCP | 3850 ± 148 |
| 9 | Pentafluorophenol | PFP | 142 ± 27 |
| 10 | Pentachlorophenol | PCP | 186 ± 64 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Alonzo, D.; De Fenza, M.; Pavone, V.; Lombardi, A.; Nastri, F. Selective Oxidation of Halophenols Catalyzed by an Artificial Miniaturized Peroxidase. Int. J. Mol. Sci. 2023, 24, 8058. https://doi.org/10.3390/ijms24098058
D’Alonzo D, De Fenza M, Pavone V, Lombardi A, Nastri F. Selective Oxidation of Halophenols Catalyzed by an Artificial Miniaturized Peroxidase. International Journal of Molecular Sciences. 2023; 24(9):8058. https://doi.org/10.3390/ijms24098058
Chicago/Turabian StyleD’Alonzo, Daniele, Maria De Fenza, Vincenzo Pavone, Angela Lombardi, and Flavia Nastri. 2023. "Selective Oxidation of Halophenols Catalyzed by an Artificial Miniaturized Peroxidase" International Journal of Molecular Sciences 24, no. 9: 8058. https://doi.org/10.3390/ijms24098058