
Autophagy and SARS-CoV-2-Old Players in New Games
 , , and
, , and 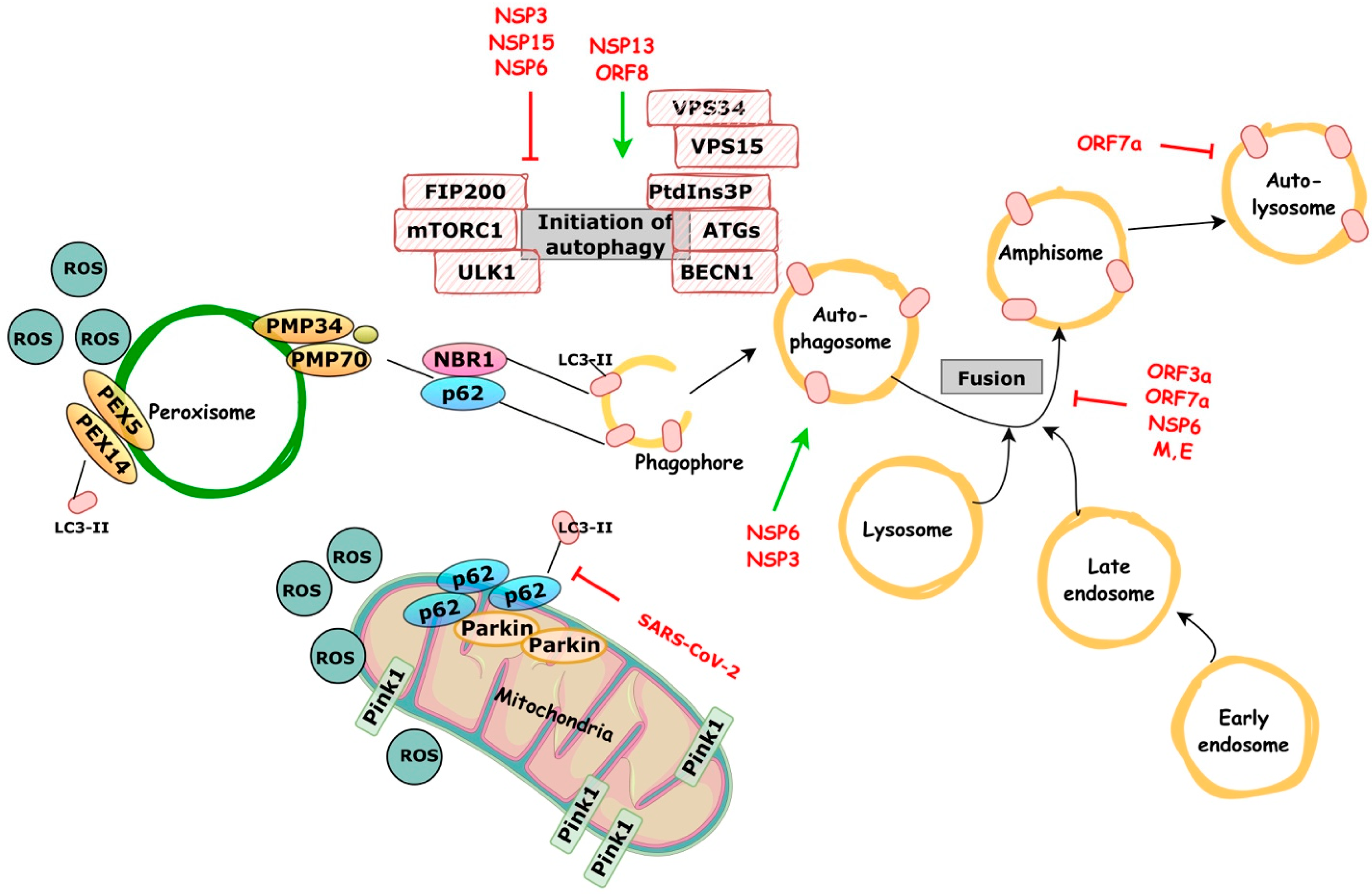{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Autophagy as a Process—Biological Importance and Types of Autophagy
3. Autophagy in Infectious Diseases
3.1. Antiviral Functions of Autophagy
3.2. Blocking of Autophagy by Viruses
3.3. Pro-Viral Role of Autophagy
4. Autophagy and COVID-19
4.1. Early Autophagy Reprogramming
4.2. Late-Stage Incomplete Autophagy
4.3. Mitophagy and Innate Immune Responses Reprograming
4.4. Putative Pexophagy Involvement
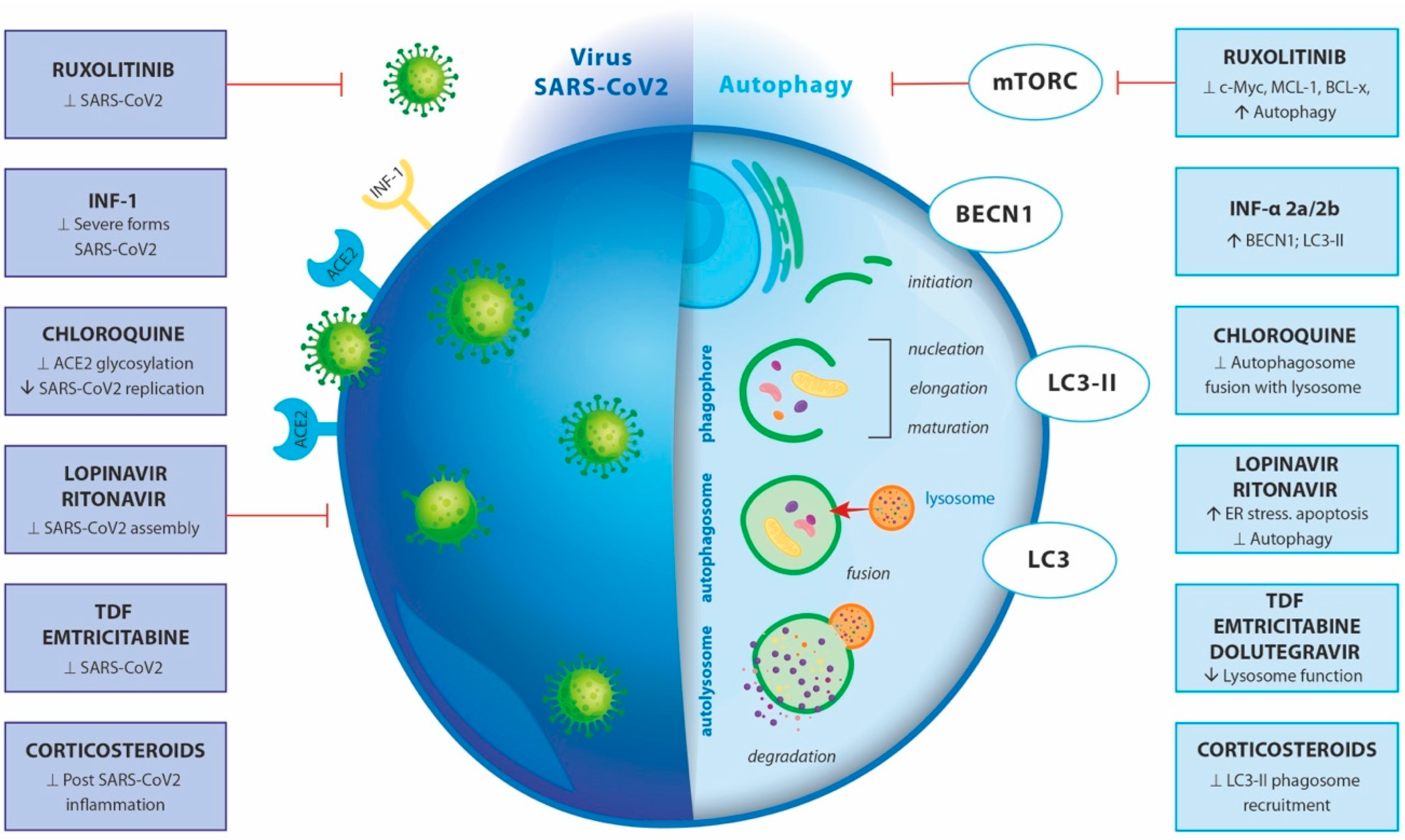
4.5. Treatment and Pharmacological Modulation
4.6. Adverse Effects of Autophagy-Targeting Drugs
5. Models to Study Autophagy in Infections
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviation
| a | |
| A549 | human lung cells |
| ACE2 | angiotensin-converting enzyme 2 |
| AKT-mTOR signaling | protein kinase B-mammalian target of rapamycin complex |
| APCs | antigen presenting cells |
| Arl8b | Arf-like GTPases |
| ARP2/3 | actin-related protein 2/3 complex |
| ARH-77 | B lymphoblast cell line |
| ASC | adaptor apoptosis-associated speck-like protein with a caspase-1 recruitment domain |
| ATG | autophagy-related genes |
| ATP6AP1 | S1 subunit of the enzyme V-type proton ATPase. |
| b | |
| BCL-xL | B-cell lymphoma-extra large |
| BECN1(Beclin-1) | mammalian ortholog of the yeast autophagy-related gene 6 |
| BORC-ARL8b | complex multisubunit complex- regulates lysosome positioning |
| c | |
| Calu-3 | human bronchial epithelial cells |
| CARD | caspase recruitment domain |
| CatB/L | cathepsin B and L |
| CAV | Coxsackie viruses |
| c-Myc | proto-oncogene |
| CNS | central nervous system |
| CXCR-4 | C-X-C chemokine receptor type 4 |
| d | |
| 3D organoids | miniature 3-dimensional structures that can mimic the structure and function of different tissues |
| DAMPs | damage-associated molecular patterns |
| DENV | Dengue virus |
| DMV | double-membrane vesicles |
| Dolutegravir | antiretroviral medication |
| e | |
| EBV | Epstein Bar virus |
| eIF2a | translation elongation factor |
| Emtricitabine | nucleoside reverse-transcriptase inhibitor |
| ER | endoplasmic reticulum |
| f | |
| FAM134 | reticulon protein- ER-phagy receptor |
| g | |
| GABARAP | GABA type receptor-associated protein |
| GSDMD | gasdermin D |
| GTPase | hydrolase enzyme binding nucleotide guanosine triphosphate |
| h | |
| HCV | hepatitis C virus |
| HEK 293T | human embryonic kidney cells |
| HIF-1 | hypoxia-induced transcription factor-1 |
| Hsc70 | heat shock cognate 71 kDa protein |
| HSV | herpes simplex virus type I |
| HOPS | homotypic fusion and protein sorting |
| i | |
| ICP34.5 | infected cell protein 34.5 |
| IFN | interferon |
| iPSCs | human induced pluripotent stem cells |
| j | |
| JAK1/2 | Janus kinase- (tyrosine kinases) |
| k | |
| KSHV | Kaposi’s sarcoma-associated herpes virus |
| l | |
| LAMP1 | lysosomal associated membrane glycoprotein 1 |
| LAMP2A | lysosomal associated membrane protein 2 |
| LC3 | microtubule associated protein 1 light chain 3B |
| LIR | LC3-interacting region |
| Lopinavir | antiretroviral protease inhibitor |
| m | |
| MAVS | mitochondrial antiviral-signaling protein |
| MCL-1 | induced myeloid leukemia cell differentiation protein |
| MHC | major histocompatibility complex |
| MHV | murine hepatitis virus |
| mTORC1/S6K/4EBP1 | mammalian target of rapamycin complex 1/p70 ribosomal S6 kinases/4E-binding protein 1 |
| n | |
| NBR1 | neighbor of BRCA1 gene |
| NCI-BL 2171 | small cell lung carcinoma |
| NDP52 | calcium binding and coiled-coil domain 2 |
| NF-kB | nuclear factor-κB (transcription factor) |
| NIC | niclosamide |
| NK | natural killer cells |
| NLRP3 | NLR family pyrin domain containing 3 inflammasome |
| NS4B | membrane-associated protein involved in viral replication or assembly |
| NSPs | non-structural proteins |
| o | |
| OPTN | Organ Procurement and Transplantation Network |
| ORFs | Open Reading Frames |
| ORF3a | encodes a viral accessory protein |
| ORF8 | encodes viral accessory protein, Betacoronavirus NS8 protein |
| p | |
| p62 | SQSTM1(sequestosome 1)- ubiquitin-binding scaffold protein |
| PAMPs | pathogen-associated molecular patterns |
| PCD | programmed cell death |
| PEX3 | peroxisomal Biogenesis Factor |
| PI3K | phosphoinositide 3-kinase, also known as VPS34. |
| PI3P | phosphatidylinositol 3-phosphate |
| PINK1 | PTEN induced kinase 1 |
| PKR | protein kinase R |
| PMP | peroxisome membrane protein |
| PP1a | protein phosphatase 1 |
| PRKN | Parkin RBR E3 ubiquitin protein ligase |
| PTEN | fosfatidilinositol-3,4,5-trisfosfato 3-fosfatasa |
| PV | Poliovirus |
| PYCARD | apoptosis-associated speck-like protein containing a CARD (or ASC) |
| PYD | pyrin domain |
| r | |
| Rab-7a | Ras-related protein late-endosomal / lysosomal GTPase |
| RCD | regulated cell death |
| RHIM | homotypic interaction motif |
| Ribavirin | antiretroviral medication inhibits- RNA/DNA viruses |
| RIP | proteins containing a receptor-interacting protein |
| Ritonavir | antiretroviral medication, protease inhibitors |
| RNP | ribonucleoprotein complexes |
| ROS | reactive oxygen species |
| RTC | replication and transcription complex |
| Ruxolitinib | Janus kinase inhibitor |
| s | |
| SMVs | single-membrane vesicles |
| SNAP | soluble N-ethylmaleimidesSensitive factor attachment proteins |
| SNAP29 | synaptosome associated protein 29, SNARE protein, in autophagy |
| SNARE | SNAP receptor |
| SNX27 | sorting nexin family member 27 |
| STX17 | Syntaxin 17, autophagosomal SNARE protein |
| t | |
| TAK1 | transforming growth factor beta-activated kinase 1 |
| Tax1BP1 | Tax1-binding protein 1 |
| TGN | trans-Golgi network |
| TNFR1 | tumor necrosis factor receptor 1 |
| TMEM41B | transmembrane protein 41B |
| TMPRSS2 | transmembrane serine 2 protease |
| Tenofovir DF | tenofovir disoproxil fumarate, HIV medication |
| v | |
| VAMP8 | vesicle-associated membrane protein 8 |
| Vero E6 | epithelial cell from African green monkey |
| Viroporin | small hydrophobic multifunctional viral proteins |
| VMP1 | vacuolar membrane protein 1 |
| VPS | vacuolar protein sorting proteins (as VPS26, VPS29 and VPS35) |
| Vps34 | class III phosphoinositide 3-kinase (PI3K) |
| VZV | Varicella zoster virus |
| w | |
| WHO | World Health Organization |
| z | |
| ZBP1 | Z-DNA binding protein |
References
- Duve, C.D.E.; Pressman, B.C.; Gianetto, R.; Wattiaux, R.; Appelmans, F. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem. J. 1955, 60, 604. [Google Scholar] [CrossRef]
- Beaufay, H.; De Duve, C. Electron Microscopy of Lysosome-Rich Fractions from Rat Liver. J. Biophys. Biochem. Cytol. 1956, 2, 179–184. [Google Scholar]
- Clark, S.L. Cellular Differentiation in the Kidneys of Newborn Mice Studied with the Electron Microscope. J. Biophys. Biochem. Cytol. 1957, 3, 349. [Google Scholar] [CrossRef] [PubMed]
- Arstila, A.U.; Trump, B.F. Studies on cellular autophagocytosis. The formation of autophagic vacuoles in the liver after glucagon administration. Am. J. Pathol. 1968, 53, 687. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2013521/ (accessed on 27 February 2023). [PubMed]
- de Duve, C. The lysosome. Sci. Am. 1963, 208, 64–72. [Google Scholar] [CrossRef]
- Ohsumi, Y. Historical landmarks of autophagy research. Cell Res. 2014, 24, 9–23. [Google Scholar] [CrossRef]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell Biol. 1992, 119, 301–312. [Google Scholar] [CrossRef]
- Tsukada, M.; Ohsumi, Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993, 333, 169–174. [Google Scholar] [CrossRef]
- Xie, Z.; Nair, U.; Klionsky, D.J. Atg8 controls phagophore expansion during autophagosome formation. Mol. Biol. Cell 2008, 19, 3290–3298. [Google Scholar] [CrossRef]
- Yorimitsu, T.; Klionsky, D.J. Atg11 links cargo to the vesicle-forming machinery in the cytoplasm to vacuole targeting pathway. Mol. Biol. Cell 2005, 16, 1593–1605. [Google Scholar] [CrossRef]
- Jiang, G.M.; Tan, Y.; Wang, H.; Peng, L.; Chen, H.T.; Meng, X.J.; Li, L.-L.; Liu, Y.; Li, W.-F.; Shan, H. The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Mol. Cancer 2019, 18, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Deng, J.; Nan, M.L.; Zhang, J.; Okekunle, A.; Li, J.Y.; Yu, X.Q.; Wang, P.H. The interplay between pattern recognition receptors and autophagy in inflammation. Adv. Exp. Med. Biol. 2019, 1209, 79–108. [Google Scholar] [CrossRef]
- Münz, C. Autophagy Beyond Intracellular MHC Class II Antigen Presentation. Trends Immunol. 2016, 37, 755–763. [Google Scholar] [CrossRef]
- Cottam, E.M.; Whelband, M.C.; Wileman, T. Coronavirus NSP6 restricts autophagosome expansion. Autophagy 2014, 10, 1426–1441. [Google Scholar] [CrossRef]
- Gassen, N.C.; Niemeyer, D.; Muth, D.; Corman, V.M.; Martinelli, S.; Gassen, A.; Hafner, K.; Papies, J.; Mösbauer, K.; Zellner, A.; et al. SKP2 attenuates autophagy through Beclin1-ubiquitination and its inhibition reduces MERS-Coronavirus infection. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Miller, S.; Krijnse-Locker, J. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 2008, 6, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Oudshoorn, D.; Rijs, K.; Limpens, R.W.A.L.; Groen, K.; Koster, A.J.; Snijder, E.J.; Kikkert, M.; Bárcena, M. Expression and cleavage of middle east respiratory syndrome coronavirus nsp3-4 polyprotein induce the formation of double-membrane vesicles that mimic those associated with coronaviral RNA replication. mBio 2017, 8, e01658-17. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Lu, K.; Mao, B.; Liu, S.; Trilling, M.; Huang, A.; Lu, M.; Lin, Y. The interplay between emerging human coronavirus infections and autophagy. Emerg. Microbes Infect. 2021, 10, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Schröder, B.A.; Wrocklage, C.; Hasilik, A.; Saftig, P. The proteome of lysosomes. Proteomics 2010, 10, 4053–4076. [Google Scholar] [CrossRef]
- Jiang, P.; Mizushima, N. Autophagy and human diseases. Cell. Res. 2014, 24, 69–79. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef] [PubMed]
- Sumpter, R.; Sirasanagandla, S.; Fernández, Á.F.; Wei, Y.; Dong, X.; Franco, L.; Zou, Z.; Marchal, C.; Lee, M.Y.; Clapp, D.W.; et al. Fanconi Anemia Proteins Function in Mitophagy and Immunity. Cell 2016, 165, 867–881. [Google Scholar] [CrossRef]
- Tiwari, S.K.; Dang, J.W.; Lin, N.; Qin, Y.; Wang, S.; Rana, T.M. Zika virus depletes neural stem cells and evades selective autophagy by suppressing the Fanconi anemia protein FANCC. EMBO Rep. 2020, 21, e49183. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Deretic, V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat. Rev. Immunol. 2007, 7, 767–777. [Google Scholar] [CrossRef]
- Schmid, D.; Pypaert, M.; Münz, C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity 2007, 26, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Sabli, I.K.; Sancho-Shimizu, V. Inborn errors of autophagy and infectious diseases. Curr. Opin. Immunol. 2021, 72, 272–276. [Google Scholar] [CrossRef]
- Prentice, E.; McAuliffe, J.; Lu, X.; Subbarao, K.; Denison, M.R. Identification and Characterization of Severe Acute Respiratory Syndrome Coronavirus Replicase Proteins. J. Virol. 2004, 78, 9977–9986. [Google Scholar] [CrossRef]
- Choi, Y.; Bowman, J.W.; Jung, J.U. Autophagy during viral infection—A double-edged sword. Nat. Rev. Microbiol. 2018, 16, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Khawar, M.B.; Abbasi, M.H.; Rafiq, M.; Naz, N.; Mehmood, R.; Sheikh, N. A Decade of Mighty Lipophagy: What We Know and What Facts We Need to Know? Oxid. Med. Cell. Longev. 2021, 2021, 5539161. [Google Scholar] [CrossRef]
- da Silva Gomes Dias, S.; Soares, V.C.; Ferreira, A.C.; Sacramento, C.Q.; Fintelman-Rodrigues, N.; Temerozo, J.R.; Teixeira, L.; Nunes da Silva, M.A.; Barreto, E.; Mattos, M.; et al. Lipid droplets fuel SARS-CoV-2 replication and production of inflammatory mediators. PLoS Pathog. 2020, 16, e1009127. [Google Scholar] [CrossRef]
- Chawla, K.; Subramanian, G.; Rahman, T.; Fan, S.; Chakravarty, S.; Gujja, S.; Demchak, H.; Chakravarti, R.; Chattopadhyay, S. Autophagy in Virus Infection: A Race between Host Immune Response and Viral Antagonism. Immuno 2022, 2, 153–169. [Google Scholar] [CrossRef]
- Bakillah, A.; Hejji, F.A.; Almasaud, A.; Jami, H.A.; Hawwari, A.; Qarni, A.A.; Iqbal, J.; Alharbi, N.K. Lipid Raft Integrity and Cellular Cholesterol Homeostasis Are Critical for SARS-CoV-2 Entry into Cells. Nutrients 2022, 14, 3417. [Google Scholar] [CrossRef] [PubMed]
- Mironov, A.A.; Savin, M.A.; Beznoussenko, G.V. COVID-19 Biogenesis and Intracellular Transport. Int. J. Mol. Sci. 2023, 24, 4523. [Google Scholar] [CrossRef]
- Zhao, H.; Lu, L.; Peng, Z.; Chen, L.L.; Meng, X.; Zhang, C.; Ip, J.D.; Chan, W.M.; Chu, A.W.; Chan, K.H.; et al. SARS-CoV-2 Omicron variant shows less efficient replication and fusion activity when compared with Delta variant in TMPRSS2-expressed cells. Emerg. Microbes Infect. 2022, 11, 277–283. [Google Scholar] [CrossRef]
- Daniloski, Z.; Jordan, T.X.; Wessels, H.H.; Hoagland, D.A.; Kasela, S.; Legut, M.; Maniatis, S.; Mimitou, E.P.; Lu, L.; Geller, E.; et al. Identification of Required Host Factors for SARS-CoV-2 Infection in Human Cells. Cell 2021, 184, 92–105.e16. [Google Scholar] [CrossRef] [PubMed]
- Puthenveedu, M.A.; Lauffer, B.; Temkin, P.; Vistein, R.; Carlton, P.; Thorn, K.; Taunton, J.; Weiner, O.D.; Parton, R.G.; Zastrow, M.V. Sequence-Dependent Sorting of Recycling Proteins by Actin-Stabilized Endosomal Microdomains. Cell 2010, 143, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Shinde, S.R.; Maddika, S. PTEN modulates EGFR late endocytic trafficking and degradation by dephosphorylating Rab7. Nat. Commun. 2016, 7, 1–11. [Google Scholar] [CrossRef]
- Snijder, E.J.; Limpens, R.W.A.L.; de Wilde, A.H.; de Jong, A.W.M.; Zevenhoven-Dobbe, J.C.; Maier, H.J.; Faas, F.F.G.A.; Koster, A.J.; Bárcena, M. A unifying structural and functional model of the coronavirus replication organelle: Tracking down RNA synthesis. PLoS Biol. 2020, 18, e3000715. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Ricciardi, S.; Guarino, A.M.; Giaquinto, L.; Polishchuk, E.V.; Santoro, M.; Di Tullio, G.; Wilson, C.; Panariello., F.; Soares, V.C.; Dias., S.S.G.; et al. The role of NSP6 in the biogenesis of the SARS-CoV-2 replication organelle. Nature 2022, 606, 761–768. [Google Scholar] [CrossRef]
- Kumar, S.; Javed, R.; Mudd, M.; Pallikkuth, S.; Lidke, K.A.; Jain, A.; Tangavelou, K.; Gudmundsson, S.R.; Ye, C.; Rusten, T.E.; et al. Mammalian hybrid pre-autophagosomal structure HyPAS generates autophagosomes. Cell 2021, 184, 5950–5969.e22. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Javed, R.; Paddar, M.A.; Eskelinen, E.-L.; Timmins, G.S.; Deretic, V. Mammalian hybrid prophagophore is a precursor to autophagosomes. Autophagy 2023, 1–2. [Google Scholar] [CrossRef]
- Tan, X.; Cai, K.; Li, J.; Yuan, Z.; Chen, R.; Xiao, H.; Xu, C.; Hu, B.; Qin, Y.; Ding, B. Coronavirus Subverts ER-phagy by Hijacking FAM134B and ATL3 into p62 Condensates to Facilitate Viral Replication. Cell Rep. 2023, 42, 112286. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Jia, Y.; Meng, Y.; Xue, Y.; Liu, K.; Li, Y.; Liu, S.; Li, X.; Cui, K.; Shang, L.; et al. SNX27 suppresses SARS-CoV-2 infection by inhibiting viral lysosome/late endosome entry. Proc. Natl. Acad. Sci. USA 2022, 119, e2117576119. [Google Scholar] [CrossRef]
- Liu, T.; Luo, S.; Libby, P.; Shi, G.P. Cathepsin L-selective inhibitors: A potentially promising treatment for COVID-19 patients. Pharmacol. Ther. 2020, 213, 107587. [Google Scholar] [CrossRef]
- Gassen, N.C.; Papies, J.; Bajaj, T.; Emanuel, J.; Dethloff, F.; Chua, R.L.; Trimpert, J.; Heinemann, N.; Niemeyer, C.; Weege, F.; et al. SARS-CoV-2-mediated dysregulation of metabolism and autophagy uncovers host-targeting antivirals. Nat. Commun. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Mizushima, N. The ATG conjugation systems in autophagy. Curr. Opin. Cell Biol. 2020, 63, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Wenling, W.; Zhidong, L.; Chaoyang, L.; Wen, W.; Fei, Y.; Baoying, H.; Zhao, L.; Wang, H.; Zhou, W.; et al. Morphogenesis and cytopathic effect of SARS-CoV-2 infection in human airway epithelial cells. Nat. Commun. 2020, 11, 1–8. [Google Scholar] [CrossRef]
- Mohamud, Y.; Xue, Y.C.; Liu, H.; Ng, C.S.; Bahreyni, A.; Jan, E.; Luo, H. The papain-like protease of coronaviruses cleaves ULK1 to disrupt host autophagy. Biochem. Biophys. Res. Commun. 2021, 540, 75–82. [Google Scholar] [CrossRef]
- Turco, E.; Witt, M.; Abert, C.; Bock-Bierbaum, T.; Su, M.Y.; Trapannone, R.; Sztacho, M.; Danieli, A.; Shi, X.; Zaffagnini, G.; et al. How RB1CC1/FIP200 claws its way to autophagic engulfment of SQSTM1/p62-ubiquitin condensates. Autophagy 2019, 15, 1475–1477. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Song, K.; Hao, W.; Wu, Y.; Patil, G.; Hua, F.; Sun, Y.; Huang, C.; Ritchey, L.; Jones, C.; et al. FIP200 restricts RNA virus infection by facilitating RIG-I activation. Commun. Biol. 2021, 4, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ohnstad, A.E.; Delgado, J.M.; North, B.J.; Nasa, I.; Kettenbach, A.N.; Schultz, S.W.; Shoemaker, C.J. Receptor-mediated clustering of FIP200 bypasses the role of LC3 lipidation in autophagy. EMBO J. 2020, 39, e104948. [Google Scholar] [CrossRef] [PubMed]
- Clarke, P.G.H.; Puyal, J. Autophagic cell death exists. Autophagy 2012, 8, 867–869. [Google Scholar] [CrossRef]
- Twu, W.I.; Lee, J.Y.; Kim, H.; Prasad, V.; Cerikan, B.; Haselmann, U.; Tabata, K.; Bartenschlager, R. Contribution of autophagy machinery factors to HCV and SARS-CoV-2 replication organelle formation. Cell. Rep. 2021, 37, 110049. [Google Scholar] [CrossRef]
- Yin, Z.; Pascual, C.; Klionsky, D.J. Autophagy: Machinery and regulation. Microb. Cell 2016, 3, 588–596. [Google Scholar] [CrossRef]
- Bello-Perez, M.; Sola, I.; Novoa, B.; Klionsky, D.J.; Falco, A. Canonical and Noncanonical Autophagy as Potential Targets for COVID-19. Cells 2020, 9, 1619. [Google Scholar] [CrossRef]
- Fung, T.S.; Liu, D.X. Human Coronavirus: Host-Pathogen Interaction. Annu. Rev. Microbiol. 2019, 73, 529–557. [Google Scholar] [CrossRef] [PubMed]
- Jounai, N.; Takeshita, F.; Kobiyama, K.; Sawano, A.; Miyawaki, A.; Xin, K.Q.; Ishii, K.J.; Kawai, T.; Akira, S.; Suzuki, K.; et al. The Atg5-Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl. Acad. Sci. USA 2007, 104, 14050–14055. [Google Scholar] [CrossRef]
- Hwang, S.; Maloney, N.S.; Bruinsma, M.W.; Goel, G.; Duan, E.; Zhang, L.; Shrestha, B.; Diamond, M.; Dani, A.; Sosnovtsev, S.; et al. Nondegradative role of Atg5-Atg12/ Atg16L1 autophagy protein complex in antiviral activity of interferon gamma. Cell. Host Microbe 2012, 11, 397–409. [Google Scholar] [CrossRef]
- Kimmey, J.M.; Huynh, J.P.; Weiss, L.A.; Park, S.; Kambal, A.; Debnath, J.; Virgin, H.W.; Stalling, C.L. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature 2015, 528, 565–569. [Google Scholar] [CrossRef]
- Zeng, Q.; Antia, A.; Puray-Chavez, M.; Kutluay, S.B.; Ding, S. Calpain-2 mediates SARS-CoV-2 entry and represents a therapeutic target. bioRxiv 2022. [Google Scholar] [CrossRef]
- Yousefi, S.; Perozzo, R.; Schmid, I.; Ziemiecki, A.; Schaffner, T.; Scapozza, L.; Brunner, T.; Simon, H.U. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 2006, 8, 1124–1132. [Google Scholar] [CrossRef]
- Jung, S.; Jeong, H.; Yu, S.W. Autophagy as a decisive process for cell death. Exp. Mol. Med. 2020, 52, 921–930. [Google Scholar] [CrossRef]
- Wang, F.; Peters, R.; Jia, J.; Mudd, M.; Salemi, M.; Allers, L.; Javed, R.; Duque, T.L.A.; Paddar, M.A.; Trosdal, E.S.; et al. ATG5 provides host protection acting as a switch in the atg8ylation cascade between autophagy and secretion. Dev. Cell, 2023; in press. [Google Scholar] [CrossRef]
- He, W.; Gao, Y.; Zhou, J.; Shi, Y.; Xia, D.; Shen, H.M. Friend or Foe? Implication of the autophagy-lysosome pathway in SARS-CoV-2 infection and COVID-19. Int. J. Biol. Sci. 2022, 18, 4690–4703. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zhao, Y.G.; Zhang, H. Endomembrane remodeling in SARS-CoV-2 infection. Cell. Insight 2022, 1, 100031. [Google Scholar] [CrossRef]
- Wang, R.; Simoneau, C.R.; Kulsuptrakul, J.; Bouhaddou, M.; Travisano, K.A.; Hayashi, J.M.; Carlson-Stevermer, J.; Zengel, J.R.; Richards, C.M.; Fozouni, P.; et al. Genetic Screens Identify Host Factors for SARS-CoV-2 and Common Cold Coronaviruses. Cell 2021, 184, 106–119.e14. [Google Scholar] [CrossRef]
- Mészáros, B.; Sámano-Sánchez, H.; Alvarado-Valverde, J.; Čalyševa, J.; Martínez-Pérez, E.; Alves, R.; Shields, D.C.; Kumar, M.; Rippmann, F.; Chemes, L.B.; et al. Short linear motif candidates in the cell entry system used by SARS-CoV-2 and their potential therapeutic implications. Sci. Signal. 2021, 14, 665. [Google Scholar] [CrossRef] [PubMed]
- Miao, G.; Zhao, H.; Li, Y.; Ji, M.; Chen, Y.; Shi, Y.; Bi, Y.; Wang, P.; Zhang, H. ORF3a of the COVID-19 virus SARS-CoV-2 blocks HOPS complex-mediated assembly of the SNARE complex required for autolysosome formation. Dev. Cell. 2021, 56, 427–442.e5. [Google Scholar] [CrossRef]
- Chen, X.; Wang, K.; Xing, Y.; Tu, J.; Yang, X.; Zhao, Q.; Li, K.; Chen, Z. Coronavirus membrane-associated papain-like proteases induce autophagy through interacting with Beclin1 to negatively regulate antiviral innate immunity. Protein Cell. 2014, 5, 912–927. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, H.; Pei, R.; Mao, B.; Li, H.; Lin, Y.; Lu, K. The SARS-CoV-2 protein ORF3a inhibits fusion of autophagosomes with lysosomes. Cell. Discov. 2021, 7, 1–12. [Google Scholar] [CrossRef]
- Chen, D.; Zheng, Q.; Sun, L.; Ji, M.; Li, Y.; Deng, H.; Zhang, H. ORF3a of SARS-CoV-2 promotes lysosomal exocytosis-mediated viral egress. Dev. Cell. 2021, 56, 3250–3263.e5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Chen, Y.; Li, Y.; Huang, F.; Luo, B.; Yuan, Y.; Xia, B.; Ma, X.; Yang, T.; Yu, F.; et al. The ORF8 protein of SARS-CoV-2 mediates immune evasion through down-regulating MHC-Ι. Proc. Natl. Acad. Sci. USA 2021, 118, e2024202118. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Cao, S.; Qiu, F.; Kang, N. Incomplete autophagy: Trouble is a friend. Med. Res. Rev. 2022, 42, 1545–1587. [Google Scholar] [CrossRef]
- Williams, C.G.; Jureka, A.S.; Silvas, J.A.; Nicolini, A.M.; Chvatal, S.A.; Carlson-Stevermer, J.; Oki, J.; Holden, K.; Basler, C.F. Inhibitors of VPS34 and fatty-acid metabolism suppress SARS-CoV-2 replication. Cell Rep. 2021, 36, 5. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2010, 12, 9–14. [Google Scholar] [CrossRef]
- Hayn, M.; Hirschenberger, M.; Koepke, L.; Nchioua, R.; Straub, J.H.; Klute, S.; Hunszinger, V.; Zech, F.; Prelli Bozzo, C.; Afta, W.; et al. Systematic functional analysis of SARS-CoV-2 proteins uncovers viral innate immune antagonists and remaining vulnerabilities. Cell Rep. 2021, 35, 7. [Google Scholar] [CrossRef]
- Foo, J.; Bellot, G.; Pervaiz, S.; Alonso, S. Mitochondria-mediated oxidative stress during viral infection. Trends Microbiol. 2022, 30, 679–692. [Google Scholar] [CrossRef]
- Jiang, H.W.; Zhang, H.N.; Meng, Q.F.; Xie, J.; Li, Y.; Chen, H.; Zheng, Y.X.; Wang, X.N.; Qi, H.; Zhang, J.; et al. SARS-CoV-2 Orf9b suppresses type I interferon responses by targeting TOM70. Cell Mol. Immunol. 2020, 17, 998–1000. [Google Scholar] [CrossRef]
- Li, X.; Hou, P.; Ma, W.; Wang, X.; Wang, H.; Yu, Z.; Chang, H.; Wang, T.; Jin, S.; Wang, X.; et al. SARS-CoV-2 ORF10 suppresses the antiviral innate immune response by degrading MAVS through mitophagy. Cell Mol. Immunol. 2021, 19, 67–78. [Google Scholar] [CrossRef]
- Hui, X.; Zhang, L.; Cao, L.; Huang, K.; Zhao, Y.; Zhang, Y.; Chen, X.; Lin, X.; Chen, M.; Jin, M.; et al. SARS-CoV-2 promote autophagy to suppress type I interferon response. Signal. Transduct. Target. Ther. 2021, 6, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, L.; Gong, J. Mitochondrial-targeted ubiquinone: A potential treatment for COVID-19. Med. Hypotheses 2020, 144, 110161. [Google Scholar] [CrossRef] [PubMed]
- Shang, C.; Zhuang, X.; Zhang, H.; Li, Y.; Zhu, Y.; Lu, J.; Ge, C.; Cong, J.; Li, T.; Li, N.; et al. SARS-CoV-2 Causes Mitochondrial Dysfunction and Mitophagy Impairment. Front. Microbiol. 2022, 12, 4159. [Google Scholar] [CrossRef]
- Thurston, T.L.M.; Wandel, M.P.; Von Muhlinen, N.; Foeglein, Á.; Randow, F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 2012, 482, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Montespan, C.; Marvin, S.A.; Austin, S.; Burrage, A.M.; Roger, B.; Rayne, F.; Faure, M.; Campell, E.M.; Schneider, C.; Reimer, R.; et al. Multi-layered control of Galectin-8 mediated autophagy during adenovirus cell entry through a conserved PPxY motif in the viral capsid. PLoS Pathog. 2017, 13, e1006217. [Google Scholar] [CrossRef]
- Pablos, I.; Machado, Y.; Ramos de Jesus, H.C.; Mohamud, Y.; Kappelhoff, R.; Lindskog, C.; Vlok, M.; Bell, P.A.; Butler, G.S.; Grin, P.M.; et al. Mechanistic insights into COVID-19 by global analysis of the SARS-CoV-2 3CLpro substrate degradome. Cell Rep. 2021, 37, 109892. [Google Scholar] [CrossRef] [PubMed]
- Bozorgmehr, N.; Mashhouri, S.; Perez Rosero, E.; Xu, L.; Shahbaz, S.; Sligl, W.; Osman, M.; Kutsogiannis, D.J.; MacIntyre, E.; O’Neil, C.R.; et al. Galectin-9, a player in cytokine release syndrome and a surrogate diagnostic biomarker in SARS-CoV-2 infection. mBio 2021, 12, 3. [Google Scholar] [CrossRef]
- Di Domizio, J.; Gulen, M.F.; Saidoune, F.; Thacker, V.V.; Yatim, A.; Sharma, K.; Nass, T.; Guenova, E.; Schaller, M.; Conrad, C.; et al. The cGAS–STING pathway drives type I IFN immunopathology in COVID-19. Nature 2022, 603, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Zhang, X.; Lei, X.; Xiao, X.; Jiao, T.; Ma, R.; Dong, X.; Jiang, Q.; Wang, W.; Shi, Y.; et al. Sensing of cytoplasmic chromatin by cGAS activates innate immune response in SARS-CoV-2 infection. Signal. Transduct. Target. Ther. 2021, 6, 382. [Google Scholar] [CrossRef]
- Su, J.; Shen, S.; Hu, Y.; Chen, S.; Cheng, L.; Cai, Y.; Wei, W.; Wang, Y.; Rui, Y.; Yu, X.F. SARS-CoV-2 ORF3a inhibits cGAS-STING-mediated autophagy flux and antiviral function. J. Med. Virol. 2023, 95, 1. [Google Scholar] [CrossRef]
- Rui, Y.; Su, J.; Shen, S.; Hu, Y.; Huang, D.; Zheng, W.; Lou, M.; Shi, Y.; Wang, M.; Chen, S.; et al. Unique and complementary suppression of cGAS-STING and RNA sensing- triggered innate immune responses by SARS-CoV-2 proteins. Signal. Transduct. Target. Ther. 2021, 6, 123. [Google Scholar] [CrossRef]
- Han, L.; Zheng, Y.; Deng, J.; Nan, M.L.; Xiao, Y.; Zhuang, M.W.; Zhang, J.; Wang, W.; Gao, C.; Wang, P.H. SARS-CoV-2 ORF10 antagonizes STING-dependent interferon activation and autophagy. J. Med. Virol. 2022, 94, 5174–5188. [Google Scholar] [CrossRef] [PubMed]
- Gioia, U.; Tavella, S.; Martínez-Orellana, P.; Cicio, G.; Colliva, A.; Ceccon, M.; Cabrini, M.; Henriques, A.C.; Fumagalli, V.; Paldino, A.; et al. SARS-CoV-2 infection induces DNA damage, through CHK1 degradation and impaired 53BP1 recruitment, and cellular senescence. Nat. Cell Biol. 2023, 25, 550–564. [Google Scholar] [CrossRef]
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef]
- Sun, X.; Liu, Y.; Huang, Z.; Xu, W.; Hu, W.; Yi, L.; Liu, Z.; Chan, H.; Zeng, J.; Liu, X.; et al. SARS-CoV-2 non-structural protein 6 triggers NLRP3-dependent pyroptosis by targeting ATP6AP1. Cell Death Differ. 2022, 29, 1240–1254. [Google Scholar] [CrossRef] [PubMed]
- Van Zutphen, T.; Veenhuis, M.; Van Der Klei, I. Pex14 is the Sole Component of the Peroxisomal Translocon That Is Required for Pexophagy. Available online: http://www.landesbioscience.com/journals/autophagy/article/5076 (accessed on 30 March 2023).
- Beltran, P.M.J.; Cook, K.C.; Hashimoto, Y.; Galitzine, C.; Murray, L.A.; Vitek, O.; Cristea, I.M. Infection-Induced Peroxisome Biogenesis Is a Metabolic Strategy for Herpesvirus Replication. Cell Host Microbe 2018, 24, 526–541.e7. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.A.; Arutyunova, E.; Lu, J.; Khan, M.B.; Rut, W.; Zmudzinski, M.; Shahbaz, S.; Iyyathurai, J.; Moussa, E.W.; Turner, Z.; et al. SARS-CoV-2 Mpro Protease Variants of Concern Display Altered Viral Substrate and Cell Host Target Galectin-8 Processing but Retain Sensitivity toward Antivirals. ACS Cent. Sci. 2023. [CrossRef]
- Li, J.; Wang, W. Mechanisms and Functions of Pexophagy in Mammalian Cells. Cells 2021, 10, 1094. [Google Scholar] [CrossRef]
- Jo, D.S.; Park, S.J.; Kim, A.K.; Park, N.Y.; Kim, J.B.; Bae, J.E.; Park, H.J.; Shin, J.H.; Chang, J.W.; Kim, P.K.; et al. Loss of HSPA9 induces peroxisomal degradation by increasing pexophagy. Autophagy 2020, 16, 1989–2003. [Google Scholar] [CrossRef]
- Kim, P.K.; Hailey, D.W.; Mullen, R.T.; Lippincott-Schwartz, J. Ubiquitin signals autophagic degradation of cytosolic proteins and peroxisomes. Proc. Natl. Acad. Sci. USA 2008, 105, 20567–20574. [Google Scholar] [CrossRef]
- Khaminets, A.; Heinrich, T.; Mari, M.; Grumati, P.; Huebner, A.K.; Akutsu, M.; Liebmann, L.; Stolz, A.; Nietzsche, S.; Koch, N.; et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 2015, 522, 354–358. [Google Scholar] [CrossRef]
- Kazi, A.W.; Summer, R.; Sundaram, B.; George, G. Lung recovery with prolonged ECMO following fibrotic COVID-19 acute respiratory distress syndrome. Am. J. Med. Sci. 2023, 365, 3. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Jiang, S.; Li, Z.; Chen, S. Metabolic Syndrome ‘Interacts’ With COVID-19. BIO Integration 2020, 1, 168–177. [Google Scholar] [CrossRef]
- Nabirotchkin, S.; Peluffo, A.E.; Bouaziz, J.; Cohen, D. Focusing on the Unfolded Protein Response and Autophagy Related Pathways to Reposition Common Approved Drugs against COVID-19. Preprints.org 2020, 2020030302. [Google Scholar] [CrossRef]
- Shojaei, S.; Suresh, M.; Klionsky, D.J.; Labouta, H.I.; Ghavami, S. Autophagy and SARS-CoV-2 infection: A possible smart targeting of the autophagy pathway. Virulence 2020, 11, 805–810. [Google Scholar] [CrossRef]
- Shojaei, S.; Koleini, N.; Samiei, E.; Aghaei, M.; Cole, L.K.; Alizadeh, J.; Islam, M.I.; Vosoughi, A.R.; Albokashy, M.; Butterfield, Y.; et al. Simvastatin increases temozolomide-induced cell death by targeting the fusion of autophagosomes and lysosomes. FEBS J. 2020, 287, 1005–1034. [Google Scholar] [CrossRef] [PubMed]
- Savarino, A.; Di Trani, L.; Donatelli, I.; Cauda, R.; Cassone, A. New insights into the antiviral effects of chloroquine. Lancet Infect. Dis. 2006, 6, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zou, Z.; Sun, Y.; Li, X.; Xu, K.F.; Wei, Y.; Jin, N.; Jiang, C. Anti-malaria drug chloroquine is highly effective in treating avian influenza A H5N1 virus infection in an animal model. Cell. Res. 2013, 23, 300–302. [Google Scholar] [CrossRef]
- Vincent, M.J.; Bergeron, E.; Benjannet, S.; Erickson, B.R.; Rollin, P.E.; Ksiazek, T.G.; Seidah, N.G.; Nichol, S.T. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol. J. 2005, 2, 1–10. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell. Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Fung, K.L.; Chan, P.L. Comment on: COVID-19: A recommendation to examine the effect of hydroxychloroquine in preventing infection and progression. J. Antimicrob. Chemother. 2020, 75, 2016–2017. [Google Scholar] [CrossRef] [PubMed]
- Keyaerts, E.; Vijgen, L.; Maes, P.; Neyts, J.; Van Ranst, M. In vitro inhibition of severe acute respiratory syndrome coronavirus by chloroquine. Biochem. Biophys. Res. Commun. 2004, 323, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Kolosenko, I.; Grander, D.; Tamm, K.P. IL-6 activated JAK/STAT3 pathway and sensitivity to Hsp90 inhibitors in multiple myeloma. Curr. Med. Chem. 2014, 21, 3042–3047. [Google Scholar] [CrossRef]
- Kusoglu, A.; Bagca, B.G.; Ay, N.P.O.; Saydam, G.; Avci, C.B. Ruxolitinib Regulates the Autophagy Machinery in Multiple Myeloma Cells. Anticancer. Agents Med. Chem. 2020, 20, 2316–2323. [Google Scholar] [CrossRef] [PubMed]
- Ishida, S.; Akiyama, H.; Umezawa, Y.; Okada, K.; Nogami, A.; Oshikawa, G.; Nagao, T.; Miura, O. Mechanisms for mTORC1 activation and synergistic induction of apoptosis by ruxolitinib and BH3 mimetics or autophagy inhibitors in JAK2-V617F-expressing leukemic cells including newly established PVTL-2. Oncotarget 2018, 9, 26834–26851. [Google Scholar] [CrossRef]
- McKee, D.L.; Sternberg, A.; Stange, U.; Laufer, S.; Naujokat, C. Candidate drugs against SARS-CoV-2 and COVID-19. Pharmacol. Res. 2020, 157, 104859. [Google Scholar] [CrossRef]
- Kumar, R.; Gupta, N.; Kodan, P.; Mittal, A.; Soneja, M.; Wig, N. Battling COVID-19: Using old weapons for a new enemy. Trop. Dis. Travel. Med. Vaccines 2020, 6, 1–10. [Google Scholar] [CrossRef]
- Chen, F.; Chan, K.H.; Jiang, Y.; Kao, R.Y.T.; Lu, H.T.; Fan, K.W.; Cheng, V.C.C.; Tsui, W.X.; Hung, I.F.; Lee, T.S.; et al. In vitro susceptibility of 10 clinical isolates of SARS coronavirus to selected antiviral compounds. J. Clin. Virol. 2004, 31, 69–75. [Google Scholar] [CrossRef]
- Hung, I.F.N.; Lung, K.C.; Tso, E.Y.K.; Liu, R.; Chung, T.W.H.; Chu, M.Y.; Ng, Y.Y.; Lo, J.; Chan, J.; Tam, A.R.; et al. Triple combination of interferon beta-1b, lopinavir–ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: An open-label, randomised, phase 2 trial. Lancet 2020, 395, 1695–1704. [Google Scholar] [CrossRef]
- Zha, B.S.; Wan, X.; Zhang, X.; Zha, W.; Zhou, J.; Wabitsch, M.; Wang, G.; Lyall, V.; Hylemon, P.B.; Zhou, H. HIV Protease Inhibitors Disrupt Lipid Metabolism by Activating Endoplasmic Reticulum Stress and Inhibiting Autophagy Activity in Adipocytes. PLoS ONE 2013, 8, e59514. [Google Scholar] [CrossRef]
- Tian, Y.; Wang, M.L.; Zhao, J. Crosstalk between Autophagy and Type I Interferon Responses in Innate Antiviral Immunity. Viruses 2019, 11, 132. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wang, M.L.; Li, Z.; Gao, D.M.; Cai, Y.; Chang, J.; Wang, S.P. Interferon-alpha-2b induces autophagy in hepatocellular carcinoma cells through Beclin1 pathway. Cancer Biol. Med. 2014, 11, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Thangaraj, A.; Chivero, E.T.; Periyasamy, P.; Callen, S.; Burkovetskaya, M.E.; Lei Guo, M.; Buch, S. Antiretroviral-Mediated Microglial Activation Involves Dysregulated Autophagy and Lysosomal Dysfunction. Cells 2019, 8, 1168. [Google Scholar] [CrossRef] [PubMed]
- Del Amo, J.; Polo, R.; Moreno, S.; Martínez, E.; Cabello, A.; Iribarren, J.A.; Curran, A.; Macías, J.; Montero, M.; Dueñas, C.; et al. Tenofovir disoproxil fumarate/emtricitabine and severity of coronavirus disease 2019 in people with HIV infection. AIDS 2022, 36, 2171–2179. [Google Scholar] [CrossRef]
- Gavriatopoulou, M.; Ntanasis-Stathopoulos, M.I.; Korompoki, E.; Fotiou, D.; Migkou, M.; Tzanninis, I.G.; Psaltopoulou, T.; Kastritis, E.; Terpos, E.; Dimopoulos, M.A. Emerging treatment strategies for COVID-19 infection. Clin. Exp. Med. 2021, 21, 167–179. [Google Scholar] [CrossRef]
- Parasher, A. COVID-19: Current understanding of its Pathophysiology, Clinical presentation and Treatment. Postgrad. Med. J. 2021, 97, 312–320. [Google Scholar] [CrossRef]
- Cochrane Haematology Group; Wagner, C.; Griesel, M.; Mikolajewska, A.; Mueller, A.; Nothacker, M.; Kley, K.; Metzendorf, M.I.; Fischer, A.L.; Kopp, M.; et al. Systemic corticosteroids for the treatment of COVID-19. Cochrane Database Syst. Rev. 2021, 8, 8. [Google Scholar] [CrossRef]
- Chaudhuri, D.; Sasaki, K.; Karkar, A.; Sharif, S.; Lewis, K.; Mammen, M.J.; Alexander, P.; Ye, Z.; Lozano, L.E.C.; Munch, M.W.; et al. Corticosteroids in COVID-19 and non-COVID-19 ARDS: A systematic review and meta-analysis. Intensive Care Med. 2021, 47, 521–537. [Google Scholar] [CrossRef]
- Kyrmizi, I.; Gresnigt, M.S.; Akoumianaki, T.; Samonis, G.; Sidiropoulos, P.; Boumpas, D.; Netea, M.G.; van de Veerdonk, F.L.; Kontoyiannis, D.P.; Chamilos, G. Corticosteroids block autophagy protein recruitment in Aspergillus fumigatus phagosomes via targeting dectin-1/Syk kinase signaling. J. Immunol. 2013, 191, 1287–1299. [Google Scholar] [CrossRef]
- Garrett, T.J.; Coatsworth, H.; Mahmud, I.; Hamerly, T.; Stephenson, C.J.; Yazd, H.S.; Ayers, J.; Miller, M.R.; Lednicky, J.A.; Dinglasan, R.R. Niclosamide reverses SARS-CoV-2 control of lipophagy. bioRxiv 2021. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef]
- Rosa, R.B.; Dantas, W.M.; Nascimento, J.C.F.D.; da Silva, M.V.; de Oliveira, R.N.; Pena, L.J. In Vitro and In Vivo Models for Studying SARS-CoV-2, the Etiological Agent Responsible for COVID-19 Pandemic. Viruses 2021, 13, 3. [Google Scholar] [CrossRef]
- Kang, I.; Smirnova, L.; Kuhn, J.H.; Hogberg, H.T.; Kleinstreuer, N.C.; Hartung, T. COVID-19—Prime time for microphysiological systems, as illustrated for the brain. ALTEX—Altern. Anim. Exp. 2021, 38, 535–549. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.M.; Hoffman, T.; Luu, B.Q.; Ashammakhi, N.; Li, S. Application of lung microphysiological systems to COVID-19 modeling and drug discovery: A review. Biodes Manuf. 2021, 4, 757–775. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.-S.; Qi, H.-Y.; Boularan, C.; Huang, N.-N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 2014, 193, 3080–3089. [Google Scholar] [CrossRef] [PubMed]
- Stukalov, A.; Girault, V.; Grass, V.; Karayel, O.; Bergant, V.; Urban, C.; Haas, D.A.; Huang, Y.; Oubraham, L.; Wang, A.; et al. Multilevel proteomics reveals host perturbations by SARS-CoV-2 and SARS-CoV. Nature 2021, 594, 246–252. [Google Scholar] [CrossRef]
- Qu, Y.; Wang, X.; Zhu, Y.; Wang, W.; Wang, Y.; Hu, G.; Liu, C.; Li, J.; Ren, S.; Xiao, M.Z.X.; et al. ORF3a-Mediated Incomplete Autophagy Facilitates Severe Acute Respiratory Syndrome Coronavirus-2 Replication. Front. Cell Dev. Biol. 2021, 9, 2012. [Google Scholar] [CrossRef]
- Bermejo, J.A.P.; Kang, S.; Rockwood, S.J.; Simoneau, C.R.; Joy, D.A.; Silva, A.C.; Ramadoss, G.N.; Flanigan, W.R.; Fozouni, P.; Li, H.; et al. SARS-CoV-2 infection of human iPSC-derived cardiac cells reflects cytopathic features in hearts of patients with COVID-19. Sci. Transl. Med. 2021, 13, 590. [Google Scholar] [CrossRef]
- Bao, L.; Deng, W.; Huang, B.; Gao, H.; Liu, J.; Ren, L.; Wei, Q.; Yu, P.; Xu, Y.; Qi, F.; et al. The pathogenicity of SARS-CoV-2 in hACE2 transgenic mice. Nature 2020, 583, 830–833. [Google Scholar] [CrossRef]
- Valbuena, G.; Halliday, H.; Borisevich, V.; Goez, Y.; Rockx, B. A Human Lung Xenograft Mouse Model of Nipah Virus Infection. PLoS Pathog. 2014, 10, e1004063. [Google Scholar] [CrossRef] [PubMed]
- Shang, C.; Zhuang, X.; Zhang, H.; Li, Y.; Zhu, Y.; Lu, J.; Ge, C.; Cong, J.; Li, T.; Li, N.; et al. Inhibition of Autophagy Suppresses SARS-CoV-2 Replication and Ameliorates Pneumonia in hACE2 Transgenic Mice and Xenografted Human Lung Tissues. J. Virol. 2021, 95, 24. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Stange, D.E.; Ferrante, M.; Vries, R.G.J.; Van Es, J.H.; Van den Brink, S.; Van Houdt, W.J.; Pronk, A.; Van Gorp, J.; Siersem, P.D.; et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivanova, T.; Mariienko, Y.; Mehterov, N.; Kazakova, M.; Sbirkov, Y.; Todorova, K.; Hayrabedyan, S.; Sarafian, V. Autophagy and SARS-CoV-2-Old Players in New Games. Int. J. Mol. Sci. 2023, 24, 7734. https://doi.org/10.3390/ijms24097734
Ivanova T, Mariienko Y, Mehterov N, Kazakova M, Sbirkov Y, Todorova K, Hayrabedyan S, Sarafian V. Autophagy and SARS-CoV-2-Old Players in New Games. International Journal of Molecular Sciences. 2023; 24(9):7734. https://doi.org/10.3390/ijms24097734
Chicago/Turabian StyleIvanova, Tsvetomira, Yuliia Mariienko, Nikolay Mehterov, Maria Kazakova, Yordan Sbirkov, Krassimira Todorova, Soren Hayrabedyan, and Victoria Sarafian. 2023. "Autophagy and SARS-CoV-2-Old Players in New Games" International Journal of Molecular Sciences 24, no. 9: 7734. https://doi.org/10.3390/ijms24097734