
A Novel Gemcitabine-Resistant Gallbladder Cancer Model Provides Insights into Molecular Changes Occurring during Acquired Resistance
 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
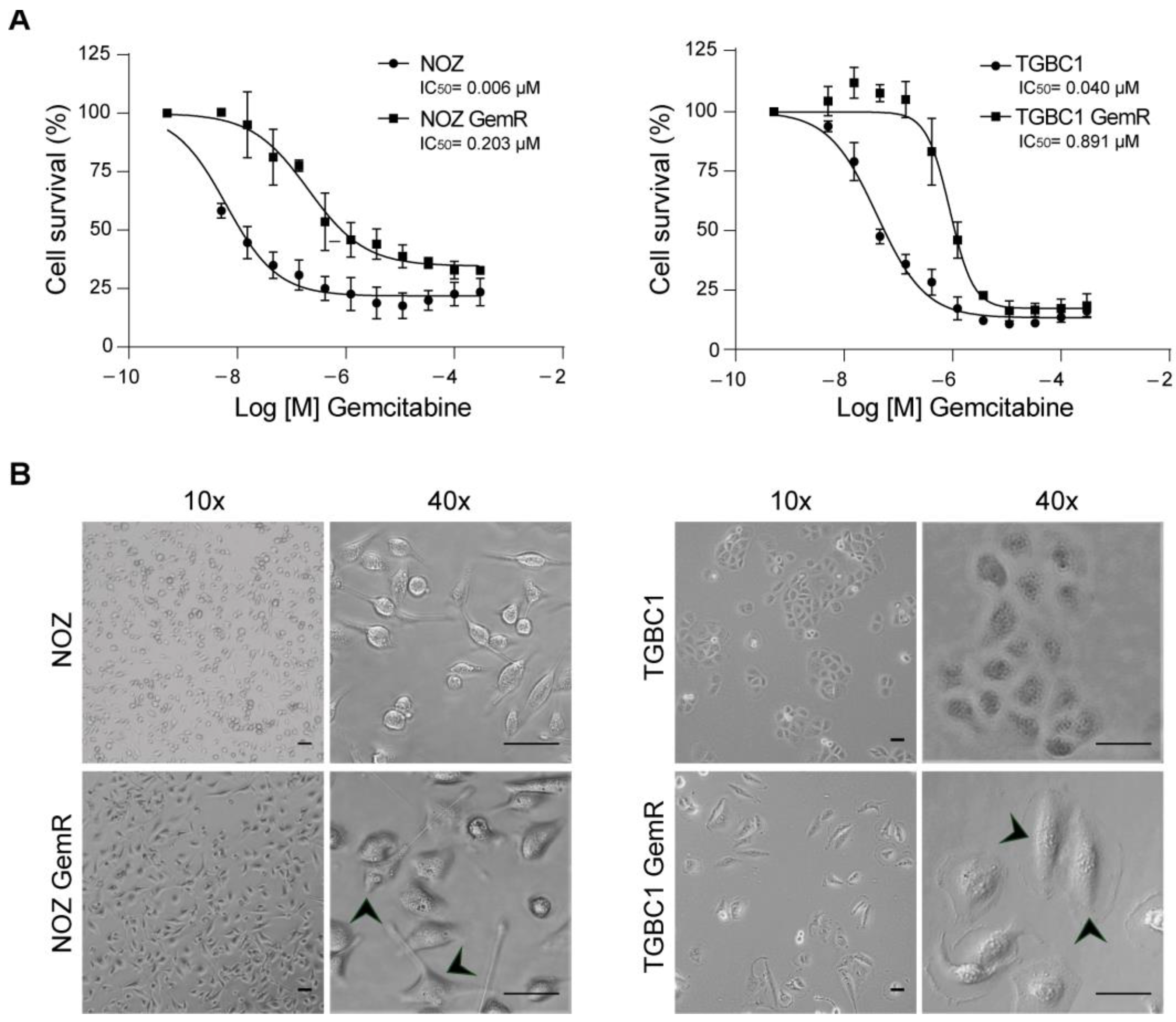
2.1. Establishment and Biological Characterization of Gemcitabine-Resistant Gallbladder Cancer Sublines
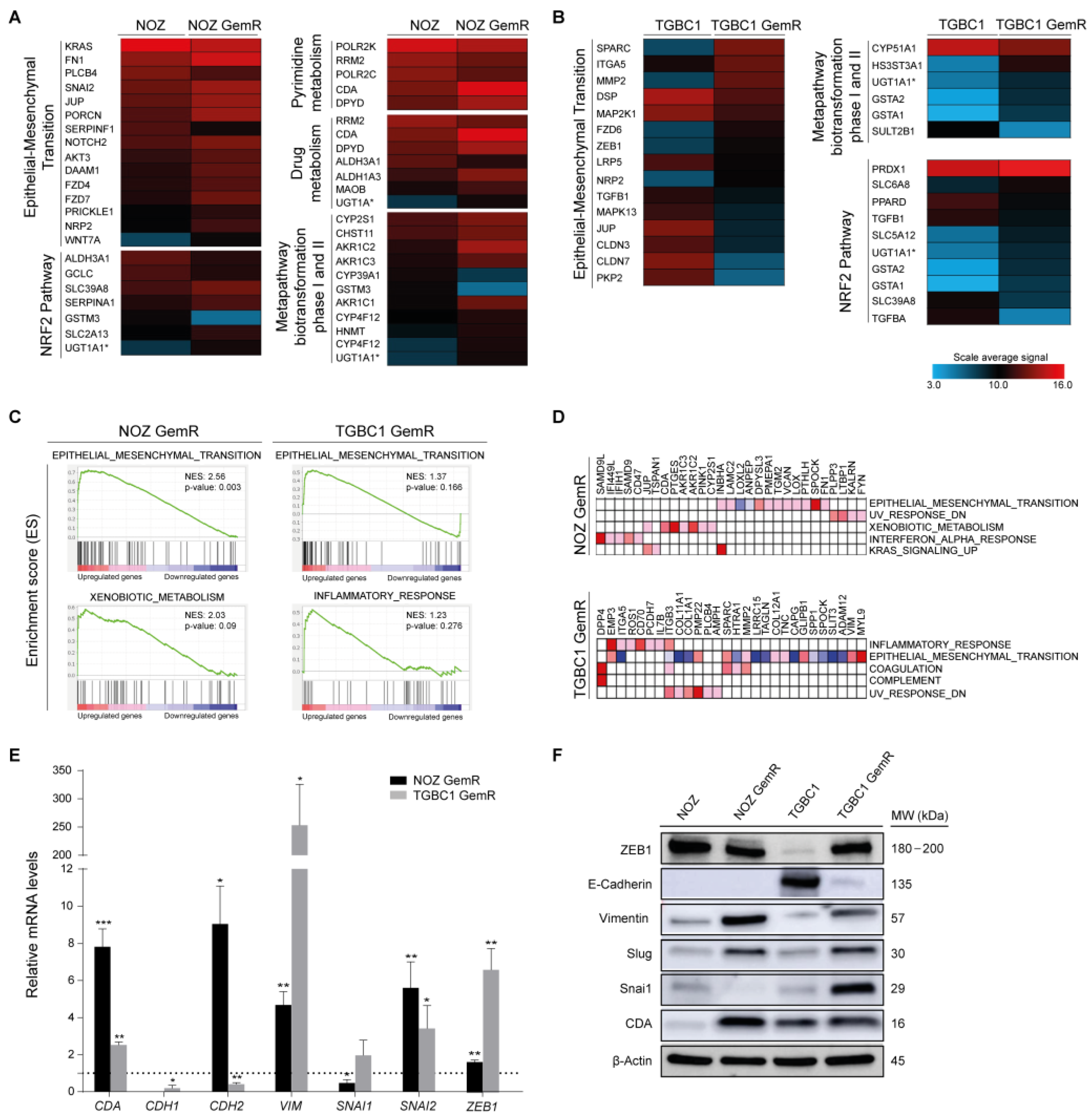
2.2. Gemcitabine-Resistant Cells Exhibit an EMT Signature and Dysregulation of Metabolic Pathways
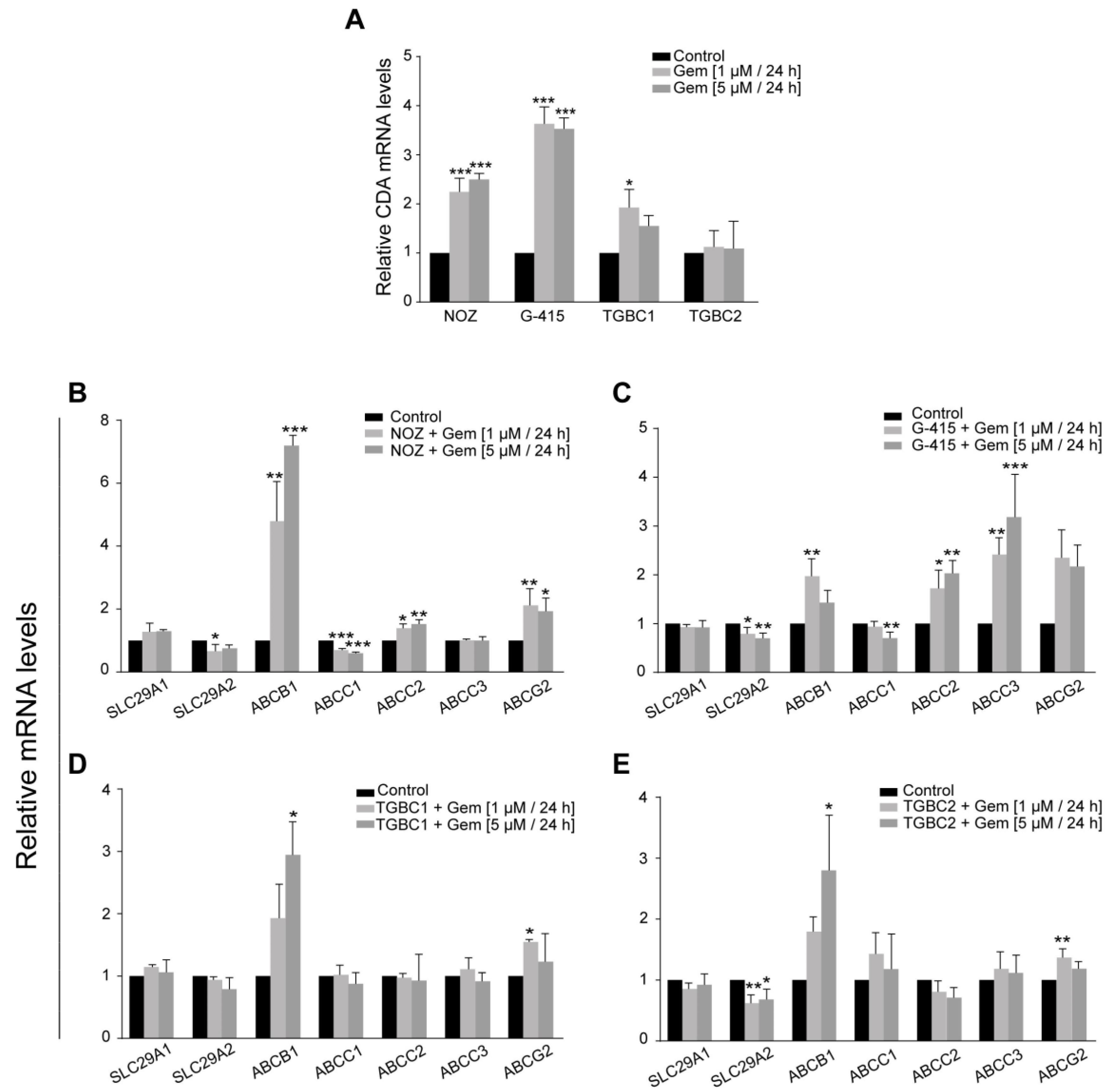
2.3. Dysregulation of Gemcitabine Metabolism-Related Genes Is an Early Adaptation of GBC Cells to Drug Toxicity
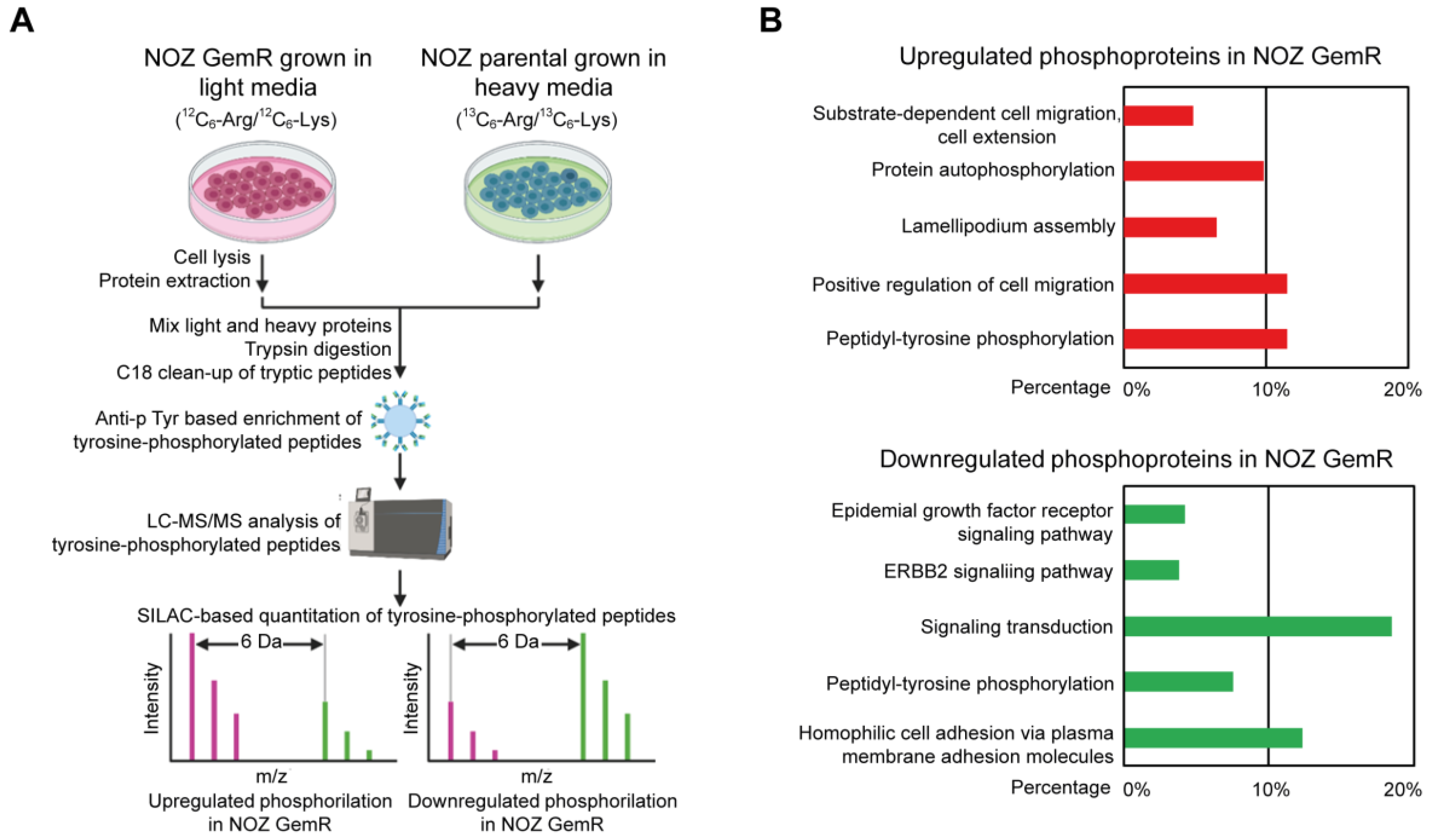
2.4. Quantitative Analysis of Tyrosine Phosphoproteome in Gemcitabine-Resistant NOZ Cells
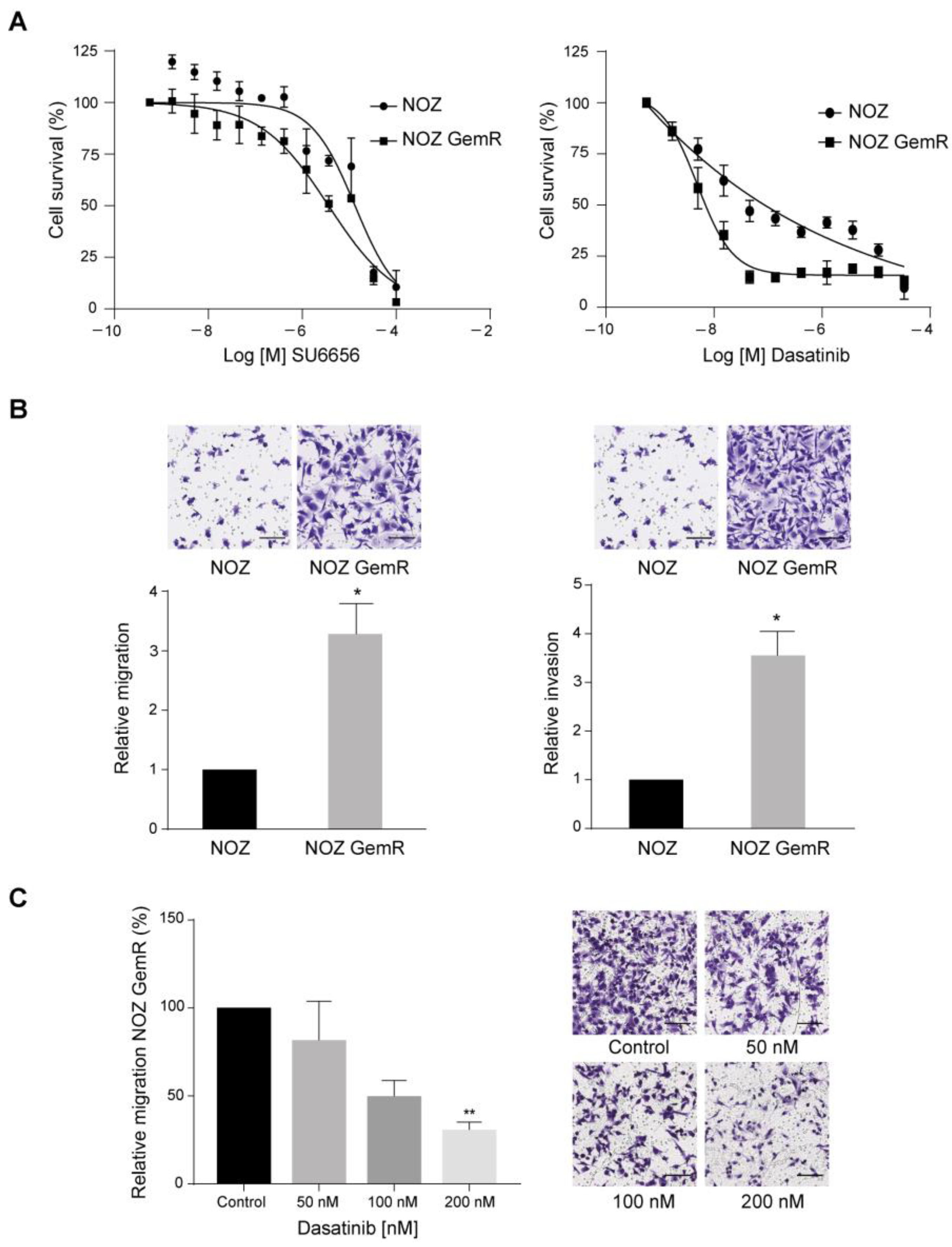
2.5. Dasatinib Selectively Killed NOZ GemR Cells and Reduced Their Migration Ability
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Establishment of Gemcitabine-Resistant Sublines
4.3. Measurement of Cell Viability
4.4. Cell Growth Characteristics
4.5. Transwell Migration and Invasion Assays
4.6. Total RNA Extraction and Microarray Hybridization
4.7. Microarray Data Analysis
4.8. Quantitative Real-Time PCR Analysis (qRT-PCR)
4.9. Western Blot Analysis
4.10. SILAC Labeling, Peptide Preparation, and Phosphopeptide Enrichment
4.11. Liquid Chromatography–Tandem Mass Spectrometry (LC–MS/MS)
4.12. Mass Spectrometric Data Analysis
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roa, J.C.; Garcia, P.; Kapoor, V.K.; Maithel, S.K.; Javle, M.; Koshiol, J. Gallbladder cancer. Nat. Rev. Dis. Prim. 2022, 8, 69. [Google Scholar] [CrossRef] [PubMed]
- Aloia, T.A.; Jarufe, N.; Javle, M.; Maithel, S.K.; Roa, J.C.; Adsay, V.; Coimbra, F.J.; Jarnagin, W.R. Gallbladder cancer: Expert consensus statement. HPB 2015, 17, 681–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czito, B.G.; Hurwitz, H.I.; Clough, R.W.; Tyler, D.S.; Morse, M.A.; Clary, B.M.; Pappas, T.N.; Fernando, N.H.; Willett, C.G. Adjuvant external-beam radiotherapy with concurrent chemotherapy after resection of primary gallbladder carcinoma: A 23-year experience. Int. J. Radiat. Oncol. Biol. Phys. 2005, 62, 1030–1034. [Google Scholar] [CrossRef] [PubMed]
- Gold, D.G.; Miller, R.C.; Haddock, M.G.; Gunderson, L.L.; Quevedo, F.; Donohue, J.H.; Bhatia, S.; Nagorney, D.M. Adjuvant therapy for gallbladder carcinoma: The Mayo Clinic Experience. Int. J. Radiat. Oncol. Biol. Phys. 2009, 75, 150–155. [Google Scholar] [CrossRef]
- Wang, S.J.; Lemieux, A.; Kalpathy-Cramer, J.; Ord, C.B.; Walker, G.V.; Fuller, C.D.; Kim, J.S.; Thomas, C.R., Jr. Nomogram for predicting the benefit of adjuvant chemoradiotherapy for resected gallbladder cancer. J. Clin. Oncol. 2011, 29, 4627–4632. [Google Scholar] [CrossRef] [Green Version]
- Okusaka, T.; Nakachi, K.; Fukutomi, A.; Mizuno, N.; Ohkawa, S.; Funakoshi, A.; Nagino, M.; Kondo, S.; Nagaoka, S.; Funai, J.; et al. Gemcitabine alone or in combination with cisplatin in patients with biliary tract cancer: A comparative multicentre study in Japan. Br. J. Cancer 2010, 103, 469–474. [Google Scholar] [CrossRef] [Green Version]
- Valle, J.; Wasan, H.; Palmer, D.H.; Cunningham, D.; Anthoney, A.; Maraveyas, A.; Madhusudan, S.; Iveson, T.; Hughes, S.; Pereira, S.P.; et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N. Engl. J. Med. 2010, 362, 1273–1281. [Google Scholar] [CrossRef] [Green Version]
- You, M.S.; Ryu, J.K.; Choi, Y.H.; Choi, J.H.; Huh, G.; Paik, W.H.; Lee, S.H.; Kim, Y.T. Therapeutic outcomes and prognostic factors in unresectable gallbladder cancer treated with gemcitabine plus cisplatin. BMC Cancer 2019, 19, 10. [Google Scholar] [CrossRef]
- Azizi, A.A.; Lamarca, A.; McNamara, M.G.; Valle, J.W. Chemotherapy for advanced gallbladder cancer (GBC): A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2021, 163, 103328. [Google Scholar] [CrossRef]
- Zhan, M.; Wang, H.; Chen, T.; Chen, W.; Yang, L.; He, M.; Xu, S.; Wang, J. NOX1 mediates chemoresistance via HIF1alpha/MDR1 pathway in gallbladder cancer. Biochem. Biophys. Res. Commun. 2015, 468, 79–85. [Google Scholar] [CrossRef]
- Wang, H.; Zhan, M.; Xu, S.W.; Chen, W.; Long, M.M.; Shi, Y.H.; Liu, Q.; Mohan, M.; Wang, J. miR-218-5p restores sensitivity to gemcitabine through PRKCE/MDR1 axis in gallbladder cancer. Cell Death Dis. 2017, 8, e2770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Zhang, W.; Lu, B.; Qian, H.; Tang, H.; Zhu, Z.; Yuan, X.; Ren, P. miR-433 accelerates acquired chemoresistance of gallbladder cancer cells by targeting cyclin M. Oncol. Lett. 2018, 15, 3305–3312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, T.; Saiki, Y.; Kudo, C.; Hirayama, A.; Mizuguchi, Y.; Fujiwara, S.; Soga, T.; Sunamura, M.; Matsumura, N.; Motoi, F.; et al. Acquisition of chemoresistance to gemcitabine is induced by a loss-of-function missense mutation of DCK. Biochem. Biophys. Res. Commun. 2015, 464, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Tang, Z.; Gong, W.; Zhang, M.; Quan, Z. Isolation and identification of tumor-initiating cell properties in human gallbladder cancer cell lines using the marker cluster of differentiation 133. Oncol. Lett. 2017, 14, 7111–7120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loria, R.; Vici, P.; Di Lisa, F.S.; Soddu, S.; Maugeri-Sacca, M.; Bon, G. Cross-Resistance Among Sequential Cancer Therapeutics: An Emerging Issue. Front. Oncol. 2022, 12, 877380. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Frances, A.; Cordelier, P. The Emerging Role of Cytidine Deaminase in Human Diseases: A New Opportunity for Therapy? Mol. Ther. 2020, 28, 357–366. [Google Scholar] [CrossRef]
- Blake, R.A.; Broome, M.A.; Liu, X.; Wu, J.; Gishizky, M.; Sun, L.; Courtneidge, S.A. SU6656, a selective src family kinase inhibitor, used to probe growth factor signaling. Mol. Cell Biol. 2000, 20, 9018–9027. [Google Scholar] [CrossRef] [Green Version]
- Lombardo, L.J.; Lee, F.Y.; Chen, P.; Norris, D.; Barrish, J.C.; Behnia, K.; Castaneda, S.; Cornelius, L.A.; Das, J.; Doweyko, A.M.; et al. Discovery of N-(2-chloro-6-methyl-phenyl)-2-(6-(4-(2-hydroxyethyl)-piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J. Med. Chem. 2004, 47, 6658–6661. [Google Scholar] [CrossRef]
- Yuan, S.; Norgard, R.J.; Stanger, B.Z. Cellular Plasticity in Cancer. Cancer Discov. 2019, 9, 837–851. [Google Scholar] [CrossRef] [Green Version]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Li, Y.; Kong, D.; Banerjee, S.; Ahmad, A.; Azmi, A.S.; Ali, S.; Abbruzzese, J.L.; Gallick, G.E.; Sarkar, F.H. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 2009, 69, 2400–2407. [Google Scholar] [CrossRef] [Green Version]
- El Amrani, M.; Corfiotti, F.; Corvaisier, M.; Vasseur, R.; Fulbert, M.; Skrzypczyk, C.; Deshorgues, A.C.; Gnemmi, V.; Tulasne, D.; Lahdaoui, F.; et al. Gemcitabine-induced epithelial-mesenchymal transition-like changes sustain chemoresistance of pancreatic cancer cells of mesenchymal-like phenotype. Mol. Carcinog. 2019, 58, 1985–1997. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuoriluoto, K.; Haugen, H.; Kiviluoto, S.; Mpindi, J.P.; Nevo, J.; Gjerdrum, C.; Tiron, C.; Lorens, J.B.; Ivaska, J. Vimentin regulates EMT induction by Slug and oncogenic H-Ras and migration by governing Axl expression in breast cancer. Oncogene 2011, 30, 1436–1448. [Google Scholar] [CrossRef] [Green Version]
- Molina-Ortiz, P.; Villarejo, A.; MacPherson, M.; Santos, V.; Montes, A.; Souchelnytskyi, S.; Portillo, F.; Cano, A. Characterization of the SNAG and SLUG domains of Snail2 in the repression of E-cadherin and EMT induction: Modulation by serine 4 phosphorylation. PLoS ONE 2012, 7, e36132. [Google Scholar] [CrossRef] [Green Version]
- Adachi, Y.; Takeuchi, T.; Nagayama, T.; Ohtsuki, Y.; Furihata, M. Zeb1-mediated T-cadherin repression increases the invasive potential of gallbladder cancer. FEBS Lett. 2009, 583, 430–436. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Wu, C.; Liang, H.; Zhao, Y.; Lin, C.; Zhang, X.; Ye, C. Knockdown of TWIST enhances the cytotoxicity of chemotherapeutic drugs in doxorubicin-resistant HepG2 cells by suppressing MDR1 and EMT. Int. J. Oncol. 2018, 53, 1763–1773. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Li, J.M.; Wei, W.; Yang, R.; Chen, D.; Ma, X.D.; Jiang, G.M.; Wang, B.L. Regulation of ATP-binding cassette subfamily B member 1 by Snail contributes to chemoresistance in colorectal cancer. Cancer Sci. 2020, 111, 84–97. [Google Scholar] [CrossRef]
- Weadick, B.; Nayak, D.; Persaud, A.K.; Hung, S.W.; Raj, R.; Campbell, M.J.; Chen, W.; Li, J.; Williams, T.M.; Govindarajan, R. EMT-Induced Gemcitabine Resistance in Pancreatic Cancer Involves the Functional Loss of Equilibrative Nucleoside Transporter 1. Mol. Cancer Ther. 2021, 20, 410–422. [Google Scholar] [CrossRef]
- Eliopoulos, N.; Cournoyer, D.; Momparler, R.L. Drug resistance to 5-aza-2′-deoxycytidine, 2′,2′-difluorodeoxycytidine, and cytosine arabinoside conferred by retroviral-mediated transfer of human cytidine deaminase cDNA into murine cells. Cancer Chemother. Pharmacol. 1998, 42, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Bjanes, T.K.; Jordheim, L.P.; Schjott, J.; Kamceva, T.; Cros-Perrial, E.; Langer, A.; Ruiz de Garibay, G.; Kotopoulis, S.; McCormack, E.; Riedel, B. Intracellular Cytidine Deaminase Regulates Gemcitabine Metabolism in Pancreatic Cancer Cell Lines. Drug Metab. Dispos. 2020, 48, 153–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, F.G.; Song, C.G.; Cao, Z.G.; Xia, C.; Chen, D.N.; Chen, L.; Li, S.; Qiao, F.; Ling, H.; Yao, L.; et al. Cytidine Deaminase Axis Modulated by miR-484 Differentially Regulates Cell Proliferation and Chemoresistance in Breast Cancer. Cancer Res. 2015, 75, 1504–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weizman, N.; Krelin, Y.; Shabtay-Orbach, A.; Amit, M.; Binenbaum, Y.; Wong, R.J.; Gil, Z. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene 2014, 33, 3812–3819. [Google Scholar] [CrossRef] [Green Version]
- Samulitis, B.K.; Pond, K.W.; Pond, E.; Cress, A.E.; Patel, H.; Wisner, L.; Patel, C.; Dorr, R.T.; Landowski, T.H. Gemcitabine resistant pancreatic cancer cell lines acquire an invasive phenotype with collateral hypersensitivity to histone deacetylase inhibitors. Cancer Biol. Ther. 2015, 16, 43–51. [Google Scholar] [CrossRef]
- Miller, A.L.; Garcia, P.L.; Gamblin, T.L.; Vance, R.B.; Yoon, K.J. Development of gemcitabine-resistant patient-derived xenograft models of pancreatic ductal adenocarcinoma. Cancer Drug Resist. 2020, 3, 572–585. [Google Scholar] [CrossRef]
- Tibaldi, C.; Camerini, A.; Tiseo, M.; Mazzoni, F.; Barbieri, F.; Vittimberga, I.; Brighenti, M.; Boni, L.; Baldini, E.; Gilli, A.; et al. Cytidine deaminase enzymatic activity is a prognostic biomarker in gemcitabine/platinum-treated advanced non-small-cell lung cancer: A prospective validation study. Br. J. Cancer 2018, 119, 1326–1331. [Google Scholar] [CrossRef] [Green Version]
- Sohal, D.; Krishnamurthi, S.; Tohme, R.; Gu, X.; Lindner, D.; Landowski, T.H.; Pink, J.; Radivoyevitch, T.; Fada, S.; Lee, Z.; et al. A pilot clinical trial of the cytidine deaminase inhibitor tetrahydrouridine combined with decitabine to target DNMT1 in advanced, chemorefractory pancreatic cancer. Am. J. Cancer Res. 2020, 10, 3047–3060. [Google Scholar]
- Ciccolini, J.; Dahan, L.; Andre, N.; Evrard, A.; Duluc, M.; Blesius, A.; Yang, C.; Giacometti, S.; Brunet, C.; Raynal, C.; et al. Cytidine deaminase residual activity in serum is a predictive marker of early severe toxicities in adults after gemcitabine-based chemotherapies. J. Clin. Oncol. 2010, 28, 160–165. [Google Scholar] [CrossRef]
- Serdjebi, C.; Seitz, J.F.; Ciccolini, J.; Duluc, M.; Norguet, E.; Fina, F.; Lacarelle, B.; Ouafik, L.; Dahan, L. Rapid deaminator status is associated with poor clinical outcome in pancreatic cancer patients treated with a gemcitabine-based regimen. Pharmacogenomics 2013, 14, 1047–1051. [Google Scholar] [CrossRef]
- Colicelli, J. ABL tyrosine kinases: Evolution of function, regulation, and specificity. Sci. Signal. 2010, 3, re6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, A.; Tripathi, R.; Turpin, C.P.; Wang, C.; Plattner, R. Abl kinase regulation by BRAF/ERK and cooperation with Akt in melanoma. Oncogene 2017, 36, 4585–4596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingley, E. Src family kinases: Regulation of their activities, levels and identification of new pathways. Biochim. Biophys. Acta 2008, 1784, 56–65. [Google Scholar] [CrossRef]
- Creeden, J.F.; Alganem, K.; Imami, A.S.; Brunicardi, F.C.; Liu, S.H.; Shukla, R.; Tomar, T.; Naji, F.; McCullumsmith, R.E. Kinome Array Profiling of Patient-Derived Pancreatic Ductal Adenocarcinoma Identifies Differentially Active Protein Tyrosine Kinases. Int. J. Mol. Sci. 2020, 21, 8679. [Google Scholar] [CrossRef] [PubMed]
- Croucher, D.R.; Hochgrafe, F.; Zhang, L.; Liu, L.; Lyons, R.J.; Rickwood, D.; Tactacan, C.M.; Browne, B.C.; Ali, N.; Chan, H.; et al. Involvement of Lyn and the atypical kinase SgK269/PEAK1 in a basal breast cancer signaling pathway. Cancer Res. 2013, 73, 1969–1980. [Google Scholar] [CrossRef] [Green Version]
- Tornillo, G.; Knowlson, C.; Kendrick, H.; Cooke, J.; Mirza, H.; Aurrekoetxea-Rodriguez, I.; Vivanco, M.D.M.; Buckley, N.E.; Grigoriadis, A.; Smalley, M.J. Dual Mechanisms of LYN Kinase Dysregulation Drive Aggressive Behavior in Breast Cancer Cells. Cell Rep. 2018, 25, 3674–3692.e3610. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.H.; Chen, X.; He, X. Platelet-derived growth factors and their receptors: Structural and functional perspectives. Biochim. Biophys. Acta 2013, 1834, 2176–2186. [Google Scholar] [CrossRef] [Green Version]
- Van Oosterwijk, J.G.; van Ruler, M.A.; Briaire-de Bruijn, I.H.; Herpers, B.; Gelderblom, H.; van de Water, B.; Bovee, J.V. Src kinases in chondrosarcoma chemoresistance and migration: Dasatinib sensitises to doxorubicin in TP53 mutant cells. Br. J. Cancer 2013, 109, 1214–1222. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Raffa, F.A.; Dai, M.; Moamer, A.; Khadang, B.; Hachim, I.Y.; Bakdounes, K.; Ali, S.; Jean-Claude, B.; Lebrun, J.J. Dasatinib sensitises triple negative breast cancer cells to chemotherapy by targeting breast cancer stem cells. Br. J. Cancer 2018, 119, 1495–1507. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Kim, J.M.; Lee, D.G.; Lee, J.; Park, J.G.; Han, T.S.; Cho, H.S.; Cho, Y.L.; Bae, K.H.; Park, Y.J.; et al. Loss of desmoglein-2 promotes gallbladder carcinoma progression and resistance to EGFR-targeted therapy through Src kinase activation. Cell Death Differ. 2021, 28, 968–984. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Weber, H.; Valbuena, J.R.; Barbhuiya, M.A.; Stein, S.; Kunkel, H.; Garcia, P.; Bizama, C.; Riquelme, I.; Espinoza, J.A.; Kurtz, S.E.; et al. Small molecule inhibitor screening identifified HSP90 inhibitor 17-AAG as potential therapeutic agent for gallbladder cancer. Oncotarget 2017, 8, 26169–26184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rush, J.; Moritz, A.; Lee, K.A.; Guo, A.; Goss, V.L.; Spek, E.J.; Zhang, H.; Zha, X.M.; Polakiewicz, R.D.; Comb, M.J. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat. Biotechnol. 2005, 23, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Kim, M.S.; Chaerkady, R.; Wu, X.; Huang, T.C.; Getnet, D.; Mitchell, C.J.; Palapetta, S.M.; Sharma, J.; O’Meally, R.N.; et al. TSLP signaling network revealed by SILAC-based phosphoproteomics. Mol. Cell Proteom. 2012, 11, 017764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huangda, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Huangda, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vergara-Gómez, L.; Bizama, C.; Zhong, J.; Buchegger, K.; Suárez, F.; Rosa, L.; Ili, C.; Weber, H.; Obreque, J.; Espinoza, K.; et al. A Novel Gemcitabine-Resistant Gallbladder Cancer Model Provides Insights into Molecular Changes Occurring during Acquired Resistance. Int. J. Mol. Sci. 2023, 24, 7238. https://doi.org/10.3390/ijms24087238
Vergara-Gómez L, Bizama C, Zhong J, Buchegger K, Suárez F, Rosa L, Ili C, Weber H, Obreque J, Espinoza K, et al. A Novel Gemcitabine-Resistant Gallbladder Cancer Model Provides Insights into Molecular Changes Occurring during Acquired Resistance. International Journal of Molecular Sciences. 2023; 24(8):7238. https://doi.org/10.3390/ijms24087238
Chicago/Turabian StyleVergara-Gómez, Luis, Carolina Bizama, Jun Zhong, Kurt Buchegger, Felipe Suárez, Lorena Rosa, Carmen Ili, Helga Weber, Javiera Obreque, Karena Espinoza, and et al. 2023. "A Novel Gemcitabine-Resistant Gallbladder Cancer Model Provides Insights into Molecular Changes Occurring during Acquired Resistance" International Journal of Molecular Sciences 24, no. 8: 7238. https://doi.org/10.3390/ijms24087238